

Abstract
We describe a general synthetic strategy for developing high affinity peptide binders against specific epitopes of challenging protein biomarkers. The epitope of interest is synthesized as a polypeptide, with a detection biotin tag and a strategically placed azide (or alkyne) presenting amino acid. This synthetic epitope (SynEp) is incubated with a library of complementary alkyne or azide presenting peptides. Library elements that bind the SynEp in the correct orientation undergo the Huisgen cycloaddition, and are covalently linked to the SynEp. Hit peptides are tested against the full-length protein to identify a best binder. We describe epitope-targeted linear or macrocycle peptide ligands against 12 different diagnostic or therapeutic analytes. The general epitope targeting capability for these low molecular weight synthetic ligands enables a range of therapeutic and diagnostic applications, similar to those of monoclonal antibodies.
Keywords: Click chemistry, Combinatorial chemistry, Macrocyclic ligands, Metathesis, Peptides
Graphical abstract
Communication: Match made in situ. We describe development of 12 peptide ligands against distinct epitopes of challenging proteins. This development is enabled by a general synthetic strategy. A synthetic epitope with an azide is screened against an alkyne containing macrocyclic library to identify the best binder. These synthetic ligands can enable antibody like therapeutic and diagnostic applications, with a small molecular footprint.
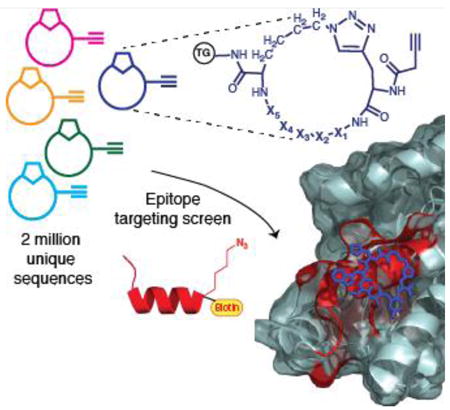
Monoclonal antibodies (mAbs) raised against a short peptide (∼10 amino acids) antigen (epitope)[1] can target protein regions inaccessible to traditional small molecule binders of proteins, which bind in well defined binding pockets. This capability has made mAbs the therapeutic platform of choice for any extracellular or membrane targets that they can readily access[2], as well as the only diagnostic reagents which can detect targets such as enzymes that are phosphorylated at specific residues[3]. Crystal structures of antibody- antigen complexes often reveal direct contacts of the antigen with ∼10 residues in the variable heavy CDR loop region, while the rest of the antibody acts as a semi-rigid scaffold holding the right residues in the correct orientation[4]. This chemical model guides our approach: we target a relatively small fragment of the protein, but we also screen that fragment against different libraries designed to explore ligand scaffold rigidity.
Our strategy mirrors that used for developing epitope targeted mAbs, but yields peptide based Protein Catalyzed Capture (PCC) agents[5] with small molecular footprints (∼1 kD). A key strategy element is in situ click chemistry, which has been demonstrated as a powerful tool for the target guided synthesis of small molecule and peptide ligands for proteins.[6] We synthesize a comprehensive One-Bead-One-Compound (OBOC) library[7] of 5-mer linear or macrocyclic peptides against a synthetic epitope (SynEp), which is a 10 to 30 amino acids long peptide representing a modified variant of the epitope of interest. The library elements are designed to present an azide (or alkyne) click handle, and a complementary alkyne (or azide) presenting amino acid is strategically substituted into the SynEp. During a screen, a library element that interacts with the SynEp in the right orientation, undergoes 1,3-dipolar cycloaddition to covalently bond to the epitope. The precise orbital alignment of terminal alkynes and azides required for the cycloaddition increases the entropic penalty of orientation, making the un-catalyzed reaction non-spontaneous under ambient conditions. This limitation is exploited here: we rely on specific interactions between the SynEp and the library element to overcome this entropic penalty so that the reaction proceeds un-catalyzed.
