

Summary
A genome-wide linkage scan of 357 European American (EA) and 72 African American (AA) pedigrees multiplex for type 2 diabetes mellitus (T2DM) was performed with multipoint nonparametric QTL linkage analysis. Four subclinical measures of cardiovascular disease (CVD): coronary artery (CCP), carotid artery (CarCP), and abdominal aortic calcified plaque (AACP) and carotid artery intima-media thickness (IMT) were mapped. Analyses were adjusted for age, gender, body mass index, and (if appropriate) ethnicity and diabetes status.
Evidence for linkage was observed in EA T2DM subjects to CarCP near 16p13 (LOD=4.39 at 8.4 cM; P = 0.00001). When all EA subjects were included, the LOD score was 2.52, suggesting an amplification of the linkage by diabetes. Linkage analysis of a principal components measure of vascular calcium (LOD = 3.85 at 9.3 cM on 16p in EA T2DM subjects) and bivariate analysis of CarCP X IMT (LOD = 3.77 at 9.3 cM on 16p in EA T2DM subjects) were consistent with this linkage. In addition, evidence for linkage was observed with CCP near D15S1515 (LOD = 2.34) in EAs. Additional loci on chromosomes 1, 2, 7, 10, 13, and 21 had LODs > 2.0. The identification of trait-determining polymorphisms underlying these linkages will help delineate risk factors for CVD in T2DM and the general population.
Keywords: type 2 diabetes mellitus, cardiovascular disease, vascular calcified plaque, carotid wall thickness, genome scan, principal components analysis
Introduction
Diabetes is an independent risk factor for the development of clinical cardiovascular disease (CVD) (Abbott et al. 1987; Haffner et al. 1998; Kannel & McGee 1979; Pan et al. 1986). For example, the relative risk of cardiovascular death was 2.1 for men and 4.9 for women comparing diabetes-affected to non-diabetic subjects in the Framingham Study (Kannel & McGee 1979). Diabetes contributes substantially to the development of premature mortality and morbidity from CVD (Miettinen et al. 1998). It has been proposed that genetic susceptibility in diabetes-affected subjects, in combination with other factors (hypertension, microalbuminuria, blood glucose control, etc.) contributes to the development of diabetic macrovascular disease (Warram et al. 1997). These complications are common, afflicting the majority of diabetes-affected individuals.
For a problem of such magnitude, little is known about the genetics underlying CVD, diabetes, or the association of diabetes and CVD. The identification of genetic components in combination with modifying non-genetic risk factors will focus both treatment and intervention strategies. Due to the high prevalence of clinical and subclinical CVD in diabetes-affected populations, families with multiple-affected diabetes members present an enriched resource for genetic analysis. Various observers (Cambien, 1997) have suggested that diabetes provides an enriched environment for the expression of CVD susceptibility genes and may be an ideal venue in which to study CVD genetics.
The Diabetes Heart Study (DHS) (Lange et al. 2002a; Wagenknecht et al. 2001) has been designed to take advantage of the high prevalence of CVD in diabetes. The DHS is a study of genetic and environmental factors of CVD in families highly enriched for type 2 diabetes mellitus (T2DM). It represents one of the few, if not the only, study in which diabetes and multiple measures of CVD, including vascular calcification and carotid wall thickness, can be evaluated simultaneously to assess the relationship between genetic and environmental contributions. Previously we have reported estimates of heritability of CVD (Lange et al. 2002a; Wagenknecht et al. 2001), race and gender interactions (Freedman et al. 2005a), studies of epidemiological relationships (Bowden et al. 2005; Freedman et al. 2005b), candidate gene identification (Burdon et al. 2006), and qualitative trait linkage analysis (Bowden et al. 2006) of subclinical CVD in the DHS. The primary focus of the Diabetes Heart Study is quantitative measures of CVD. In this report we describe linkage analysis of quantitative measures of subclinical CVD: coronary, carotid, and aortic vascular calcified plaque measured by computed tomography, and carotid wall thickness measured by B-mode ultrasound in families from the DHS.
Materials and Methods
Subjects
The Diabetes Heart Study is being conducted in Forsyth County, North Carolina to study the genetic and epidemiological origins of cardiovascular disease in families affected with T2DM. Siblings concordant for diabetes were recruited from internal medicine clinics, endocrinology clinics, and community advertising. T2DM was defined as a clinical diagnosis of diabetes after the age of 34 years, in the absence of historical evidence of diabetic ketoacidosis, and active treatment at the time of examination. Unaffected siblings, similar in age to the siblings with T2DM, were also invited to participate, as were any additional diabetes-affected siblings. The sample includes European American (EA) and African-American (AA; approximately 15% of the total) participants. The results reported here are from 973 European American subjects from 357 pedigrees with at least two individuals with T2DM (653 T2DM relative pairs; 1191 total relative pairs) and 175 African American subjects from 72 pedigrees with at least two individuals with T2DM (107 T2DM relative pairs; 155 total relative pairs). Thus a total of 760 T2DM sibling pairs and 1346 relative pairs were included in the analysis. The family structures of the subjects (both affected and unaffected in each family) included in the genome scan ranged from simple T2DM-affected sib pairs (60% of the families) to one family with 10 subjects: 215 EA and 53 AA families with 2 sibs, 79 EA and 8 AA families with 3 subjects, 35 EA and 10 AA families with 4 subjects, 14 EA and 1 AA families with 5 subjects, 7 EA families with 6 subjects, 4 EA families with 7 subjects, 2 EA families with 8 subjects, and one EA family with 10 subjects. For the 215 families with 2 sibs, each sib has T2DM. The larger families have combinations of T2DM-affected and unaffected relatives, but each has at least one pair of T2DM-affected sibs.
Individuals with serious health conditions, e.g., renal replacement therapy, were not eligible to participate. Recruitment was based upon family structure and there were no inclusions/exclusions based on prior or current evidence of prevalent CVD at the time of recruitment.
Clinical Evaluation
The participant examinations were conducted in the General Clinical Research Center of the Wake Forest University Baptist Medical Center and included interviews for medical history and health behaviors, anthropometric measures, resting blood pressure, a fasting blood draw and a spot urine collection. Laboratory assays included urine albumin and creatinine, total cholesterol, non-high density lipoprotein (HDL) cholesterol, low density lipoprotein (LDL), HDL, triglycerides, glycated hemoglobin, fasting glucose and blood chemistries. A detailed medical history was collected with emphasis on CVD. In addition, a resting 12-lead electrocardiogram (ECG) was performed to assess history of clinically significant (past or present) cardiovascular disease. 96 subjects (8.5%) had Minnesota codes 1.1 – 1.2 (except 1.2.8) which capture patterns of Q-wave MI. 59 of these subjects (61%) reported prior myocardial infarctions (MI).
Coronary calcified plaque (CCP), carotid calcified plaque (CarCP) and abdominal aortic calcified plaque (AACP) were measured with single and multidetector cardiac CT systems using a standardized protocol based on those currently implemented in the National Heart, Lung and Blood Institute’s (NHLBI’s) CARDIA and MESA studies for measuring the coronary arteries (Carr et al. 2005; Detrano et al. 2005). We have described previously the methods for measuring the carotid bifurcation and have used a comparable approach in the abdominal aorta (Carr et al. 2000). The vascular segments of the carotid were based on those used in the Atherosclerosis Risk In Communities (ARIC) carotid ultrasound protocol. Specifically, 15 mm of internal and external carotid above the bifurcation and 30 mm of carotid bulb and common carotid artery below the bifurcation were measured and the vascular segments summed for each carotid. For the abdominal aorta, the proximal segment (suprarenal abdominal aorta) was defined as starting 25 mm proximal to the superior mesenteric artery. The entire juxtrarenal and infrarenal aorta were measured and 25 mm length of the common iliac arteries below the aortic bifurcation was measured. The SmartScores software package (General Electric Medical Systems, Waukesha, WI, USA) was used to analyze the image data by experienced analysts, producing an Agatston score corrected for slice thickness for each arterial territory. The slice thickness was 2.5–3.0 mm for all scans, and the number of adjacent pixels used to define a calcified plaque was one, resulting in a minimum lesion size of 1 mm2. The reproducibility of CAP scores in the coronary and carotid arteries was assessed by obtaining duplicate scans with an inter- and intraobserver variability exceeding 0.96.
