

Abstract
Purpose
We delineated the functions of the HIF1α target NADH Dehydrogenase (Ubiquinone) 1 alpha subcomplex 4-like 2 (NDUFA4L2) in ccRCC and characterized NDUFA4L2 as a novel molecular target for ccRCC treatment.
Experimental Design
We evaluated normal kidney and ccRCC patient microarray and RNAseq data from Oncomine and The Cancer Genome Atlas (TCGA) for NDUFA4L2 mRNA levels and the clinical implications of high NDUFA4L2 expression. Additionally, we examined normal kidney and ccRCC patient tissue samples, human ccRCC cell lines, and murine models of ccRCC for NDUFA4L2 mRNA and protein expression. Utilizing shRNA, we performed NDUFA4L2 knockdown experiments and analyzed the proliferation, clonogenicity, metabolite levels, cell structure, and autophagy in ccRCC cell lines in culture.
Results
We found that NDUFA4L2 mRNA and protein are highly expressed in ccRCC samples but undetectable in normal kidney tissue samples, and that NDUFA4L2 mRNA expression correlates with tumor stage and lower overall survival. Additionally, we demonstrated that NDUFA4L2 is a HIF1α target in ccRCC and that NDUFA4L2 knockdown has a profound anti-proliferative effect, alters metabolic pathways, and causes major stress in cultured RCC cells.
Conclusions
Collectively, our data show that NDUFA4L2 is a novel molecular target for ccRCC treatment.
Keywords: NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4-like 2 (NDUFA4L2), clear cell renal cell carcinoma (ccRCC), kidney cancer, carcinogenesis, tumor metabolism, hypoxia inducible factor 1α (HIF1α), von Hippel-Lindau (VHL), mitochondria, autophagy
Introduction
Clear cell renal cell carcinoma (ccRCC) is the most common form of kidney cancer, representing more than 80% of all diagnoses (1). ccRCC is a disease of altered cell metabolism (2), and many genes known to contribute to the development and/or progression of ccRCC are involved in sensing oxygen, iron, nutrients, and energy (3). Altered metabolism is now a widely accepted hallmark of cancer (4), and metabolic alterations greatly influence tumor development and progression (5). Thus, targeting metabolic abnormalities in ccRCC may provide novel opportunities for developing more effective treatments (2).
The majority of ccRCCs possess an inactivating mutation in the von Hippel-Lindau (VHL) tumor suppressor gene, which leads to the stabilization of hypoxia inducible factors (HIFs) (6, 7). HIF1α and HIF2α are transcription factors that influence expression of many genes involved in altered metabolic pathways in cancer, including glucose uptake, enhanced glycolytic metabolism, and decreased mitochondrial respiration (8). We previously demonstrated that expression of a constitutively active HIF1α mutant protein exclusively in kidney proximal tubule cells is sufficient to drive a program of early tumorigenesis in a murine model of ccRCC called the TRACK (TRAnsgenic model of Cancer of the Kidney) model (9). TRACK mice exhibit increased expression of HIF1α target genes that are associated with a shift in metabolism from mitochondrial oxidative phosphorylation to increased aerobic glycolysis and lactate production, similar to what is observed in human ccRCC (10). Additionally, our metabolomics data show that TRACK kidneys and human ccRCC samples have increased levels of glycolytic intermediates and lactate, and decreased levels of metabolites of the TCA cycle (10). Similar to our findings in the TRACK kidneys, Arreola et al. found that stable expression of HIF1α in primary kidney epithelial cells increased expression of key glycolytic enzymes and favored pyruvate conversion into lactate under nutrient poor conditions (11). In contrast, stable expression of HIF2α in primary kidney epithelial cells did not appear to mediate glycolysis or inhibit oxidative phosphorylation (11). Indeed, HIF2α had minimal influence on glycolysis and was found to support oxidative phosphorylation in a nutrient rich environment (11).
In the current study we evaluated the levels and role of the gene NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4-like 2 (NDUFA4L2, Gene ID: 56901). NDUFA4L2 is one of the most highly expressed genes in almost all ccRCC samples compared to healthy kidney tissue, according to several sets of microarray data from the Oncomine database (12–15) and RNA sequencing (RNAseq) data from The Cancer Genome Atlas (TCGA) (16, 17), and NDUFA4L2 expression is similarly increased by >50 fold in TRACK kidneys (10, 16). Hypoxia induces NDUFA4L2 expression via a mechanism involving HIF1α, but not HIF2α (18). Blocking HIF1α by RNA interference abrogated NDUFA4L2 expression in a VHL negative ccRCC cell line, whereas blocking HIF2α expression by RNA interference did not affect the expression of NDUFA4L2 (18). Under hypoxic conditions NDUFA4L2 is involved in lowering mitochondrial oxygen consumption and Complex I mitochondrial activity, which in turn causes a shift from mitochondrial respiration to anaerobic glycolysis and reduces ROS production (18). Thus, NDUFA4L2 plays a role in altering cellular metabolism when HIF1α is stabilized and active within a cell. In the current study we analyzed the functions of NDUFA4L2 in ccRCC.
Materials and Methods
Analysis of published data
The Oncomine database was used to compare microarray data from human ccRCC samples and matched normal kidney tissue (19). We analyzed the most highly expressed transcripts in tumors compared to normal kidney tissue in four studies (12–15).
Clinical parameters and next generation RNA sequencing data (RNAseqv2) were obtained from TCGA ccRCC and matched normal kidney tissue (KIRC) sample set. Normalized gene counts (gene counts re-scaled according to library size) from each patient were compiled into a tab-delimited file for downstream analysis by EdgeR software (20) and hierarchical cluster analysis completed using Cluster3.0 (21). Clinical data used for these studies include the American Joint Committee on Cancer (AJCC) stage and Fuhrman grade of the sequenced tumors and the survival status of the patient from which the tumor was removed.
We used the integrated genome viewer to analyze a published HIF1α ChIPseq (chromatin immune-precipitation coupled with next generation sequencing) dataset (GEO: GSE39089) in the context of the HG18 genome build (22). These data were performed to assess HIF1α binding in human umbilical vein endothelial cells in hypoxic and normoxic conditions.
