

Abstract
CaV1.3 channels are a major class of L-type Ca2+ channels which contribute to the rhythmicity of the heart and brain. In the brain, these channels are vital for excitation-transcription coupling, synaptic plasticity, and neuronal firing. Moreover, disruption of CaV1.3 function has been associated with several neurological disorders. Here, we focus on the de novo missense mutation A760G which has been linked to autism spectrum disorder (ASD). To explore the role of this mutation in ASD pathogenesis, we examined the effects of A760G on CaV1.3 channel gating and regulation. Introduction of the mutation severely diminished the Ca2+-dependent inactivation (CDI) of CaV1.3 channels, an important feedback system required for Ca2+ homeostasis. This reduction in CDI was observed in two major channel splice variants, though to different extents. Using an allosteric model of channel gating, we found that the underlying mechanism of CDI reduction is likely due to enhanced channel opening within the Ca2+-inactivated mode. Remarkably, the A760G mutation also caused an opposite increase in voltage-dependent inactivation (VDI), resulting in a multifaceted mechanism underlying ASD. When combined, these regulatory deficits appear to increase the intracellular Ca2+ concentration, thus potentially disrupting neuronal development and synapse formation, ultimately leading to ASD.
L-type voltage-gated Ca2+ channels are critical conduits for Ca2+ entry into many excitable cells. The CaV1.3 channel represents a distinctive subtype of these channels, important in neurological1,2,3,4, cardiac3,4,5, and endocrine4,6,7 function. The biophysical properties of these channels are thus precisely tuned to this function, as they are activated at relatively hyperpolarized potentials compared to other L-type voltage-gated Ca2+ channels3,8,9,10,11,12 and undergo distinct forms of negative feedback regulation3,13,14.
CaV1.3 channels employ two major forms of feedback regulation, voltage-dependent inactivation (VDI) and Ca2+-dependent inactivation (CDI)14. These two regulatory processes are controlled within each cell type, utilizing splice variation3,15,16,17, RNA editing18,19, and auxiliary subunit pairing20,21 to tune the inactivation properties of the channel to specific cellular functions. In particular, both splice variation and RNA editing are able to modulate both CDI3,10,17,18,19,22,23,24 and channel open probability15 by tailoring the components contained within the channel carboxy tail. In addition, channel beta subunits are known to both traffic channels to the membrane25,26 and alter their voltage inactivation properties21,26,27,28.
The precise control of these regulatory processes are a vital component of normal physiology and disruption of this regulation has been linked to multiple human disorders including autism3,29,30,31, auditory deficits32,33, and hyperaldosteronism34,35. In mice, knockout of CaV1.3 results in profound deafness and severe bradycardia33,36, while in humans a similar phenotype is observed in patients harboring a 3-base pair insertion in exon 8b32. This insertion abolishes channel conduction, resulting in sinoatrial node dysfunction and deafness (SANDD) syndrome, a phenotype similar to that described in CaV1.3-knockout mice. Moreover, multiple gain-of-function mutations have been linked to patients with hyperaldosteronism34,35. Finally, two gain-of-function mutations in CaV1.3 (G407R and A749G) have been linked to autism spectrum disorders (ASD)30,31,37. Prior studies of these two mutations demonstrated alterations in channel gating including a hyperpolarizing shift in channel activation and inactivation curves31, but the differential effects on CDI versus VDI have yet to be determined. Discerning these precise effects may be highly relevant to understanding the mechanism of pathogenesis, as disruption of each of these components in the related CaV1.2 L-type channel has been shown to underlie Timothy syndrome (a severe multisystem disorder including autism and cardiac deficits)38,39,40, as well as long-QT syndrome associated with mutations in calmodulin41. It is interesting to note that, unlike the CaV1.2 channelopathies, CaV1.3 mutations have often been associated with single-system phenotypes30,37, despite the multi-system distribution of CaV1.3 channels. This isolation of symptoms is curious and requires further mechanistic investigation.
Here, we examine the underlying channel regulatory deficits of the autism-associated A760G mutation in rat CaV1.3 (equivalent to the A749G31 or A769G30 mutation in the human, depending on the channel backbone), focusing on the specific biophysical alterations produced by the mutation. We find that the mutation causes a significant reduction of CDI and a delay in channel deactivation in two major channel splice variants. In addition, we utilize an allosteric model of channel gating to gain insight into the underlying mechanism of this CDI deficit. Further examination of the biophysical defects of this mutation also revealed a beta subunit-dependent increase in VDI, an effect which would oppose the Ca2+ overload due to the decrease in CDI and a delay in channel deactivation. Thus the severe effects of this ‘gain-of-function’ mutation could be mitigated by a ‘loss-of-function’ effect on VDI.
Results
A760G significantly decreases CDI and alters CaV1.3 channel gating
Voltage-gated Ca2+ channel α1-subunits are composed of four domains, each containing six transmembrane α -helices (Fig. 1A). The four S6 helices line the channel pore through which Ca2+ enters the cell. The intracellular portion of these S6 helices form the activation gate of the channel, and mutations within this region are known to alter channel activation31,42,43,44,45,46. Moreover, the S6 helices are known to contribute to VDI and CDI in many CaV channels, including CaV1.315,40,42,43,44,45,46,47,48,49,50. Thus, the effect of the A760G mutation on channel activation and inactivation31 may be explained by its location in the S6 helix of domain II (IIS6).