High-resolution B-mode carotid ultrasonography was performed as described previously (Lange et al. 2002a) using a 7.5-MHz transducer and a Biosound Esaote (AU5) machine (Biosound Esaote, Inc., Indianapolis, IN, USA). Scans were performed of the near and far walls of the distal 10-mm portion of the common carotid artery (CCA) at 5 predefined interrogation angles on each side. The mean value of up to 20 CCA IMT values was reported.
All protocols were approved by the Institutional Review Board of Wake Forest University School of Medicine, and all participants gave informed consent.
Genotyping
DNA extraction was performed using the PureGene system (Gentra Systems, Minneapolis, MN, USA). A genome-wide scan was completed by the Mammalian Genotyping Service (MGS, Marshfield, WI, USA). 1,177 DNA samples were genotyped with 411 polymorphic markers. Markers were taken from Screening Set 13 (Ghebranious et al. 2003) with an average spacing of 9.3cM, and no inter-marker gaps greater than 17.5 cM.
Analysis
Each pedigree was examined for consistency of familial relationships using PREST (Pedigree RElationship Statistical Test) (McPeek & Sun 2000). When the self-reported familial relationships were inconsistent with that determined from the observed genotypic data for that pedigree, then 1) the pedigree was modified when the identity by descent (IBD) statistics suggested a very clear alternative, or 2) a minimal set of genotypic data was converted to missing. Each genetic marker was also examined for Mendelian inconsistencies using PedCheck (O’Connell & Weeks 1998), and sporadic problem genotypes converted to missing.
Ethnic-specific maximum likelihood allele frequencies were used to estimate the single-point and multipoint identity by descent (IBD) statistics using LOKI (Heath, 1997). The LOKI runs were repeated four times using different seeds to check for consistency and convergence of the results. LOKI was used to facilitate a combined ethnicity analysis using ethnic-specific allele frequencies. To further verify the results, multipoint IBD estimates using ethnic-specific allele frequencies were computed using the exact methods implemented in the software MERLIN.(Abecasis et al. 2002). The resulting ethnic-specific linkage analyses were fully comparable to the corresponding LOKI-based linkage analyses. Finally, the results from the linkage analyses were converted from the Haldane to Kosambi genetic map to be consistent with the Marshfield maps.
To determine the contribution of genetic factors to the primary traits (CCP, CarCP, AACP, IMT), we analyzed the transformed data obtained on family members using the SOLAR software package (Almasy & Blangero 1998). SOLAR performs a variance components analysis of family data that decomposes the total variance of the phenotype (e.g. CCP) into components that are due to genetic (polygenic) effects (additive genetic variance), measured covariates, and random environmental effects. The relative contribution of genetic factors to trait variation is then estimated by the heritability (h2), defined by the ratio of the genetic variance component to the residual (after removal of covariates) phenotypic variance. A series of models were developed that incorporated an increasing number of covariates related to each trait, in order to determine the extent of genetic factors contributing to the variation in the trait independent of the measured risk factors. This has been described in detail elsewhere (Wagenknecht et al. 2001). Heritability estimates presented here are adjusted for the covariates age, gender, BMI, diabetes status, and race (as appropriate). These covariates explain a significant amount of the variance in these traits as summarized in Supplementary Table S1. Significance of the estimated heritabilities was determined by likelihood ratio tests, in which the likelihood of the models with the additive genetic variance component and covariates was compared to the model with the likelihood in which the additive genetic variance component was constrained to be zero.
For estimation of genetic and phenotypic variance of traits, the overall phenotypic variation was partitioned into common genetic (ρg) and environmental (ρe) correlations between traits which were calculated using QUANTBIV, a bivariate extension of the SOLAR software (Almasy & Blangero 1998). Statistical significance was assessed via maximum likelihood ratio tests. A principal component analysis was computed based on the genetic correlations among the vascular calcium and IMT measures to provide a measure of common variation across the three vascular calcium beds and IMT. The first principal component was effectively the mean of CCP, CarCP and AACP, (explaining 61% of the variation) and the second was IMT (explaining 23% of the variation). The construction of the principal components was based on the genetic correlation matrix calculated in SOLAR after adjusting for the covariates in the mixed model. The eigenvectors of the first principal component were comparable across ethnicities, yielding effectively the mean of the log of the vascular calcium measures within an individual. Thus, what is reported as the principal component is the mean of the natural logarithm and is consistent across ethnicities. As noted in the text, the actual linkage analysis is adjusted for covariates as well. A principal component is a score within the same individual, and the first principal component was effectively the mean of the natural logarithm in both ethnicities. In addition, we used linear regression to impute the predicted value of a missing vascular bed measure when the other two beds were successfully measured. For example, if aortic calcium score was missing but there were scores for carotid and coronary vascular calcium the predicted value for aortic calcium was imputed based on the predicted value from the linear regression model that contained carotid and coronary vascular calcium scores. If only one of the three vascular beds had a score, no imputation was attempted. A total of 288 (AACP = 217, CCP = 62 CarCP = 9) individuals in the EA sample and 43 (AACP = 32, CCP = 9, CarCP = 2) individuals in the AA sample had imputed values analyzed. The regression coefficients and proportion of variation explained by the regression lines used in the imputation were relatively consistent across ethnicities and vascular beds (AACP: , ; CCP: , ; CarCP: , . Supplementary Table S2 provides a summary of the imputations.
Both univariate and bivariate quantitative trait linkage analyses were performed using the variance component approach adjusting for the covariates age, gender, BMI, ethnicity (where appropriate), and diabetes status using the SOLAR software (Almasy & Blangero 1998). The proportion of variation of subclinical cardiovascular disease measures that can be explained by demographic and physiological characteristics has been evaluated in the DHS (data not shown). When age, gender and BMI are included, other measures have limited additional influence on the proportion of variation, and, consistent with this observation, have little influence in the linkage analysis. The phenotypes IMT, CCP and CarCP were natural log transformed [log(x+1)] and AACP was square root transformed to best approximate the distributional assumptions of the variance component QTL linkage analysis. Five linkage analyses were completed: 1) entire sample, 2) EA pedigrees, 3) AA pedigrees; 4) diabetic members of EA pedigrees and 5) diabetic members of AA pedigrees. For bivariate analysis the primary measures of subclinical CVD were evaluated pairwise: CCP X CarCP, CCP X IMT, etc. Trait-specific LOD scores reported are empirical LOD scores determined using the lodadj procedure within SOLAR. Although the distribution of each phenotype analyzed for linkage has been carefully examined for homogeneity of variance, outliers, and conditional normality (conditional on the covariates in the model), empirical LOD scores were computed using the lodadj option in SOLAR (Blangero et al. 2000). For each phenotype and each chromosome the lodadj procedure conducts a simulation under the null hypothesis of no linkage (e.g., 100,000 replications), using a fully informative marker and the existing pedigree structure, to obtain a correction factor for the LOD score. Bivariate analyses in SOLAR do not support a lodadj analysis and large-sample LOD scores are reported for bivariate analyses.
Several participants in the study have prior interventions that are relevant to the data analysis. Individuals with prior endarterectomies were excluded from the analysis of CarCP and IMT and those with coronary artery bypass graft were excluded from CCP analysis. 151 subjects have prior angioplasty procedures. These subjects were included in the analysis to avoid truncating the distributions of the quantitative traits. Incorporating angioplasty as a covariate in the analyses did not materially change the inferences of the study (data not shown).
Results
Table 1 summarizes characteristics of 1148 EA and AA subjects with phenotypic data from the genome scan. 85% of the subjects are EA. The AA sample has little power to detect AA-specific linkage signals but provides some ability to assess whether there are common contributors to linkage in the two ethnic groups. Overall the subjects in the DHS have biometric and clinical characteristics consistent with a diabetes-enriched family population: older (mean age of 61) and obese (mean BMI of 32). Diabetes-affected subjects have lower cholesterol and LDL cholesterol than their unaffected relatives. This is likely due to a treatment affect since 41% of the T2DM-affecteds are treated with statins (Bowden et al. 2005).
Table 1.
