

Abstract
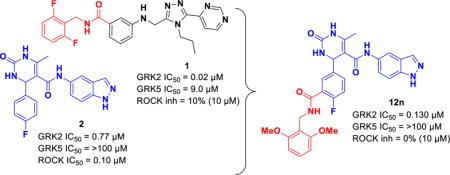
G protein-coupled receptors (GPCRs) are central to many physiological processes. Regulation of this superfamily of receptors is controlled by GPCR kinases (GRKs), some of which have been implicated in heart failure. GSK180736A, developed as a Rho-associated coiled-coil kinase 1 (ROCK1) inhibitor, was identified as an inhibitor of GRK2, and co-crystallized in the active site. Guided by its binding pose overlaid with the binding pose of a known potent GRK2 inhibitor, Takeda103A, a library of hybrid inhibitors was developed. This campaign produced several compounds possessing high potency and selectivity for GRK2 over other GRK subfamilies, PKA, and ROCK1. The most selective compound, 12n (CCG-224406), had an IC50 for GRK2 of 130 nM, greater than 700-fold selectivity over other GRK subfamilies, and no detectable inhibition of ROCK1. Four of the new inhibitors were crystallized with GRK2 to give molecular insights into the binding and kinase selectivity of this class of inhibitors.
Keywords: G protein-coupled receptor kinases, G protein-coupled receptors, β-adrenergic receptors, protein crystallography, inhibitors, kinases, paroxetine, Takeda103A, GSK180736A, CCG-215022, CCG-224406
TOC image
INTRODUCTION
Heart failure is characterized by an inability of the heart to produce a myocardial contraction strong enough to effectively pump blood throughout the body. To compensate for the reduced output, the sympathetic nervous system increases the levels of catecholamines such as norepinephrine and epinephrine.1–3 These catecholamines bind to the β-adrenergic receptors (βARs) on the exterior of the cell, leading to an increase in cyclic adenosine 3′, 5′-monophosphate (cAMP) inside the cell, which in turn mediates the positive inotropic effect needed for enhanced contraction.4, 5 Once stimulated, the βARs are then desensitized via phosphorylation of serine and threonine residues on their cytoplasmic loops or tails by G-protein coupled receptor kinases (GRKs).6, 7 Chronic heart failure leads to increased desensitization and uncoupling of the βARs,8 which can ultimately lead to death.9, 10 Current therapeutics, such as β-blockers, target this mechanism by antagonizing βARs and reducing blood pressure, but take four to six weeks to ameliorate dysregulation of βARs and produce cardio protective effects.11 Furthermore, long term use of β-blockers leads to a myriad of side effects resulting from bradycardia such as dizziness, weakness, and low blood pressure.12
Inhibition of GRKs is another route by which heart failure might be addressed. The seven GRKs found in vertebrates are grouped into three subfamilies, represented by GRK1, GRK2, and GRK4. GRK2, a member of the GRK2 subfamily, and GRK5, a member of the GRK4 subfamily, are known to play significant roles in the heart and have shown promise as alternative therapeutic targets for heart failure.7, 13 GRK2 is overexpressed during, and can serve as a biomarker for, heart failure.3, 9, 13 Inhibition of GRK2 disrupts internalization of βARs, leading to an increase in activated βARs and other GPCRs on the cell membrane that will allow the heart to remain responsive to the sympathetic nervous system.4, 5 The effectiveness of targeting GRK2 has been demonstrated in mice wherein cardiac-specific inhibition of GRK2 using adeno-associated viral delivery of βARKct, a portion of the c-terminus of GRK2 that acts as a dominant negative, following myocardial infarction led to improvement in both cardiac function and survival.14 Furthermore, βARKct mediated GRK2 inhibition in several animal models, including swine, indicates that inhibition of GRK2 activity improves contractile function, reduces catecholamine levels, and reverses cardiac remodeling.9 Notably, both GRK2 and GRK5 also act in non-GPCR pathways that are beneficial to the heart.15, 16 GRK2 influences cardiac glucose uptake leading to abnormal cardiac metabolism when GRK2 is upregulated, effecting the growth of new cardiomyocytes.16 Uniquely, GRK5 becomes localized to the cell nuclei of cardiomyocytes upon GPCR activation17, 18 where it phosphorylates histone deacetylase 5 (HDAC5).15 This phosphorylation event activates myocyte enhancer factor-2, which regulates the stress and fetal gene response underlying maladaptive cardiac hypertrophy.19, 20 Thus, in addition to GRK2, GRK5 may also serve as a promising target for heart failure treatment.
GRKs belong to the protein kinase A, G and C (AGC) family and their kinase domains share ~32% sequence identity with that of protein kinase A (PKA). When targeting GRKs, it is imperative to identify potent chemical probes that are highly selective against other AGC kinases as they play key roles in regulating many vital physiological processes, including inotropy. There is, however, precedence for both potent and selective chemical probes. The natural product balanol (Figure 1) inhibits GRK2 with an IC50 of 50 nM (at 3 μM ATP), but also potently inhibits PKA and PKC.17, 18 Takeda Pharmaceuticals developed a series of compounds, including Takeda103A (1) (Figure 1), that are highly selective for GRK2.19, 20 Compound 1, under our assay conditions, has an IC50 for GRK2 of 20 nM (at 5 μM ATP) — 50-fold more potent than other tested AGC kinases. Despite their impressive in vitro profile, these compounds never advanced to clinical trials, presumably due to poor bioavailability.
Figure 1.
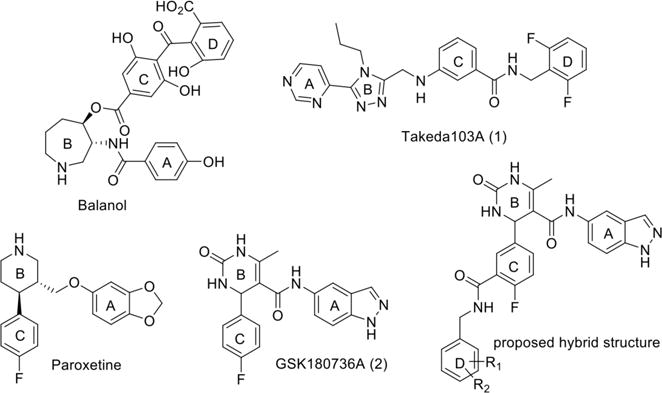
Known GRK2 inhibitors. The A, B, C and D rings pack in the adenine, ribose, polyphosphate, and hydrophobic subsites of the kinase domain, respectively. That is, the A ring interacts with the hinge, whereas the D ring interacts in a pocket defined by the P-loop, the αB and αC helices in the small lobe, and by the DFG loop in the large loop.
The selective serotonin reuptake inhibitor paroxetine (Figure 1) was later identified as a modestly potent inhibitor of GRK2 with an IC50 of 1.1 μM (at 5 μM ATP) and greater than 6-fold selectivity over other GRKs.21, 22 The fact that paroxetine is an FDA-approved drug allowed therapeutic potential to be readily assessed and it was shown to improve left ventricular activity as well as decreased cardiac remodeling in a mouse model of myocardial infarction.23 Most recently, GSK180736A (2), a compound structurally similar to paroxetine that was developed as a Rho-associated, coiled-coil containing kinase 1 (ROCK1) inhibitor24 (Figure 1), was shown to be an even more potent and selective inhibitor of GRK2 with an IC50 of 0.77 μM25 and >100-fold selectivity over other GRKs. It is a weak inhibitor of PKA with an IC50 of 30 μM, but highly potent against ROCK1 (IC50 = 100 nM). Therefore its use as a GRK2 inhibitor is limited by its lack of selectivity versus ROCK1 as well as its poor bioavailability.24
Structural and functional analysis of the GRK2·1 complex (PDB entry 3PVW) suggests that the selectivity of compound 1 for GRK2 results from its ability to recognize a conformation that is more readily accessible to GRK2 than to the other GRKs.19 Docking of 1 into the inactive conformations of GRK1 and GRK6 predicts steric clashes of the terminal difluorophenyl ring of 1 with residues that form what is referred to as the hydrophobic subsite (Figure 2), a pocket positioned adjacent to the γ phosphate in the ATP binding site.19 Alignment of the crystal complexes of 1, balanol, and 2 in GRK2 (Figure 2) revealed that although the compounds are chemically unrelated, they occupy the adenine, ribose, and P-loop subsites of the ATP-binding pocket in remarkably similar ways. However, unlike compound 1 and balanol, 2 does not have a substituent that can occupy the hydrophobic subsite.25 We hypothesized that extending the scaffold of 2 into the hydrophobic subsite would increase potency and, potentially, selectivity against other AGC kinases such as PKA and ROCK1.26 Utilizing a hybrid approach, we added moieties to the position ortho to the fluorine of the C ring of 2 through an amide linker appending a fourth, so-called D ring (Figure 1). We have thereby successfully generated more potent inhibitors that are highly selective for GRK2 as well as inhibitors that are potent for both GRK2 and GRK5. Crystal structures of four representative compounds in complex with GRK2 give insights into the molecular basis for the improved potency and selectivity of these compounds.
Figure 2.
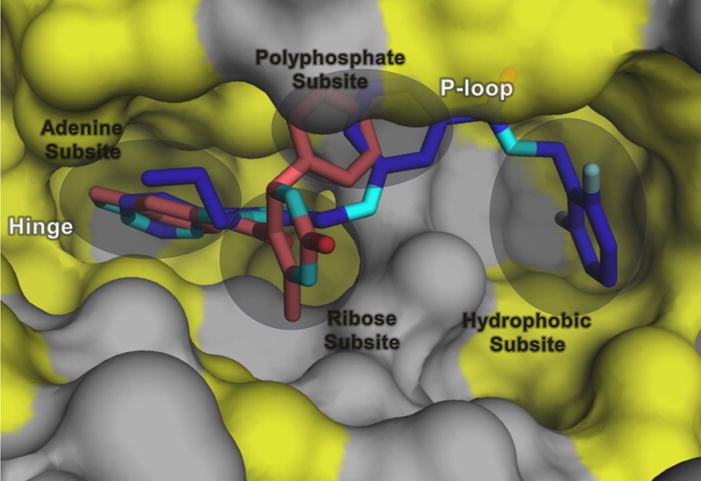
The hydrophobic subsite is unexploited in the GRK2 inhibitor 2 complex. Shown is a superposition of the small lobes of GRK2 in complex with 2 (salmon) and 1 (purple) (PDB entries 4PNK and 3PVW, respectively). Hydrophobic surfaces are colored yellow. The D-ring of balanol (Figure 1) also extends into the hydrophobic subsite.
RESULTS AND DISCUSSION
Chemistry
Hybrid analogs 12a-r and 13a-c were prepared as described in Scheme 1. Synthesis began with Fischer esterification of 4-fluoro-3-methylbenzoic acid 3 followed by benzylic bromination under radical conditions to give the methyl ester 4. After reduction of the ester, displacement of the bromine using sodium cyanide afforded the nitrile 5. The nitrile was hydrolyzed under basic conditions to yield the carboxylic acid 6. Oxidation of the benzylic alcohol of 6 using 2-iodoxybenzoic acid or under Swern conditions proved unsuccessful. Fortunately, Parikh Doering oxidation yielded the aldehyde 7 cleanly and in high yield. In a converging pathway, treatment of 5-aminoindazole 8 with 2,2,6-trimethyl-4H-1,3-dioxin-4-one resulted in the acetylated compound 9.27 Through a Biginelli cyclization, catalyzed by ytterbium triflate, either 2-fluoro-5-formylbenzoic acid or intermediate 7 was condensed with urea and 9 to yield 10 or 11, respectively.28 Derivatives were then introduced via amidation resulting in the final compounds 12a-r and 13a-c (summarized in Table 1).
Scheme 1.
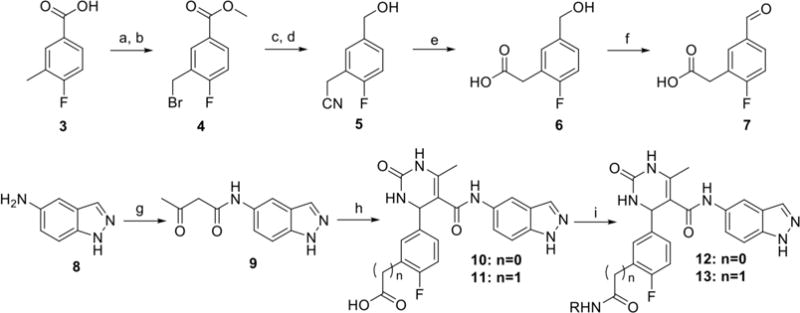
Reagents and conditions: a) cat. H2SO4, MeOH, b) (BzO)2, NBS, CCl4, 80°C, 25 min, c) DIBAL, toluene, d) NaCN, DMSO, e) NaOH, reflux, f) SO3·pyridine, DMSO, Et3N, g) 2,2,6-trimethyl-4H-1,3-dioxin-4-one, CH3CN, 100°C, h) 2-fluoro-5-formylbenzoic acid or 7, Yb(OTf)3, urea, CH3CN, 100°C, i) HATU, DIEA, RNH2, DMF
Table 1.
Kinase inhibitory activity of new analogs of 12 and 13.