Tissue samples
Human tissue samples were obtained from patients who underwent radical or partial nephrectomy at Weill Cornell Medical College (WCMC). The pathologist selected samples of tumor and normal kidney tissue to be used for research. The Institutional Review Board at WCMC approved this study.
Male C57BL/6 wild type mice were obtained from Jackson Laboratories and TRACK mice were generated as described (9). All procedures involving the use of mice were approved by the WCMC Institutional Animal Use and Care Committee.
Cell Proliferation and Colony Formation Assays
RCC4 (ATCC) and SKRC48 (Sloan Kettering Institute, not authenticated) cell lines were cultured in DME medium (MP Biomedicals; 1033122) containing 10% fetal bovine serum (Gibco, Life Technologies; 10439-016), Caki1 cells (ATCC) were grown in McCoy’s 5a Medium Modified (Gibco; 16600-082) containing 10% fetal bovine serum, and 786-O and HK2 cells (ATCC) were grown in RPMI-1640 medium (Gibco; 11875-093) containing 10% fetal bovine serum. Cells were cultured in a humidified tissue culture incubator at 37°C in a 10% CO2 atmosphere.
For proliferation assays, cells were plated at 2.5 × 104 per well in multi-well culture plates. Cell numbers were counted in triplicate using an electron particle counter (Coulter Z; Beckman Coulter) after 1, 3, 5, and 7 d of culture. For colony formation assays, cells were plated at 500 per 6-well plate. After approximately 14 days, cells were stained with crystal violet. The number of colonies was quantified using ImageJ software (National Institutes of Health).
Knockdown of NDUFA4L2 using shRNA
We produced shRNA containing lentiviral particles as described (23). We used two pLKO shNDUFA4L2 vectors directed specifically towards human NDUFA4L2 (Sigma Aldrich; TRCN0000046592 and TRCN0000046589) or a pLKO non-targeting shRNA plasmid (control) (Supplementary Table S1). RCC4, SKRC48, Caki1, 786-O, and HK2 cell lines were infected with shRNA lentiviral particles, and 4 μg/mL polybrene (Aldrich; 107689) for 24 hours. After infections, cells were treated with 2 μg/mL (SKRC48, Caki1, and HK2) or 4 μg/mL (RCC4 and 786-O) puromycin (Sigma; P8833) for 4 days and knockdown was assessed by Western blotting.
Western blot analysis
Proteins were extracted in SDS final sample buffer, boiled, separated on SDS-PAGE gels, and transferred onto nitrocellulose membranes. Membranes were probed with primary antibodies (Supplementary Table S2) overnight at 4°C, and further incubated with secondary antibody at 22°C for 1 h. Chemiluminescence was recorded with a quantitative gel imaging station (Bio-Rad ChemiDoc) and data were analyzed using Image Lab software (Bio-Rad).
RNA isolation and PCR reactions
Total RNA was isolated using TRIzol reagent (Ambion, Life Technologies; 15596018). mRNA (5 μg) was reverse transcribed using the qScript cDNA SuperMix (Quanta Biosciences; 95048) (24). Semi-quantitative RT-PCR reactions were performed with 2 μg cDNA as template using a BioRad iCycler (25). All primers were designed around introns (Supplementary Table S3).
Metabolomics analysis
Cells were plated on 10-cm plates and metabolites were extracted following established protocols using 80% methanol (cooled to −80°C) (10, 26). Metabolites were dried using a SpeedVac (Savant) and stored at −80°C until being shipped on dry ice to the Metabolomics Core Facility at Beth Israel Deaconess Medical Center (BIDMC). Targeted mass spectrometry was performed by the BIDMC core facility to analyze metabolites (26).
Metabolomics data were processed, normalized to protein, and analyzed using the MetaboAnalyst program (27) For statistical analyses, one-way ANOVA was performed on parental RCC4 vs. shNDUFA4L2.1 treated cells and on shCTL treated vs. shNDUFA4L2.1 treated cells. Metabolites that were significantly altered (p<0.05) in shNDUFA4L2.1 treated cells compared to both parental and shCTL treated cells were evaluated and used for pathway analysis.
Electron microscopy
Cells were plated on a 6-well plate and grown to approximately 90% confluence. Cells were fixed in 2.5% glutaraldehyde, 4% paraformaldehyde, and 0.02% picric acid in 0.1M sodium cacodylate buffer, pH 7.3 for at least 1 hour at 4°C (28). The plates were processed and embedded in EMbed 812 resin (Electron Microscopy Sciences) as previously described (29). Images were taken by the WCMC Electron Microscopy & Histology core facility on a JSM 100-CX transmission electron microscope (JEOL USA, Inc.).
For each cell type, three images were taken of five different cells (15 images total/cell type). Analyses of images were performed in a blinded manner with respect to sample identity. Swollen mitochondria were defined as large, round in shape with few, disorganized cristae. Improved mitochondria were contracted and regular in shape, with more organized cristae. Autophagosomes were defined as vacuoles with double membranes and/or undigested materials, and autolysosomes were defined as vacuoles with single membranes and amorphous electron-dense regions (30).
Statistical analyses
Student’s t-test was performed on at least three separate, independent experiments (n=3 or >3) using the Graph Pad Prism 6.0 software. Metabolomics data were analyzed with MetaboAnalyst (27) and RNAseq data were analyzed with EdgeR software (Bioconductor) (20).
Results
NDUFA4L2 mRNA and protein levels are increased in human ccRCC
To identify potentially important genes that contribute to renal carcinogenesis, we first searched the Oncomine database of published microarray data from matched malignant and non-malignant samples (19). We analyzed four studies that in total compared gene expression data from 69 ccRCC samples and 45 adjacent, non-tumor kidney samples (12–15) (Table 1). Additionally, we collected TCGA RNAseqV2 data from 530 ccRCC samples and 72 adjacent non-tumor samples (Table 1) (17). In each of these studies, one gene in particular, NDUFA4L2, stood out as having a strikingly large fold increase in ccRCC. Indeed, Yusenko et al. (14) found that NDUFA4L2 transcripts are increased by nearly 90-fold in ccRCC compared to non-tumor kidney samples (Table 1). Additionally, ~90% of the 530 ccRCC samples for which RNAseq data is available in TCGA possessed elevated NDUFA4L2 expression (17). Although NDUFA4L2 mRNA is considerably increased in ccRCC compared to normal kidney tissue according to microarray and RNAseq data, this gene has not been well characterized and its role in ccRCC has not been thoroughly defined.