Characteristics of the study sample
| Phenotype | European American
|
African American
|
Combined
|
||
|---|---|---|---|---|---|
| T2DM-affected (N = 811) Mean±Std Dev (Median) |
Unaffected (N = 162) Mean±Std Dev (Median) |
T2DM-affected (N = 157) Mean±Std Dev (Median) |
Unaffected (N = 18) Mean±Std Dev (Median) |
N = 1148 Mean Std±Dev (Median) |
|
| Gender (% male) | 49.2% | 36.4% | 33.1% | 27.8% | 44.9% |
| Age | 62.0±9.2 (62.1) | 59.6±10.2 (59.5) | 59.0±9.0 (58.3) | 57.2±10.6 (57.8) | 61.1±9.5 (61.0) |
| T2DM Duration | 10.4±7.2 (8.0) | NA | 10.7±7.9 (8.0) | NA | 10.4±7.3 (8.0) |
| BMI (kg/m2) | 32.3±6.7 (31.2) | 28.8±5.2 (28.1) | 34.0±7.2 (33.4) | 31.8±6.7 (31.8) | 32.0±6.7 (30.9) |
| Systolic Blood | 139.7±19.1 (138.0) | 134.9±19.7 (133.0) | 143.0±20.7 (143.0) | 140.8±19.4 (139.5) | 139.5±19.5 (138.5) |
| Pressure (mm Hg) | |||||
| Diastolic Blood | 72.8±10.4 (72.0) | 74.2±10.5 (74.0) | 76.5±11.7 (76.0) | 76.6± 10.3 (76.8) | 73.5±10.7 (73.0) |
| Pressure (mm Hg) | |||||
| Diagnosis of hypertension (%) | 88.4% | 66.7% | 87.9% | 72.2% | 85.0% |
| Use of hypertension medication | 79.3% | 45.6% | 79.9% | 66.7% | 74.7% |
| HbA1c (% glycated hemoglobin) | 7.70±1.79 (7.40) | 5.56±0.51 (5.50) | 8.93±2.72 (8.10) | 5.70±0.53 (5.60) | 7.53±2.05 (7.10) |
| Fasting Glucose (mg/dL) | 150.6±58.5 (138.0) | 93.3±11.3 (93.0) | 160.2±79.3 (145.0) | 93.0±12.1 (94.0) | 143.0±61.4 (128.0) |
| Total Cholesterol (mg/dL) | 187.2±43.7 (183.0) | 194.9±34.5 (194.0) | 190.5±38.8 (189.5) | 193.8±28.7 (196.0) | 188.8±41.7 (185.0) |
| HDL Cholesterol (mg/dL) | 42.2±12.0 (41.0) | 48.1±13.7 (46.0) | 49.6±14.9 (47.5) | 57.2±12.2 (56.5) | 44.3±13.1 (42.0) |
| LDL Cholesterol (mg/dL) | 103.8±31.7 (101.0) | 114.2±29.3 (112.0) | 113.2±32.1 (114.0) | 117.3±26.9 (119.0) | 106.9±31.7 (105.0) |
| Statin use (%) | 45.0% | 28.6% | 35.9% | 22.2% | 41.1% |
| Coronary Calcified Plaque | 1428±2656 (364.5) | 476.2±1040 (39.0) | 752±1649 (81.5) | 570.5±1272 (10.0) | 1167±2355 (222.8) |
| CCP>0 (%) | 95.0% | 83.8% | 92.9% | 77.8% | 92.7% |
| Carotid Calcified Plaque | 375.3±731.8 (84.5) | 172.0±467.9 (4.0) | 213.9±651.3 (16.3) | 30.2±64.9 (0.0) | 318.0±688.2 (45.5) |
| CarCP>0 (%) | 78.3% | 57.7% | 65.5% | 44.4% | 73.0% |
| Abdominal Aortic Calcified Plaque | 3965±4776 (2003) | 2431±4132 (811.5) | 1680±3198 (486) | 1518±2035 (626.0) | 3341±4527 (1425) |
| AACP>0 (%) | 95.1% | 84.2% | 85.7% | 76.5% | 91.7% |
| Intima Media | 0.68±0.14 (0.66) | 0.64±0.12 (0.62) | 0.70±0.13 (0.66) | 0.61±0.09 (0.60) | 0.68±0.13 (0.66) |
| Thickness (mm) | |||||
| Smoking (%) | |||||
| Current | 16.2% | 21.1% | 25.8% | 22.2% | 18.3% |
| Past | 43.3% | 34.2% | 36.1% | 38.9% | 41.0% |
| Never | 40.5% | 44.7% | 38.1% | 38.9% | 40.7% |
| Self-reported clinical CVD1 | 40.7% | 19.8% | 26.8% | 11.1% | 35.4% |
Angina, Stroke, Heart Attack, CABG, Coronary Angioplasty, or Carotid Endarterectomy
Both T2DM-affected and unaffected subjects have significant amounts of vascular calcified plaque in each of the 3 arterial beds evaluated here (92.7%, 73%, and 91.7% respectively for CCP, CarCP, and AACP). A characteristic of T2DM patients is the high prevalence of CVD, and specifically vascular calcification, which is clearly reflected in this study where 95% of the EA T2DM-affected subjects have detectable CCP with a mean value of 1428 and where 93% of the AA T2DM-affected subjects have detectable CCP with a mean value of 752. As it appears in Table 1 and reported previously (Freedman et al. 2005a; Freedman et al. 2005b; Wagenknecht et al. 2004), AA subjects have lower levels of vascular calcification and greater IMTs. It is striking that unaffected relatives also have substantial levels of vascular calcification, e.g. 84% of T2DM-unaffected EA subjects with a mean CCP of 476.
In an earlier phase of this study heritabilities for CCP and IMT were reported (Lange et al. 2002a; Wagenknecht et al. 2001). Table 2 summarizes estimates of the heritabilities of calcified plaque in each vascular bed (CCP, CarCP, AACP), the principal component of vascular calcification (VCP-PC), and IMT in the completed dataset. In the combined EA and AA samples the heritabilities range from 0.36 to 0.52 and are highly significant. These estimates are driven by the EA subjects which make up 85% of the sample and in which heritabilities range from 0.38 (IMT) to 0.58 (CCP). With the exception of IMT, heritability estimates in the AA subjects were not significant, reflecting the small sample size.
Table 2.
Heritability estimates for vascular calcium and IMT in the Diabetes Heart Study adjusted for age, gender, BMI, race (where appropriate), and diabetes status
| Trait1 | Sample | h2±SE(h2) | P-value | Proportion of Variance Due to Covariates |
|---|---|---|---|---|
| CCP | All | 0.51±0.08 | 3.2×10−12 | 0.30 |
| European American | 0.58±0.09 | 4.1×10−13 | 0.31 | |
| African American | 0.22±0.20 | 0.13 | 0.20 | |
| CarCP | All | 0.36±0.07 | 3.6×10−8 | 0.29 |
| European American | 0.40±0.08 | 1.6×10−8 | 0.29 | |
| African American | 0.10±0.19 | 0.25 | 0.25 | |
| AACP | All | 0.50±0.08 | 3.2×10−12 | 0.34 |
| European American | 0.50±0.09 | 4.1×10−13 | 0.34 | |
| African American | 0.59±0.20 | 0.13 | 0.25 | |
| Vascular Calcium PC | All | 0.52±0.07 | 1.2×10−15 | 0.38 |
| European American | 0.56±0.08 | 4.1×10−16 | 0.39 | |
| African American | 0.27±0.21 | 0.09 | 0.27 | |
| IMT | All | 0.41±0.08 | 1.8×10−8 | 0.26 |
| European American | 0.38±0.08 | 6.0×10−7 | 0.27 | |
| African American | 0.72±0.22 | 0.00085 | 0.23 |
transformations. CCP, CarCP, IMT: log; AACP: sqrt
There are several hypotheses for the relationship among CVD, T2DM and their risk factors. Among these hypotheses is pleiotropy, i.e., a polymorphism may influence multiple traits. As shown in Table 3, the measures of subclinical CVD: CCP, CarCP, AACP, and IMT are significantly correlated. These correlations can be partitioned into genetic and environmental correlations. Strong genetic correlations were observed in vascular calcium in all 3 vascular beds. The significant genetic correlations between the vascular calcified plaque measures in the three vascular beds suggest that some genes may contribute to systemic vascular calcification. Although both the estimated environmental and genetic correlations among CCP, CarCP and AACP are highly significant, their genetic correlations are greater than their environmental correlations. A principal component (PC) analysis was computed based on the genetic correlations among the vascular calcium and IMT measures to provide a measure of common variation across the three vascular calcium beds and IMT. Two PCs explained 84% of the variation in these four measures. The first PC was effectively the mean of CCP, CarCP and AACP, (loadings: 0.49, 0.58, 0.58, 0.29; explaining 61% of the variation) and the second was IMT (−0.32, 0.05, −0.22, explaining 23% of the variation). −0.93; Thus, we computed the mean of CCP, CarCP and AACP as the first PC, and IMT as the second PC.