We provide a detailed description of the screening process and demonstrate its generality through the identification of 12 epitope targeted PCC agents. These ligands fulfill very challenging targeting aims such as selective detection of a phosphorylated epitope[8], a single amino acid point mutation[9], and detection of sequences within malarial protein biomarkers that distinguish specific species of the Plasmodium genus or, for a different malarial biomarker, small regions of the protein that are geographically conserved. The development of the PCC agents against the malarial biomarker proteins are elaborated to illustrate the technique. Macrocyclic peptide libraries have yielded superior performing PCC agents, and so are described in detail.
The various proteins and epitopes targeted, along with the amino acid sequence of the best PCC binder, are given in Table 1. The SynEp is a 9-30 amino acid long fragment of the target protein. In general, PCCs developed against shorter (9-12-mer) epitopes and longer (20-30-mer) SynEps have similar affinity and selectivity.
Table 1. Epitope targeted PCC Agents.
| Protein and description | SynEp (corresponding residues in full length protein) | Binder Sequence | EC50 or KD (bold) |
|---|---|---|---|
| I. PfLDH Plasmodium falciparum lactate dehydrogenase (malaria diagnostics) | LISDAELEAIFD-Az4 – PEG5- Biotin (amino acids 218-229 of PfLDH) | Cy(HWSAN-click) | 23.4 nM (Fig S1) 40.6 nM (Fig 2D) |
| II. PxLDH Plasmodium (x = many species) lactate dehydrogenase (general malaria diagnostics) | Biotin-W-PEG5-GVEQV-Pra-ELQLN (amino acids 297-308 of PxLDH) | hevwh (Li) | 1.7 μM (Fig S2) |
| III. PfHRP2 Plasmodium falciparum histidine rich protein-2 (malaria diagnostics) epitope A (C terminal of PfHRP-2) | AHHATDAHHAAAHHEAATHC -Pra – PEG5- Biotin | Cy(GSTEWL-rcm) | 20 nM (Fig S3) 54.2 nM (Fig 2H) |
| IV. PfHRP2 epitope B (variant of type 2 repeat of PfHRP2) | Biotin-PEG5–AHHAADAHHA–Pra | Cy(Y-4F-Phe-YRV-click) | 538 pM (Fig S4) |
| V. PfHRP2 epitope C (type 2 repeat of PfHRP2) | Biotin-PEG5-AHHAHHAAD-Pra | Cy(YKYYR-click) | 218 nM (Fig S5) |
| VI. PfHRP2 epitope D (N terminal repeat of PfHRP2) | Biotin-PEG5-LHETQAHVDD-Pra | Cy(RYKHY-click) | 4 nM (Fig S6) |
| VII. L1R vaccinia virus L1R myristyl protein (closely related to variola) | Biotin-PEG5–KALMQLTTKATQIA-Pra-PKQVAGTGVQ | Cy(DARNI-click) | 875 nM (Fig S7) |
| VIII. IL17-F (Interleukin member of the highly homologous IL-17 family) | Biotin-PEG3-FFQ[Az4]PPVPGGS (amino acids 40-54 of IL17-F) | Cy(RRATS-click) | 66 nM (Fig S8) |
| IX. p-Akt2 (phospho serine / threonine kinase; strategy targets p-Ser474 epitope) | Biotin-PEG5-ITPPDRYDSLGLL ELQRTHFPQF(pS)YSASIRE (amino acids 450-481 of pAkt2) | Cy(YYTYT-click) | 122 nM (Fig S9) |
| X. p-Akt2 (Protein kinase B2; strategy targets adjacent to p-Ser474 epitope)[8] | ITPPDRYDSLGLLELQRTHFPQF[pS-(Zn2L)]YSASIRE (amino acids 450-481 of pAkt2) | wkvkl (Li) | 3.6 μM |
| XI. Akt1E17K (Protein kinase B with oncogenic point mutation)[9] | Biotin-PEG5-PEVAIVKEGWLKKRGK Y[Pra]KT WRPRYFLLKNDG | yleaf (Li) | 61 nM 54 nM |
| XII. BoNT A LC (Botulinum neurotoxin serotype A light chain)[10] | Az4-SFGHEVLNLTRN-PEG4-Biotin (amino acids 166-179) | Cy(NYRWL-click) | 68 nM 70 nM |
Table 1 description. For each entry, the sequence of the SynEp is given, indicating the substitution position for the click handle and the biotin handle, and, in parentheses, the position of the epitope in the full-length protein. Macrocycle PCCs are indicated by the prefix ‘cy’, while linear PCCs are indicated by the suffix (Li). Macrocycles closed using triazoles or metathesis chemistry are indicated by ‘click’ or ‘rcm.’ PCC IX is obtained through a variation of the epitope targeting strategy. PCC X was developed by screening against the epitope chelated to a dinuclear zinc ligand tagged with an azide handle and biotin.