Table 1.
Summary of four studies that collected microarray data and TCGA RNAseq data from ccRCC samples and adjacent non-tumor samples.
| Reference | ccRCC samples | Non-tumor samples | NDUFA4L2 fold change in ccRCC | p-Value |
|---|---|---|---|---|
| Gumz et al (12) | 10 | 10 | 53.935 | 2.60 × 10−11 |
| Jones et al (15) | 23 | 23 | 8.361 | 7.32 × 10−8 |
| Yusenko et al (14) | 26 | 3 | 86.564 | 7.04 × 10−15 |
| Lenburg et al (13) | 9 | 9 | 23.340 | 5.51 × 10−9 |
| TCGA (17) | 530 | 72 | 49.121 | 3.06 × 10−105 * |
For TCGA data, q-value is listed instead of p-value.
Following this analysis of published mRNA expression data, we evaluated both NDUFA4L2 mRNA and protein levels in human ccRCC tumors and matched non-tumor kidney tissue by performing semi-quantitative PCR and Western blot analyses, respectively. We found that NDUFA4L2 transcripts were highly elevated in tumors compared to normal, adjacent kidney tissue (Fig. 1A, left panel). Tumor 1 has an approximately 20-fold and tumor 2 a ~30-fold increase in NDUFA4L2 transcripts compared to non-malignant kidney tissue samples from the same patients. Additionally, we show that NDUFA4L2 protein (~13 kDa) is highly expressed in tumor samples from patients 3, 4, and 5 (Fig. 1A, right panel). Quantitation of bands indicates that NDUFA4L2 protein is increased by 10 to 30-fold in tumors compared to adjacent non-malignant kidney tissue. These mRNA and protein data from human patient samples are striking, as NDUFA4L2 is highly expressed in each of the five tumor samples, and is barely detectable in each of the non-malignant renal tissue samples under our conditions for PCR and western blotting (Fig. 1A). These data also suggest that elevated expression of NDUFA4L2 is a common event in ccRCC and that expression is restricted to the tumors.
Figure 1. NDUFA4L2 expression is elevated in ccRCC and is clinically significant.

(A) NDUFA4L2 mRNA levels (left panel) and NDUFA4L2 and NDUFA4 protein levels (right panel) in human ccRCC samples vs. matched normal kidney tissue. (B) Levels of NDUFA4L2 mRNA (left panel) and NDUFA4L2 and NDUFA4 protein (right panel) in several human ccRCC cell lines and in HK2 cells, a normal, non-transformed, proximal tubule cell line. For semiquantitative RT-PCR, HPRT mRNA is used as a loading control. For Western blot analysis β-actin is the loading control. Results are representative of experiments performed on three separate occasions with fresh cells (n=3) with similar results. (C) NDUFA4L2 mRNA levels in normal kidney tissue and in each American Joint (AJCC) stage of ccRCC as reported by TCGA. (D) NDUFA4L2 mRNA levels in normal kidney tissue and in each Fuhrman grade of ccRCC as reported by TCGA. The expression values are the normalized NDUFA4L2 counts. The results are presented as mean ± standard error of the mean (SEM). Data were analyzed in EdgeR to generate a q-values. (E) A Kaplan-Meier curve to compare the overall survival of patients with low NDUFA4L2 expression (normalized count < 60,000, n = 448) versus high expression (normalized count > 60,000, n =60). The p-value was generated using the log-rank test.
To identify relevant cell line models to utilize in further experimentation, we evaluated NDUFA4L2 at the mRNA and protein levels in various human clear cell renal cell carcinoma cell lines and a normal, non-transformed human proximal tubule cell line, HK2 (Fig. 1B). RCC4 and SKRC48 ccRCC cell lines have activated HIF1α and thus we detected NDUFA4L2 mRNA and protein in these cells (Fig. 1B). We did not detect NDUFA4L2 mRNA in the ccRCC cell lines Caki1 and 786-O, in which HIF1α is not active, nor in the normal proximal tubule cell line HK2 (Fig. 1B). Based on high NDUFA4L2 expression, we chose to use the RCC4 and SKRC48 cell lines as in vitro ccRCC models for further studies.
The commercially available antibodies targeting NDUFA4L2 (Supplementary Table S1) also detect NDUFA4 (~10 kDa) (Fig. 1A,B, right panels). NDUFA4L2 and NDUFA4 have high homology at the C-terminus, the region of the NDUFA4L2 protein that the antibodies recognize. NDUFA4 was recently shown to be a subunit of mitochondrial Complex IV (cytochrome c oxidase) (31). NDUFA4 protein is decreased in the tumor samples by approximately 10 to 30-fold (Fig. 1A, right panel) and by approximately 5-fold in RCC4 and SKRC48 cells compared to Caki1 cells (Fig. 1B, right panel).
Increased NDUFA4L2 expression correlates with more advanced human ccRCC stage, grade, and reduced survival
To assess if increased NDUFA4L2 expression in ccRCC had any clinical relevance, we evaluated TCGA RNAseq data and corresponding clinical parameters (17). For our study, level 2 RSEM normalized expression count data were obtained from the TCGA. With regard to available clinical information, we prioritized tumor AJCC stage, Fuhrman grade, and survival data from patients with histologically confirmed ccRCC. AJCC staging is the most important factor predicting prognosis and it is based on tumor size and metastatic involvement (32). Patient:stage distributions were: stage I (n=253), stage II (n=56), stage III (n=125) and stage IV (n=81). Stage III ccRCCs have a ~25% increase in NDUFA4L2 mRNA expression compared to stages I and II (Fig. 1C). Fuhrman grading, which refers to how closely the cancer cells look like normal kidney cells under a microscope, is another important prognosis factor. Patient:grade distributions were: grade I (n=11), grade II (n=229), grade III (n=203), grade IV (n=75). Grade III ccRCCs have a ~35% increase in NDUFA4L2 mRNA expression compared to stage II (Fig. 1D).