Table 3.
Genetic and environmental correlations.
| Environmental Correlation
|
|||||
|---|---|---|---|---|---|
| CCP | CarCP | AACP | IMT | ||
| Genetic Correlation | CCP | 0.37 ± 0.07 | 0.42 ± 0.10 | 0.13 ± 0.09 | |
| CarCP | 0.52 ± 0.11 | 0.45 ± 0.10 | 0.19 ± 0.09 | ||
| AACP | 0.61 ± 0.10 | 0.80 ± 0.08 | 0.34 ± 0.10 | ||
| IMT | 0.15 ± 0.15 | 0.38 ± 0.16 | 0.19 ± 0.15 | ||
Bold: P<0.05
Table 4 presents a summary of LOD scores greater than 2.0 from the variance components linkage analysis. In univariate analysis a locus on the distal arm of chromosome 16p has the strongest evidence of linkage with a maximum LOD score of 4.39 with CarCP at 8.4 cM as illustrated in Figure 1A. This evidence of linkage was observed when analysis was restricted to EA-T2DM affected subjects. When all EA subjects were included in the analysis the maximum LOD score was 2.52 (at 2.8 cM; Fig. 1A). If one accepts that vascular calcified plaque is a measure of systemic atherosclerotic burden, more systemic measures of calcified plaque should show evidence of linkage to the 16p locus. Evidence was also strong for 16p linkage to the vascular calcified plaque principal component (VCP-PC) with a LOD score of 3.85 at 9.3 cM in EA-T2DM affecteds (Fig. 1A). In an effort to assess pleiotropic effects, bivariate analyses were performed. Bivariate LOD scores greater than 3.0 were observed for CCPXCarCP, CarCPXIMT, and CarCPXAACP in EA-T2DM subjects, but these were less than the maximum LOD for CarCP alone. Some evidence for coincident linkage on chromosome 16p should be apparent for the other measures of vascular calcified plaque: CCP and AACP if the 16p locus influences systemic vascular plaque. CCP, AACP, and, in addition, IMT independently showed modest evidence of linkage to the 16p locus also (CCP LOD = 0.88; AACP LOD = 1.15 in EA-T2DM sample; data not shown). In addition, bivariate analysis of CCPXAACP in EA-T2DM subjects on 16p resulted in a LOD score of 1.46 at 11 cM.
Table 4.
Summary of LOD scores ≥2.0 for subclinical CVD phenotypes in the EA Diabetes Heart Study population. Bold indicates LOD scores ≥3.0
| Chromosome | Trait1 | Sample | Position (cM) | LOD | LOD-1 Interval (cM) | p-value | Nearest Marker |
|---|---|---|---|---|---|---|---|
| 1 | IMT | EA | 52.0 | 2.18 | 39–65 | 0.0015 | D1S3720 |
| 1 | IMTXAACP | EA | 52.0 | 2.03 | 39–64 | 0.0023 | D1S3720 |
| 1 | CCPXIMT | All | 53.0 | 2.18 | 39–67 | 0.0016 | D1S3720 |
| 1 | CCPXAACP | All | 183.0 | 2.11 | 175–191 | 0.0018 | D1S3720 |
| 2 | AACP | EA-T2DM | 147.0 | 2.38 | 135–175 | 0.00094 | D2S1334 |
| 2 | CarCPXAACP | EA-T2DM | 147.0 | 2.09 | 131–161 | 0.0019 | D2S1334 |
| 2 | IMTXAACP | EA-T2DM | 147.0 | 2.06 | 136–164 | 0.0021 | D2S1334 |
| 2 | IMTXAACP | All | 149.0 | 2.30 | 138–163 | 0.0012 | D2S1334 |
| 7 | CCPXIMT | AA | 114.0 | 2.31 | 104–124 | 0.0011 | D7S1799 |
| 10 | CCPXCarCP | EA | 3.8 | 2.68 | tel-12 | 0.00045 | D10S1435 |
| 13 | IMT | EA-T2DM | 61.0 | 2.27 | 50–76 | 0.0012 | D13S800 |
| 15 | CCPXAACP | All | 119.0 | 3.27 | 109-tel | 0.0001 | D15S1515 |
| 15 | CCP | EA | 120.0 | 2.34 | 110-tel | 0.00140 | D15S1515 |
| 15 | CCPXCarCP | All | 120.0 | 2.31 | 110-tel | 0.0011 | D15S1515 |
| 15 | CCPXIMT | EA | 120.0 | 2.35 | 109-tel | 0.001 | D15S1515 |
| 15 | CCPXAACP | EA | 120.0 | 2.71 | 109-tel | 0.00042 | D15S1515 |
| 16 | CarCP | EA-T2DM | 8.4 | 4.39 | tel-15 | 0.00001 | D16S2616 |
| 16 | VCP-PC | EA-T2DM | 9.3 | 3.85 | 3.8–15 | 0.00003 | D16S2616 |
| 16 | CCPXCarCP | EA-T2DM | 10.0 | 3.90 | 2–15 | 0.00005 | D16S2616 |
| 16 | CarCPXIMT | EA-T2DM | 9.3 | 3.77 | tel-15 | 0.00003 | D16S2616 |
| 16 | CarCPXAACP | EA-T2DM | 8.4 | 3.58 | tel-15 | 0.00005 | D16S2616 |
| 16 | CarCP | EA | 2.8 | 2.52 | tel-12 | 0.00067 | D16S2616 |
| 16 | VCP-PC | EA | 7.5 | 2.36 | tel-15 | 0.00098 | D16S2616 |
| 21 | IMT | EA-T2DM | 25.0 | 2.33 | tel-29 | 0.0011 | D21S2052 |
VCP-PC: vascular calcified plaque principal component
Figure 1.
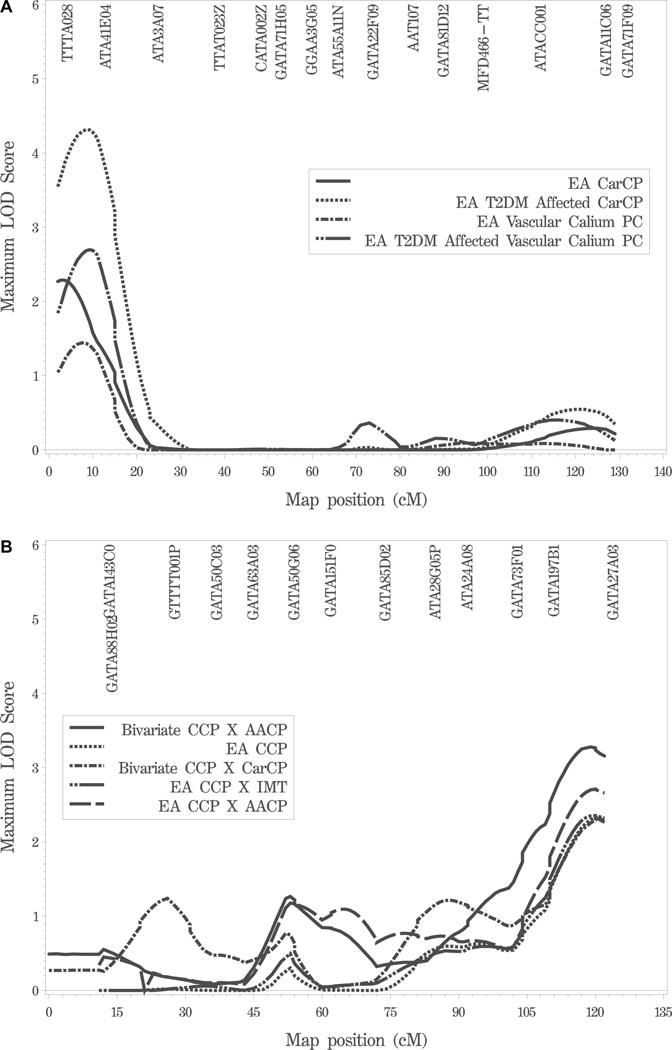
Variance components linkage analysis on chromosomes 16 and 15. 1A. Chromosome 16 univariate linkage analysis of CarCP in the EA families and EA-T2DM subjects and VCP-PC EA families and EA-T2DM subjects. 1B. Chromosome 15 univariate linkage analysis of CCP in the EA families and bivariate linkage analysis of CCPXAACP and CCPXIMT in EA families, and CCPXAACP and CCPXCarCP in the combined EA and AA samples.