The SynEp is prepared with a terminally-appended biotin assay label, as well as the click handle substitution, which can be appended at the C or N terminus of the SynEp (Table 1, entries IV-VI), or it can be substituted for specific natural residues.
We have replaced arginine and lysine residues with Az4 (Table 1, entries I, VIII, XII) and leucine and isoleucine with Pra (Table 1, entries II, III, VII, XI). To develop binders that detect single point mutations (Akt1 E17K) or a post-translational modification (Akt2 pS474), separating the click handle by 3-4 residues from the key residue is an effective strategy.
The epitope targeted in situ click screen is a single generation screen, with results that are filtered through one or more anti-screens. The OBOC peptide libraries,[7] which are comprehensive in 18 amino acids (∼2 million sequences), are screened against a biotin tagged scrambled sequence of the same length as the SynEp, or an off target peptide representing a different epitope of the same protein (Table S1). Non-specific binders from the anti-screen are identified colorimetrically by treatment of the screened library with anti-biotin mAb - alkaline phosphatase (anti-biotin-AP) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP). Scheme 1 illustrates a screen such as that used against PfLDH (Table 1, Entry I). The binders against a scrambled epitope are eliminated, and the rest of the library is washed with protein denaturizing solutions. After re-equilibration in the screening buffer, the pre-cleared library is incubated with the SynEp, which, for PfLDH, was based upon a unique falciparum-specific sequence at residues 218 – 229 (Table 1). For this screen, the library is washed with protein denaturizing solution to remove any non-covalently bound SynEp, prior to enzymatic development to identify hit beads. The hit beads (typically about 2-5 per 106 screened peptides) are synthesized in bulk without the terminal alkyne, and tested for binding to full-length protein and, for the case of PfLDH, to the targeted epitope (without the click handle) and to non-falciparum variants of LDH. The best binder (Table 1 and Scheme 1) is selected based on these and similar selectivity and affinity assays.
Scheme 1. An epitope targeted in situ click screen.

A (macrocycle) peptide library is first screened against a scrambled variant of the SynEp. The library elements that bind to that variant are detected by utilizing the biotin label (yellow) to execute an enzymatic assay that changes the color of reactive beads. The remaining library is washed, resuspended in buffer, and screened against the target SynEp, and again thoroughly washed to remove non-covalently bound copies of the SynEp. Following treatment with anti-biotin-AP and its BCIP substrate, hit beads are picked for sequencing. Candidate ligands are tested against the full-length protein to identify the best binder.
A result from Table 1, and from previous work[5], is the superior performance of the macrocycles relative to the linear PCCs. This is anticipated[11]. Macrocycles yield an average –log[KD (or EC50] value of >7, while for linear PCCs, that value is <6. The macrocyclic libraries used here are designed for these screens, and so we turn to a discussion of those libraries.