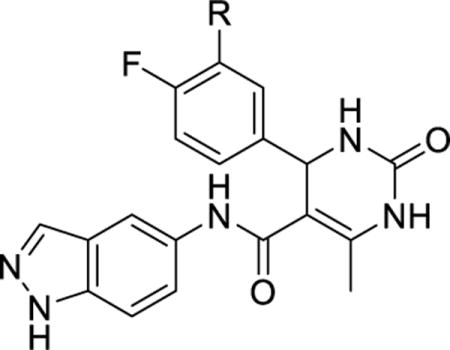
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | GRK2 IC50 (μM) |
GRK1 IC50 (μM) |
GRK5 IC50 (μM) |
PKA IC50 (μM) |
ROCK IC50 (μM) |
| Paroxetine | – | 1.38±1.00 | >100 | >100 | >100 | 10%* |
| 1 | – | 0.02±0.001 | 9.0±3.2 | 2.2±0.92 | ND | 10%* |
| 2 | H | 0.77±0.5 | >100 | >100 | 30±19 | 0.10±0.09 |
| 10 | COOH | 20±10 | >100 | >100 | 78.1±72 | 0.19±0.14 |
| 12a | CONHMe | 4.3±0.7 | >100 | >100 | >100 | 0.56±0.68 |
| 12b | CONHBn | 0.69±0.3 | 71±2 | 4.7±1.9 | >100 | 0.069±0.044 |
| 12c |
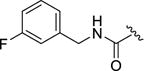
|
0.20±0.1 | 11.9±2.3 | 0.80±0.0.1 | >100 | 0.021±0.13 |
| 12d |
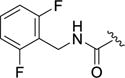
|
0.22±0.2 | >100 | 5.0±1.2 | >100 | 0.11±0.084 |
| 12e |
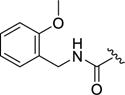
|
0.060±0.03 | 16±6.4 | 2.3±2.7 | >100 | 0.057±0.044 |
| 12f |
|
0.42±0.05 | >100 | 3.8±0.9 | >100 | 0.097±0.076 |
| 12g |
|
0.46±0.3 | >100 | 8.1±4.7 | >100 | 0.050±0.011 |
| 12h |
|
0.15±0.07 | 3.9±1.0 | 0.38±0.06 | >100 | 0.011±0.013 |
| 12i |
|
0.28±0.1 | >100 | 6.2±3.8 | >100 | 0.023±0.022 |
| 12j |
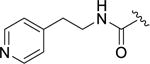
|
4.8±1.9 | >100 | 40.0±14 | >100 | 0.084±0.009 |
| 12k |
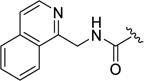
|
0.28±0.06 | 0.10±0.05 | 1.8±0.54 | ND | 0.012±0.004 |
| 12l |
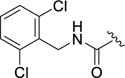
|
0.13±0.03 | >100 | >100 | >100 | 6.7±8.2 |
| 12m |
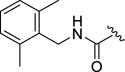
|
0.07±0.01 | >100 | 63±32 | >100 | 5.8±5.5 |
| 12n |
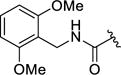
|
0.13±0.03 | >100 | >100 | >100 | 0%* |
| 12o |
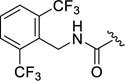
|
1.2±0.7 | >100 | >100 | >100 | 22%* |
| 12p |
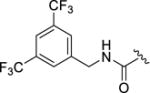
|
2.7±2.3 | >100 | >100 | >100 | 1.9±0.25 |
| 12q |
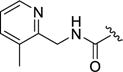
|
1.9±1.3 | >100 | 68±26 | >100 | 0.15±0.05 |
| 13a |
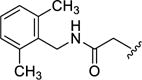
|
25±12 | >100 | 88±60 | >100 | 0.45±.28 |
| 13b |
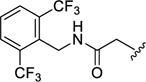
|
>100 | >100 | >100 | >100 | 0.40±0.32 |
| 13c |
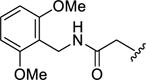
|
0.45±0.29 | >100 | >100 | >100 | 0.20±0.05 |
| 12r |
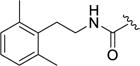
|
0.23±0.05 | >100 | 58±13 | >100 | 0.29±0.033 |
All IC50 measurements are an average of three separate experiments run in duplicate.
Errors shown represent standard error of the mean.
Percent inhibition at 10 μM inhibitor concentration.
Synthesis of the N-methylated hybrid molecule 20 commenced with acylation of 5-aminoindazole 8 using trifluoroacetic anhydride to give 14.29 Attempts at selective methylation of the acetylated amine through the use of varying basic conditions led to competing N-methylation of the indazole. Therefore, protection of the indazole nitrogen was necessary, and was accomplished using Boc-anhydride to give two regioisomers of 15. Methyl iodide in the presence of sodium hydride then furnished the methylated amide 16. Removal of the trifluoroacetate and Boc under refluxing basic conditions afforded the monomethylated amine 17 which was then acylated with 2,2,6- trimethyl-4H-1,3-dioxin-4-one to give 18. Final analogue 20 was completed using Biginelli cyclization followed by amide coupling as described previously in Scheme 1.
Incorporation of the benzodioxole ring was undertaken as shown in Scheme 3. Synthesis began with alkylation of 3,4-(methylenedioxy)aniline through heating with ethyl acetoacetate and pyridine to give ketoamide 22. Biginelli cyclization followed by HATU-mediated amidation furnished the hybrid compound 24.
Scheme 3.
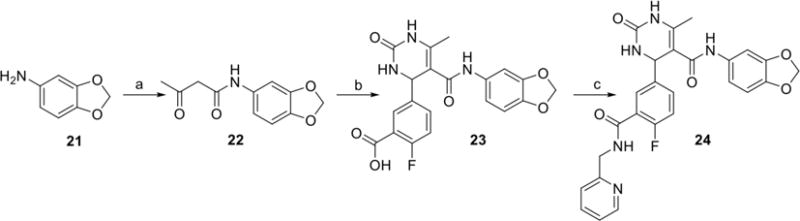
Reagents and conditions: a) ethyl acetoacetate, pyridine, 100°C, b) 2-fluoro-5-formylbenzoic acid, Yb(OTf)3, Urea, CH3CN, 100°C c) HATU, DIEA, 2-(aminomethyl)pyridine, DMF
Structure–Activity Relationships
Our hybrid design utilized an amide linker between the template of 2 and the new substituents intended to fill the hydrophobic subsite of GRK2. Although it was hypothesized that addition of a methyl amide group alone would establish an extra hydrogen bond interaction with the P-loop of GRK2 similar to that observed in the crystal complex with 1, prototype compound 12a exhibited a 5-fold loss in potency against GRK2. However, the compound maintained selectivity against GRK1 and GRK5. It is possible that the additional hydrogen bond formed by the amide with the enzyme cannot overcome a desolvation penalty or an entropic penalty for fixing the conformation of the P-loop. Notably, incorporation of the methyl amide did result in a favorable loss in potency against PKA (IC50 > 100 μM). The carboxylic acid derivative 10, on the other hand, gave a dramatic 20-fold decrease in potency for GRK2 and only a 2-fold loss against PKA.
As both 1 and balanol place an aromatic ring into the hydrophobic pocket of GRK2, we introduced a benzyl amide (12b), which was essentially equipotent with the lead (2) with respect to GRK2 and ROCK1, but gained substantial potency for GRK5.17, 19 Adding a single fluorine atom in the meta position of the benzyl (12c) resulted in a “pan” GRK inhibitor, in that it inhibited GRK1, 2 and 5 but lacked PKA activity. The results with both 12b and 12c suggest that addition of lipophilicity to the amide appendage is a path to higher potency, but not necessarily GRK2 selectivity. Incorporation of the 2,6-difluorobenyzl amide (12d), the same D ring as in Takeda103A, did not improve upon the GRK2 potency of the 3-fluorobenzyl amide, but restored selectivity for GRK2 versus GRK1 and GRK5, providing a clue that the size of the amide substituent may be an important selectivity determinant among GRKs.
Next, the effect of adding polar groups to the D-ring extension was explored as the D-ring of balanol has two polar groups in the 2 and 6 positions. The 2-methoxy benzyl amide 12e gave a 13-fold increase in potency against GRK2 (IC50 = 60 nM) with selectivity of 270- and 38-fold over GRK1 and GRK5, respectively. Unfortunately, 12c retained potency against ROCK1 (IC50 = 57 nM). Movement of the methoxy substituent around the benzyl ring to the meta (12f) and para (12g) positions resulted in a significant loss of potency compared to the 2-methoxy benzyl amide (12e) for GRK2 and GRK1, but was more tolerated in GRK5 and ROCK1. Further investigation of the ortho position of the benzyl ring led to the 2-pyridylmethyl amide analog 12h which showed little selectivity among the GRKs and ROCK1 (thus a pan-inhibitor) having sub-μM affinity for all 3 GRKs and exhibiting a 10-fold increase in potency over 2 for ROCK1. Extending the linker between the amide bond and the terminal pyridine ring by one carbon (12i) substantially improved selectivity in comparison to 12h against GRK1 and GRK5 but somewhat reduced inhibition of GRK2. The 4-pyridyl ethyl amide (12j) was not well tolerated by any of the GRKs, suggesting the importance of the 2-pyridyl nitrogen to binding.
We next investigated the potential of increasing the size of the pyridine of 12h to an isoquinoline (12k), which improved selectivity against GRK5 6.4-fold (versus 2.5-fold for 12h). However, there was no change in ROCK1 inhibition between 12h and 12k (both ~10 nM). Interestingly, 12k achieved the highest potency and selectivity for GRK1 with an IC50 of 100 nM – a 50-fold increase in comparison to the 2-pyridylmethyl analog 12h and a greater than 100-fold increase in comparison to the parent compound 2.
As the 2,6-difluorobenzyl (12d) showed marginally improved selectivity in comparison to the unsubstituted benzyl 12a, the potential for achieving greater selectivity for GRK2 by further increasing the size of the D-ring amide substituents was explored. Both the 2,6-dichloro (12l) and the 2,6-dimethyl (12m) resulted in dramatic improvements in GRK2 selectivity. Both analogs had over 770-fold selectivity against GRK1 and GRK5, as well as 50-fold (12l) and 80-fold (12m) selectivity against ROCK1. Thus, for the first time we observed that ROCK inhibition can be mitigated via the introduction of steric bulk into the hydrophobic subsite. Extending this line of reasoning to the larger dimethoxy (12n) and di-trifluoromethyl (12o) analogs essentially eliminated all kinase inhibitory activity except for GRK2. The dimethoxy analog 12n retains excellent potency against GRK2 compared to the parent compound (0.13 versus 0.77 μM) and is our most selective GRK2 inhibitor to date. Compared to the 2-methoxy benzylamide analogue 12e, addition of the second methoxy in 12n was clearly a key step in building in selectivity for GRK2 (over 175-fold decrease in potency at ROCK1, 6-fold at GRK1, and 43-fold at GRK5). Moving the added bulk to the meta positions (with the 3,5-bis(trifluoromethyl)benzyl amide 12p) produced a profound loss in GRK2 potency revealing that the 2,6 disubstitution pattern of the D ring is critical for potency and selectivity. The remarkable impact of sterics on GRK2 selectivity is further highlighted by comparing 2-pyridyl amide 12h with the corresponding ortho-methyl analog 12q. The latter compound, although less potent at GRK2, had increased selectivity versus GRK1 (>50-fold) and GRK5 (36-fold) versus 12h (33-fold and 2-fold, respectively).
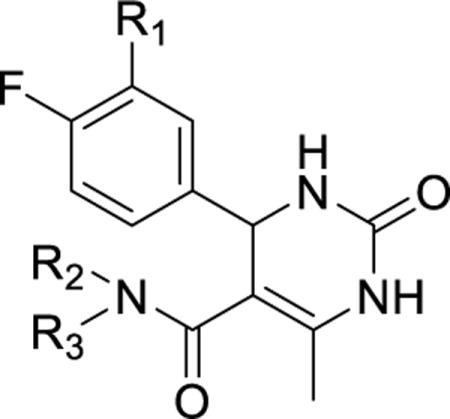
In general, homologating the amide substituent by insertion of a methylene between the amide and the aromatic ring led to decreases in potency and selectivity. This was particularly evident with 13a and 13b. In each case, GRK2 potency dropped precipitously and, oddly, sub-μM ROCK1 activity returned. Homologation of 12n (13c) resulted in a less dramatic decrease in GRK2 potency, but also restored ROCK1 inhibition. This interesting pattern was also observed with ethyl homolog 12r, which has decreased GRK2 potency and increased ROCK1 potency versus the shorter 12m. We briefly explored replacing the indazole moiety of 12h, which binds to the hinge of the kinase domain, with the benzodioxole group of paroxetine (24, Table 2).22 Because paroxetine has excellent selectivity for GRK2 versus ROCK1, we anticipated that the benzodioxole might increase selectivity. Despite the potent pan-kinase inhibition of 12h, the corresponding benzodioxole analog 24 was completely inactive. Several other benzodioxole analogs of other hybrids were similarly inactive (data not shown).
Table 2.
Kinase inhibitory activity of compounds 20 and 24.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | GRK2 IC50 (μM) |
GRK1 IC50 (μM) |
GRK5 IC50 (μM) |
PKA IC50 (μM) |
ROCK IC50 (μM) |
| 24 |
|
H |
|
>100 | >100 | >100 | >100 | ND |
| 20 |
|
Me |
|
4.6±3.0 | >100 | >100 | >100 | 2.1±1.3 |
All IC50 measurements are an average of three separate experiments run in duplicate.
Errors are standard error of the mean.
In a final attempt to further design out ROCK1 affinity, the nitrogen of the amide bond linking the indazole and the dihydropyrimidone core was methylated. Prior crystallization of a close analog of 2 into ROCK1 revealed that the amide nitrogen forms a water-mediated hydrogen bond to ROCK1-Asp216,24 and therefore, methylation of the amide would be expected to weaken its affinity. The methylated amide of potent hybrid 12m was thus synthesized. The resulting compound 20 however showed ROCK1 potency similar to 12m having an IC50 of 2.1 μM, and a nearly 70-fold decrease in potency at GRK2, discouraging us from pursuing additional N-methyl analogues.
GRK2–Gβγ·Inhibitor Crystal Structures
To gain insight into the molecular basis for the relative potency and selectivity of this series of compounds, 12h, 12k, 12n, and 12r were each co-crystallized with GRK2–Gβγ and their resulting X-ray crystal structures were determined (Figure 3, Supplemental Table 1). All four complexes crystallized in space group C2221 with nearly identical unit cell constants with resolution ranges of 2.56 Å – 3.26 Å. In comparison to the previously reported structure of 2 in complex with GRK2–Gβγ (PDB entry 4PNK)25, all four inhibitor complexes exhibit nearly identical kinase domain conformations with differences in relative rotations between the large and small lobes no greater than 1°. This result is consistent with the idea that the indazole, which occupies the adenine subsite and forms two hydrogen bonds with the hinge of the kinase domain, dictates the overall conformation of the large and small lobes.25, 26
Figure 3.
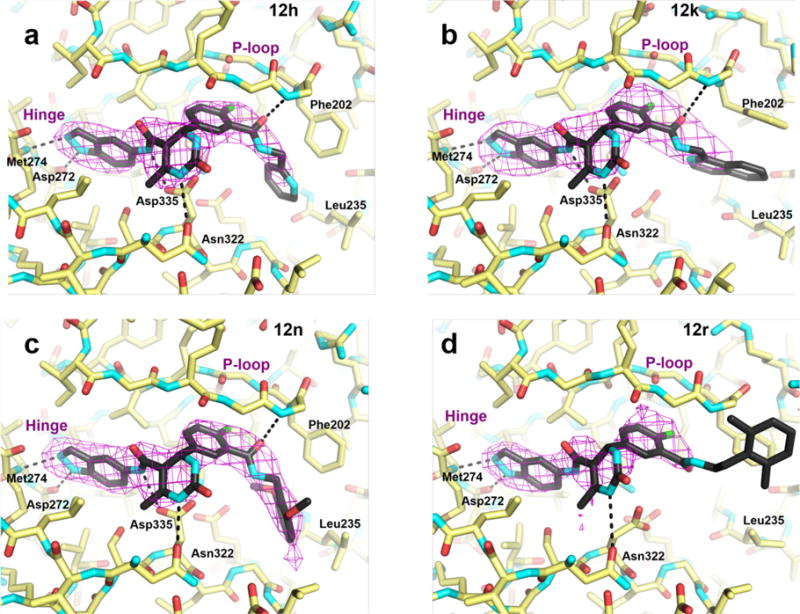
Co-crystal structures reveal that the inhibitors bind in the ATP-binding pocket in a similar conformation as the compound 2 parent structure. 3σ |Fo| – |Fc| omit maps of compounds 12h (a), 12k (b), 12n (c), and a 2σ |Fo| – |Fc| omit map of 12r (d) are represented as magenta wire cages superimposed onto the refined X-ray crystal structures. Hydrogen bonds with the labeled GRK2 residues are shown as black dashed lines. The P-loop and hinge region are indicated for reference.