With the individual traits no other LOD score exceeded 2.50 (Table 4), though there are several loci with LOD scores greater than 2.0. For example, IMT shows LODs greater than 2.0 on chromosomes 1, 13, and 21 and CCP shows a LOD of 2.34 on chromosome 15 at 120 cM in the combined EA analysis (Fig. 1B). The 15q locus has the strongest evidence for linkage after the 16p locus. In bivariate analysis of CCPXAACP in the entire dataset, the maximum bivariate LOD score is 3.27 at 119 cM on chromosome 15 (Table 4; Fig. 1B), which is substantially higher than the univariate results. Several additional bivariate loci with LODs greater than 2.0 are also observed on chromosomes 1, 2 (around 147–149 cM), 7 (CCPXIMT in AA pedigrees), and 10 (CCPXCarCP in EA pedigrees) (Table 4).
Discussion
We have recruited and comprehensively phenotyped T2DM-enriched families in the Diabetes Heart Study to create a resource for genetic and epidemiological studies of subclinical cardiovascular disease.
As part of these studies we have previously estimated heritability of CCP and IMT (Lange et al. 2002a; Wagenknecht et al. 2001). Here we report the heritabilities of CCP and IMT in the completed dataset and, in addition, report the heritabilities of calcified plaque in two other vascular beds, CarCP and AACP, and heritability of the principal component of vascular calcified plaque from the three vascular beds (Table 2). Heritability estimates for CCP and IMT are very similar to those previously reported (Lange et al. 2002a; Wagenknecht et al. 2001). Each measure of subclinical CVD is moderately heritable (h2 ranging from 0.35 to 0.58) and highly significant in the EA sample. To our knowledge this is the only report of heritability for CarCP and the second report for AACP. The AACP heritability is comparable to that previously reported (O’Donnell et al. 2002). We have further partitioned the genotypic and phenotypic variance of these quantitative traits into genetic and environmental correlations between the traits (Table 3). Vascular calcified plaque in the three vascular beds exhibited high genetic correlations suggesting that there are significant common genetic contributors to these phenotypes. This observation is consistent with the view that vascular calcified plaque is representative of systemic vascular disease. In addition, genetic correlations between vascular calcium (CCP, CarCP and AACP) and IMT are modest, suggesting the possibility that different genes contribute to vascular calcification and IMT. We previously reported the low correlation of CCP and IMT (Wagenknecht et al. 2004).
While we previously reported the results of a qualitative trait linkage analysis in the DHS (Bowden et al. 2006), the primary focus of the overall study has been analysis of quantitative measures of subclinical CVD. Analysis of the DHS genome scan data provides strong evidence for linkage of CarCP to a locus on chromosome 16p13.13-p13.3 with a maximum LOD score of 4.39 in T2DM-affected EA subjects (Table 4; Fig. 1A). Bivariate (e.g., CarCP X CCP, LOD = 3.90 at 10.0 cM on 16p) and principal components based analyses (LOD = 3.85 at 9.3 cM on 16p; Table 4; Fig. 1A) are also consistent with a vascular calcified plaque locus. Thus, three related analyses (single trait, principal component, and bivariate analyses) give similar results. This would be consistent with the high genetic correlation of the vascular calcium measures. The magnitude of LOD scores for the bivariate and principal components analyses, however, do not exceed the maximum LOD observed with CarCP alone, suggesting that the evidence for linkage is driven primarily by CarCP alone. As with all such studies, the ultimate identification of the trait-defining gene will be necessary in order to resolve the basis for this linkage.
In addition to the 16p locus, suggestive evidence of linkage (LOD > 2.0) was observed with IMT, CCP, and AACP on several other chromosomes (Table 4). Of these, the evidence for linkage on chromosome 15q is the strongest, and, in addition, the bivariate LOD score of 3.27 for CCPXAACP at the same locus is substantially higher than LODs with univariate traits. We note that the AA sample is small and thus the evidence for linkage of CCPXIMT on chromosome 7 in AAs alone should be viewed with caution. For each of these loci, analyses with other traits or subsets of the study subjects (e.g. EA total versus EAT2DM) provided linkage evidence consistent with, but of lesser magnitude, than these results shown in Table 4 (data not shown). In addition, Supplementary Figure S1 provides figures of the linkage results for the primary traits in all chromosomes.
As with any comprehensive genetic analysis, multiple tests of linkage have been computed. The genetic correlations and principal components analysis show that the vascular calcium measures are highly correlated and IMT is much less correlated with them (Table 3). In addition, we have calculated empirical LOD scores using the lodadj procedure within SOLAR. Even with these efforts, the magnitude of the linkage results reported here should be judged in the context of the overall study and the number of analyses that were performed. Overall the linkage data reported here should help inform other studies such as genome-wide association studies.
The 16p LOD-1 interval encompasses 12.1 Mb containing 329 genes. Several candidate CVD genes are in the region: ABCA3 (ATP-binding cassette, sub-family A member 3), SEPX1 (selenoprotein X), and SOCS1 (suppressor of cytokine signaling 1). The LOD score in T2DM-affected EA subjects was substantially higher than in all EA subjects, suggesting an interaction of this locus with diabetes status. The introduction of fasting glucose, HbA1c, or duration of diabetes as covariates into the analysis has, however, little effect on the linkage results (data not shown). It should be noted that both the chromosome 16p and 15q linkage peaks are near the ends of chromosomes: mapping in these regions sometimes is not robust. In both cases there are markers distal to the LOD maximum. In addition, we have carried out extensive further studies of the 16p locus and evidence of linkage continues to be strong (Lehtinen, in preparation).
CVD clusters in families, but CVD and surrogate measures of CVD such as subclinical disease do not segregate as Mendelian traits. Risk of CVD is significantly increased in people with T2DM. It is likely that the combination of genetic liability interacting with behavioral, environmental and other metabolic risk factors (e.g. insulin resistance) ultimately defines CVD susceptibility. Diabetes-associated CVD is a significant public health burden accounting for 65% of deaths in diabetes-affected individuals. We have hypothesized that the diabetic environment provides a physiologic environment that magnifies the effects of some vascular disease genes, and that may facilitate identification of such genes. It is noteworthy that 95% of the diabetes affected subjects have detectable CCP (Table 1). It could be questioned that the hyperglycemic environment could obscure expression of CVD genes, but measures of glycemic control provide little additional contribution to variance in the measures of subclinical CVD (Supplementary Table S1). Equally, approximately 40% of the subjects were on lipid lowering medications at the time of examination, but, again, we observed no significant contribution of this medication class to the variance in subclinical CVD measures (Supplementary Table S1). We do note that even if genes located using this approach solely influence CVD in diabetes, the significance in this population alone would justify the effort.
This study has focused on subclinical CVD in diabetes-enriched families by the analysis of quantitative measures of vascular calcified plaque and carotid atherosclerosis. CCP measured by CT has been demonstrated to predict hard and soft coronary heart disease (CHD), as well as total CVD, and all cause mortality (Greenland et al. 2004; Vliegenthart et al. 2005). In these prospective studies, the predicative ability of the calcium score was demonstrated to add significantly to traditional cardiovascular risk factors incorporated in the Framingham Risk index and to outperform C-reactive protein in predicting events. While consensus on the predictive value of vascular calcification measures has not been reached, numerous reports suggest that vascular calcification, with an emphasis on CCP is an excellent surrogate marker of CVD (Greenland et al. 2004; Terry et al. 2005; Vliegenthart et al. 2005; Walsh et al. 2002).