Figure 1. A760G decreases CDI and promotes channel activation.
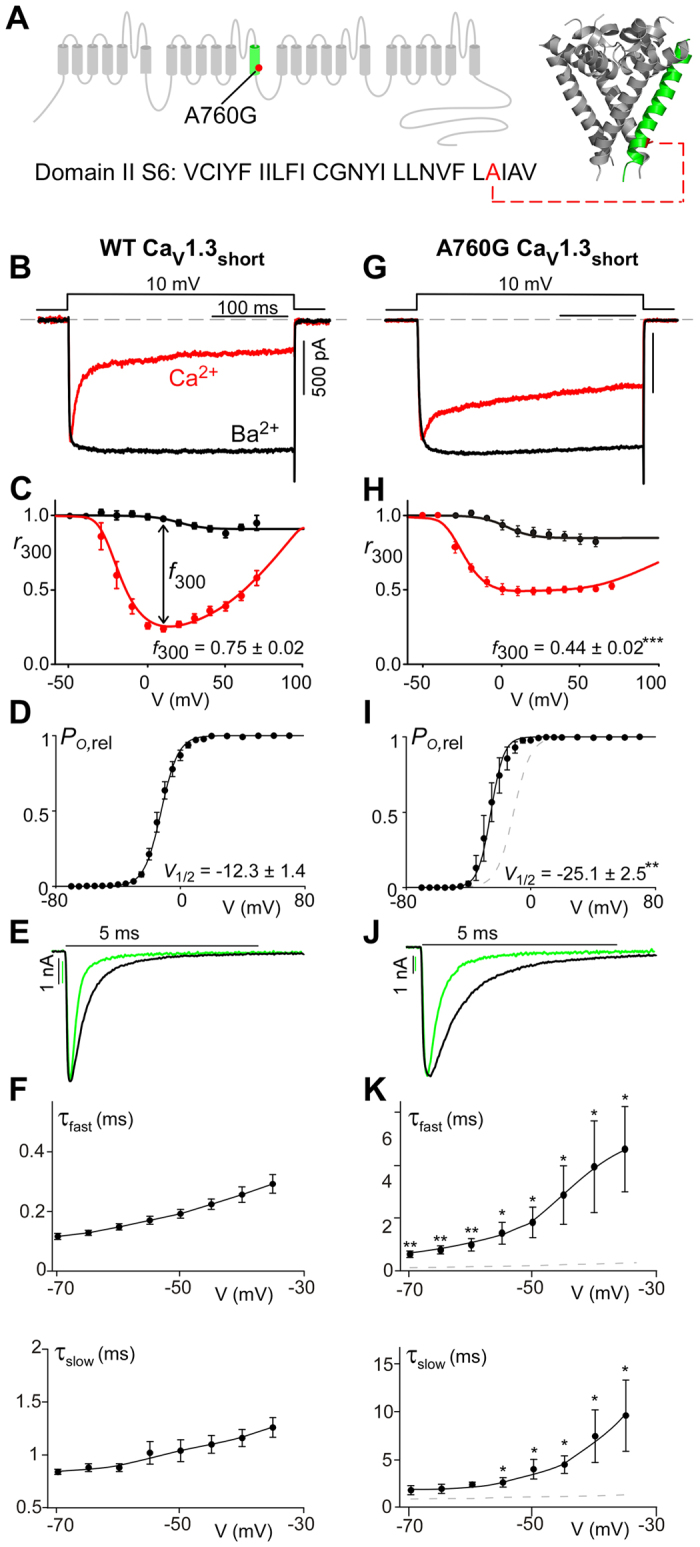
(A) Cartoon representing Cav1.3 and the location of the autism-associated mutation A760G. Left, the A760G missense mutation resides in the S6 helix of domain II. Right, structural homology model of Cav1.3 S5/S6 segments, based on NaVAb with the A760G highlighted in red. (B) Exemplar Ca2+ (red) and Ba2+ (black) current traces through WT CaV1.3short evoked by a 10-mV depolarizing step. Currents are normalized for comparison. Scale bar corresponds to the Ca2+ trace. (C) Population data of fraction of current remaining after 300 ms (r300) for Ca2+ (red) and Ba2+ (black). f300 determined at 10 mV (n = 12). Data are plotted as mean ± SEM here and throughout. (D) The activation curve for WT CaV1.3short obtained via a tail activation protocol with Ba2+ as the charge carrier (n = 6). (E) Exemplar Ba2+ tail currents obtained from a transition from 80 mV to −40 mV (black) and −60 mV (green). Traces are normalized to one another. Scale bars correspond to the traces of the same color. (F) Population data of the fast (top) and slow (bottom) deactivation time constants (τ) plotted as a function of voltage (n = 5). (G) Exemplar Ca2+ (red) and Ba2+ (black) current traces through A760G CaV1.3short evoked by a 10-mV depolarizing step. Compared to that of WT (B), Ca2+ current through A760G channels display significantly less CDI. (H) Population data of r300 for Ca2+ (red) and Ba2+ (black) currents through A760G channels. CDI is significantly smaller (***p < 0.001, n = 6) than that of the WT channels. (I) The activation curve of A760G CaV1.3short channels (black) shows a 13-mV hyperpolarizing shift (**p < 0.01, n = 4) compared to WT (reproduced in gray). (J) Exemplar Ba2+ tail currents for the A760G channel, obtained from a transition from 80 mV to −40 mV (black) and −60 mV (green). (K) Population data of the fast (top) and slow (bottom) deactivation time constants (τ) plotted as a function of voltage for the A760G channel. Deactivation is significantly slowed as compared to WT reproduced as the gray dashed line (*p < 0.05, **p < 0.01, n = 4).