Macrocyclic peptide libraries prepared using phage display are typically cyclized through a disulfide linkage originating from two cysteine residues[12], and are susceptible to a number of physical and (bio)chemical processes [13] which can confound screening results. Peptide sequences from such libraries are obtained via DNA sequencing. We sought to develop a macrocycle library which was stably cyclized, and which could be sequenced using standard methods such as Edman degradation. We use the Cu(I)-promoted alkyne/azide Cycloaddition (CuAAC) reaction[14] and the Ru(IV) catalyzed Ring-Closing Metathesis reaction (RCM)[15] to create OBOC macrocyclic libraries on tentagel (TG) beads for sequencing via Edman degradation. CuAAC cyclized libraries start with the synthesis of a linear library Pra-X1X2X3X4X5-L-Az4-TG using standard Solid Phase Peptide Synthesis (SPPS). X1-X5 comprises the variable region and artificial amino acids are readily incorporated. The library is subjected to the CuAAC reaction (Fig 1). The 4-carbon side chain of Az4 on the C-terminal is optimal for intramolecular cyclization with Pra at the N-terminus. Alkyne containing amino acids at the N-terminus give higher cyclization yields[16]. A dimethylformamide (DMF) solution of sodium diethyldithiocarbamate is used to remove the adsorbed Cu(I) from the library. IR spectroscopy is used to verify on bead cyclization for one representative case. The uncyclized peptide has an asymmetric NNN stretch (∼ 2100 cm-1), while the isobaric cyclized peptide does not (Figure S10). The CuAAC reaction typically yields monomer as the major product, with small amounts of dimer and trimer formation (Figure S11).
Figure 1. Synthesis and sequencing of a CuAAC cyclized peptide library.
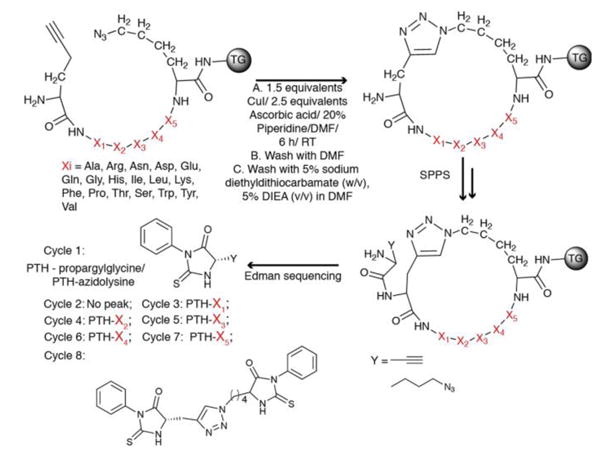
A linear peptide library (top left) is subjected to Cu catalyzed Azide Alkyne Cycloaddition (CuAAC) reaction. An azide or alkyne containing amino acid is incorporated after the cycloaddition and its protecting group removed, following standard SPPS protocol. The library is treated with a TFA cocktail to remove side chain protecting groups and stored in buffer. For sequencing, a single bead is loaded into a cartridge of an Edman Peptide Sequencer. The first cycle yields the phenylthiohydantoin (PTH) derivative of the azide or alkyne handle containing amino acid. The next Edman cycle yields no peak as it cleaves the amide bond forming the cycle but the PTH derivative is bound to the resin. Successive cycles yield PTH derivatives of component amino acids.
For RCM stapled peptides, a linear library of the form R8-GX1X2X3X4X5-S5-linker-TG, (where R8 =(R)-2-(7′-octenyl)alanine, S5 = (S)-2-(4′-pentenyl)alanine) is synthesized. On bead peptides are cyclized using 1st generation Grubb's catalyst[15] in dichloroethane (DCE), twice for 6 hours under argon. Excess catalyst is removed by washing with 5% triphenyl phosphine in dichloromethane (DCM) and 5% oxine solutions in DMF (Figure S12). The amino acid pair R8 and S5 are chosen based upon a literature report[17]. Following RCM cyclization of the linear peptides, High Pressure Liquid Chromatography (HPLC) purification shows the major peak either as the monomer (Figure S13A), or as the dimer with traces of monomer (Figure S13B). This is confirmed via mass spectral analysis. The relative yields of the cyclic monomer or dimer appear to be dependent upon the position of the PEG linker and biotin handle (Figure S13C).
Single beads are sequenced by Edman degradation (Figures S14, S15), each yielding the characteristic pattern described in Fig. 1 (bottom left).
We describe PCCs developed for binding to the Plasmodium falciparum-specific malarial biomarker proteins PfHRP2 and PfLDH. Precise epitope targeting can significantly help to identify reagents for the universal, selective capture of these two proteins. For example, PfLDH has high sequence homology with PxLDH, where x represents malaria species other than falciparum. PfHRP2 is an unstructured protein that exhibits broad sequence variations across the geographic regions in which the parasite is found[18]. The few regions of PfHRP2 that are conserved are represented by short sequences.