Coronary atherosclerosis has long been considered a primary determinant of CVD and carotid vascular calcified plaque (CarCP) has predictive value in cerebrovascular disease (Hollander et al. 2002; Vliegenthart et al. 2002). As a result, vascular calcification measurements serve as a standard gauge of atherosclerotic burden, predicting prevalent CVD and total mortality in asymptomatic individuals (Greenland et al. 2004; Raggi et al. 2001; Shemesh et al. 2004; Vliegenthart et al. 2002). Pathological studies indicate that CVD is a systemic disease and it is unusual to have disease localized to a single vascular bed. In addition to CCP, calcified lesions are commonly seen in the carotid artery and the aorta (Simon et al. 1995), and the extent of this peripheral arterial calcification appears to correlate with CCP. The technology used here to image calcified plaque cannot differentiate between medial as opposed to the intimal association of atherosclerosis of vascular plaque. Medial calcification is most commonly associated with the peripheral vasculature in the lower extremities in individuals with diabetes. There is limited evidence of medial calcification in the carotid and coronary arteries. In addition, medial calcification is most prominently associated with renal failure and it should be noted that preserved renal function was one of the criteria for recruitment in the DHS. While we cannot exclude the possibility that patterns of calcified plaque in our subjects differ from a non-diabetic population, Lehto et al. reported evidence of high correlation of medial and intimal calcification in the femoral arteries of T2DM patients, and, moreover, showed that medial calcification was a strong predictor of CVD mortality (Lehto et al. 1996).
Assessment of the carotid arteries complements that of the coronary arteries, and provides a full cardiovascular risk profile. Intimal-medial thickening (IMT) of the carotid artery is widely accepted as a quantitative measure of atherosclerosis that has a graded, predictive relationship to overt CVD (O’Leary et al. 1999). In fact, anatomical manifestations of carotid disease are among the strongest predictors of clinical events. Specifically, IMT serves as a reflection of CVD risk factors (Zanchetti et al. 1998), an indicator of atherosclerotic burden (Persson et al. 1992), a predictor of subsequent events (Hodis et al. 1998), and is recognized by the Food & Drug Administration as a measure of atherosclerosis. Although plaque as a carotid atherosclerosis phenotype is not studied as frequently as IMT, several studies indicate that CarCP alone is a strong risk factor for CVD events and CVD death (Ebrahim et al. 1999; Spence et al. 2002). Likewise, individuals with measurable AACP are at an increased risk of CVD and ultimately, CVD mortality (Wilson et al. 2001; Witteman et al. 1986).
Relatively few reports have described results from genome scans for the subclinical CVD phenotypes mapped here, and no previous reports have addressed vascular calcified plaque in multiple arterial beds and carotid wall thickness in the same analysis. In addition, no other studies have focused on the study of diabetes families for these phenotypes. Genome scans for CCP as a discrete (Lange et al. 2002b) and continuous trait (Turner et al. 2006) in subjects from the Family Blood Pressure Program, and as a quantitative trait in abstract form from the Family Heart Study (Carr et al. 2004) have been described. Genome scans for carotid IMT (Fox et al. 2004; Wang et al. 2005; Wang et al. 2004) and carotid artery plaque detected by ultrasound (Pankow et al. 2004) have also been reported. There is limited consistency of results across these previous studies, and with the results presented here. Lange et al. reported evidence of linkage on 6p21.3 (maximum LOD score = 2.22) and 10q21.3 (maximum LOD score = 3.24), and on chromosome 17p with a LOD of 1.13 at 29.4 cM for linkage to CCP in European Americans from the Family Blood Pressure Program (Lange et al. 2002b). While the chromosome 6 and 10 loci do not correspond to peaks in this study, there is a modest peak (LOD = 1.12) at 27 cM on 17p for linkage to CCP in the EAs in this study. In addition, Wang et al. recently reported evidence for association of polymorphisms in a gene, vitamin K epoxide reductase complex subunit 1 (VKORC1), on 16p with increased risk for vascular diseases, including stroke, coronary heart disease, and aortic dissection (Wang et al. 2006). VKORC1 is, however, located in chromosome 16p11.2, centromeric to the 16p13.13-p13.3 linkage interval for CarCP/vascular calcification identified in this study. The lack of consistency between the limited number of mapping studies is not surprising given the different study designs and use of diverse analytical approaches, combined with recruitment of different populations from different geographic locations and (in some cases) different ethnicities. The study reported here is built around recruitment of diabetes-enriched families and the most compelling linkage result, linkage to 16p of CarCP, is amplified in the diabetes-only sample. It is now widely recognized that linkage analysis has limited power to detect modest genetic effects. Consequently, the numerous differences between these studies could mask consistent evidence of linkage since most descriptions do not report minor (but potentially relevant) linkage peaks.
In addition, there have been several recent reports of the results from genome-wide association studies (GWAS) of CHD (e.g., McPherson et al. 2007; Helgadottir et al. 2007; Samani et al. 2007; Larson et al. 2007), and, in one case, vascular calcium (O’Donnell et al. 2007). Although none of these GWAS identify significant loci on chromosome 16p, the results of the genome scan linkage analysis in the Diabetes Heart Study do show some overlap with the significant results of the Framingham Heart Study GWAS (Larson et al. 2007; O’Donnell et al. 2007). Of particular interest are the results from both studies for chromosome 15. The Framingham Heart Study identified polymorphisms on chromosome 15 that were associated with carotid artery IMT (O’Donnell et al. 2007) and major CHD (Larson et al. 2007). These polymorphisms are located within the LOD-1 support interval for chromosome 15 linkage to CCP in EAs from the Diabetes Heart Study. The results from chromosomes 1 and 2 are also broadly consistent between the Framingham Heart Study and the Diabetes Heart Study. It is interesting to note that the region consistently replicated in CHD GWAS (i.e., polymorphisms located near the CDKN2A and CDKN2B genes on chromosome 9p21) does not appear to influence vascular calcification or IMT in the Diabetes Heart Study population.
In conclusion, the Diabetes Heart Study is a comprehensively phenotyped sample which comprises an excellent resource for the study of the genetic component of CVD in diabetes. The evidence presented here suggests that several loci, in particular a locus on 16p, harbor genes contributing to subclinical CVD.
Supplementary Material
Supplementary Figure S1. Graphical displays of the LOD score plots for each autosome and each trait are provided. (A) CCP, (B) CarCP, (C) AACP, (D) VCP-PC, and (E) IMT. Each figure plots LOD scores for the combined EA and AA analysis, EA subjects alone, AA subjects alone, EA subjects with T2DM, and AA subjects with T2DM.
Supplementary Table S1. Proportion of Variation of Subclinical Cardiovascular Disease Explained by Demographic and Physiological Characteristics in the Diabetes Heart Study.
Supplementary Table S2. Vascular Calcium Imputation for DHS CVD genome scan.
Acknowledgments
This study was supported in part by the General Clinical Research Center of the Wake Forest University School of Medicine grant M01 RR07122, R01 AR48797 (JJC), and NHLBI R01 HL67348 (DWB). The genome scan was performed by the NHLBI Mammalian Genotyping Service, Center for Human Genetics, Marshfield Clinic Research Foundation, Marshfield, WI (Contract Number HV48141).