In order to study the effects of the A760G mutation on Ca2+ regulation of channels, we undertook whole-cell voltage clamp recordings of CaV1.3 channels heterologously expressed in HEK293 cells. To minimize the possible confounding effects on CDI measurements, the channels were co-expressed with the β2a subunit which is known to minimize VDI21,26,28,51,52. In addition, an internal solution containing 10 mM BAPTA was utilized to restrict Ca2+ elevation to only the nanodomain of the channel, thus diminishing cell-to-cell variability53,54. Figure 1B shows exemplar current traces for wild-type (WT) CaV1.3. The sharp decay of Ca2+ current (red) evoked by a 10-mV depolarizing step depicts CDI, while the Ba2+ current trace (black) exhibits only VDI, which is mostly absent here due to the choice of β subunit. This robust CDI can be observed in the population data, where the fraction of current remaining after 300-ms depolarization (r300) is displayed as a function of voltage (Fig. 1C), and the U-shaped dependence on voltage recapitulates a classic hallmark of CDI55,56. Here, the difference between the r300 values for Ca2+ and Ba2+, normalized by the Ba2+ r300, gauges the magnitude of CDI (f300). However, when the A760G mutation is introduced into these channels, a drastic reduction in the speed and magnitude of CDI is observed (Fig. 1G, Supplementary Figure 1). This effect is further evident in the population data across multiple voltages (Fig. 1H) illustrating a clear CDI deficit due to the autism-associated A760G mutation.
Beyond CDI, S6 mutations are likely to affect channel activation31,42,43,44,45,46. We therefore probed the effect of the A760G on the voltage dependence of CaV1.3 channel activation. Here, we measured the relative open probability (Po,rel) of the channels across voltages via a tail activation protocol optimized for CaV1.3 channels54 (Fig. 1D). Introduction of the A760G mutation produced a significant hyperpolarizing shift (13 mV; p < 0.01) in channel activation (Fig. 1I), consistent with previous studies of this channel31. Moreover, analysis of the deactivation kinetics during the tail activation protocol revealed a marked effect of A760G on channel deactivation. Evaluation of the deactivation kinetics during a transitions from 80 mV to multiple voltages near the foot of the activation curve provided an estimation of the kinetics of channels transitioning from fully open to closed (Fig. 1E,F). A double exponential function was used to quantify a fast and slow component of channel deactivation (τfast, τslow respectively). The A760G mutation significantly increased both time constants across voltages, indicating a considerable slowing of channel closing (Fig. 1J,K).
L-type channel S6 mutations have previously been shown to affect CDI via modulation of modal channel gating14,38,57. We therefore consider the underlying mechanism linking channel activation and CDI. An allosteric model of channel gating (Fig. 2A) is known to describe CDI of CaV1.3 channels well14. Within this model, channels initially open within the mode 1 regime, which is characterized by a relatively large open probability (PO/mode1). Upon channel opening, Ca2+ influx drives channels into the mode Ca regime where channels maintain the ability to open, but with a significantly reduced PO (PO/modeCa). It is this reduction in PO that results in the CDI seen in whole-cell currents, such that
Figure 2. An allosteric mechanism underlying the CDI reduction.

(A) Diagram representing an allosteric model of CDI. Channels transition from mode 1 with high PO, to mode Ca2+ with lower PO, in response to Ca2+ entry. Equilibrium constants QEFF (concerted movement of S1–S4 segments), L (S6 movement), and a (mutation effect) govern transitions between open and closed channel configurations14,38, while the effective equilibrium constant J(Ca2+) governs entry into mode Ca2+. The parameter f (0 < f < 1) scales the PO in mode Ca2+ resulting in CDI. State transitions expected to be effected by the A760G are shaded pink. (B) Total CDI (black), the product of FCDI (blue) and CDImax (green), is plotted as a function of ∆∆Ga for the model shown in panel A. The A760G mutation left-shifts voltage activation (∆∆Ga < 0, a > 1), predicting a decrease in CDI due to a decrease in CDImax. (C) Exemplar Ca2+ current traces through WT CaV1.3short channels. Larger current amplitudes (ii, iii) allow a greater influx of Ca2+, enhancing entry into mode Ca and thus increasing CDI as compared to diminutive Ca2+ currents (i). (D) f300 values for individual cells expressing WT CaV1.3short are plotted as a function of current density. The curve saturates at CDImax ~0.9 (red dashed line). Traces in C correspond to i–iii. (E) Exemplar Ca2+ current traces through A760G CaV1.3short channels. Similar to that of WT (C), larger current amplitude increases f300 values. (F) Population data representing CDI of A760G CaV1.3short channels. f300 values saturate at CDImax ~0.7 (red dashed line), significantly lower than WT.