As expected, the four inhibitors bind in the ATP pocket of GRK2 in essentially the same manner as the parent compound 2, with the exception that the amide bond connecting the indazole and the dihydropyrimidine is flipped relative to the model of 2 (Figure 3). However, the electron density for this amide in the complex with 2 is ambiguous and hence the linker may adopt multiple configurations in the previous structure. As noted above, the indazole rings bind in the adenine subsite forming two hydrogen bonds with backbone atoms of the hinge residues Asp272 and Met274, and the dihydropyrimidine and fluorophenyl rings fill the ribose and polyphosphate subsites, respectively. However, the presence of the D-rings, and presumably their interactions in the hydrophobic subsite, seems to alter the conformation of the A-C rings to some extent among the four complexes (Figure 4). Compared to the GRK2–Gβγ·2 complex, the 12h, 12k, and 12n compounds form additional hydrogen bonds in the ribose subsite with Asp335 and Asn322. As predicted, their variable amide-linked D-rings occupy the hydrophobic subsite of GRK2, and the carbonyls of the amide bond linker ortho to the fluorine atom in the C-ring form a hydrogen bond with the backbone nitrogen of Phe202 in the P-loop. The D-ring of 12r, however, flips out of the hydrophobic site towards the solvent, and there is no interpretable electron density beyond the amide linker.
Figure 4.
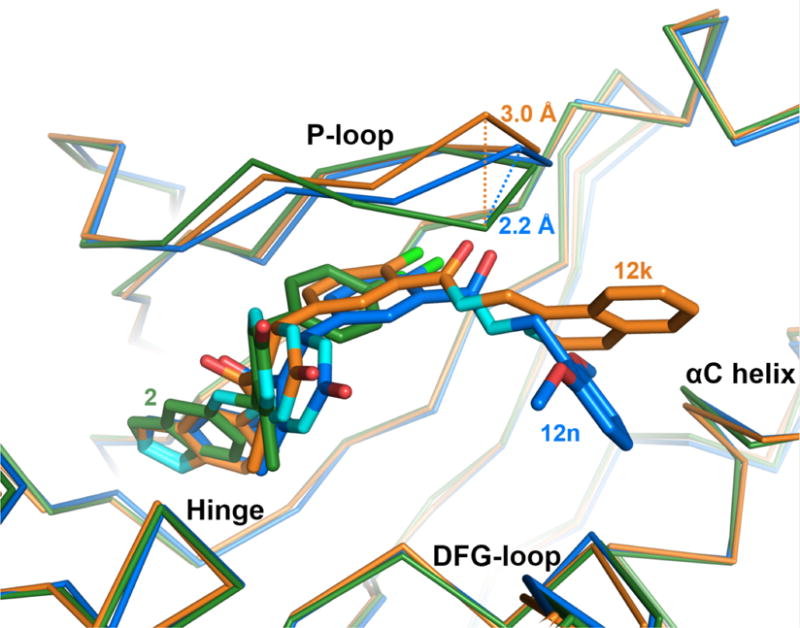
Adaptive structural changes in the GRK2 P-loop. Compared to the P-loop conformation when bound to compound 2 (green), the Cα carbon of Gly201 shifts away from the binding site by 2.2 Å when bound to compound 12n (blue), 12h or 12r (not shown), and by 3.0 Å when bound to 12k (orange). The magnitude of the shift thus appears to depend on the size of the D-ring.
The largest conformational changes induced by the various inhibitors occur in the P-loop (Figure 4). Relative to 2, each of the four inhibitors causes the P-loop to shift away from the polyphosphate subsite as if to accommodate the terminal aromatic rings. Compounds 12h, 12n, and 12r each have a maximum P-loop shift of 2.2 Å at the Gly201 Cα relative to the parent structure (2). Compound 12k demonstrates the largest P-loop shift of 3.0 Å, possibly because it has the bulkiest substituent. In addition, the benzene ring of Phe202 rotates to allow space for the terminal aromatic substituents depending on their orientation (Figure 3). Notably, AST loop residues 487–493, which are typically ordered in active conformations of AGC kinases, but are disordered in most GRK2 kinase domain structures to date, are visible in the 12n electron density map and pack on top of the P-loop. The reason for these residues being more ordered in the 12n complex relative to the others is unclear, but the density may simply reflect the higher quality of this particular crystallographic data set (Supplemental Table 1).
Molecular Basis for Potency and Selectivity
Analysis of these four crystal structures in comparison to structures of other AGC kinase domains provides insight into the molecular basis for their relative potencies and selectivities. Consistent with previous studies,30–32 the number of hydrogen bonds does not correlate well with inhibitor binding affinity in AGC kinases (Figure 5). For example, compound 12r has the second highest affinity of the four crystallized compounds from this study (12h, 12k, 12n, 12r,) and the parent compound 2, but only forms three hydrogen bonds. Plotting the Ki of 10 potent GRK2 inhibitors (calculated from the IC50 values using the Cheng-Prusoff transformation) versus both the number of hydrogen bonds and the buried accessible surface area (Figure 5) exhibited no correlation between the number of hydrogen bonds and Ki but did show a correlation between buried surface area and Ki. The buried accessible surface area is the most consistent determinants of inhibitor potency, as previously noted for GRK2.32
Figure 5.
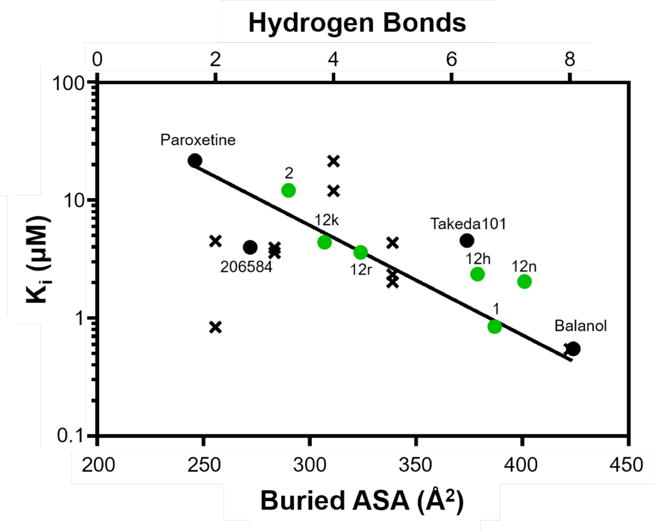
Inhibitor potency for GRK2 correlates with buried ASA (R2=0.8) but not the number of hydrogen bonds (R2=0.01). Ki values were calculated from experimentally determined IC50 values of the GRK2 inhibitors for which there is a crystal structure (Supplementary Table 2) using the Cheng-Prusoff transformation and plotted as a function of both their buried ASA (circles) and the number of hydrogen bonds they form with GRK2 (x’s). Buried ASA for inhibitors from this study are shown in green.
Interestingly, neither 2 nor any of its derivative compounds reported here inhibit PKA below concentrations of 30 μM despite its kinase domain being highly homologous to those of GRKs and ROCK1. Comparison to both the PKA·AMPPNP substrate analog structure (PDB entry 4HPT) and the PKA-balanol complex (PDB entry 1BX6) indicates that the derivatives (of compound 2) should not be sterically blocked from binding in the PKA binding site. However, the hinge in the structures of PKA is shifted 1.5–1.7 Å away from the adenine subsite relative to the position of the hinge in both GRK2 and GRK5 (Figure 6). This difference may prevent the formation of favorable contacts between hinge backbone atoms and the indazole ring common to 2 and all of its derivatives.
Figure 6.
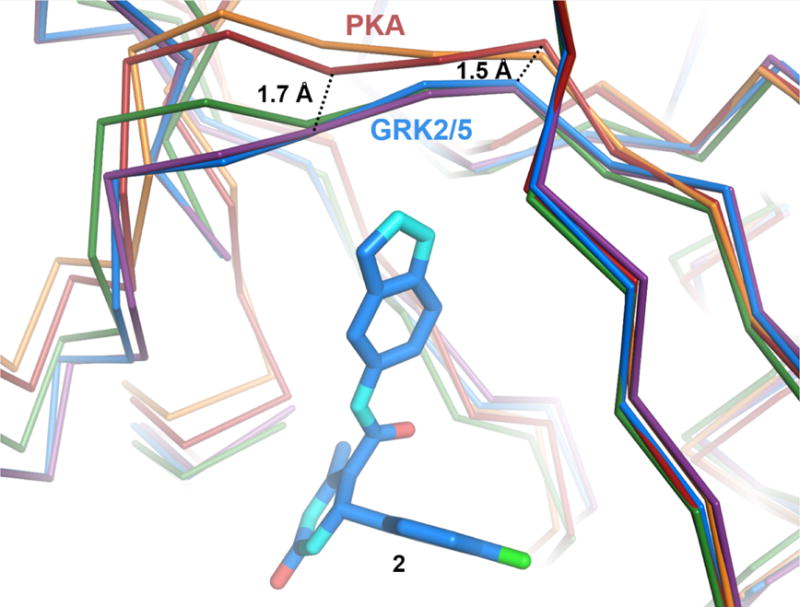
Structural differences in the hinge regions of PKA, GRK2, and GRK5. Cα traces of PKA bound to AMPPNP (PDB entry 4HPT, red) or balanol (PDB entry 1BX6, orange), superimposed onto GRK2·compound 2 (PDB entry 4PNK, blue). GRK2·12n (purple) and GRK5·12h (green) are also shown for comparison. Hinge residues that form hydrogen bonds with the indazole nitrogens of compound 2 and its derivatives are 1.5–1.7 Å closer to the inhibitor in the structures of GRK2 and GRK5 relative to those of PKA.
Compounds 2, 12n, and 12r do not bind tightly to either GRK1 or GRK5, but 12h and 12k inhibit these kinases with potencies in the low μM or nM range. To explore the molecular basis of this selectivity among GRK subfamilies, the structures of GRK5·12h and GRK2·12h were compared (Figure 7). The hydrophobic subsite in GRK5 is deeper and narrower than that of GRK2.26 Thus, one would predict that compounds with larger D-ring substituents would tend to be excluded from the hydrophobic subsite of GRK5. In the GRK5·12h complex, a hydrogen bond is formed between the catalytic lysine (Lys215 in GRK5) and the pyridine nitrogen of the inhibitor. In the crystal structure of GRK2·12h, the pyridine nitrogen most likely faces the solvent. The fact that 12h buries nearly identical surface area in each complex (only 5 Å2 additional buried surface area when bound to GRK5) is consistent with its similar potencies against GRK2 and GRK5 and its behavior as a pan-GRK inhibitor.
Figure 7.
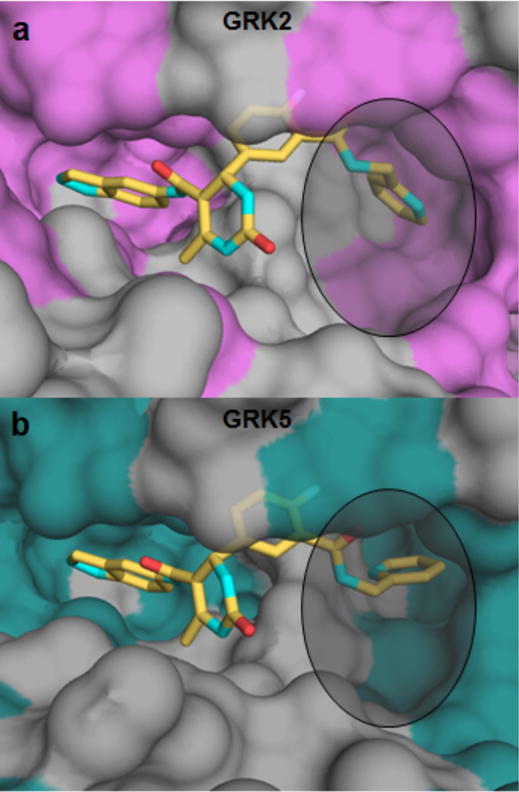
Comparison of GRK2 and GRK5 hydrophobic binding pockets when bound to 12h (yellow) (a) GRK2 has a much wider and shallower binding pocket (non-polar residues highlighted in purple) and (b) GRK5 has a deeper, narrower, and overall smaller binding pocket (non-polar residues are highlighted in teal).
Based on the hydrophobic subsite hypothesis above, the isoquinoline ring of 12k was predicted to select against GRK5 and its close homolog GRK1. However, it was an efficacious inhibitor of all three GRKs as well as ROCK1, with 56-fold and 1000-fold increases in potency against GRK5 and GRK1 relative to the parent compound 2. The crystal structure shows that the isoquinoline reorganizes local structure in the hydrophobic subsite of GRK2, in particular the P-loop, such that its benzyl ring forms a π-π stacking interaction with the side chain of Phe202 (Figure 3b). Superimposing 12k from the GRK2-Gβγ structure in the active sites of GRK1 and GRK5 illustrates that the isoquinoline group would be compatible with their hydrophobic subsites as well, provided their P-loops can similarly reorganize. This modeling exercise also suggests that the nitrogen of the isoquinoline ring could form an additional hydrogen bond with the active site lysine as 12h does in GRK5. A greater degree of kinase domain closure in GRK1 and GRK5 relative to GRK2 may account for why enhancement of potency is higher for GRK1 and GRK5.
The dimethoxybenzyl D-ring of the most selective inhibitor, 12n, binds snugly in the hydrophobic subsite of GRK2 with one methoxy group packing deep in the pocket, and the other projecting towards solvent (Figures 3c, 8a). Docking the compound in the active site of GRK5 (Figure 8b) demonstrates that the 2,6-dimethoxybenzyl substituent of 12n would collide with the DFG loop, which is shifted towards the hydrophobic subsite due to a greater degree of kinase domain closure in GRK5 than in GRK2. This collision also likely explains the GRK2 selectivity seen with the similar, but not quite as bulky, hybrids 12l and 12m. The packing of 12n is likely mimicked by the analogous potent ortho methoxy hybrid 12e. Movement of the methoxy substituent to the meta and para positions (as in 12f and 12g) would cause the methoxy to collide with the αC-helix or DFG loop, respectively, explaining their less favorable binding to GRK2.