Next, we evaluated overall survival based on NDUFA4L2 mRNA levels (Fig. 1E). Notably, nearly all ccRCC samples (~90%) have high NDUFA4L2. Consequently, we defined two patient subgroups based on normalized NDUFA4L2 expression, (i) patients with lower NDUFA4L2 expression (counts < 60,000; n= 448), and (ii) patients with higher NDUFA4L2 expression (normalized count > 60,000; n= 60), which is >90-fold higher NDUFA4L2 expression than in normal kidney tissue. We found that patients with the highest NDUFA4L2 mRNA levels exhibited lower overall survival compared to patients with lower NDUFA4L2 mRNA (log rank test; p= 0.0011) (Fig. 1E). For patients with lower NDUFA4L2 expression, the median survival is 2454 days, and for patients with the highest NDUFA4L2 expression the median survival is 1111 days (Fig. 1E).
NDUFA4L2 is HIF1α regulated in ccRCC
It was previously reported that NDUFA4L2 is a direct HIF1α target under hypoxic conditions (18). To validate HIF1α regulation of NDUFA4L2 in ccRCC we assessed NDUFA4L2 expression in the kidneys of the TRACK murine model of ccRCC, as well as in HIF2αM3 transgenic positive (TG+) and transgenic negative (TG−) mice. TRACK mice express a constitutively active form of HIF1α specifically in proximal tubule cells and HIF2αM3 TG+ mice express a constitutively active form of HIF2α specifically in proximal tubule cells (9, 33). We previously reported that TRACK mice display a tumorigenesis program that more closely resembles early human ccRCC than HIF2αM3 TG+ mice (9, 10, 16, 33, 34). We found that NDUFA4L2 expression is regulated by HIF1α and not HIF2α, as NDUFA4L2 transcripts and protein are highly expressed in the TRACK kidneys, but not in HIF2αM3 TG+ or TG− mouse kidneys (Fig. 2A). Additionally, western blot analyses of different tissues from TRACK mice indicate that NDUFA4L2 expression is unique to the kidneys where constitutively active HIF1α is specifically expressed (Fig. 2B). NDUFA4L2 protein cannot be detected in the normal pancreas, liver, heart, lung, testes, or intestines of TRACK mice (Fig. 2B).
Figure 2. NDUFA4L2 expression is mediated specifically by HIF1α in TRACK kidneys.
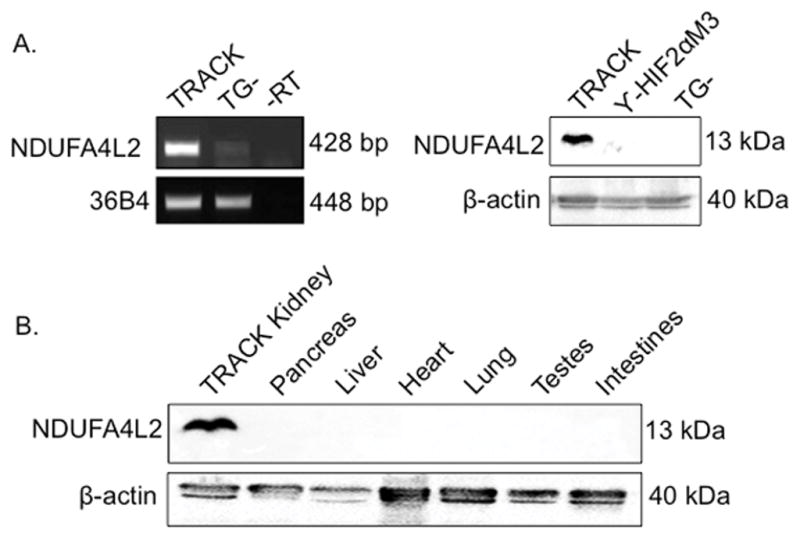
(A) Expression of NDUFA4L2 mRNA (left panel) and protein (right panel) in kidney tissue from the TRACK model, γ-HIF2αM3 TG+ mice, and TG− mice. (B) NDUFA4L2 protein expression in various tissues of the TRACK model. For semiquantitative RT-PCR, 36B4 mRNA is used as a loading control. For Western blot analysis β-actin is the loading control. Western blot and RT-PCR were performed on three or more mice (n≥3) and results are representative of triplicate experiments with similar results.
To determine whether HIF1α plays a role in direct regulation of NDUFA4L2, we used the integrated genome viewer to analyze a published HIF1α ChIPseq (chromatin immune-precipitation coupled with next generation sequencing) dataset (GEO: GSE39089) (22). These data indicate that HIF1α is recruited to the NDUFA4L2 locus in hypoxic human umbilical vein endothelial cells (Supplementary Fig. S1A). It is therefore likely that HIF1α plays a similar role in kidney proximal tubule epithelial cells. Additionally, hierarchical clustering analysis of the TCGA ccRCC dataset shows that NDUFA4L2 clusters with other known HIF1α target genes in ccRCC, such as VEGFA, HK2, and SLC16A3 (Supplementary Fig. S1B).
NDUFA4L2 knockdown impairs cell proliferation and colony formation in ccRCC cell lines
To assess the functional significance of NDUFA4L2 in ccRCC, we performed knockdown of NDUFA4L2 with two independent shRNAs via lentivirus-mediated infection in RCC4 and SKRC48 ccRCC cells that express NDUFA4L2 (Fig. 1B). We confirmed NDUFA4L2 knockdown by >90% at the protein level with both NDUFA4L2 shRNA targeting constructs, shNDUFA4L2.1 and shNDUFA4L2.2, by Western blotting. No changes in NDUFA4L2 levels were observed when RCC4 and SKRC48 cells were infected with a non-targeted control hairpin (shCTL) (Fig. 3A).
Figure 3. Effective knockdown of NDUFA4L2 (13 kDa) in RCC4 and SKRC48 cells with shRNA impairs cellular proliferation and colony formation.