 |
where FCDI is the fraction of channels within mode Ca. Therefore at a saturating level of Ca2+, FCDI will approach unity as virtually all channels will reside within mode Ca. Under this condition, a maximal level of CDI (CDImax) is achieved:
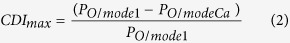 |
We now consider the effects of the S6 mutation A760G within our model. As we and others31 have shown that this mutation causes a hyperpolarizing shift in channel activation, the variable a was introduced to account for the altered free energy (∆∆Ga) required to open the mutant channels14. For a hyperpolarizing mutation such as A760G, ∆∆Ga will be negative, indicating decreased energy required to open the mutant channel. This decrease in free energy will result in increased channel opening in both mode 1 and mode Ca, thus decreasing CDImax (Equation 2, Fig. 2B, green). This reduction of CDImax could account for the overall decrease in CDI (Fig. 2B, black) observed in whole-cell experiments (Fig. 1), despite the increased FCDI (Fig. 2B, blue) due to increased Ca2+ influx.
To test the hypothesis that the CDI deficit due to A760G is primarily a result of a decrease in CDImax, we undertook whole-cell patch clamp recordings in which conditions enabled a sustained saturating level of Ca2+ at the mouth of the channel. By significantly reducing the intracellular Ca2+ buffer (0.5 mM EGTA), the accumulation of Ca2+ within the cell should overpower the nanodomain Ca2+ signal, thus raising Ca2+ to saturating levels as whole cell current increases58 and providing an estimation of CDImax54. For WT CaV1.3 channels, exemplar Ca2+ traces illustrate a saturating amount of CDI as a function of current density (Fig. 2C,D). The lack of additional CDI accumulation beyond a current density of 50 pA/pF indicates that we reached CDImax at a value of ~0.9 for WT channels (Fig. 2D, red dashed line). A760G channels, however, demonstrate a significant reduction in CDImax to ~0.7 (Fig. 2E,F), which can be observed at multiple test potentials (Supplementary Figure 2), confirming the underlying mechanism of CDI loss (Fig. 2B).
The A760G mutation differentially affects CaV1.3 splice variants
CaV channels are a critical conduit for Ca2+ entry into multiple cell types and must therefore be precisely tuned for specific cellular functions. Nature employs multiple mechanisms with which to accomplish such fine tuning, including modulation of channel splice patterns3,17,22,23,59. An example of such splicing in CaV1.3 channels results from inclusion of exon 42 or 42a, yielding a channel with a long versus short C-terminus59. The long channel variant activates at a somewhat more positive potential3,24, has a lower open probability15, and exhibits dramatically reduced CDI as compared to the short splice variant3,16. Due to these distinct properties, we examined the effect of the A760G mutation in both relevant splice variants. Having already demonstrated a significant reduction in CDI within the short channel variant, the isoform with more robust CDI3,59 (Fig. 1), we next undertook a similar approach within the long CaV1.3 splice variant (CaV1.3long).
Examination of CDI within the WT CaV1.3long channel under high buffering conditions (10 mM BAPTA) demonstrated significantly smaller, yet appreciable CDI (Fig. 3A,B), as compared to the short channel variant (Fig. 1B,C). Introduction of the A760G mutation blunted this CDI (Fig. 3F,G), though to a lesser extent as compared to the short variant. We next examined the activation of WT and A760G CaV1.3long channels. The resulting Po,rel versus voltage relationships (Fig. 3C,H) demonstrated an 11 mV hyperpolarizing shift due to the introduction of A760G (p < 0.01), similar to that observed in the short variant (Fig. 1D,I). Likewise, analysis of the deactivation kinetics also revealed a significant slowing of channel closing across multiple voltages (Fig. 3D,E,I,J). Thus, the biophysical deficits produced by the A760G mutation are qualitatively similar within each relevant channel backbone, although the magnitude of the CDI effect is somewhat decreased in the long splice variant.
Figure 3. CDI reduction due to A760G mutation within the CaV1.3 long variant.