Footnotes
Supplementary Material
The following material is available for this article online:
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abbott RD, Donahue RP, MacMahon SW, Reed DM, Yano K. Diabetes and the risk of stroke. The Honolulu Heart Program. Jama. 1987;257:949–952. [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangero J, Williams JT, Almasy L. Robust LOD scores for variance component-based linkage analysis. Genet Epidemiol. 2000;19:S8–S14. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bowden DW, Lange LA, Langefeld CD, Brosnihan KB, Freedman BI, Carr JJ, Wagenknecht LE, Herrington DM. The relationship between C-reactive protein and subclinical cardiovascular disease in the Diabetes Heart Study (DHS) Am Heart J. 2005;150:1032–1038. doi: 10.1016/j.ahj.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Bowden DW, Rudock M, Ziegler J, Lehtinen AB, Xu J, Wagenknecht LE, Herrington D, Rich SS, Freedman BI, Carr JJ, Langefeld CD. Coincident linkage of type 2 diabetes, metabolic syndrome, and measures of cardiovascular disease in a genome scan of the Diabetes Heart Study. Diabetes. 2006;55:1985–1994. doi: 10.2337/db06-0003. [DOI] [PubMed] [Google Scholar]
- Burdon KP, Bento JL, Langefeld CD, Campbell JK, Carr JJ, Wagenknecht LM, Herrington DM, Freedman BI, Rich SS, Bowden DW. Association of protein tyrosine phosphatase-N1 polymorphisms with coronary calcified plaque in the Diabetes Heart Study. Diabetes. 2006;55:651–658. doi: 10.2337/diabetes.55.03.06.db05-0058. [DOI] [PubMed] [Google Scholar]
- Cambien F. Genetics of atherosclerosis and its complications in type 2 diabetes; Presented at Proceedings of Atherosclerotic Vascular Diseases in Diabetes; Lappeenranta, Finland. Jul 25–27, 1997. [Google Scholar]
- Carr JJ, Lange LA, Province M, North KE, Lewis C, Myers RH, Hunt SC, Hixson J, Pankow J, Eckfeldt JH. Genome scan for calcified atherosclerotic plaque in the coronary arteries: evidence for linkage to chromosomes 4q and 10p from the NHLBI Family Heart Study. Circulation. 2004;110:823. [Google Scholar]
- Carr JJ, Nelson JC, Wong ND, McNitt-Gray M, Arad Y, Jacobs DR, Jr, Sidney S, Bild DE, Williams OD, Detrano RC. Calcified coronary artery plaque measurement with cardiac CT in population-based studies: standardized protocol of Multi-Ethnic Study of Atherosclerosis (MESA) and Coronary Artery Risk Development in Young Adults (CARDIA) study. Radiology. 2005;234:35–43. doi: 10.1148/radiol.2341040439. [DOI] [PubMed] [Google Scholar]
- Carr JJ, Wagenknecht LE, Bowden DW, Langefeld C, Freedman BI, Burdette JH. Carotid calcium as a measure of atherosclerosis:methodology and reproducibility. Radiology. 2000;217:244. [Google Scholar]
- Detrano RC, Anderson M, Nelson J, Wong ND, Carr JJ, McNitt-Gray M, Bild DE. Coronary calcium measurements: effect of CT scanner type and calcium measure on rescan reproducibility–MESA study. Radiology. 2005;236:477–484. doi: 10.1148/radiol.2362040513. [DOI] [PubMed] [Google Scholar]
- Ebrahim S, Papacosta O, Whincup P, Wannamethee G, Walker M, Nicolaides AN, Dhanjil S, Griffin M, Belcaro G, Rumley A, Lowe GD. Carotid plaque, intima media thickness, cardiovascular risk factors, and prevalent cardiovascular disease in men and women: the British Regional Heart Study. Stroke. 1999;30:841–850. doi: 10.1161/01.str.30.4.841. [DOI] [PubMed] [Google Scholar]
- Fox CS, Cupples LA, Chazaro I, Polak JF, Wolf PA, D’Agostino RB, Ordovas JM, O’Donnell CJ. Genomewide linkage analysis for internal carotid artery intimal medial thickness: evidence for linkage to chromosome 12. Am J Hum Genet. 2004;74:253–261. doi: 10.1086/381559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman BI, Hsu FC, Langefeld CD, Rich SS, Herrington DM, Carr JJ, Xu J, Bowden DW, Wagenknecht LE. The impact of ethnicity and sex on subclinical cardiovascular disease: the Diabetes Heart Study. Diabetologia. 2005a;48:2511–2518. doi: 10.1007/s00125-005-0017-2. [DOI] [PubMed] [Google Scholar]
- Freedman BI, Langefeld CD, Lohman KK, Bowden DW, Carr JJ, Rich SS, Wagenknecht LE. Relationship between albuminuria and cardiovascular disease in Type 2 diabetes. J Am Soc Nephrol. 2005b;16:2156–2161. doi: 10.1681/ASN.2004100884. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, Vaske D, Yu A, Zhao C, Marth G, Weber JL. STRP screening sets for the human genome at 5 cM density. BMC Genomics. 2003;4:6. doi: 10.1186/1471-2164-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenland P, LaBree L, Azen SP, Doherty TM, Detrano RC. Coronary artery calcium score combined with Framingham score for risk prediction in asymptomatic individuals. Jama. 2004;291:210–215. doi: 10.1001/jama.291.2.210. [DOI] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- Heath SC. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet. 1997;61:748–760. doi: 10.1086/515506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson D, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- Hodis HN, Mack WJ, LaBree L, Selzer RH, Liu CR, Liu CH, Azen SP. The role of carotid arterial intima-media thickness in predicting clinical coronary events. Ann Intern Med. 1998;128:262–269. doi: 10.7326/0003-4819-128-4-199802150-00002. [DOI] [PubMed] [Google Scholar]
- Hollander M, Bots ML, Del Sol AI, Koudstaal PJ, Witteman JC, Grobbee DE, Hofman A, Breteler MM. Carotid plaques increase the risk of stroke and subtypes of cerebral infarction in asymptomatic elderly: the Rotterdam study. Circulation. 2002;105:2872–2877. doi: 10.1161/01.cir.0000018650.58984.75. [DOI] [PubMed] [Google Scholar]
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. Jama. 1979;241:2035–2038. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002a;33:1876–1881. doi: 10.1161/01.str.0000019909.71547.aa. [DOI] [PubMed] [Google Scholar]
- Lange LA, Lange EM, Bielak LF, Langefeld CD, Kardia SL, Royston P, Turner ST, Sheedy PF, 2nd, Boerwinkle E, Peyser PA. Autosomal genome-wide scan for coronary artery calcification loci in sibships at high risk for hypertension. Arterioscler Thromb Vasc Biol. 2002b;22:418–423. doi: 10.1161/hq0302.105721. [DOI] [PubMed] [Google Scholar]
- Larson MG, Atwood LD, Benjamin EJ, Cupples LA, D’Agostino RB, Sr, Fox CS, Govindaraju DR, Guo CY, Heard-Costa NL, Hwang SJ, Murabito JM, Newton-Cheh C, O’Donnell CJ, Seshadri S, Vasan RS, Wang TJ, Wolf PA, Levy D. Framingham Heart study 100K project: genome-wide associations from cardiovascular disease outcomes. BMC Med Genet. 2007;8(Suppl 1):S5. doi: 10.1186/1471-2350-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto S, Niskanen L, Suhonen M, Ronnemaa T, Laakso M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1996;16:978–983. doi: 10.1161/01.atv.16.8.978. [DOI] [PubMed] [Google Scholar]
- McPeek MS, Sun L. Statistical tests for detection of misspecified relationships by use of genome-screen data. Am J Hum Genet. 2000;66:1076–1094. doi: 10.1086/302800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen H, Lehto S, Salomaa V, Mahonen M, Niemela M, Haffner SM, Pyorala K, Tuomilehto J. Impact of diabetes on mortality after the first myocardial infarction. The FIN-MONICA Myocardial Infarction Register Study Group. Diabetes Care. 1998;21:69–75. doi: 10.2337/diacare.21.1.69. [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell CJ, Chazaro I, Wilson PW, Fox C, Hannan MT, Kiel DP, Cupples LA. Evidence for heritability of abdominal aortic calcific deposits in the Framingham Heart Study. Circulation. 2002;106:337–341. doi: 10.1161/01.cir.0000022663.26468.5b. [DOI] [PubMed] [Google Scholar]