(A) Western blot analysis of NDUFA4L2 levels in RCC4 and SKRC48 parental cell lines and cells treated with NDUFA4L2 specific shRNA (see Materials and Methods for sequences) for 24 hours followed by a 96 hour selection with puromycin. NDUFA4L2 (13 kDa) is knocked down by greater than 95% in RCC4 cells and greater than 98% in SKRC48 cells following treatment with shNDUFA4L2 constructs 1 and 2 and is unchanged when cells are infected with a non-targeting construct (shCTL). β-actin was used as a loading control. (B) Cell proliferation assays of RCC4 and SKRC48 cells after viral infection with shRNA and selection. Data are the average of three independent experiments (n=3) and error bars represent SEM. (C) Colony formation of RCC4 and SKRC48 parental cells and cells treated with the indicated viral shRNA for 24 hours followed by selection with puromycin for 96 hours. Representative plates of experiments performed at least three times (n=3) with fresh cells are shown. (D) Graph of the number of colonies formed (expressed as mean ± SEM). ** p < 0.01; *** p < 0.005; **** p < 0.0001.
We next analyzed the growth characteristics of ccRCC cells that lack NDUFA4L2 expression. Both RCC4 and SKRC48 cells infected with shNDUFA4L2 constructs showed reduced proliferation over 7 days compared to the parental or shCTL-treated cells (Fig. 3B). In both RCC4 and SKRC48 cell lines, shNDUFA4L2.1 treatment reduced cell proliferation by more than 60% and shNDUFA4L2.2 treatment reduced cell proliferation by approximately 90% (Fig. 3B).
As further confirmation of the importance of NDUFA4L2 for cell proliferation, we assayed for clonogenic growth in RCC4 and SKRC48 cells lacking NDUFA4L2 expression. Consistent with the proliferation assays, NDUFA4L2 knockdown greatly diminished the capacity of cells to form colonies (Fig. 3C), reducing the numbers of colonies by > 90% (Fig. 3D). Collectively, the proliferation and colony formation assays show that NDUFA4L2 expression promotes cell proliferation and clonogenic growth of VHL-negative ccRCC cells.
We also performed viral infection with the shCTL, shNDUFA4L2.1, and shNDUFA4L2.2 constructs in Caki1 and 786-O ccRCC cell lines and HK2, a non-transformed proximal tubule cell line, which do not express NDUFA4L2 (Fig. 1B). We found that treatment with the shCTL and shNDUFA4L2.1 constructs had no effect on proliferation and clonogenicity (Supplementary Fig. S2A,B). Treatment with the shNDUFA4L2.2 construct impaired proliferation by ~30–50% in the Caki1 and 786-O cell lines and impaired clonogenicity by ~80% in Caki1 cells (Supplementary Fig. S2A,B). However, the shNDUFA4L2.2 did not impair proliferation in the HK2 normal proximal tubule cell line (Supplementary Fig. S2A). These data suggest that the Caki1 and 786-O cell lines have low levels of NDUFA4L2 not detectable in our assays or that shNDUFA4L2_2 has an additional off-target effect.
NDUFA4L2 knockdown alters metabolic pathways
To evaluate why NDUFA4L2 is critical for proliferation and clonogenic growth of RCC4 and SKRC48 cells, we next evaluated changes in metabolite levels upon NDUFA4L2 knockdown. Under hypoxic conditions, NDUFA4L2 inhibits Complex I of oxidative phosphorylation and thus mediates a cellular shift to glycolysis for generating ATP (18). ccRCC is widely characterized as a metabolic cancer that relies on glycolysis instead of oxidative phosphorylation to produce ATP (3), which is an adaptive measure to facilitate the uptake and incorporation of nutrients into biomass in order to produce new cells (35).
A metabolomics analysis demonstrated that RCC4 cells treated with shNDUFA4L2.1 showed increases in the levels of several tricarboxylic acid (TCA) cycle intermediates and decreases in the levels of several pentose phosphate pathway intermediates compared to parental and shCTL treated cells (Fig. 4A,B). With respect to the TCA cycle, we observed 2-fold or greater increases in the levels of citrate/isocitrate, α-ketoglutarate, succinate, and oxaloacetate (Fig. 4A). In contrast, we saw marked decreases in the pentose phosphate pathway intermediates ribose 5-phosphate, sedoheptulose 1,7-bisphosphate, glyceraldehyde 3-phosphate, and fructose 6-phosphate (Fig. 4B). These data suggest that upon NDUFA4L2 knockdown, pyruvate generated by the glycolysis pathway is converted to acetyl-CoA to fuel the TCA cycle and as such, glycolytic intermediates are not shunted into the pentose phosphate pathway (Fig. 4C). This is important because proliferating cancer cells shunt glycolytic metabolites into the pentose phosphate pathway to make ribose-5-phosphate, which is needed for nucleic acid production (35). Ribose 5-phosphate is 1.6-fold lower in shNDUFA4L2.1 treated RCC4 cells compared to shCTL treated cells, and 2.2-fold lower compared to parental RCC4s (Fig. 4B). Additionally, a principal component analysis showed that RCC4 parental and shCTL treated cells cluster distinctly from RCC4 cells treated with shNDUFA4L2.1 (Supplementary Fig. S3A), and pathway analysis of significantly altered metabolites in the RCC4 cells treated with shNDUFA4L2.1 compared to the parental and shCTL treated cells demonstrated that two of the most significantly altered metabolic pathways were metabolism of pyrimidine and purine nucleotides (Supplementary Fig. S3B). Thus, the metabolomics data demonstrate that NDUFA4L2 knockdown alters metabolism in a manner such that the pentose phosphate pathway is down-regulated and less ribose 5-phosphate is being produced, which in turn likely reduces purine and pyrimidine synthesis.
Figure 4. Knockdown of NDUFA4L2 causes metabolic alterations.

(A) Comparison of the levels of the TCA cycle intermediates citrate/isocitrate, α-ketoglutarate, succinate, and oxaloacetate in parental RCC4 cells and cells infected with the indicated shRNA constructs for 24 hours followed by puromycin for 96 hours. (B) Comparison of the levels of the pentose phosphate pathway intermediates ribose 5-phosphate, sedoheptulose 7-phosphate, glyceraldehyde 3-phosphate, and fructose 6-phosphate in parental RCC4 cells and cells infected with the indicated shRNA constructs for 24 hours followed by puromycin for 96 hours. (C) Schematic overview of the changes in the TCA cycle and pentose phosphate pathway upon treatment with shNDUFA4L2.1 in RCC4 cells. Arrows next to metabolites indicate that the metabolite levels are changed in shNDUFA4L2.1 treated cells compared to parental and shCTL treated cells. Metabolomics analysis was performed on samples in triplicate (n=3 for each group) and data are graphed as the mean ± SEM. ** p < 0.01; **** p < 0.0001.