(A) Exemplar Ca2+ (red) and Ba2+ (black) current traces through the alternate splice variant CaV1.3long illustrating decreased CDI as compared to the short splice variant (Fig. 1B). (B) Despite the reduction in magnitude, significant CDI is demonstrated by the difference in r300 for Ca2+ (red) and Ba2+ (black), plotted across multiple voltages. (f300 = 0.28 ± 0.04, n = 3). (C) Voltage activation curve for Ba2+ current through WT CaV1.3long channels (V1/2 = −1.0 ± 1.8 mV, n = 5). (D) Exemplar Ba2+ tail currents obtained from a transition from 80 mV to −40 mV (black) and −60 mV (green). Traces are normalized to one another such that the scale bars correspond to the traces of the same color. (E) Population data of the fast (top) and slow (bottom) deactivation time constants (τ) plotted as a function of voltage. Error bars indicate ± SEM, n = 5. (F) Exemplar current traces through A760G CaV1.3long channels depicting diminished CDI as compared to WT channels. (G) Population data for r300 (red = Ca2+, black = Ba2+) plotted across multiple voltages (f300 = 0.10 ± 0.03; n = 4, *p < 0.05). (H) The activation curve of A760G CaV1.3long channels (black, V1/2 = −12.0 ± 2.1 mV, n = 4) shows an 11-mV hyperpolarizing shift (**p < 0.01) compared to WT (reproduced in gray for reference). (I) Exemplar Ba2+ tail currents for the A760G CaV1.3long channel, obtained from a transition from 80 mV to −40 mV (black) and −60 mV (green). Traces are normalized to one another such that the scale bars correspond to the traces of the same color. (J) Population data of the fast (top) and slow (bottom) deactivation time constants (τ) plotted as a function of voltage for the A760G CaV1.3long channel. Deactivation is significantly slowed as compared to WT reproduced as the gray dashed line (*p < 0.05, **p < 0.01). Error bars indicate ± SEM, n = 4.
Opposing VDI changes may mitigate the detrimental effects of A760G
In addition to the critical Ca2+-dependent feedback, VDI also plays a major role in controlling Ca2+ entry through CaV1 channels. The underlying structural components for VDI have been shown to involve the linker region between domains I and II (I-II linker), which acts as a hinged-lid to close the pore following depolarization14,48. This process is known to be variably modulated by the binding of different isoforms of channel β subunits to the I-II linker14,26. For example, when coexpressed with β2a, CaV1 channels display little VDI due to restricted movement caused by palmitoylation, and thus membrane anchoring, of the β2a (Fig. 4A, blue)28,51,52. On the other hand, if the channels are coexpressed with the β1b subunit, most CaV1 isoforms will display strong VDI due to the lack of the palmitoylation site22,28. CaV1.3 channels, however, are unique in that their S6 helices have been shown to act as a shield (Fig. 4A,D, red) to prevent closing of the I-II linker lid, thus, endowing the channels with minimal VDI regardless of β subunit isoform co-expressed.
Figure 4. A760G increases VDI.

(A) Cartoon depicting a β2a subunit (blue) interacting with the VDI hinged lid (green) of a CaV1.3short channel. With palmitoylation, the β2a subunit is anchored to the plasma membrane (black coil) restricting the movement of the channel hinged lid. CaV1.3short channels also possess a ‘VDI shield’ (red). The A760G mutation disrupts this shield (bottom). (B) Exemplar Ba2+ current through WT (black) and A760G (blue) CaV1.3short channels shows re-emergence of VDI even in the presence of β2a. (C) Population data displaying Ba2+ r300 values as a function of voltage for WT (black) and A760G (blue). At 10 mV, WT: r300 = 0.98 ± 0.01; n = 10; A760G: r300 = 0.89 ± 0.03, n = 6. VDI is significantly increased in A760G channels (p < 0.01). (D) Cartoon depicting a β1b subunit (blue) interacting with the hinged lid (green) of a CaV1.3 channel. Note the absence of palmitoylation of this β subunit which allows the VDI hinged lid to move freely. (E) Exemplar Ba2+ current trace through WT (black) and A760G (blue) CaV1.3short channels in the presence of β1b. Note the absence of VDI in WT channels despite the lack of β subunit palmitoylation due to the presence of a VDI shield. A760G causes a pronounced re-emergence of VDI under these conditions. (F) The increase in VDI is confirmed in population data where Ba2+ r300 values for WT (black) and A760G (blue) are plotted as a function of voltage. At 10 mV, WT: r300 = 0.98 ± 0.01, n = 3; A760G: r300 = 0.77 ± 0.05, n = 4. VDI is significantly increased for A760G channels (p < 0.01).
A close inspection of the Ba2+ current through the A760G CaV1.3 channels reveals a slight, but significant re-emergence of VDI despite the presence of the β2a subunit (Fig. 4B,C). Importantly, this mutation-induced VDI can be discerned in either of the two channel variants (Fig. 1G versus B, Fig. 3F versus A). This re-emergence of VDI is accentuated when A760G CaV1.3 is coexpressed with β subunits lacking a palmitoylation site. Figure 4E,F demonstrate a significant increase in VDI in channels harboring the A760G mutation in the presence of the β1b subunit. Note that the WT CaV1.3 channels (black) have minimal VDI even in the presence of the β1b subunit due to the existence of the ‘shield’. Thus the re-emergence of VDI in these channels indicates that the A760G mutation may disrupt the VDI shield (Fig. 4A,D, bottom). This increase in VDI may act in opposition to the loss of CDI within these mutant channels, thus mitigating the detrimental effects of increased Ca2+ flux into cells.