Figure 8.
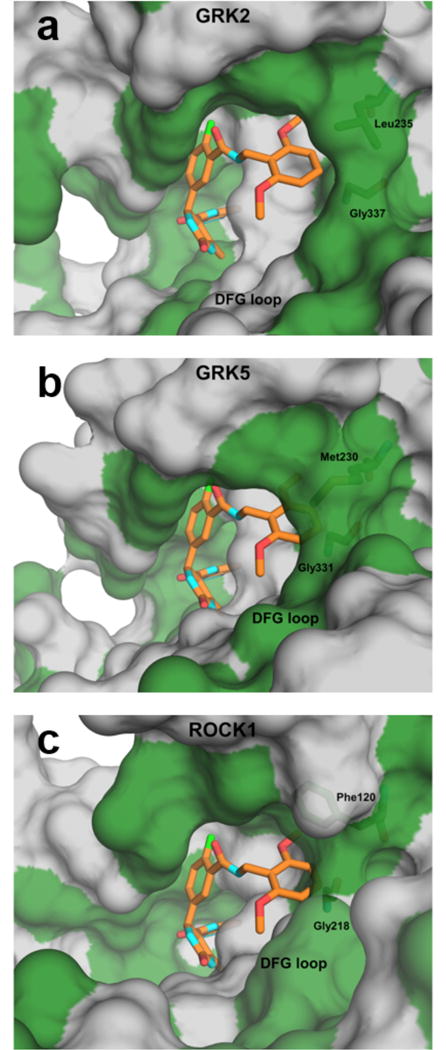
Molecular origins of selectivity for compound 12n. Surface representations of GRK2·12n (a), GRK5 (PDB entry 4WNK, (b), and ROCK1 (PDB entry 3V8S, (c) with hydrophobic and polar/charged residues colored green and gray, respectively. 12n is superimposed onto GRK5 and ROCK1 to demonstrate potential clashes. (b) In GRK5, 12n appears to clash with both Met230 from the αC helix (Leu235 in GRK2) and Gly331 from the DFG loop (Gly337 in GRK2). (c) In ROCK1, 12n may clash with Phe120 (Leu-235 in GRK2) and backbone atoms of Gly218 in the DFG loop (Gly337 in GRK2). For this modeled complex, ROCK1-Asp216 was changed to the rotamer of the analogous residue in the GRK2·12r complex (Asp335).
Of the four inhibitors structurally characterized here, only 12n achieves selectivity over both the GRK1 and GRK4 subfamilies and ROCK1. Upon superimposing these inhibitor structures on ROCK1 in complex with an indazole derivative similar to 2 (3V8S)33, it is apparent that ROCK1-Phe120 (Leu235 in GRK2) sterically clashes with each of the four characterized inhibitor D-rings. However, ROCK1-Phe120 must be able to adopt a rotamer more similar to that of GRK2-Leu235 because 12h, 12k, and 12r all inhibit ROCK1 with high potency. Compound 12n, however, also sterically clashes with the backbone carbonyl of ROCK1-Gly218 (Gly337 in GRK2, Figure 8c) in the DFG loop, which is shifted towards the hydrophobic subsite by 2.9 Å in ROCK1 relative to GRK2. The other inhibitors avoid generating this collision but 12n cannot, as a consequence of its two methoxy substituents, which greatly restrict its ability to alter its conformation within the hydrophobic subsite. The analogous ethylene linked inhibitor 13c has low nanomolar affinity for ROCK1 despite having a bulky 2,6-dimethoxyphenyl substituent. It is likely that the extra carbon extension allows the inhibitor to pack outside of the hydrophobic pocket to avoid steric clashes, as does 12r.
Electron density for the 2,6-dimethyl phenethyl moiety of 12r is not evident after the amide linker (Figure 3d), suggesting that this arm of the inhibitor is flexible while bound to GRK2. This flexibility likely results from the extra degree of rotational freedom conferred by the longer ethylene linker and its inability to be accommodated within the hydrophobic subsite. For the same reasons, the D-ring of 12r likely would not be able to occupy the hydrophobic subsites of GRK1 and GRK5. Thus, the compound would bind no better than 2 alone, consistent with the relatively poor binding of 12r to GRK1 and GRK5 (Table 1).
Contractility in Mouse Cardiomyocytes
To ascertain the ability of these inhibitors to produce a myocardial effect, they were incubated with cardiomyocytes isolated from adult mice, and then stimulated with the βAR agonist isoproterenol as described previously.26 In the presence of a GRK2 inhibitor, the strength of the resulting contraction should increase as there is more βAR signaling. Previously, it was shown that paroxetine induces a significant increase in contractility in comparison to the DMSO control when dosed at 10 μM.22 In comparison, 2 showed similar efficacy as paroxetine when dosed at 1 μM (Figure 9), consistent with the increased potency of this compound of GRK2 relative to paroxetine.
Figure 9.
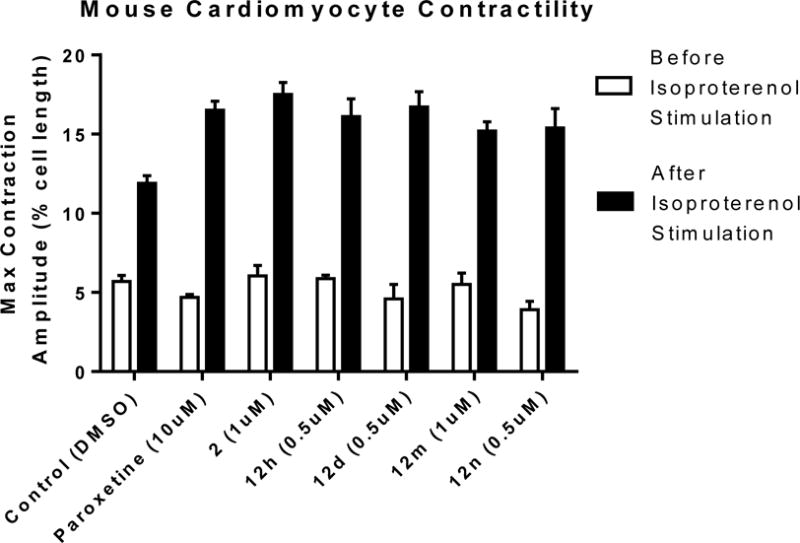
Maximum contraction amplitudes of the known GRK2 inhibitor paroxetine, the lead 2, 12h, 12d,12m, and 12n before and after isoproterenol stimulation. Doses shown are the minimum inhibitory concentrations that exhibit a p-value <0.05 versus Control. Values represent the mean ± SEM for 8–10 cardiomyocytes.
Four of the hybrid molecules were evaluated in cardiomyocytes: 12d, 12h, 12m, and 12n (Figure 8). All of the hybrids were equivalent to or slightly more potent than the lead 2 at stimulating cardiomyocyte contraction. Unfortunately, it is not possible to draw any firm conclusions from this limited study regarding whether different levels of selectivity for GRK2 vs GRK5 lead to greater or lesser efficacy in the cardiomyocytes. The GRK2 selectivities in this group ranged from 2-fold (12h) to >700-fold (12m and 12n), but no clear differences in cardiomyocyte potency are apparent. Interpretation of these results is further complicated by unknown differences in cell permeability of the hybrid analogs. Future studies may require the careful determination of EC50 values with probes of known equivalent cell permeabilities to provide a definitive answer to what GRK inhibition selectivity profile is optimum for achieving maximum efficacy in cardiomyocytes.
CONCLUSIONS
Utilizing a hybrid approach we envisioned development of selective and potent GRK2 inhibitors constructed from the potent ROCK1 inhibitor 2 and the selective GRK2 inhibitor 1. Overlaying the binding poses of the two compounds (Figure 1) in the active site of GRK2 revealed that it may be possible to utilize the hydrophobic binding site of GRK2 to achieve selectivity over other AGC kinases. A small library of compounds was thus synthesized in which a variety of amide substituents were appended to the fluoro-aromatic ring of 2 to mimic the difluorobenzyl amide of 1. The major SAR findings from this study were that small benzyl amide substituents lead to significant inhibition of GRK5 in addition to GRK2, and that addition of steric bulk to the benzyl amides favors GRK2 selectivity over all of the other AGC kinases tested. This ultimately led to the identification of 12n, a 2,6-dimethoxybenzylamide with remarkable potency against and selectivity for GRK2 and complete loss of the ROCK inhibitory activity of the lead 2. We also identified some potent pan-inhibitors of GRKs 1, 2 and 5, including 2-pyridyl amide 12h. Collectively these new hybrid analogs represent an important start in the design of small molecule probes that can be used to explore the physiological roles of the individual GRKs.
Co-crystal structures of several representative analogs bound to GRK2 were solved to help define the molecular basis for their differing selectivities. Comparison of the GRK2-12h structure with the previously determined GRK5-12h crystal structure showed that the hydrophobic subsite of GRK5 is much narrower and deeper than the shallower, wider subsite of GRK2, helping to rationalize the increased GRK2 selectivity seen with bulkier D-ring appendages. In addition, three other GRK2 inhibitor complexes were crystallized: GRK2-12k, GRK2-12n, and GRK2-12r. Overall, the structures confirm that the size and shape of the hydrophobic subsite, which is in part dependent on the degree of kinase domain closure, puts constraints on the chemical nature of the D-ring amide substituent that can be accommodated. Larger substituents, such as the 2,6-dimethoxy benzyl of 12n, can fit into GRK2 more readily than the other AGC kinases tested in this study. These conclusions were supported by overlays of the bound poses of our analogs in GRK2 with other published kinase structures. Based on the published GRK5-12h structure and the selectivity data we observed, we also conclude that smaller hydrogen-bonding benzyl amides can interact effectively with the hydrophobic subsite of GRK5, conferring potent inhibitory activity to the GRK5-inactive lead 2.
Finally, evaluation of selected inhibitors in cardiomyocytes suggests that pan-GRK inhibition or dual GRK2/GRK5 inhibition is just as effective as GRK2 inhibition alone at stimulating contraction in the failing heart, but further studies will be needed to confirm this hypothesis. In the future we plan to evaluate the pharmacokinetics of these lead compounds with an eye towards their future development as potential heart failure therapeutics.
Experimental Section
Chemistry
All reagents were used without further purification as received from commercial sources unless noted otherwise. 1H NMR and 13C NMR spectra were taken in DMSO-d6 or MeOD at room temperature on Varian Inova 400 MHz or Varian Inova 500 MHz instruments. Reported chemical shifts for the 1H NMR and 13C NMR spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Mass spectrometry data was measured using a Waters Corporation LCT. HPLC was used to determine purity of biologically tested compounds on an Agilent 1100 series with an Agilent Zorbax Eclipse Plus–C18 column. A gradient of 10–90% acetonitrile/water over 13 min was used with detection at 254 nm. All tested compounds had purity >95%. Solvent abbreviations used: MeOH (methanol), DCM (dichloromethane), EtOAc (ethyl acetate), hex (hexanes), DMSO (dimethylsulfoxide), DMF (dimethylformamide), H2O (water), THF (tetrahydrofuran). Reagent abbreviations used: DIBAL (diisobutylaluminum hydride), HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate), DIEA (diisopropylethylamine), MgSO4 (magnesium sulfate), NaHCO3 (sodium bicarbonate), Na2CO3 (sodium carbonate), NH4Cl (ammonium chloride), NaCl (sodium chloride), K2CO3 (potassium carbonate), H2SO4 (sulfuric acid), and HCl (hydrogen chloride).
Methyl 4-fluoro-3-methylbenzoate (3b)
To a 100 mL round bottom flask with 40 mL of methanol was added 4-fluoro-3-methyl benzoic acid (4.0 g, 25.96 mmol) followed by concentrated H2SO4 (1.0 mL). The reaction vessel was equipped with a condenser and refluxed overnight at 65 °C. Cooled down reaction and diluted with ether and NaHCO3. The organic layer was washed with brine (3x), dried over MgSO4, and concentrated at room temperature under reduced pressure to give 3b as a clear liquid (98% yield, 4.38g). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 7.5, 2.2 Hz, 1H), 7.83 (ddd, J = 7.9, 5.0, 2.3 Hz, 1H), 7.27 (t, J = 9.1 Hz, 1H), 3.83 (s, 3H), 2.28 (s, 3H). HPLC purity: 100%.
Methyl 3-(bromomethyl)-4-fluorobenzoate (4)
To a 250 mL closed pressure vessel was added 3 (4.02g, 23.91 mmol) dissolved in anhydrous carbon tetrachloride (40 mL). N-bromosuccinimide (3.83g, 21.50 mmol) was then added followed by benzoyl peroxide (0.290g, 1.20 mmol). The reaction stirred for 1 hour at 65 °C then cooled and filtered through celite with dichloromethane and concentrated. Purified using 0%–1% EtOAc/Hexanes using flash chromatography to give 4 as a clear oil (82% yield, 4.31g). 1H NMR (400 MHz, DMSO-d6) δ 8.19 (dd, J = 7.4, 2.3 Hz, 1H), 8.00 (ddd, J = 8.7, 5.0, 2.3 Hz, 1H), 7.41 (dd, J = 9.8, 8.7 Hz, 1H), 4.79 (d, J = 1.1 Hz, 2H), 3.87 (s, 3H).
(3-(Bromomethyl)-4-fluorophenyl)methanol (4b)
To a dry 250 mL flask was added toluene (30 mL) followed by 1M DIBAL in toluene (23.16 mL, 23.16mmol). The flask was then cooled to 0 °C and then 4 (2.73 g, 11.05 mmol) in toluene (5 mL) was added. The reaction stirred for 2 hours and was then quenched (on ice) with 2N HCl to a pH of 2. The reaction was extracted with ethyl acetate (2x), the organic layers were combined and washed with NaCl (2x), dried over MgSO4, and concentrated to give 4b as a white solid (94% yield, 2.09g). 1H NMR (500 MHz, DMSO-d6) δ 7.46 (dd, J = 7.5, 2.2 Hz, 1H), 7.32 (ddd, J = 7.8, 5.1, 2.2 Hz, 1H), 7.18 (dd, J = 10.0, 8.4 Hz, 1H), 5.28 (s, 1H), 4.69 (s, 2H), 4.46 (s, 2H).