NDUFA4L2 knockdown improves mitochondrial morphology and induces autophagy
Cancer cells that have a reduced capacity for aerobic respiration also exhibit abnormalities in both the content and composition of their mitochondria (36). We hypothesized that NDUFA4L2 inhibits aerobic respiration in ccRCC, and that elevated expression of NDUFA4L2 contributes to abnormal mitochondrial structure. When we analyzed RCC4 parental, shCTL-treated, and NDUFA4L2 knockdown cells by transmission electron microscopy (TEM), we found that RCC4 parental and shCTL treated cells had many swollen mitochondria with disorganized cristae (Fig. 5A, filled arrows). However, RCC4 cells treated with both shNDUFA4L2 constructs had more contracted mitochondria with improved cristae (Fig. 5A, open arrows). Indeed, approximately 50% of the mitochondria in parental and shCTL treated RCC4 cells are swollen with disorganized cristae, while less than 10% display this morphology in cells treated with shNDUFA4L2.1 and shNDUFA4L2.2 (Fig. 5B). Though the morphology of the mitochondria is changed when NDUFA4L2 levels are reduced, the numbers of mitochondria remain the same (Fig. 5C).
Figure 5. NDUFA4L2 knockdown improves mitochondrial morphology and induces autophagy in RCC4 cells.

(A) Representative images from transmission electron microscopy of RCC4 parental cells and cells infected with the indicated shRNA constructs for 24 hours followed by puromycin for 96 hours. Filled arrows point to swollen mitochondria with disorganized cristae, open arrows point to contracted mitochondria with improved cristae, filled stars indicate autophagosomes, and open stars indicate autolysosomes. Magnification, 25000 x. Scale bar on the bottom left represents 2 μm. (B) Quantification of the percentages of swollen mitochondria with disorganized cristae compared to the percentages of contracted mitochondria with improved cristae. (C) Quantification of the number of mitochondria per 100 μm2. (D) Quantitation of the number of autophagosomes and autolysosomes per 100 μm2 in RCC4 cells. (E) Western blot analysis of LC3 in RCC4 and SKRC48 cell lines following 24 hours of shRNA treatment, 96 hours of puromycin selection, and a 5 day, untreated, incubation period. β-actin was used as loading control. Quantification of TEM data was performed in a blinded manner. Data were collected from three images from five different cells (15 images/cell type) and graphed as mean ± SEM. ** p < 0.01; *** p< 0.005; **** p < 0.0001; n.s. p>0.05.
NDUFA4L2 knockdown also promotes autophagy, a process by which cytosolic components are engulfed in double-membrane autophagosomes that fuse with lysosomes for degradation (37), and mitochondrial turnover is dependent on autophagy (38). In RCC4 cells treated with both shNDUFA4L2 constructs, we visualized many autophagosomes (Fig. 5A, filled stars) as defined by vacuoles with double membranes and/or undigested materials, and autolysosomes (Fig. 5A, open stars) as defined by vacuoles with single membranes and amorphous electron-dense regions (30). We detected a 3.8 (±1.3) -fold increase in autophagosomal vesicles in RCC4 cells treated with shNDUFA4L2.1 and a 5.1 (±1.6) –fold increase in shNDUFA4L2.2 treated cells compared to parental cells (Fig. 5D). Additionally, we counted more autolysosomes than autophagosomes in RCC4 cells treated with shNDUFA4L2.1 and shNDUFA4L2.2 (Fig. 5D).
LC3, or tubule-associated protein 1/A/1B-light chain 3, is a biomarker of autophagy. During autophagy, the cytosolic form of LC3 (LC3-I) is converted to its lipidated form, LC3-II, which is recruited to autophagosomal membranes. We detected a >8-fold induction of LC3-II in both RCC4 and SKRC48 lines treated with shNDUFA4L2.1 and shNDUFA4L2.2 compared to the parental and shCTL-treated cells (Fig. 5E). Together, the improved mitochondrial structures and the induction of autophagy upon NDUFA4L2 knockdown suggest that NDUFA4L2 knockdown promotes mitochondrial turnover via autophagy.
We also analyzed Caki1 cells, which do not have elevated expression of NDUFA4L2 (Fig. 1B), to determine if shRNA treatment improves mitochondria morphology and/or induces autophagy. We found that mitochondria in Caki1 parental and Caki1 cells treated with shCTL, shNDUFA4L2.1, and shNDUFA4L2.2 have contracted mitochondria with typical cristae (Supplementary Fig. S4A). There was no improvement in mitochondrial morphology in Caki1 upon treatment with either shNDUFA4L2 construct (Supplementary Fig. S4A).
Next, we evaluated the numbers of autophagic vacuoles in Caki1 parental and shRNA treated cells. We found that Caki1 cells treated with shNDUFA4L2.2 had more autophagic vacuoles than Caki1 parental cells and cells treated with shCTL and shNDUFA4L2.1 (Supplementary Fig. S4B). However, the increase in autophagic vacuoles seen in Caki1 cells treated with shNDUFA4L2.2 was not as high as in RCC4 cells treated with shNDUFA4L2.1 or shNDUFA4L2.2 (Supplementary Fig. S4B and Fig. 5D). Additionally, we did not detect an increase in LC3-II levels in Caki1, 786-O, or HK2 cells treated with either shNDUFA4L2 construct (Supplementary Fig. S5).