PfLDH is distinguished from the 90% homologous PvLDH (v=vivax) at residues 218-229[19],[20]. The epitope (highlighted in blue, Fig. 2A), screened according to Scheme 1, yields six ligand sequences (Table S2). The best binder is chosen based on binding to both the epitope (Fig. S16), and the full-length protein (Fig. S17). The molecular structure of the best binder 1, and associated single point ELISA assay results showing selectivity, are provided in Figures 2B and 2C. Biotin tagged 1 is titrated against 100 nM recombinant Glutathione-S-Transferase (GST) tagged PfLDH, PvLDH and hLDH (h=human), to test selectivity. Subsequent treatment with an anti-GST antibody - Horse Radish Peroxidase conjugate (anti-GST-HRP) and a chromogenic substrate shows selective binding of 1 to the PfLDH. The fluorescence polarization (FP) of fluorescein tagged 1 against PfLDH yields a KD of 40.6 ± 9.3 nM (Fig 2D). The KD value is close to the the EC50 value of 23.4 ± 6.4 nM (Figure S1).
Figure 2. Two distinct cases of epitope targeting.
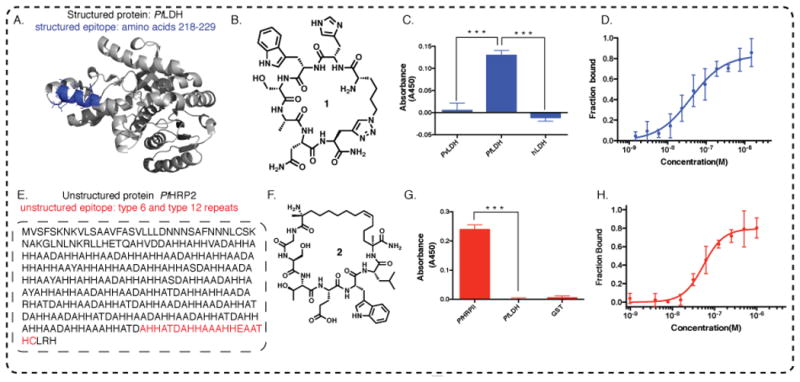
A. A region from the structured PfLDH protein that uniquely distinguishes PfLDH relative to other LDH variants, is highlighted in blue. B. The molecular structure of 1, the consensus PCC developed to bind to the blue highlighted epitope of PfLDH; C. Peptide 1 selectively distinguishes PfLDH from PvLDH (v=vivax) and hLDH (h=human) in single point ELISA (proteins spiked into buffer solution at 100 nM concentration). D. Fluorescein tagged 1 is titrated with recombinant PfLDH and the fluorescence polarization is measured, yielding a KD of 40.63 ± 9.27 nM. E. Targeting a universally conserved epitope of intrinsically unstructured PfHRP2 protein. One recombinant (r) PfHRP2 amino acid sequence is shown. The epitope targeted is highlighted in red. F. Molecular structure of 2, the consensus PCC developed to bind to the highlighted epitope from PfHRP2. G. Selective binding of 2 to 20 nM GST-PfHRP2, showing negligible cross-reactivity with 20 nM GST-PfLDH, a related target from Plasmodium falciparum species, and with 20 nM GST protein; H. Peptide 2 binds to recombinant PfHRP2 with a KD of 54.26 ± 12.05 nM, as measured by fluorescence polarization.
PfHRP2, which is localized in the cytoplasm and at the cell membrane of infected erythrocytes, is secreted into the serum by erythrocytes[21] and is a Pf specific biomarker for both acute and chronic falciparum infection[22]. PfHRP2 consists of varying numbers of repeats of alanine and histidine rich motifs (Fig. 2E)[18]. The type 6 repeat (sequence AHHATD) and the type 12 repeat (sequence AHHAAAHHEAATH) of PfHRP2 are near universally conserved[18]. Thus, we developed a PCC against a SynEp representing the C-terminus of the protein and containing both conserved regions (Table 1, Entry III). The molecular structure of the consensus PCC (see Table S3, Fig. S18, Fig. S19 for tested candidates), is depicted in Fig. 2F. The biotin tagged ligand shows no cross-reactivity with a related LDH protein from the same Plasmodium falciparum species (Fig. 2G), and binds selectively to rPfHRP2 in 1% serum (Fig. S20). The ligand was titrated with varying concentrations of the GST tagged rPfHRP2 and treated with anti-GST-HRP to obtain an EC50 of 20.5 ± 2 nM (Fig. S3), which is close to the KD (54.3 ± 12.1 nM) determined by FP (Fig. 2H).