2-(2-Fluoro-5-(hydroxymethyl)phenyl)acetonitrile (5)
To a 250 mL round bottom flask was added 4b (2.06 g, 10.26 mmol) dissolved in DMSO (40 mL) followed by sodium cyanide (1.51 g, 30.77 mmol). After stirring at room temperature for 2 hours the reaction was diluted with H2O and ethyl acetate. The layers were separated and the aqueous layer was further extracted with ethyl acetate (2x). The organic layers were combined, washed 3x with NaCl, dried over MgSO4, and concentrated. Using flash chromatography (15% – 30% EtOAc/Hexanes) the residue was purified to obtain 5 as a clear liquid (1.50 g, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.42 (dd, J = 7.5, 2.1 Hz, 1H), 7.32 (ddd, J = 7.7, 5.1, 2.1 Hz, 1H), 7.21 (dd, J = 9.9, 8.4 Hz, 1H), 5.31 (t, J = 5.7 Hz, 1H), 4.48 (d, J = 5.6 Hz, 2H), 4.06 (s, 2H).
2-(2-Fluoro-5-(hydroxymethyl)phenyl)acetic acid (6)
To a 100 mL round bottom flask was added 5 (0.881 g, 5.33 mmol) followed by 2N NaOH (20 mL). The reaction vessel was equipped with a condenser and refluxed overnight. After cooling the reaction down HCl was added to reach a pH of 3. The reaction was extracted with ethyl acetate (2x). The organic layers were combined, washed with NaCl (2x), dried over MgSO4, and concentrated in vacuo to give 6 as a white solid (0.969 g, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.45 (s, 1H), 7.28 – 7.20 (m, 2H), 7.11 (dd, J = 9.7, 8.5 Hz, 1H), 5.22 (s, 1H), 4.45 (s, 2H), 3.60 (s, 2H).
2-(2-Fluoro-5-formylphenyl)acetic acid (7)
To a 100 mL round bottom flask at room temperature was added 6 (1.26g, 7.58 mmol), triethylamine (8.46 mL, 60.66 mmol), and DMSO (20 mL). Sulfur trioxide pyridine complex (4.83 g, 30.33 mmol) in DMSO (20 mL) was then added to the reaction and the reaction was stirred for 20 minutes. The reaction was diluted with Na2CO3 then extracted with ether. To the aqueous layer 2N HCl was added to give a pH of 3. The aqueous layer was then extracted with ethyl acetate (2x). The ethyl acetate layers were combined and washed with NaCl (2x), dried over MgSO4, and concentrated in vacuo to give 7 as a white solid (1.24g, 100% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.49 (s, 1H), 9.96 (s, 1H), 7.98 – 7.85 (m, 2H), 7.43 (dd, J = 9.6, 8.3 Hz, 1H), 3.76 (d, J = 1.4 Hz, 2H).
N-(1H-Indazol-5-yl)-3-oxobutanamide (9)
To a 50 mL pressure vessel was added 1H-indazol-5-amine (0.800 g, 6.00 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (1.57 g, 12.00 mmol), sodium acetate (0.492 g, 6.00 mmol), and acetonitrile (5 mL). The reaction was stirred overnight at 100°C. The reaction was diluted with citric acid (10%) and EtOAc. After separating, the organic layer was washed 2x with NaCl, dried over MgSO4, and concentrated to give a brown residue. The residue was then triturated with 2:1 dichloromethane:ethyl acetate and filtered off to give a light brown solid. The solid was then purified on flash chromatography in a gradient of 0% to 3% MeOH/DCM to afford 9 as an off white solid (0.901 g, 69% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.99 (s, 1H), 10.08 (s, 1H), 8.12 (s, 1H), 8.02 (s, 1H), 7.49 (d, J = 8.9 Hz, 1H), 7.38 (d, J = 9.2 Hz, 1H), 3.56 (s, 2H), 2.23 (d, J = 1.6 Hz, 3H).
5-(5-((1H-Indazol-5-yl)carbamoyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-4-yl)-2-fluorobenzoic acid (10)
To a 100 mL pressure vessel equipped with a stir bar was added 2-fluoro-5-formylbenzoic acid (0.682 g, 4.054 mmol), 9 (0.801 g, 3.686 mmol), urea (0.332 g, 5.529 mmol), ytterbium trifluormethanesulfonate (0.229 g, 0.3686 mmol) and acetonitrile (15.0 mL). The reaction mixture was heated to 100 °C and stirred. The reaction mixture went from a white suspension to a clear solution, and then back to a white suspension. After four hours, the reaction was diluted with 2 mL of water then ethyl acetate (~60 mL). The resulting white precipitate was filtered off and washed with ethyl acetate followed by diethyl ether yielding 10 as a white powder (84% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H), 9.59 (s, 1H), 8.79 (s, 1H), 7.98 (t, J = 2.1 Hz, 2H), 7.88 – 7.78 (m, 1H), 7.64 (s, 1H), 7.50 (s, 1H), 7.46 – 7.33 (m, 2H), 7.33 – 7.22 (m, 1H), 5.42 (s, 1H), 2.06 (s, 3H).
2-(5-(5-((1H-Indazol-5-yl)carbamoyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-4-yl)-2-fluorophenyl)acetic acid (11)
Compound 11 was prepared as described for compound 10, replacing 2-fluoro-5-formylbenzoic acid with 7 (72% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.80 (s, 1H), 12.53 (s, 1H), 9.54 (s, 1H), 8.69 (d, J = 2.0 Hz, 1H), 7.99 – 7.92 (m, 2H), 7.58 (s, 1H), 7.42 – 7.33 (m, 2H), 7.26 – 7.16 (m, 2H), 7.11 (t, J = 9.1 Hz, 1H), 5.38 (s, 1H), 3.56 (s, 2H), 2.04 (s, 3H).
4-(4-Fluoro-3-(methylcarbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12a)
Compound 10 (0.100 g, 0.244 mmol), HATU (0.185 g, 0.489 mmol), DIEA (0.085 mL, 0.489 mmol), and 2N methylamine (0.245 mL, 0.489 mmol) were added to 5 mL of DMF in a 25 mL round bottom flask and allowed to stir overnight at room temperature. The reaction was diluted with water and ethyl acetate giving a white suspension in the organic layer. The layers were separated and the organic layer was washed once with Na2CO3 and twice with NaCl. The organic suspension was then filtered off and washed with water giving a white solid. The solid was then washed with dichloromethane and purified using flash chromatography (0–15% MeOH/DCM) yielding 12a as a white solid: 80.7 mg (78%). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.58 (s, 1H), 8.75 (d, J = 1.9 Hz, 1H), 8.20 (t, J = 1.3 Hz, 1H), 7.98 (d, J = 7.1 Hz, 2H), 7.63 – 7.57 (m, 2H), 7.44 – 7.35 (m, 3H), 7.24 (dd, J = 10.3, 8.6 Hz, 1H), 5.43 (d, J = 2.9 Hz, 1H), 3.17 (d, J = 4.6 Hz, 1H), 2.75 (d, J = 4.6 Hz, 3H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.92, 163.71, 152.24, 140.48, 138.29, 136.77, 133.18, 131.90, 130.01, 128.19, 123.70, 122.50, 120.85, 115.97, 115.79, 110.25, 109.72, 104.96, 54.32, 26.17, 16.98. HPLC purity: 96%; MS (ESI+) m/z: 445.1 (M+1).
4-(3-(Benzylcarbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12b)
Compound 12b was prepared as described for 12a, replacing methylamine with benzylamine. Yield: 21.0 mg, 17% yield). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.59 (s, 1H), 8.83 (td, J = 5.4, 1.5 Hz, 1H), 8.77 (d, J = 1.4 Hz, 1H), 7.98 (d, J = 18.6 Hz, 2H), 7.65–7.60 (m, 2H), 7.44 – 7.37 (m, 3H), 7.32 – 7.20 (m, 6H), 5.45 (d, J = 2.9 Hz, 1H), 4.45 (d, J = 6.1 Hz, 2H), 2.08 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.92, 163.46, 159.13, 157.15, 152.26, 140.51, 139.09, 138.40, 136.78, 133.18, 131.91, 130.12, 128.14, 126.95, 126.62, 123.71, 123.59, 122.51, 120.85, 116.06, 115.87, 110.26, 109.72, 104.91, 54.28, 42.52, 16.99. HPLC purity: 95%; MS (ESI+) m/z: 499.1 (M+1).
4-(4-Fluoro-3-((3-fluorobenzyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12c)
Compound 12c was prepared as described for 12a, replacing methylamine with 3-fluorobenzylamine. Yield: 46.8 mg (37%). 1H NMR (500 MHz, DMSO-d6) δ 12.90 (s, 1H), 9.58 (s, 1H), 8.88 (s, 1H), 8.76 (s, 1H), 7.97 (d, J = 18.0 Hz, 2H), 7.63 (s, 2H), 7.46 – 7.31 (m, 4H), 7.27 (t, J = 9.4 Hz, 1H), 7.12 (dd, J = 15.2, 9.0 Hz, 2H), 7.08 – 7.02 (m, J = 11.8, 5.3 Hz, 1H), 5.44 (s, 1H), 4.46 (d, J = 5.8 Hz, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO) δ 171.71, 165.49, 164.12, 163.62, 161.69, 159.73, 157.75, 152.83, 142.48, 141.11, 139.00, 132.47, 130.85, 130.67, 128.82, 124.02, 123.50, 123.06, 121.44, 116.57, 114.18, 113.97, 110.85, 110.36, 105.45, 54.85, 42.65, 17.56. HPLC purity: 99%; MS (ESI+) m/z: 516.9 (M+1), 538.8 (M+Na+).
4-(3-((2,6-Difluorobenzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12d)
Compound 12d was prepared as described for 12a, replacing methylamine with 2,6-difluorobenzylamine. Yield: 60.1 mg (31%). 1H NMR (400 MHz, DMSO-d6) δ 12.91 (s, 1H), 9.55 (s, 1H), 8.74 (s, 1H), 8.70 (s, 1H), 7.96 (d, J = 10.5 Hz, 2H), 7.59 (s, 1H), 7.52 (d, J = 6.6 Hz, 1H), 7.42–7.35 (m, 4H), 7.19 (t, J = 9.5 Hz, 1H), 7.05 (t, J = 7.8 Hz, 2H), 5.40 (s, 1H), 4.47 (d, J = 4.9 Hz, 2H), 2.05 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.90, 163.22, 152.25, 140.41, 138.43, 136.78, 133.17, 131.89, 130.11, 129.72, 128.12, 123.47, 122.50, 120.87, 115.96, 115.78, 113.79, 111.43, 111.23, 110.28, 109.72, 109.42, 104.88, 54.23, 31.19, 16.99. HPLC purity: 98%; MS (ESI+) m/z: 534.9 (M+1), 556.8 (M+Na+).
4-(4-Fluoro-3-((2-methoxybenzyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12e)
Compound 12e was prepared as described for 12a, replacing methylamine with 2-methoxybenzylamine. Yield: 120 mg (46%). 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 9.60 (s, 1H), 8.78 (s, 1H), 8.64 (s, 1H), 8.01 (s, 1H), 7.97 (s, 1H), 7.67 – 7.61 (m, 2H), 7.45 – 7.36 (m, 3H), 7.31 – 7.17 (m, 3H), 6.98 (d, J = 8.0 Hz, 1H), 6.88 (t, J = 7.4 Hz, 1H), 5.45 (s, 1H), 4.42 (d, J = 6.0 Hz, 2H), 3.81 (s, 3H), 2.08 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.92, 163.45, 163.44, 157.21, 156.46, 152.27, 140.52, 138.43, 136.78, 133.18, 131.91, 130.14, 128.26, 127.88, 127.05, 126.29, 123.63, 122.51, 120.86, 119.99, 115.96, 110.34, 109.57, 104.91, 55.20, 54.28, 37.75, 16.99. HPLC purity: 95%; MS (ESI+) m/z: 528.8 (M+1), 550.8 (M+Na+).
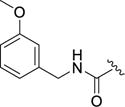
4-(4-Fluoro-3-((3-methoxybenzyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12f)
Compound 12f was prepared as described for 12a, replacing methylamine with (3-methoxyphenyl) methanamine. Yield: 74.3 mg (58%). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.58 (s, 1H), 8.82 (broad s, 1H), 8.76 (s, 1H), 7.98 (d, J = 16 Hz, 2H), 7.65 – 7.57 (m, 2H), 7.46 – 7.34 (m, 3H), 7.32 – 7.17 (m, 2H), 6.91 – 6.84 (m, 2H), 6.80 (dt, J = 8.2, 1.4 Hz, 1H), 5.45 (d, J = 2.8 Hz, 1H), 4.43 (d, J = 6.1 Hz, 2H), 3.72 (s, 3H), 2.07 (s, 3H). HPLC purity: 95%; MS (ESI+) m/z: 529.1 (M+1), 551.1 (M+Na+).
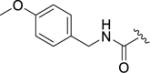
4-(4-Fluoro-3-((4-methoxybenzyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12g)
Compound 12g was prepared as described for 12a, replacing methylamine with 4-methoxybenzylamine. Yield: 10.7 mg (8%). 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 9.60 (s, 1H), 8.77 (s, 2H), 7.98 (d, J = 14.1 Hz, 2H), 7.63 (s, 1H), 7.60 (dd, J = 6.9, 2.3 Hz, 1H), 7.44 – 7.35 (m, 3H), 7.27 (d, J = 10.2 Hz, 1H), 7.22 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 5.44 (s, 1H), 4.37 (d, J = 6.0 Hz, 2H), 3.71 (s, 3H), 2.07 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.47, 163.86, 158.60, 157.43, 152.83, 138.93, 137.32, 133.74, 132.46, 131.57, 128.91, 128.73, 125.28, 124.22, 123.06, 121.40, 116.62, 116.39, 114.09, 110.80, 110.29, 105.47, 55.47, 54.81, 42.53, 17.55. HPLC purity: 99%; MS (ESI-) m/z: 527.0 (M−1).