The A760G mutation can cause an increase in intracellular Ca2+
We now know that the A760G mutation has a significant effect on CDI, VDI, and deactivation. Given the known link between excess cytosolic Ca2+ and severe disease states39,60,61, we wondered if Ca2+ overload due to an increased Ca2+ influx may play a role in the phenotype of A760G patients. However, the effects of the A760G mutation on Ca2+ entry are multi-fold, such that a decrease in CDI and slowing of channel deactivation are opposed by an increase in VDI. We therefore sought to confirm the cumulative effect of the A760G mutation on overall Ca2+ entry and intracellular Ca2+ concentration ([Ca2+]i).
To this end, we stimulated HEK293 cells expressing CaV1.3 channels with a 1-Hz train of neuronal action potentials (Fig. 5A, top) and recorded both the Ca2+ current passing through the channels as well as the intracellular Ca2+ level. This stimulation protocol was chosen as it matches well with the spontaneous neuronal firing frequencies in some populations of hippocampal neurons, thus mimicking a basal activity level62,63. When this protocol was applied to WT CaV1.3 channels, a small decrease in peak current amplitude was observed over time as CDI accumulated (Fig. 5A, middle panel). This amplitude decay matches well with the relatively small increase in [Ca2+]i, (Fig. 5A, bottom panel). When the A760G mutation was introduced into the channel, however, a significant increase in both the rate and extent of intracellular Ca2+ accumulation was observed (Fig. 5B–D, blue) due to an increased duration of Ca2+ entry during each action potential (Fig. 5B, left). This increased Ca2+ entry is likely a net result of decreased channel inactivation and a slowing of channel deactivation. This excess [Ca2+]i increased the extent of CDI observed in the current recordings over time, although this decrease in current entry was not sufficient to counteract the Ca2+ overload within the cytosol. Overall, the cumulative effect was a steady state Ca2+ level approaching 0.8 uM (Fig. 5C), substantially larger than the normal resting [Ca2+]i of a neuron64. This excess cytosolic Ca2+ due to A760G could be a significant contributing factor to the disease pathogenesis.
Figure 5. Effects of A760G on cytosolic Ca2+.

(A) Exemplar Ca2+ currents in response to a 1-Hz train of neuronal action potentials delivered to HEK293 cells expressing WT CaV1.3short channels. On the left, a single action potential and corresponding Ca2+ current is displayed on an expanded time course for resolution. Each action potential (top) and corresponding current response (middle) is magnified for display purposes and represents a 23 ms interval. The peak of each action potential is aligned with the time course displayed on the bottom panel. Note the gradual decrease in peak current as the level of cytosolic Ca2+ (bottom right panel) rises. (B) Exemplar Ca2+ currents in response to a 1-Hz train of neuronal action potentials delivered to HEK293 cells expressing A760G CaV1.3short channels. On the left, a magnified view demonstrates an increased duration of Ca2+ entry (blue) during a single action potential as compared to WT (A). At a comparable current density, A760G causes considerably more cytosolic Ca2+ accumulation (bottom right panel) as compared to WT channels. This increased cytosolic Ca2+ persists despite causing an enhancement of CDI (middle right panel). (C) Steady state level of intracellular Ca2+ in response to a 1-Hz train of action potentials. [Ca2+]ss is measured after 60 s of stimulation. A760G CaV1.3 channels display significantly higher levels of [Ca2+]ss than WT channels (WT: 0.28 ± 0.05, n = 7; A760G: 0.73 ± 0.13, n = 8; p < 0.01). (D) Average [Ca2+]i as a function of time as HEK293 cells expressing WT or A760G CaV1.3short channels are stimulated by a train of 1-Hz action potential. Error bars indicate ± SEM, n = 7, 8 for WT and A760G respectively.
Discussion
We have demonstrated a significant effect of the autism-associated mutation A760G on the gating of CaV1.3 such that channel activation is significantly left-shifted, CDI is decreased, and deactivation is slowed, resulting in excess Ca2+ entry through channels. However, these effects are mitigated by an increase in VDI. Importantly, the balance between these opposing mechanisms may be dependent on the specific properties of the particular channel complex harboring the mutation. In particular, we have demonstrated that two major CaV1.3 splice variants respond to the introduction of the A760G mutation with distinct levels of CDI disruption (Figs 1 and 3). As each of these splice variants has a significantly different affinity for commonly used L-type channel blockers59, such a variation in CDI effects may have important implications on the response of patients to treatment. Moreover, these two splice variants represent only a subset of a panoply of channel isoforms found across different tissues, each with uniquely tuned channel gating and feedback regulation3,11,17,18,19,59. Like the short versus long splice isoforms, it is possible that the A760G mutation may differentially alter the biophysical properties of these variants. Thus the extent of channel alteration may depend on the expression pattern within each specific cell type resulting in variable phenotypes across different tissues.
The original case report for A760G describes a patient exhibiting primarily neurological deficits classified as ASD30. Upon further examination of the Simons Simplex Collection database, the classification of ASD in this proband appeared to be of a relatively milder nature on the autism spectrum (pervasive developmental disorder not otherwise specified or PDD NOS) without additional non-neurological symptoms. Such a narrow symptom profile without any cardiac or hearing deficits stands in contrast to the broad tissue distribution of CaV1.3. Moreover, this lack of severe multisystem characteristics is unusual for autism-related Ca2+ channelopathies29,39,40,65. The multitude of CaV1.3 channel variants across different tissues17,22,23 may account for some of this lack of a multi-system phenotype. The relatively moderate symptoms of this proband may be due, in part, to differential effects of the A760G mutation on specific channel variants expressed in each system. In addition, the VDI effects of the A760G mutation could also contribute to the milder phenotype displayed by the proband. In particular, the increased Ca2+ entry due to altered CDI and channel activation/deactivation may be partially offset by increased VDI. As this VDI enhancement is accentuated in the presence of select beta subunits (Fig. 4), expression patterns of different beta subunits66 may further increase the variability of A760G effects across systems.