- O’Donnell CJ, Cupples LA, D’Agostino RB, Fox CS, Hoffmann U, Hwang SJ, Ingellson E, Liu C, Murabito JM, Polak JF, Wolf PA, Demissie S. Genome-wide association study for subclinical atherosclerosis in major arterial territories in the NHLBI’s Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S4. doi: 10.1186/1471-2350-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK., Jr Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999;340:14–22. doi: 10.1056/NEJM199901073400103. [DOI] [PubMed] [Google Scholar]
- Pan WH, Cedres LB, Liu K, Dyer A, Schoenberger JA, Shekelle RB, Stamler R, Smith D, Collette P, Stamler J. Relationship of clinical diabetes and asymptomatic hyperglycemia to risk of coronary heart disease mortality in men and women. Am J Epidemiol. 1986;123:504–516. doi: 10.1093/oxfordjournals.aje.a114266. [DOI] [PubMed] [Google Scholar]
- Pankow JS, Heiss G, Evans GW, Sholinsky P, Province MA, Coon H, Ellison RC, Miller MB, Qaqish B. Familial aggregation and genome-wide linkage analysis of carotid artery plaque: the NHLBI family heart study. Hum Hered. 2004;57:80–89. doi: 10.1159/000077545. [DOI] [PubMed] [Google Scholar]
- Persson J, Stavenow L, Wikstrand J, Israelsson B, Formgren J, Berglund G. Noninvasive quantification of atherosclerotic lesions. Reproducibility of ultrasonographic measurement of arterial wall thickness and plaque size. Arterioscler Thromb. 1992;12:261–266. doi: 10.1161/01.atv.12.2.261. [DOI] [PubMed] [Google Scholar]
- Raggi P, Cooil B, Callister TQ. Use of electron beam tomography data to develop models for prediction of hard coronary events. Am Heart J. 2001;141:375–382. doi: 10.1067/mhj.2001.113220. [DOI] [PubMed] [Google Scholar]
- Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh J, Morag-Koren N, Goldbourt U, Grossman E, Tenenbaum A, Fisman EZ, Apter S, Itzchak Y, Motro M. Coronary calcium by spiral computed tomography predicts cardiovascular events in high-risk hypertensive patients. J Hypertens. 2004;22:605–610. doi: 10.1097/00004872-200403000-00024. [DOI] [PubMed] [Google Scholar]
- Simon A, Giral P, Levenson J. Extracoronary atherosclerotic plaque at multiple sites and total coronary calcification deposit in asymptomatic men. Association with coronary risk profile. Circulation. 1995;92:1414–1421. doi: 10.1161/01.cir.92.6.1414. [DOI] [PubMed] [Google Scholar]
- Spence JD, Eliasziw M, DiCicco M, Hackam DG, Galil R, Lohmann T. Carotid plaque area: a tool for targeting and evaluating vascular preventive therapy. Stroke. 2002;33:2916–2922. doi: 10.1161/01.str.0000042207.16156.b9. [DOI] [PubMed] [Google Scholar]
- Terry JG, Carr JJ, Tang R, Evans GW, Kouba EO, Shi R, Cook DR, Vieira JL, Espeland MA, Mercuri MF, Crouse JR., 3rd Coronary artery calcium outperforms carotid artery intima-media thickness as a noninvasive index of prevalent coronary artery stenosis. Arterioscler Thromb Vasc Biol. 2005;25:1723–1728. doi: 10.1161/01.ATV.0000173418.42264.19. [DOI] [PubMed] [Google Scholar]
- Turner ST, Peyser PA, Kardia SL, Bielak LF, Sheedy PF, 3rd, Boerwinkle E, de Andrade M. Genomic loci with pleiotropic effects on coronary artery calcification. Atherosclerosis. 2006;185:340–346. doi: 10.1016/j.atherosclerosis.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Vliegenthart R, Hollander M, Breteler MM, Van Der Kuip DA, Hofman A, Oudkerk M, Witteman JC. Stroke is associated with coronary calcification as detected by electron-beam CT: the Rotterdam Coronary Calcification Study. Stroke. 2002;33:462–465. doi: 10.1161/hs0202.103071. [DOI] [PubMed] [Google Scholar]
- Vliegenthart R, Oudkerk M, Hofman A, Oei HH, van Dijck W, van Rooij FJ, Witteman JC. Coronary calcification improves cardiovascular risk prediction in the elderly. Circulation. 2005;112:572–577. doi: 10.1161/CIRCULATIONAHA.104.488916. [DOI] [PubMed] [Google Scholar]
- Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes. 2001;50:861–866. doi: 10.2337/diabetes.50.4.861. [DOI] [PubMed] [Google Scholar]
- Wagenknecht LE, Langefeld CD, Carr JJ, Riley W, Freedman BI, Moossavi S, Bowden DW. Race-specific relationships between coronary and carotid artery calcification and carotid intimal medial thickness. Stroke. 2004;35:e97–99. doi: 10.1161/01.STR.0000127081.99767.1d. [DOI] [PubMed] [Google Scholar]
- Walsh CR, Cupples LA, Levy D, Kiel DP, Hannan M, Wilson PW, O’Donnell CJ. Abdominal aortic calcific deposits are associated with increased risk for congestive heart failure: the Framingham Heart Study. Am Heart J. 2002;144:733–739. doi: 10.1067/mhj.2002.124404. [DOI] [PubMed] [Google Scholar]
- Wang D, Yang H, Quinones MJ, Bulnes-Enriquez I, Jimenez X, De La Rosa R, Modilevsky T, Yu K, Li Y, Taylor KD, Hsueh WA, Hodis HN, Rotter JI. A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke. 2005;36:540–545. doi: 10.1161/01.STR.0000155746.65185.4e. [DOI] [PubMed] [Google Scholar]
- Wang Q, Rao S, Shen GQ, Li L, Moliterno DJ, Newby LK, Rogers WJ, Cannata R, Zirzow E, Elston RC, Topol EJ. Premature myocardial infarction novel susceptibility locus on chromosome 1P34–36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–271. doi: 10.1086/381560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang W, Zhang Y, Yang Y, Sun L, Hu S, Chen J, Zhang C, Zheng Y, Zhen Y, Sun K, Fu C, Yang T, Wang J, Sun J, Wu H, Glasgow WC, Hui R. VKORC1 haplotypes are associated with arterial vascular diseases (stroke, coronary heart disease, and aortic dissection) Circulation. 2006;113:1615–1621. doi: 10.1161/CIRCULATIONAHA.105.580167. [DOI] [PubMed] [Google Scholar]
- Warram JH, Kopczynski J, Janka HU, Krolewski AS. Epidemiology of non-insulin-dependent diabetes mellitus and its macrovascular complications. A basis for the development of cost-effective programs. Endocrinol Metab Clin North Am. 1997;26:165–188. doi: 10.1016/s0889-8529(05)70239-5. [DOI] [PubMed] [Google Scholar]
- Wilson PW, Kauppila LI, O’Donnell CJ, Kiel DP, Hannan M, Polak JM, Cupples LA. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation. 2001;103:1529–1534. doi: 10.1161/01.cir.103.11.1529. [DOI] [PubMed] [Google Scholar]
- Witteman JC, Kok FJ, van Saase JL, Valkenburg HA. Aortic calcification as a predictor of cardiovascular mortality. Lancet. 1986;2:1120–1122. doi: 10.1016/s0140-6736(86)90530-1. [DOI] [PubMed] [Google Scholar]
- Zanchetti A, Bond MG, Hennig M, Neiss A, Mancia G, Dal Palu C, Hansson L, Magnani B, Rahn KH, Reid J, Rodicio J, Safar M, Eckes L, Ravinetto R. Risk factors associated with alterations in carotid intima-media thickness in hypertension: baseline data from the European Lacidipine Study on Atherosclerosis. J Hypertens. 1998;16:949–961. doi: 10.1097/00004872-199816070-00008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Graphical displays of the LOD score plots for each autosome and each trait are provided. (A) CCP, (B) CarCP, (C) AACP, (D) VCP-PC, and (E) IMT. Each figure plots LOD scores for the combined EA and AA analysis, EA subjects alone, AA subjects alone, EA subjects with T2DM, and AA subjects with T2DM.
Supplementary Table S1. Proportion of Variation of Subclinical Cardiovascular Disease Explained by Demographic and Physiological Characteristics in the Diabetes Heart Study.
Supplementary Table S2. Vascular Calcium Imputation for DHS CVD genome scan.