Discussion
Altered metabolism is a major characteristic of ccRCC. We recently reported on the importance of HIF1α in mediating altered tumor metabolism in the TRACK model of ccRCC, similar to the changes observed in human ccRCC (10, 16). Targeting pathways that mediate altered metabolism may provide novel and more effective treatments (2, 17). In this study, we demonstrated that a HIF1α target gene involved in mediating glycolysis, NDUFA4L2, is highly expressed at the mRNA and protein levels in human ccRCC, yet is not expressed in normal kidney tissue (Table 1 and Fig. 1). We also showed that NDUFA4L2 mRNA levels are greater in the more advanced staged and graded ccRCCs (Fig. 1C, D), and that patients with the highest expression of NDUFA4L2 in their tumors exhibit a lower overall survival rate (Fig. 1E). Moreover, we found that NDUFA4L2 knockdown leads to a profound decrease in proliferation and colony formation In HIF1α positive cell lines (Fig. 3). Collectively, these data demonstrate NDUFA4L2 is an attractive therapeutic target to treat ccRCC.
Previous work on NDUFA4L2 is limited. In the one major study charactering NDUFA4L2, Tello et al demonstrated that NDUFA4L2 is a direct HIF1α target gene (18). By assessing NDUFA4L2 expression in our various murine models, we confirmed that NDUFA4L2 is regulated by HIF1α, but not HIF2α (Fig. 2). Although deletion of HIF1α has previously been reported in ccRCC (39, 40), the results shown here and available in the TCGA-KIRC RNAseq expression data support an important role for HIF1α, acting in part via NDUFA4L2, in the majority of human ccRCCs (34).
We and others have shown that HIF1α mediates the Warburg effect in ccRCC, a phenomenon in which cancer cells rely on aerobic glycolysis to generate ATP instead of the TCA cycle and oxidative phosphorylation (10, 16, 41). Tello et al showed that NDUFA4L2 inhibits complex I of oxidative phosphorylation to mediate a shift to glycolysis (18). Thus, it is likely that NDUFA4L2 has a role in mediating the Warburg effect in ccRCC. Interestingly, aerobic glycolysis is a less efficient method for producing ATP. One explanation for why aerobic respiration is favored is that proliferating cancer cells not only require ATP, but also need nucleotides, fatty acids, and proteins (35). Reprogrammed metabolism supports the synthesis of these macromolecules required for rapid proliferation (35). We demonstrated that NDUFA4L2 is important for supporting ccRCC metabolism to generate nucleic acids (Fig. 4). Nucleic acids, which include DNA and RNA, are vital building blocks for cell growth and division. Thus, it is likely that NDUFA4L2 knockdown impairs proliferation in ccRCC cells, at least in part, by disrupting the synthesis of critical macromolecules, DNA and RNA.
Cancer cells that have reduced aerobic respiration frequently display abnormalities in both the content and composition of their mitochondria (36). In parental RCC4 and RCC4 shCTL treated cells we visualized swollen mitochondria with abnormal, disorganized cristae (Fig. 5). As mitochondrial morphology and function are closely linked (42), these data suggest that these cells have diminished mitochondrial capacity. However, when NDUFA4L2 levels are reduced, we found that the mitochondrial structures improve and the cristae become less disorganized (Fig. 5). These findings, and the metabolomics data indicating that TCA cycle intermediates are increased upon NDUFA4L2 knockdown (Fig. 3), suggest that mitochondrial bioenergetics are improved when NDUFA4L2 levels are reduced. The restoration of mitochondrial bioenergetics is currently being pursued as an approach for treating a variety of different diseases, including aging-related diseases. SS-31, a mitochondrial-targeted peptide, is one example of a compound that protects mitochondrial cristae and promotes ATP synthesis by interacting with cardiolipin, an inner mitochondrial membrane phospholipid that has a central role in in the structure and organization of cristae (43). Interestingly, SS-31 was shown to protect the kidneys of rats after ischemia (44). Similar to our data, treatment with SS-31 resulted in more normal and elongated mitochondria with finely stacked cristae membranes (44). Targeting mitochondrial biogenesis for the treatment of ccRCC has not been well studied, and SS-31 has not been tested in the context of this disease. However, based on our data from knocking down NDUFA4L2 in ccRCC cells and the success of SS-31 in protecting rat kidneys from ischemia, further research into improving mitochondria bioenergetics in the context of active HIF1α as a treatment for ccRCC is warranted.
Mitochondrial turnover is dependent on the intracellular degradation process autophagy (38), which we detected in the ccRCC cells uponNDUFA4L2 knockdown (Fig. 5). Thus it is possible that autophagy is induced in NDUFA4L2 knockdown cells to remove the damaged mitochondria in order to generate more functional mitochondria. Another potential explanation for the induction of autophagy in the RCC4 and SKRC48 NDUFA4L2 knockdown cells is that the cells are nutrient-deprived when NDUFA4L2 levels are diminished. It is well established that autophagy is induced under starvation or serum deprivation (38), and although nutrients are readily available to NDUFA4L2 knockdown cells in culture, their ability to metabolize nutrients is altered (Fig. 4). While further experiments are required to delineate the exact influence of NDUFA4L2 knockdown on autophagy, it is clear that NDUFA4L2 knockdown causes major stress on RCC4 and SKRC48 cells.
ccRCC is characterized as a highly metabolic disease and alterations in metabolism are likely to be fundamental to the development of advanced ccRCC (3). Thus, targeting the metabolic basis of ccRCC could be an effective strategy for treating the disease (17, 45–47).
Targeting NDUFA4L2 for inhibition to treat ccRCC has several advantages. To start, NDUFA4L2 is an appealing target because it is so frequently and highly expressed in ccRCC but is not normally expressed in kidney tissue or other tissues of the body. Additionally, targeting NDUFA4L2 should not disrupt glycolysis in normal cells. Many of the other genes involved in increasing the rate of glycolysis in cancer cells encode enzymes that also play critical roles in non-transformed cells, and thus inhibiting them could harm healthy, normal cells. For these reasons, and because of our data showing that NDUFA4L2 knockdown profoundly inhibits ccRCC proliferation, we propose that NDUFA4L2 is a novel molecular target for inhibition to be pursued for the treatment of ccRCC.
Supplementary Material
Translational Relevance.