The reported epitope targeting strategy is conceptually similar to the approach used for developing monoclonal antibodies, in that PCCs can apparently target most parts of the protein, and don't require hydrophobic binding pockets. PCC reagents, however, maintain many advantages of small molecules. A primary consideration in developing the PCCs of Table 1 was the value of the targeted epitope within the context of the ultimate application. For example, the ability to target conserved regions of otherwise genetically varying proteins, or regions of protein targets that differentiate those targets from close analogues, makes this approach a compelling strategy for many challenging diagnostic targets. Depending upon the final required metrics, the PCCs of Table 1 may be viewed as starting point, similar to how an initial small molecule drug candidate is viewed. Both types of ligands can be improved via chemical iterations to improve avidity and/or stability characteristics.[23]
A limitation of the approach described here is that libraries above a few million in size are difficult to manage. While other ‘artificial antibody’ approaches (add 2 refs given at end) can support larger libraries, the synthetic flexibility used here to enable the in situ click screen, or to incorporate non-natural amino acids, is tough to achieve with biological methods. Epitope targeting could likely be done with aptamers or peptide display methods, but the advantage of the in situ click screen in directly identifying hits via the formation of an on-bead reaction product (rather than a simple binding assay) is significant and yields viable binders from a single generation screen. However done, the general ability to target specific epitopes on specific proteins is enabling. For example, the broad flexibility of the epitope targeting strategy is already permitting exploitation of the tertiary structure of drug targets as a scaffold for developing highly potent inhibitors[10].
Supplementary Material
Acknowledgments
The various PCCs and methods reported here were developed under funding from the Bill and Melinda Gates Foundation, the Institute for Collaborative Biotechnologies (W911NF-09-0001) from the U.S. Army Research Office, the Defense Advanced Research Projects Agency (DARPA) through the Cooperative Agreement HR0011-11-2-0006, the Jean Perkins Foundation, and that National Cancer Institute through grant #5U54 CA119347 (JRH PI).
References
- 1.Hancock DC, OReilly NJ. Immunochemical Protocols. Humana Press; 2005. pp. 13–25. [Google Scholar]
- 2.Chames P, Van Regenmortel M, Weiss E, Baty D. Brit J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. Structure. 1998;6:1153–1167. doi: 10.1016/s0969-2126(98)00116-6. [DOI] [PubMed] [Google Scholar]; b) Thomas GV, Horvath S, Smith BL, Crosby K, Lebel LA, Schrage M, Said J, De Kernion J, Reiter RE, Sawyers CL. Clin Cancer Res. 2004;10:8351–8356. doi: 10.1158/1078-0432.CCR-04-0130. [DOI] [PubMed] [Google Scholar]
- 4.a) Robin G, Sato Y, Desplancq D, Rochel N, Weiss E, Martineau P. J Mol Biol. 2014;426:3729–3743. doi: 10.1016/j.jmb.2014.08.013. [DOI] [PubMed] [Google Scholar]; Da Silva GF, Harrison JS, Lai JR. Biochemistry. 2010;49:5464–5472. doi: 10.1021/bi100293q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Clackson T, Wells JA. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 5.a) Agnew HD, Rohde RD, Millward SW, Nag A, Yeo WS, Hein JE, Pitram SM, Tariq AA, Burns VM, Krom RJ, Fokin VV, Sharpless KB, Heath JR. Angew Chem Int Ed Engl. 2009;48:4944–4948. doi: 10.1002/anie.200900488. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Millward SW, Agnew HD, Lai B, Lee SS, Lim J, Nag A, Pitram S, Rohde R, Heath JR. Integr Biol (Camb) 2012 doi: 10.1039/c2ib20110k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mocharla VP, Colasson B, Lee LV, Roper S, Sharpless KB, Wong CH, Kolb HC. Angew Chem Int Ed Engl. 2004;44:116–120. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 7.Lam KS, Lehman AL, Song A, Doan N, Enstrom AM, Maxwell J, Liu R. Methods Enzymol. 2003;369:298–322. doi: 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- 8.Nag A, Das S, Yu MB, Deyle KM, Millward SW, Heath JR. Angew Chem Int Ed Engl. 2013;52:13975–13979. doi: 10.1002/anie.201305882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deyle KM, Farrow B, Qiao Hee Y, Work J, Wong M, Lai B, Umeda A, Millward SW, Nag A, Das S, Heath JR. Nat Chem. 2015;7:455–462. doi: 10.1038/nchem.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrow B, Wong M, Malette J, Lai B, Deyle KM, Das S, Nag A, Agnew HD, Heath JR. Angew Chem Int Ed Engl. 2015;54:7114–7119. doi: 10.1002/anie.201502451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giebel LB, Cass RT, Milligan DL, Young DC, Arze R, Johnson CR. Biochemistry. 1995;34:15430–15435. doi: 10.1021/bi00047a006. [DOI] [PubMed] [Google Scholar]
- 12.Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, Parry T, Kent R, Enright J, Wu Ql, Conley G, DeOliveira D, Morganelli L, Ducar M, Wescott CR, Ladner RC. J Biol Chem. 2003;278:15532–15540. doi: 10.1074/jbc.M212934200. [DOI] [PubMed] [Google Scholar]
- 13.a) Chandrasekhar S, Epling DE, Sophocleous AM, Topp EM. Journal of Pharmaceutical Sciences. 2014;103:1032–1042. doi: 10.1002/jps.23906. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Winther JR, Thorpe C. Biochimica et Biophysica Acta. 2014;1840:838–846. doi: 10.1016/j.bbagen.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Miller SJ, Blackwell HE, Grubbs RH. Journal of the American Chemical Society. 1996;118:9606–9614. [Google Scholar]
- 16.Punna S, Kuzelka J, Wang Q, Finn MG. Angew Chem Int Ed Engl. 2005;44:2215–2220. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]
- 17.Schafmeister CE, Po J, Verdine GL. Journal of the American Chemical Society. 2000;122:5891–5892. [Google Scholar]
- 18.Baker J, McCarthy J, Gatton M, Kyle DE, Belizario V, Luchavez J, Bell D, Cheng Q. J Infect Dis. 2005;192:870–877. doi: 10.1086/432010. [DOI] [PubMed] [Google Scholar]
- 19.Hurdayal R, Achilonu I, Choveaux D, Coetzer THT, Goldring JPD. Peptides. 2010;31:525–532. doi: 10.1016/j.peptides.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Turgut-Balik D, Akbulut E, Shoemark DK, Celik V, Moreton KM, Sessions RB, Holbrook JJ, Brady RL. Biotechnology Letters. 2004;26:1051–1055. doi: 10.1023/B:BILE.0000032958.78158.10. [DOI] [PubMed] [Google Scholar]
- 21.Benedetti CE, Kobarg J, Pertinhez TA, Gatti RM, de Souza ON, Spisni A, Meneghini R. Mol Biochem Parasitol. 2003;128:157–166. doi: 10.1016/s0166-6851(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 22.Kifude CM, Rajasekariah HG, Sullivan DJ, Jr, Stewart VA, Angov E, Martin SK, Diggs CL, Waitumbi JN. Clinical and vaccine immunology : CVI. 2008;15:1012–1018. doi: 10.1128/CVI.00385-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Pfeilsticker JA, Umeda A, Farrow B, Hsueh CL, Deyle KM, Kim JT, Lai BT, Heath JR. PLoS One. 2013;8:e76224. doi: 10.1371/journal.pone.0076224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Farrow B, Hong SA, Romero EC, Lai B, Coppock MB, Deyle KM, Finch AS, Stratis-Cullum DN, Agnew HD, Yang S, Heath JR. ACS Nano. 2013;7:9452–9460. doi: 10.1021/nn404296k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.