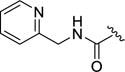
4-(4-Fluoro-3-((pyridin-2-ylmethyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12h)
Compound 12h was prepared as described for 12a, replacing methylamine with 2-(aminomethyl)pyridine. Yield: 41.2 mg (20%) 1H NMR (400 MHz, DMSO-d6) δ ppm 12.93, 9.59, 8.87, 8.77, 8.50, 7.98, 7.73, 7.69, 7.63, 7.46–7.37, 7.33–7.24, 5.46, 4.56, 2.08. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 9.64 (s, 1H), 8.91 (q, J = 5.3 Hz, 1H), 8.76 (d, J = 2.0 Hz, 1H), 8.48 (d, J = 4.3 Hz, 1H), 7.99 (s, 1H), 7.94 (s, 1H), 7.71 (td, J = 7.7, 1.8 Hz, 1H), 7.66 (dd, J = 6.9, 2.4 Hz, 1H), 7.62 (t, J = 2.4 Hz, 1H), 7.47 – 7.36 (m, 3H), 7.33 – 7.20 (m, 3H), 5.45 (d, J = 2.9 Hz, 1H), 4.53 (d, J = 5.9 Hz, 2H), 2.06 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.89, 164.024, 158.56, 157.90, 152.83, 149.28, 141.14, 138.93, 137.17, 133.74, 132.47, 130.94, 128.94, 123.77, 123.65, 122.55,121.43, 121.12, 116.71, 116.52, 110.83, 110.30, 105.49, 54.88, 45.15, 17.55. HPLC purity: 97%; MS (ESI+) m/z: 500.3 (M+1), 522.2 (M+Na+).
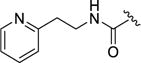
4-(4-Fluoro-3-((2-(pyridin-2-yl)ethyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide
Compound 12i was prepared as described for 12a, replacing methylamine with 2-pyridin-2-ylethylamine. Yield: 38 mg (20%). 1H NMR (400 MHz, DMSO) δ ppm 1H NMR (400 MHz, DMSO-d6) δ 12.92 (s, 1H), 9.58 (s, 1H), 8.77 (s, 1H), 8.47 (d, J = 4.0 Hz, 1H), 8.37 (dd, J = 8.2, 5.1 Hz, 1H), 7.97 (d, J = 13.9 Hz, 2H), 7.67 (td, J = 7.6, 1.8 Hz, 1H), 7.62 (s, 1H), 7.57 (dd, J = 6.9, 2.3 Hz, 1H), 7.43 – 7.34 (m, 3H), 7.26–7.15 (m 3H), 5.42 (d, J = 2.1 Hz, 1H), 3.57 (dd, J = 13.2, 6.9 Hz, 2H), 2.94 (t, J = 7.2 Hz, 2H), 2.05 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.91, 163.16, 159.33, 158.86, 156.86, 152.26, 148.93, 140.49, 138.32, 136.75, 136.34, 133.18, 131.90, 130.03, 128.17, 123.05, 122.50, 121.40, 120.84, 116.02, 110.238, 109.74, 104.93, 54.31, 36.90, 16.99. HPLC purity: 95%; MS (ESI+) m/z: 514.0 (M+1), 536.0 (M+Na+).
4-(4-Fluoro-3-((2-(pyridin-4-yl)ethyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide
Compound 12j was prepared as described for 12a, replacing methylamine with 2-(4-pyridyl)ethyl amine. Yield: 95.8 mg (51%). 1H NMR (400 MHz, DMSO) δ 12.94 (s, 1H), 9.59 (s, 1H), 8.78 (s, 1H), 8.45 (d, J = 5.5 Hz, 2H), 8.38 (s, 1H), 7.99 (d, J = 13.9 Hz, 2H), 7.63 (s, 1H), 7.55 (d, J = 4.7 Hz, 1H), 7.47 – 7.34 (m, 3H), 7.29 – 7.18 (m, J = 7.5 Hz, 3H), 5.44 (s, 1H), 3.50 (dd, J = 12.8, 6.6 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.48, 163.94, 152.83, 149.87, 148.68, 141.05, 138.95, 137.34, 133.75, 132.47, 130.63, 128.61, 124.50, 123.07, 121.42, 116.56, 116.37, 114.22, 110.82, 110.30, 105.48, 54.86, 39.47, 34.50, 17.55. HPLC purity: 99%; MS (ESI+) m/z: 514.1 (M+1), 536.1 (M+Na+).
4-(4-Fluoro-3-((isoquinolin-1-ylmethyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12k)
Compound 12k was prepared as described for 12a, replacing methylamine with isoquinolin1-ylmethylamine. Yield: 5.3 mg (4%). 1H NMR (400 MHz, DMSO-d6) δ ppm 12.92 (s, 1H), 9.58 (s, 1H), 8.87 (d, J = 5.7 Hz, 1H), 8.77 (s, 1H), 8.42 (d, J = 5.7 Hz, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 7.3 Hz, 2H), 7.95 (s, 1H), 7.83–7.73 (m, 3H), 7.69 (t, J = 7.6 Hz, 1H), 7.63 (s, 1H), 7.47–7.34 (m, 3H), 7.28 (t, J = 9.5 Hz, 1H), 5.46 (s, 1H), 5.13 (d, J = 5.0 Hz, 2H), 2.06 (s, 3H). MS (ESI+) m/z: 550.1 (M+1). HPLC purity: 98%.
4-(3-((2,6-Dichlorobenzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12l)
Compound 12l was prepared as described for 12a, replacing methylamine with 2,6-dichlorobenzylamine. Yield: 68.0 mg (49%). 1H NMR (500 MHz, DMSO-d6) δ 12.91 (s, 1H), 9.56 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.55 (t, J = 5.3 Hz, 1H), 7.98 (s, 1H), 7.95 (s, 1H), 7.61 – 7.59 (m, 1H), 7.52 (dd, J = 6.7, 2.4 Hz, 1H), 7.45 (s, 1H), 7.43 (s, 1H), 7.41 – 7.35 (m, 3H), 7.33 (dd, J = 8.5, 7.6 Hz, 1H), 7.18 (dd, J = 10.0, 8.7 Hz, 1H), 5.41 (d, J = 2.2 Hz, 1H), 4.70 – 4.61 (m, 2H), 2.05 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.90, 163.37, 152.28, 140.30, 138.50, 136.78, 135.55, 133.18, 132.88, 131.91, 130.07, 128.38, 128.02, 123.83, 123.71, 122.51, 120.88, 115.92, 115.73, 110.28, 109.71, 104.88, 54.20, 38.91, 16.99. MS (ESI+) m/z: 568.1 (M+1). HPLC purity: 97%;
4-(3-((2,6-Dimethylbenzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5 carboxamide (12m)
Compound 12m was prepared as described for 12a, replacing methylamine with 2,6-dimethylbenzylamine. Yield: 82.9 mg (65%). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.58 (s, 1H), 8.76 (d, J = 1.6 Hz, 1H), 8.41 (t, J = 4.6 Hz, 1H), 7.99 (d, J = 18.7 Hz, 2H), 7.51 (dd, J = 6.7, 2.3 Hz, 1H), 7.43 – 7.34 (m, 3H), 7.19 (dd, J = 9.9, 8.7 Hz, 1H), 7.10 – 7.03 (m, 1H), 6.99 (d, J = 7.5 Hz, 2H), 5.43 (d, J = 2.4 Hz, 1H), 4.43 (t, J = 4.7 Hz, 2H), 2.32 (s, 6H), 2.07 (s, 3H). HPLC purity: 96%; MS (ESI+) m/z: 527.1 (M+1).
4-(3-((2,6-Dimethoxybenzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12n)
Compound 12n was prepared as described for 12a, replacing methylamine with 2,6-dimethoxybenzylamine. Yield: 86.0 mg (32%). 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 9.58 (s, 1H), 8.77 (d, J = 1.9 Hz, 1H), 8.03 – 7.93 (m, 3H), 7.62 (dt, J = 5.0, 2.4 Hz, 2H), 7.45 – 7.32 (m, 3H), 7.29 – 7.14 (m, 2H), 6.65 (d, J = 8.4 Hz, 2H), 5.43 (d, J = 2.9 Hz, 1H), 4.48 (d, J = 4.9 Hz, 2H), 3.77 (s, 6H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO) δ 164.89, 158.21, 152.26, 140.43, 138.40, 136.76, 133.68, 133.17, 131.91, 128.98, 128.59, 123.41, 123.30, 122.50, 120.85, 115.93, 115.74, 112.95, 110.24, 109.71, 104.95, 103.99, 55.68, 54.28, 32.25, 16.98. HPLC purity: 97%; MS (ESI+) m/z: 559.1 (M+1), 581.1 (M+Na+).
4-(3-((2,6-Bis(trifluoromethyl)benzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12o)
Compound 12o was prepared as described for 12a, replacing methylamine with 2,6 bis(trifluoromethyl)benzylamine. Yield: 82.5 mg (53%). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.57 (s, 1H), 8.76 (s, 1H), 8.61 (s, 1H), 8.10 (d, J = 8.0 Hz, 2H), 7.97 (d, J = 10.9 Hz, 2H), 7.80 (t, J = 8.0 Hz, 1H), 7.62 (s, 1H), 7.51 (dd, J = 6.7, 2.4 Hz, 1H), 7.45 – 7.34 (m, 3H), 7.19 (dd, J = 9.9, 8.6 Hz, 1H), 5.43 (d, J = 2.9 Hz, 1H), 4.73 – 4.59 (m, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.44, 163.55, 159.45, 157.54, 152.90, 140.89, 139.16, 137.34, 133.98, 133.69, 132.44, 130.09, 128.33, 125.24, 125.14, 123.05, 121.41, 116.50, 116.24, 110.96, 110.31, 105.40, 54.65, 37.73, 17.54. HPLC purity: 95%; MS (ESI+) m/z: 635.0 (M+1), 657.0 (M+Na+).
4-(3-((3,5-Bis(trifluoromethyl)benzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12p)
Compound 12p was prepared as described for 12a, replacing methylamine with 3,5 bis(trifluoromethyl)benzylamine. Yield:24.6 mg (16%). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.58 (s, 1H), 9.05 (s, 1H), 8.77 (s, 1H), 8.03 (s, 2H), 8.00 (d, J = 4.3 Hz, 2H), 7.95 (s, 1H), 7.65 – 7.59 (m, 2H), 7.46 – 7.28 (m, 4H), 5.44 (s, 1H), 4.64 (d, J = 5.9 Hz, 2H), 2.07 (s, 3H). HPLC purity: 95%; MS (ESI+) m/z: 635.0 (M+1), 657.0 (M+Na+).
4-(4-Fluoro-3-(((3-methylpyridin-2-yl)methyl)carbamoyl)phenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12q)
Compound 12q was prepared as described for 12a, replacing methylamine with (3-methylpyridin-2-yl)methanamine. Yield: 29.8 mg (24%). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.60 (s, 1H), 8.87 (broad s, 1H), 8.77 (s, 1H), 8.35 – 8.29 (m, 2H), 7.99 (d, J = 16 Hz, 2H), 7.65 – 7.61 (m, 2H), 7.47 – 7.36 (m, 3H), 7.30 (dd, J = 10.3, 8.5 Hz, 1H), 7.17 (d, J = 5.0 Hz, 1H), 5.46 (s, 1H), 4.43 (d, J = 5.9 Hz, 2H), 2.28 (s, 3H), 2.08 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 164.28, 157.75, 155.04, 152.83, 150.47, 147.76, 146.21, 139.12, 137.25, 133.59, 132.48, 131.28, 130.82, 128.71, 123.94, 123.07, 121.40, 116.63, 116.41, 110.74, 110.19, 105.43, 98.78, 54.74, 40.56, 17.45, 15.74. HPLC purity: 97%; MS (ESI+) m/z: 514.1 (M+1), 536.1 (M+Na+).
4-(3-(2-((2,6-Dimethylbenzyl)amino)-2-oxoethyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (13a)
Compound 13a was prepared as described for 12a, replacing methylamine with 2,6-dimethylbenzylamine and 10 with 11. Yield: 52.3 mg (68%). 1H NMR (400 MHz, DMSO-d6) δ 12.98 (s, 1H), 9.61 (s, 1H), 8.70 (s, 1H), 8.15 (s, 1H), 7.99 (d, J = 21.7 Hz, 2H), 7.60 (s, 1H), 7.41 (s, 2H), 7.28 – 7.15 (m, 2H), 7.15 – 6.96 (m, 4H), 5.40 (s, 1H), 4.25 (s, 2H), 3.45 (s, 2H), 2.28 (s, 6H), 2.06 (s, 3H). 13C NMR (126 MHz, DMSO) δ 169.05, 165.61, 159.38, 152.90, 140.63, 138.38, 138.38, 137.69, 137.34, 135.06, 133.71, 132.53, 130.25, 128.34, 127.62, 123.83, 121.47, 115.41, 110.85, 110.25, 109.99, 105.84, 55.02, 37.86, 35.77, 19.83, 17.52. HPLC purity: 95%
4-(3-(2-((2,6-Bis(trifluoromethyl)benzyl)amino)-2-oxoethyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (13b)
Compound 13b was prepared as described for 13a, replacing 2,6-dimethylbenzylamine with 2,6 – bis(trifluoromethyl)benzylamine. Yield: 15.9 mg (17%). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.55 (s, 1H), 8.68 (d, J = 2.0 Hz, 1H), 8.35 (t, J = 3.9 Hz, 1H), 8.13 (d, J = 8.0 Hz, 2H), 8.00 (d, J = 1.4 Hz, 1H), 7.96 (s, 1H), 7.82 (t, J = 8.0 Hz, 1H), 7.58 (t, J = 2.5 Hz, 1H), 7.44 – 7.36 (m, 2H), 7.26 – 7.16 (m, 2H), 7.10 (t, J = 7.5 Hz, 1H), 5.40 (d, J = 2.9 Hz, 1H), 4.48 (d, J = 3.7 Hz, 2H), 3.43 (s, 2H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.04, 165.15, 158.92, 152.44, 140.10, 137.99, 136.88, 134.15, 133.24, 132.05, 130.872, 130.03, 126.54, 124.68, 123.01, 122.56, 121.03, 114.92, 114.75, 110.42, 109.77, 105.34, 54.52, 38.57, 36.23, 17.04. HPLC purity: 95%
4-(3-(2-((2,6-Dimethoxybenzyl)amino)-2-oxoethyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (13c)
Compound 13c was prepared as described for 13a, replacing 2,6-dimethylbenzylamine with 2,6 – dimethoxybenzylamine. Yield: 30.2 mg (56%). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.56 (s, 1H), 8.67 (d, J = 2.0 Hz, 1H), 7.98 (d, J = 20.4 Hz, 2H), 7.70 (t, J = 4.6 Hz, 1H), 7.57 (s, 1H), 7.45 – 7.35 (m, 2H), 7.27 – 7.15 (m, 3H), 7.09 (t, J = 9.1 Hz, 1H), 6.64 (d, J = 8.4 Hz, 2H), 5.40 (s, 1H), 4.24 (d, J = 4.6 Hz, 2H), 3.74 (s, 6H), 3.42 (s, 2H), 2.06 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.73, 165.52, 159.29, 158.82, 152.86, 140.57, 138.21, 137.35, 133.68, 132.46, 130.37, 129.42, 126.78, 123.86, 123.00, 121.51, 115.26, 113.65, 110.81, 110.16, 105.92, 104.46, 56.10, 54.77, 35.41, 32.35, 17.22. HPLC purity: 97%
4-(3-((2,6-Dimethylphenethyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (12r)
Compound 12r was prepared as described for 12a, replacing methylamine with 2-(2,6-dimethylphenyl)ethanamine. (Yield: 89.7 mg, 93%). 1H NMR (500 MHz, DMSO-d6) δ 12.93 (s, 1H), 9.59 (s, 1H), 8.77 (s, 1H), 8.50 (dd, J = 8.4, 5.6 Hz, 1H), 7.98 (d, J = 19.6 Hz, 2H), 7.66 (dd, J = 7.0, 2.3 Hz, 1H), 7.63 (broad s, 1H), 7.45–7.36 (m, 3H), 7.26 (dd, J = 10.3, 8.6 Hz, 1H), 6.98 (d, J = 1.1 Hz, 3H) 5.45 (d, J = 1.8 Hz, 1H), 3.32 – 3.23 (m, 2H), 2.82 (dd, J = 10.0, 6.5 Hz, 2H), 2.33 (s, 6H), 2.08 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.49, 163.73, 157.76, 152.81, 141.14, 138.84, 137.35, 136.67, 136.04, 133.73, 132.45, 130.78, 128.91, 128.33, 126.37, 124.01, 123.07, 121.45, 116.50, 110.88, 110.28, 105.53, 54.94, 38.91, 30.03, 19.84, 17.54. HPLC purity: 97%; MS (ESI+) m/z: 541.1 (M+1), 563.1 (M+Na+).