The A760G mutation is capable of substantially raising cytosolic Ca2+ concentration when overexpressed in HEK293 cells (Fig. 5). Of note, this effect was achieved even at a relatively slow 1-Hz pacing rate, comparable to the spontaneous firing rate of some hippocampal neurons62,63. The experimental conditions utilized here were optimized for maximal resolution of the A760G effect. However, under physiological heterozygous expression levels, the effect of the A760G channels will likely be considerably less. Nonetheless, the idea of excessive Ca2+ entry underlying ASD is not unprecedented as the ASD phenotype has been linked to Ca2+ overload through a myriad of Ca2+ handling molecules67,68,69, including multiple voltage-gated Ca2+ channels39,40,65,70. Overall, it seems plausible that the gating defects of the CaV1.3 channels harboring the A760G mutation may result in excess Ca2+ entry, which in turn may be over-activating the downstream Ca2+ signaling pathways involved in neural development and plasticity67,68,69,70. While the mechanisms underlying ASD remain elusive, the identification of mutations such as A760G, hint at important contributing factors.
Methods
Molecular Biology
The point mutation (A760G) was introduced into rat CaV1.3 short and long splice variants (gifts from Dr. Tuck Wah Soong59) in the homologous position to that found in humans using QuikChange™ site-directed mutagenesis (Agilent). The equivalent human mutation was found in patients at A769G in chromosome 3, position 5376449330 and corresponds to the A749G mutation previously described in an alternate human splice variant31.
Transfection of HEK293 cells
HEK293 cells were cultured on glass coverslips in 10-cm dishes and WT or mutant CaV1.3 channels, along with their auxiliary subunits, were transiently transfected using a standard calcium phosphate method71. 8 μg of rat CaV1.3 was co-expressed with 8 μg of rat brain β2a (M80545) or β1b (NM_017346), 8 μg of rat brain α2δ (NM012919.2) subunits, and 2 μg of simian virus 40 T antigen cDNA. Expression of all constructs was driven by a cytomegalovirus promoter and β subunits were contained within an EGFP-IRES bicistronic vector to allow visualization of transfected cells.
Whole Cell Electrophysiology
Whole-cell voltage-clamp recordings of HEK293 cells were done 1–2 days after transfection at room temperature. Recordings were obtained using an Axopatch 200B amplifier (Axon Instruments). Whole-cell voltage-clamp records were low pass filtered at 2 kHz, and then digitally sampled at 10 kHz. P/8 leak subtraction was used, with series resistances of 1–2 MΩ. For voltage-clamp experiments, internal solutions contained (in mM): CsMeSO3, 114; CsCl, 5; MgCl2, 1; MgATP, 4; HEPES (pH 7.3), 10; and either BAPTA, 10 or EGTA, 0.5; at 295 mOsm adjusted with CsMeSO3. External solutions contained (in mM): TEA-MeSO3, 140; HEPES (pH 7.4), 10; and CaCl2 or BaCl2, 40; at 300 mOsm, adjusted with TEA-MeSO3.
For simultaneous ratiometric Ca2+ measurements and current recordings, a fixed ratio of two Ca2+-sensitive dyes (Fluo-2 high affinity, TEFLabs; Fluo-2 low affinity, TEFLabs) and Alexa568 (Invitrogen) were added into a 0.5-mM EGTA internal solution. Two Ca2+ indicators at different binding affinity were chosen to accurately measure both baseline and peak Ca2+ concentrations. The dye mixture was calibrated to obtain absolute Ca2+ concentrations72. The external solution (Tyrode’s solution) contained (in mM): NaCl, 135; KCl, 5.4; CaCl2, 1.8; MgCl2, 0.33; NaH2PO4, 0.33; HEPES, 5; glucose, 5 (pH 7.4). During current recordings, Ca2+ concentration was measured by exciting dyes using a 514-nm Argon laser, via a 545DCLP dichroic mirror and either a 545/40BP (Fluo) or 580LP (Alexa568) filter. Cells were held at −80 mV and a 1-Hz train of neuronal action potential recordings from cortical neurons of E18 mouse embryos were used as voltage stimulus.