In this study, we show that NADH Dehydrogenase (Ubiquinone) 1 alpha subcomplex 4-like 2 (NDUFA4L2) is a novel molecular target for clear cell renal cell carcinoma (ccRCC) treatment. We demonstrate that NDUFA4L2 is highly expressed at the mRNA and protein levels in human ccRCC, yet is not expressed in normal, healthy kidney tissue. We also show that NDUFA4L2 mRNA expression is high across all stages of ccRCC compared to normal kidney, with an even greater expression in higher stage ccRCC. Additionally, patients with the highest expression of NDUFA4L2 in their tumors exhibit a lower overall survival. Knockdown of NDUFA4L2 in cell culture models of ccRCC greatly impairs proliferation, perturbs metabolic pathways, and causes major stress on RCC cells.
Acknowledgments
Financial Support
This research was supported by WCMC, the Turobiner Kidney Cancer Research Fund, the Weiss Family, and the Genitourinary Oncology Research Fund. DRM was supported by NIH T32 Training Grant (5T32CA062948). LF holds the Robert H. McCooey Genitourinary Oncology Research Fellowship and was supported in part by NIH T32 Training Grant (5T32CA062948).
We thank Sagit Goldenberg and Dr. Brian Robinson for the human ccRCC samples, and the Gudas and Nanus laboratories for thoughtful discussion.
Footnotes
Conflicts of Interests
None.
References
- 1.Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003;27:612–24. doi: 10.1097/00000478-200305000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–85. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linehan WM, Ricketts CJ. The metabolic basis of kidney cancer. Semin Cancer Biol. 2013;23:46–55. doi: 10.1016/j.semcancer.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondo K, Kaelin WG. The von Hippel-Lindau tumor suppressor gene. Exp Cell Res. 2001;264:117–25. doi: 10.1006/excr.2000.5139. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 9.Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Generation of a mouse model of Von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1 α. Cancer Res. 2011;71:6848–56. doi: 10.1158/0008-5472.CAN-11-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minton DR, Fu L, Chen Q, Robinson BD, Gross SS, Nanus DM, et al. Analyses of the Transcriptome and Metabolome Demonstrate That HIF1 α Mediates Altered Tumor Metabolism in Clear Cell Renal Cell Carcinoma. PLoS One. 2015;10:e0120649. doi: 10.1371/journal.pone.0120649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arreola A, Cowey CL, Coloff JL, Rathmell JC, Rathmell WK. HIF1α and HIF2 α exert distinct nutrient preferences in renal cells. PLoS One. 2014;9:e98705. doi: 10.1371/journal.pone.0098705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrand SN, et al. Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:4740–9. doi: 10.1158/1078-0432.CCR-07-0143. [DOI] [PubMed] [Google Scholar]
- 13.Lenburg ME, Liou LS, Gerry NP, Frampton GM, Cohen HT, Christman MF. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data. BMC Cancer. 2003;3:31. doi: 10.1186/1471-2407-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yusenko MV, Kuiper RP, Boethe T, Ljungberg B, van Kessel AG, Kovacs G. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152. doi: 10.1186/1471-2407-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones J, Otu H, Spentzos D, Kolia S, Inan M, Beecken WD, et al. Gene signatures of progression and metastasis in renal cell cancer. Clin Cancer Res. 2005;11:5730–9. doi: 10.1158/1078-0432.CCR-04-2225. [DOI] [PubMed] [Google Scholar]
- 16.Fu L, Minton DR, Zhang T, Nanus DM, Gudas LJ. Genome-Wide Profiling of TRACK Kidneys Shows Similarity to the Human ccRCC Transcriptome. Mol Cancer Res. 2015;13:870–8. doi: 10.1158/1541-7786.MCR-14-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Network CGAR. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordóñez Á, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–79. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 19.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 22.Mimura I, Nangaku M, Kanki Y, Tsutsumi S, Inoue T, Kohro T, et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol Cell Biol. 2012;32:3018–32. doi: 10.1128/MCB.06643-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beauséjour M, Beaulieu JF, et al. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res. 2013;319:1463–70. doi: 10.1016/j.yexcr.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynertson KA, Charlson ME, Gudas LJ. Induction of murine embryonic stem cell differentiation by medicinal plant extracts. Exp Cell Res. 2011;317:82–93. doi: 10.1016/j.yexcr.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laursen KB, Wong PM, Gudas LJ. Epigenetic regulation by RAR α maintains ligand-independent transcriptional activity. Nucleic Acids Res. 2012;40:102–15. doi: 10.1093/nar/gkr637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7:872–81. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–60. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito S, Karnovsky MJ. Formaldehyde-glutaraldeyde fixatives containing trinitro compounds. Journal of Cell Biology. 1968;39 [Google Scholar]
- 29.Cohen-Gould L. Handling cell culture monolayers for transmission electron microscopy. Microscopy Today. 2013;21:36–9. [Google Scholar]
- 30.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landázuri MO, et al. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012;16:378–86. doi: 10.1016/j.cmet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. 7. New York: Springer-Verlag; 2009. [Google Scholar]
- 33.Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Activation of HIF2α in Kidney Proximal Tubule Cells Causes Abnormal Glycogen Deposition but not Tumorigenesis. Cancer Res. 2013;73:2916–25. doi: 10.1158/0008-5472.CAN-12-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gudas LJ, Fu L, Minton DR, Mongan NP, Nanus DM. The role of HIF1α in renal cell carcinoma tumorigenesis. J Mol Med (Berl) 2014;92:825–36. doi: 10.1007/s00109-014-1180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in human cancer. Int J Biochem Cell Biol. 2009;41:2062–8. doi: 10.1016/j.biocel.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–36. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- 39.Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:222–35. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–81. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39:231–4. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- 42.Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) 2010;7:7. doi: 10.1186/1743-7075-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–28. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–61. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricketts CJ, Linehan WM. Intratumoral heterogeneity in kidney cancer. Nat Genet. 2014;46:214–5. doi: 10.1038/ng.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinthus JH, Whelan KF, Gallino D, Lu JP, Rothschild N. Metabolic features of clear-cell renal cell carcinoma: mechanisms and clinical implications. Can Urol Assoc J. 2011;5:274–82. doi: 10.5489/cuaj.10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaravinos A, Pieri M, Mourmouras N, Anastasiadou N, Zouvani I, Delakas D, et al. Altered metabolic pathways in clear cell renal cell carcinoma: A meta-analysis and validation study focused on the deregulated genes and their associated networks. Oncoscience. 2014;1:117–31. doi: 10.18632/oncoscience.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.