2,2,2-Trifluoro-N-(1H-indazol-5-yl)acetamide (14)
To a dry 100mL flask at 0 °C was added 5-amino-1H-indazole (2.0g, 15.02 mmol) followed by THF (40 mL) and triethylamine (3.36 mL, 24.03 mmol). Then trifluoroacetic anhydride (2.54 mL, 18.02 mmol) was added dropwise. The reaction warmed to room temperature and stirred for two hours. Using water the reaction was quenched then diluted with ethyl acetate and brine. After separating the layers the aqueous layer was extracted 1x with EtOAc. The organic layers were combined and then washed with NaCl 2x, dried over MgSO4, and concentrated to give as a slightly red solid 14 with > 95% purity by HPLC (3.302 g, 96% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 11.26 (s, 1H), 8.12 (d, J = 6.3 Hz, 2H), 7.62 – 7.47 (m, 2H).
Tert-butyl 5-(2,2,2-trifluoroacetamido)-1H-indazole-1-carboxylate (15)
THF (40 mL) was added to a 250 mL flask with 14 (3.30 g, 14.40 mmol) at 0 °C. Then 4-dimethylaminopyridine (0.176g, 1.44 mmol) and N,N-diisopropylethylamine (4.28 mL, 24.48 mmol) were added, followed, lastly, by boc anhydride (4.71g, 21.60 mmol). The reaction was allowed to warm to room temperature and stir 1.5 hours. Water was added to quench the reaction. The reaction was then extracted with ethyl acetate 2x, washed with NaCl 2x, dried over MgSO4, and concentrated to give 15 as a white powder. Took as is to next step without further purification.
Tert-butyl 5-(2,2,2-trifluoro-N-methylacetamido)-1H-indazole-1-carboxylate (16)
To a 250 mL flask at 0 °C was added sodium hydride (60% on mineral oil) (1.02g, 25.51 mmol) and DMF (15mL). Then 15 (5.60g, 17.01 mmol) in DMF (15 mL) was added. The reaction bubbled and turned green. Once bubbling ceased ~15 minutes, methyl iodide (1.38 mL, 22.11 mmol) was added dropwise at 0 °C and the reaction was stirred for two hours. The reaction was then quenched slowly with water (10 mL). Diluted further with water, extracted with EtOAc 2x, washed organic layers with NaCl 2x, dried over MgSO4, and concentrated to give as an amorphous solid 16. Took as is to next step with no further purification.
N-Methyl-1H-indazol-5-amine (17)
To a 250 mL flask was added 16 (17.01 mmol, 5.84g), potassium carbonate (85.05 mmol, 11.75g), methanol (30 mL), and water (15 mL). The reaction was heated to 85 °C and stirred overnight with a condenser. Cooled down reaction and neutralized with 1N HCl. Extracted the aqueous layer with ethyl acetate 2x, washed with NaCl 2x, dried over MgSO4, and concentrated. Purified the residue with normal phase using 30% EtOAc/Hexanes to give 17 as a white solid (1.189 g, 56% over three steps). 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H), 7.76 (s, 1H), 7.27 (d, J = 8.8 Hz, 1H), 6.79 (dd, J = 8.9, 2.1 Hz, 1H), 6.56 (s, 0H), 5.39 (q, J = 5.3 Hz, 1H), 2.68 (d, J = 5.2 Hz, 3H).
N-(1H-Indazol-5-yl)-N-methyl-3-oxobutanamide (18)
To a 50 mL pressure vessel was added 17 (0.800g, 5.44 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (0.78 mL, 5.98 mmol), and acetonitrile (7 mL). The reaction was then heated to 100 °C and stirred overnight. The reaction was then concentrated and purified using 2% MeOH/DCM to afford 18 as an off-white solid (1.25g, 98% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.25 (s, 1H), 8.11 (t, J = 1.3 Hz, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.25 (dd, J = 8.7, 1.9 Hz, 1H), 3.26 (s, 2H), 3.20 (s, 3H), 1.95 (s, 3H).
5-(5-((1H-Indazol-5-yl)(methyl)carbamoyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-4-yl)-2-fluorobenzoic acid (19)
To a 100 mL pressure vessel equipped with a stir bar was added 2-fluoro-5-formylbenzoic acid (0.800 g, 4.757 mmol), 18 (1.00 g, 4.324 mmol), urea (0.429 g, 7.135 mmol), ytterbium trifluormethanesulfonate (0.295 g, 0.476 mmol) and acetonitrile (15.0 mL). The reaction mixture was heated to 100 °C and stirred. The reaction mixture went from a white suspension to a clear solution. After four hours the reaction was not complete and was stirred overnight. The solvent was evaporated off and the resulting residue was then washed with water and purified using 20% MeOH (1%TFA)/DCM to give 19 as a white powder (140mg, 8% yield). 1H NMR (500 MHz, DMSO-d6) δ 13.17 (s, 2H), 8.44 (s, 1H), 8.03 (s, 1H), 7.64 (d, J = 6.9 Hz, 1H), 7.51 (d, J = 8.9 Hz, 1H), 7.39 – 7.31 (m, 2H), 7.24 (t, J = 9.2 Hz, 1H), 7.16 (s, 1H), 7.11 (d, J = 9.0 Hz, 1H), 4.73 (s, 1H), 3.14 (s, 3H), 1.75 (s, 3H).
4-(3-((2,6-Dimethylbenzyl)carbamoyl)-4-fluorophenyl)-N-(1H-indazol-5-yl)-N,6-dimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (20)
Compound 19 (0.140 g, 0.331 mmol), HATU (0.251 g, 0.661 mmol), DIEA (115.0 μL, 0.661 mmol) and 2,6-dimethylbenzylamine (0.089 g, 0.661 mmol) were added to a 10 mL round bottom flask with 3.0 mL DMF and stirred at room temperature overnight. The reaction was quenched with water and diluted with ethyl acetate. The organic layer was then washed 1x with brine, 1x with 10% citric acid, 1x with saturated NaHCO3, and then 2x with NaCl. The organic layer was then dried over MgSO4, concentrated, and purified using a gradient of 2–7% MeOH/DCM on flash chromatography to give, as a white solid, 20 (40.9 mg, 23% yield). 1H NMR (500 MHz, MeOD) δ ppm 8.07 (s, 1H), 7.60–7.56 (m, 2H), 7.50 (dd, J1=10.0 Hz, J2=0 Hz, 1H), 7.37 (bs, 1H), 7.24–7.01 (m, 6H), 4.63 (d, J=5.0 Hz, 2H), 3.24 (s, 3H), 2.41 (s, 6H), 1.93 (s, 3H). 13CNMR (500 MHz, MeOD) δ ppm 170.74, 166.64, 161.99, 159.50, 140.71, 138.80, 137.71, 134.67, 131.98, 131.89, 129.90, 129.44, 129.38, 128.87, 126.67, 125.04, 124.89, 124.35, 119.21, 117.70, 117.47, 112.18, 57.16, 39.76, 19.92, 17.03; HPLC: 89% purity; MS (ESI+) m/z: 541.2 (M+1).
N-(Benzo[d][1,3]dioxol-5-yl)-3-oxobutanamide (22)
To a 25 mL round bottom flask was added ethylacetoacete (0.775 mL, 6.08 mmol) and pyridine (0.881 mL, 10.94 mmol). The reaction was refluxed at 120 °C while stirring for three hours. 3,4-(methylenedioxy)aniline (1.0 g, 7.29 mmol) was then added in one portion and the reaction continued to reflux overnight at 120 °C. Cooled reaction down and concentrated in vacuo to give a black oil. Purified using a gradient of 15%-40% EtOAc/Hexanes to give 22 as a light brown crystalline solid (0.536g, 40% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 7.28 (d, J = 2.0 Hz, 1H), 6.93 (dd, J = 8.4, 2.0 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 5.98 (s, 2H), 3.50 (s, 2H), 2.20 (s, 3H).
5-(5-(Benzo[d][1,3]dioxol-5-ylcarbamoyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-4-yl)-2-fluorobenzoic acid (23)
Prepared as described for compound 10, replacing compound 9 with 22 (90% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 9.49 (s, 1H), 8.80 (s, 1H), 7.79 (d, J = 6.8 Hz, 1H), 7.64 (s, 1H), 7.47 (s, 1H), 7.28 (t, J = 9.7 Hz, 1H), 7.19 (s, 1H), 6.97 – 6.74 (m, 2H), 5.95 (s, 2H), 5.39 (s, 1H), 2.02 (s, 3H).
N-(Benzo[d][1,3]dioxol-5-yl)-4-(4-fluoro-3-((pyridin-2-ylmethyl)carbamoyl)phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (24)
To a 15 mL flask with DMF (5 mL) was added 23 (0.125 g, 0.300 mmol), 2-(aminomethyl)pyridine (0.062 mL, 0.600 mmol), HATU (0.228g, 0.600 mmol), and DIEA (0.105 mL, 0.600 mmol). The reaction was stirred overnight at room temperature then diluted with EtOAc and brine and separated. Washed organic layer with sodium carbonate 1x, then NaCl 1x, and concentrated. Purified the residue using a gradient of 5% – 8% MeOH/DCM to give compound 24 as a white solid (112 mg, 74% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 8.89 (td, J = 5.8, 3.1 Hz, 1H), 8.79 (d, J = 1.9 Hz, 1H), 8.55 – 8.47 (m, 1H), 7.76 (td, J = 7.7, 1.8 Hz, 1H), 7.68 – 7.61 (m, 2H), 7.40 (ddd, J = 7.8, 4.9, 2.4 Hz, 1H), 7.36 – 7.23 (m, 4H), 7.21 (d, J = 2.0 Hz, 1H), 6.93 (dd, J = 8.4, 2.1 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 5.94 (q, J = 1.0 Hz, 2H), 5.40 (d, J = 2.9 Hz, 1H), 4.56 (d, J = 5.9 Hz, 2H), 2.04 (s, 3H).13C NMR (101 MHz, DMSO) δ 164.71, 163.44, 158.00, 157.08, 152.21, 148.73, 146.74, 142.73, 140.50, 138.59, 136.62, 133.33, 130.31, 128.28, 123.23, 123.09, 122.00, 120.54, 116.19, 115.95, 112.38, 107.72, 104.74, 101.76, 100.74, 54.19, 44.58, 40.01, 39.80, 39.59, 39.38, 39.17, 38.96, 38.75, 16.97. HPLC purity: 97%; MS (ESI+) m/z: 504.0 (M+1), 526.0 (M+Na+).
Protein Purification
The S670A mutant of either bovine or human full length GRK2 containing a C-terminal hexahistidine tag was expressed in High Five cells using the Bac to Bac insect cell expression system (Life Technologies) and harvested 48 h post-infection. GRK2 was purified from clarified lysate using nickel-nitrilotriacetic acid affinity and cation exchange chromatography as described previously for GRK1535-His634 and subsequently gel filtered into 20 mM HEPES pH 7.5, 100 mM NaCl, and 1 mM DTT using a Sephadex 200 (S200) column. Soluble human Gβ1γ2 containing an N-terminally hexahistidine-tagged β subunit and the C68S mutation was expressed using a dual promoter insect cell expression vector (a gift from Dr. Brian Kobilka) in High Five cells and harvested 48 h post-infection. Gβ1γ2 (C68S) was purified from clarified lysate using nickel-nitrilotriacetic acid affinity, and anion exchange chromatography as described previously.35 Fractions containing Gβ1γ2 (C68S) were subsequently pooled and gel filtered into 20 mM HEPES pH 8.0, 100 mM NaCl, and 1 mM DTT using a S200 column. Purified proteins were concentrated to 10–12 mg/mL as determined by Bradford analysis in a 30kD cut-off Amicon Ultra-15 Centrifugal Filter Unit, flash frozen in liquid nitrogen, and stored at −80 °C until future use.
Crystal Structure Determination
For crystallization trials, GRK2 and Gβ1γ2 were mixed in a 1.2:1 molar ratio of GRK2: Gβγ with a final total protein concentration of 9–11 mg/mL. A final concentration of 500 μM inhibitor was added from a 25 mM stock in 100% DMSO. A final concentration of 2 mM MgCl2 was then added from a 120 mM stock. The complex was stored on ice for 30 min prior to filtration through a 0.2 μm Nanosep Centrifugal Device. All inhibitor complexes were crystallized at 4 °C by vapor diffusion using hanging drops consisting of 0.8 μL GRK2-Gβγ-inhibitor complex and 0.8 μL reservoir solution which contained 50 mM MES pH 6.0, 0.8–1.2 M NaCl, and 8–16% PEG 3350. Crystals appeared after three days and continued to grow for 1–2 wk. Crystals were harvested in a cryoprotectant solution containing the contents of the reservoir solution supplemented with 25% ethylene glycol and 500 μM inhibitor and were flash frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source on the LS-CAT beamline 21-ID-G at a wavelength of 0.97857 Å. Data reduction was performed using HKL2000.36 Phases for each of the structures were initially estimated using molecular replacement in the Phaser module of CCP437 with PDB ID 4PNK25 as the search model. Refinement was conducted by alternating between manual real-space refinement and reciprocal space refinement using both the Refmac5 module of CCP438 and the phenix.refine program of the PHENIX suite.39 Model building was performed using Coot.40 The final models were validated with MolProbity41 prior to deposition into the Protein Data Bank under accession codes 5HE0, 5HE1, 5HE2, and 5HE3.
Enzymatic Assays
PKA and ROCK1 inhibition assays were performed using the ADP-Glo Kinase Assay system (Promega) in accordance with the manufacturer’s recommendations and as described previously.25 Luminescence was measured using a BMG Labtech PHERAstar imaging system, and inhibition curves were analyzed using GraphPad Prism. Kinetic activity of the inhibitors with respect to the GRKs were run in a buffer comprised of 20 mM HEPES (pH 7.0), 2 mM MgCl2, and 0.025% DDM with 50 nM GRK and 500 nM tubulin. ATP (500 μCi, 5μM) was then added to initiate the kinetic reactions which were then run for 5 min, at which time SDS loading buffer was added to quench the reactions and they were then separated via SDS-PAGE. The resulting gels were dried using a vacuum gel drying system, and exposed with a phosphorimaging screen. The images were then scanned and quantification via Typhoon imager and Image Quant respectively, as previously reported.25, 26 Data were analyzed and inhibition curves were fit via GraphPad Prism with a three variable dose-inhibitor response curve and a fixed Hill slope of 1.
Myocyte Shortening Measurements
Cardiac myocytes were isolated from LV free wall and septum of C57/Bl6 mice as previously described.42 All cells were used within 2–8 h of isolation. Myocytes were plated on laminin-coated coverslips and were bathed in HEPES-buffered (20 mM, pH 7.4) medium 199 containing 1.8 mM extracellular Ca2+. When recording, coverslips containing myocytes were mounted in the Dvorak-Stotler chamber and bathed in 0.7 mL of fresh medium. Cells were paced at 1 Hz and imaged with a variable field-rate camera (Zeiss IM35, Ionoptix) by both edge detection and sarcomere length. Peak contraction was measured as the percentage of cell shortening. Cells were treated with isoproterenol (Iso, 0.5 μM) for 2 min for the recording of contraction, with pretreatment of either PBS as vehicle or paroxetine (10 μM), 215022 (0.1 μM, 0.5 μM, 1 μM, 10 μM), 215023 (0.1 μM, 0.5 μM, 1 μM, 10 μM), 224064 (0.1 μM, 0.5 μM, 1 μM, 10 μM), and GSK180736A (0.5 μM, 1 μM), for 10 min.22
Supplementary Material
Scheme 2.
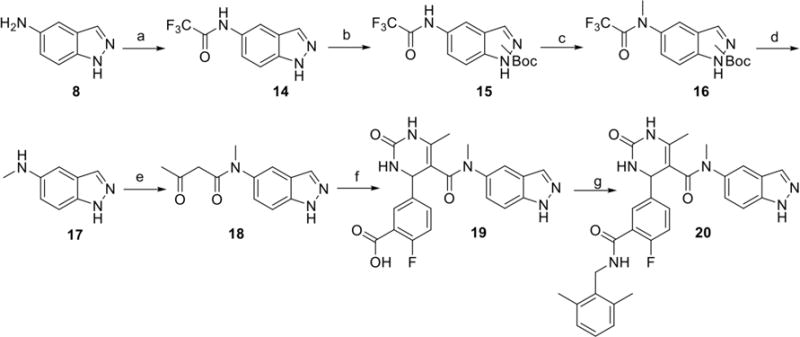
Reagents and conditions: a) TFAA, Et3N, Toluene, 0°C, b) Boc2O, DIEA, DMAP, THF, c) NaH, MeI, DMF, 0°C to rt, d) K2CO3, H2O, MeOH, reflux, e) 2,2,6-trimethyl-4H-1,3-dioxin-4-one, CH3CN, 100°C, f) 2-fluoro-5-formylbenzoic acid, Yb(OTf)3, urea, CH3CN, 100°C, j) HATU, DIEA, 2,6 – dimethyl benzylamine, DMF
Acknowledgments
This work was supported by NIH grants HL071818 and HL086865 (to J.T.), and American Heart Association grants N014938 (to K.T.H.) and 15PRE22730028 (to H.V.W.). Work at Temple (to W.J.K) was supported by NIH grants R37 HL061690, P01 HL075443 (Project 2), P01 HL108806 (Project 3) and P01 HL091799. J.T. and S.D.L. were supported by grants from the Center for Discovery of New Medicine, University of Michigan. M.C.C. and O.C. acknowledge training grant support from the University of Michigan Chemistry-Biology Interface (CBI) training program (NIH grant 5T32GM008597). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357, and use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant [085P1000817].
ABBREVIATIONS
- ASA
Accessible Surface Area
- GRK
G-protein coupled receptor kinase
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS publication website. Crystal refinement statistics, tabulation and comparison of Ki values, H-bonds, and buried surface area of select compounds, complete data for the cardiomyocyte contractility experiments at varying concentrations
Author Contributions
The manuscript was written primarily by H.V.W., M.C.C, J.J.G.T., and S.D.L. H.V.W. synthesized all compounds and performed biochemical assays. K.T.H. purified Gβγ, GRKs and PKA, performed biochemical assays, and crystallized and collected diffraction data for the GRK2–Gβγ·12k complex. O.C. conducted ROCK1 inhibition assays and trained M.C.C. M.C.C. and J.W.-S. purified and crystallized the GRK2–Gβγ complexes with 12h, 12n, and 12r, and collected diffraction data. M.C.C. and J.J.G.T. refined models for each crystal structure. M.C.C performed ROCK and PKA inhibition assays. A.C., J. S., J.Y.C and W.J.K. isolated adult cardiomyocytes from mice and performed contractility assays. All authors have given approval to the final version of the manuscript.
Accession Codes
5HE0 (GRK2-Gβγ·12h), 5HE1 (GRK2-Gβγ·12k), 5HE2 (GRK2-Gβγ·12n), and 5HE3 (GRK2-Gβγ·12i)
References
- 1.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart-failure. N England J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 2.Port JD, Bristow MR. Altered Beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol. 2001;33:887–905. doi: 10.1006/jmcc.2001.1358. [DOI] [PubMed] [Google Scholar]
- 3.Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- 4.Lefkowitz RJ, Stadel JM, Caron MG. Adenylate cyclase=coupled beta-adrenergic receptors – structure and mechanisms of activation and desensitization. Annu Rev Biochem. 1983;52:159–186. doi: 10.1146/annurev.bi.52.070183.001111. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland EW, Robison GA, Butcher RW. Some Aspects of the Biological Role of Adenosine 3′,5′-monophosphate (Cyclic AMP) Circulation. 1968;37:279–306. [Google Scholar]
- 6.Baameur F, Morgan DH, Yao H, Tran TM, Hammitt RA, Sabui S, McMurray JS, Lichtarge O, Clark RB. Role for the Regulator of G-Protein Signaling Homology Domain of G Protein-Coupled Receptor Kinases 5 and 6 in β2-Adrenergic Receptor and Rhodopsin Phosphorylation. Mol Pharmacol. 2010;77:405–415. doi: 10.1124/mol.109.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 8.Eschenhagen T. beta-adrenergic signaling in heart failure – adapt or die. Nat Med. 2008;14:485–487. doi: 10.1038/nm0508-485. [DOI] [PubMed] [Google Scholar]
- 9.Raake PW, Vinge LE, Gao EH, Boucher M, Rengo G, Chen XW, DeGeorge BR, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, Koch WJ. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta-1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 11.Satwani S, Dec GW, Narula J. β-Adrenergic Blockers in Heart Failure: Review of Mechanisms of Action and Clinical Outcomes. J Cardiovas Pharmacol Ther. 2004;9:243–255. doi: 10.1177/107424840400900404. [DOI] [PubMed] [Google Scholar]
- 12.Packer M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001;110:81–94. doi: 10.1016/s0002-9343(01)00676-3. [DOI] [PubMed] [Google Scholar]
- 13.Huang ZM, Gold JI, Koch WJ. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci Landmark Ed. 2011;16:3047–3060. doi: 10.2741/3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raake PWJ, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, Pleger S, Mier W, Haberkorn U, Koch WJ, Katus HA, Most P, Muller OJ. AAV6.beta ARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J. 2013;34:1437–1447. doi: 10.1093/eurheartj/ehr447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martini JS, Raake P, Vinge LE, DeGeorge BR, Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. PNAS. 2008;105:12457–12462. doi: 10.1073/pnas.0803153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW, Trimarco B, Iaccarino G, Koch WJ. G Protein–Coupled Receptor Kinase 2 Activity Impairs Cardiac Glucose Uptake and Promotes Insulin Resistance After Myocardial Ischemia. Circulation. 2011;123:1953–1962. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tesmer JJG, Tesmer VM, Lodowski DT, Steinhagen H, Huber J. Structure of Human G Protein-Coupled Receptor Kinase 2 in Complex with the Kinase Inhibitor Balanol. J Med Chem. 2010;53:1867–1870. doi: 10.1021/jm9017515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Setyawan J, Koide K, Diller TC, Bunnage ME, Taylor SS, Nicolaou KC, Brunton LL. Inhibition of protein kinases by balanol: Specificity within the serine/threonine protein kinase subfamily. Mol Pharmacol. 1999;56:370–376. doi: 10.1124/mol.56.2.370. [DOI] [PubMed] [Google Scholar]
- 19.Thal DM, Yeow RY, Schoenau C, Huber J, Tesmer JJG. Molecular Mechanism of Selectivity among G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol Pharmacol. 2011;80:294–303. doi: 10.1124/mol.111.071522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda S, K M, Fujiwara S, et al. WO2007034846. Cardiotonic agent comprising GRK inhibitor. 2007 Mar 29;
- 21.Mayer G, Wulffen B, Huber C, Brockmann J, Flicke B, Neumann L, Hafenbradl D, Klebl BM, Lohse MJ, Krasel C, Blind M. An RNA molecule that specifically inhibits G-protein-coupled receptor kinase 2 in vitro. RNA. 2008;14:524–534. doi: 10.1261/rna.821908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, Chuprun JK, Song J, Gao E, Cheung JY, Sklar LA, Koch WJ, Tesmer JJG. Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem Biol. 2012;7:1830–1839. doi: 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, GTesmer JJ, Koch WJ. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7:27731. doi: 10.1126/scitranslmed.aaa0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sehon CA, Wang GZ, Viet AQ, Goodman KB, Dowdell SE, Elkins PA, Semus SF, Evans C, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Wright LL, Srnith GK, Behm DJ, Bentley R, Doe CP, Hu E, Lee D. Potent, Selective and Orally Bioavailable Dihydropyrimidine Inhibitors of Rho Kinase (ROCK1) as Potential Therapeutic Agents for Cardiovascular Diseases. J Med Chem. 2008;51:6631–6634. doi: 10.1021/jm8005096. [DOI] [PubMed] [Google Scholar]
- 25.Homan KT, Larimore KM, Elkins JM, Szklarz M, Knapp S, Tesmer JJG. Identification and Structure–Function Analysis of Subfamily Selective G Protein-Coupled Receptor Kinase Inhibitors. ACS Chem Biol. 2014;10:310–319. doi: 10.1021/cb5006323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Homan KT, Waldschmidt HV, Glukhova A, Cannavo A, Song J, Cheung JY, Koch WJ, Larsen SD, Tesmer JJG. Crystal Structure of G Protein-Coupled Receptor Kinase 5 in Complex with a Rationally Designed Inhibitor. J Biol Chem. 2015;290:20646–20659. doi: 10.1074/jbc.M115.647370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sridharan V, Ruiz M, Menéndez JC. Mild and High-Yielding Synthesis of β-Keto Esters and β-Ketoamides. Synthesis. 2010:1053–1057. [Google Scholar]
- 28.Drewry DHE Brian, Goodman Krista B, Green Darren, Victor Steven, Jung David Kendall, Lee Denis, Stavenger Robert A, Wad Sjoerd Nocolaas, Indazolo-tetrahydropyrimidine-carboxamide derivative kinase inhibitors WO2004/112719. 2004 Dec 29; [Google Scholar]
- 29.Houlihan WJ, Cooke G, Denzer M, Nicoletti J. 1-Alkyl-4-aryl-3,4-dihydro-2(1H)-quinazolinones and thiones. Synthesis and 1H-NMR spectra. J Heterocycl Chem. 1982;19:1453–1456. [Google Scholar]
- 30.Jacobs M, Hayakawa K, Swenson L, Bellon S, Fleming M, Taslimi P, Doran J. The Structure of Dimeric ROCK I Reveals the Mechanism for Ligand Selectivity. J Biol Chem. 2006;281:260–268. doi: 10.1074/jbc.M508847200. [DOI] [PubMed] [Google Scholar]
- 31.Breitenlechner C, Gaßel M, Hidaka H, Kinzel V, Huber R, Engh RA, Bossemeyer D. Protein Kinase A in Complex with Rho-Kinase Inhibitors Y-27632, Fasudil, and H-1152P. Structure. 2003;11:1595–1607. doi: 10.1016/j.str.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Homan KT, Tesmer JJG. Molecular basis for small molecule inhibition of G protein-coupled receptor kinases. ACS Chem Biol. 2015;10:246–256. doi: 10.1021/cb5003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li R, Martin MP, Liu Y, Wang B, Patel RA, Zhu J-Y, Sun N, Pireddu R, Lawrence NJ, Li J, Haura EB, Sung S-S, Guida WC, Schonbrunn E, Sebti SM. Fragment-Based and Structure-Guided Discovery and Optimization of Rho Kinase Inhibitors. J Med Chem. 2012;55:2474–2478. doi: 10.1021/jm201289r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJG. Structures of Rhodopsin Kinase in Different Ligand States Reveal Key Elements Involved in G Protein-coupled Receptor Kinase Activation. J Biol Chem. 2008;283:14053–14062. doi: 10.1074/jbc.M708974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozasa T. Purification of G Protein Subunits from Sf9 Insect Cells Using Hexahistidine-Tagged α and βγ Subunits. In: Smrcka A, editor. G Protein Signaling. Vol. 237. Humana Press; 2004. pp. 21–38. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Methods in Enzymology. Vol. 276. Academic Press; 1997. [20] Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song J, Zhang X-Q, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange versus Na+-K+-ATPase. Am J Physiol: Heart and Circulatory Physiology. 2008;295:H1615–H1625. doi: 10.1152/ajpheart.00287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Homan KT, Wu E, Wilson MW, Singh P, Larsen SD, Tesmer JJG. Structural and Functional Analysis of G Protein–Coupled Receptor Kinase Inhibition by Paroxetine and a Rationally Designed Analog. Mol Pharmacol. 2014;85:237–248. doi: 10.1124/mol.113.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.