Generation of a CaV1.3 Homology Model
We used MODELLER v9.1473 to build a homology model of CaV1.3 pore regions (Fig. 1A) based on the crystal structure of the bacterial Na channel NaVAb (PDB accession code: 4EKW)74 as previously described14. Briefly, we generated 10 decoy models from which a model with lower objective function was chosen. The four domains of CaV1.3 were constrained to adopt a clockwise orientation when viewed from the extracellular surface by analogy to the orientation of the related voltage-gated sodium channels75. The alignment used for the various pore subsegments are as follows:
S5 Segment
NaVAb SVAALLTVVFYIAAVMATNLYGATFP
CaV1.3 Domain I HIALLVLFVIIIYAIIGLELFIGKMH
CaV1.3 Domain II SLLLLLFLFIIIFSLLGMQLFGGKFN
CaV1.3 Domain II INIMIVTTLLQFMFACIGVQLFKGKFY
CaV1.3 Domain IV YVALLIAMLFFIYAVIGMQMFGKVAM
P Loop
NaVAb EWFGDLSKSLYTLFQVMTLESWSMGIVRPVMNV
CaV1.3 Domain I TNFDNFAFAMLTVFQCITMEGWTDVLYWVNDAI
CaV1.3 Domain II STFDNFPQALLTVFQILTGEDWNAVMYDGIMAY
CaV1.3 Domain III FNFDNVLSAMMVLFTVSTFEGWPALLYKAIDSN
CaV1.3 Domain IV NNFQTFPQAVLLLFRCATGEAWQEIMLACLPGK
S6 Segment
NaVAb HPNAWVFFIPFIMLTTFTVLNLFIGII
CaV1.3 Domain I WEWPWVYFVSLIILGSFFVLNLVLGVL
CaV1.3 Domain II GMIVCIYFIILFICGNYILLNVFLAIA
CaV1.3 Domain III RVEISIFFIIYIIIVAFFMMNIFVGFV
CaV1.3 Domain IV SNFAIVYFISFYMLCAFLIINLFVAVI
Data Analysis and Statistics
The fraction of current remaining after 300 ms of channel activation (r300) was calculated as:
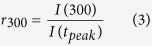 |
and CDI measurements were corrected for VDI effects by calculating a metric for pure CDI (f300) as follows:
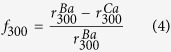 |
r300 and f300 values were reported at 10 mV for the CaV1.3short channel and at 0 mV for CaV1.3long. The relative open probability (PO,rel) was determined by a tail activation protocol54 where channels are fully activated at 80 mV prior to stepping to variable test potentials. The ratio of peak and steady state currents then represent the relative PO,rel of each voltage. The voltage activation curve was fit by the Boltzmann equation:
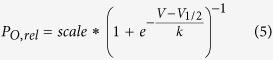 |
where V1/2 and k represent the half activation voltage and slope factor, respectively.
Time constants for channel deactivation (τfast and τslow in Figs 1F,K and 3E,J) were calculated by fitting the deactivating Ba2+ tail currents resulting from a transition from 80 mV (channels maximally open based on the activation curve, Figs 1D,I and 3C,H) to multiple voltages near the base of the activation curve (channels closed) with the equation:
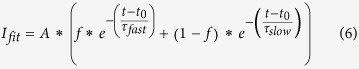 |
where f is the fraction of the faster portion of the current decay, and time constants τfast and τslow represent the fast and slow components of the current decay.
All data are presented as mean ± SEM. Statistical significance for variability was determined by a two-tailed student’s t-test.
Additional Information
How to cite this article: Limpitikul, W. B. et al. An autism-associated mutation in CaV1.3 channels has opposing effects on voltage- and Ca2+-dependent regulation. Sci. Rep. 6, 27235; doi: 10.1038/srep27235 (2016).
Supplementary Material
Acknowledgments
We are grateful to Dr. Evan E. Eichler for investigating the previously unreported clinical phenotype of the proband and generously sharing relevant results with us. We thank Wanjun Yang for dedicated technical support and Ingie Hong for providing us with neuronal stimulus waveforms and experimental input. We also thank Gordon Tomaselli for providing valuable advice and discussions, and members of the Calcium Signals Lab for ongoing feedback. This work was supported by grants NIMH R01MH065531 and NHLBI 5R37HL076795 (D.T.Y.).
Footnotes
Author Contributions W.B.L. contributed to all aspects of the experiments and data analysis. M.B.-J. assisted with the initial experimental design and homology modeling of the channel. W.B.L. and I.E.D wrote the manuscript with notes from D.T.Y. and input from all authors. D.T.Y. conceived the project and supervised the work.
References
- Berger S. M. & Bartsch D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 357, 463–476, 10.1007/s00441-014-1936-3 (2014). [DOI] [PubMed] [Google Scholar]
- Pinggera A. & Striessnig J. Ca 1.3 (CACNA1D) L-type Ca channel dysfunction in CNS disorders. J. Physiol., 10.1113/JP270672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J., Pinggera A., Kaur G., Bock G. & Tuluc P. L-type Ca channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3, 15–38, 10.1002/wmts.102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W., Striessnig J., Koschak A. & Dolphin A. C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 67, 821–870, 10.1124/pr.114.009654 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P., Torrente A. G. & Mangoni M. E. Functional role of voltage gated Ca(2+) channels in heart automaticity. Front. Physiol. 6, 19, 10.3389/fphys.2015.00019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett P. Q. et al. Role of voltage-gated calcium channels in the regulation of aldosterone production from zona glomerulosa cells of the adrenal cortex. J. Physiol., 10.1113/JP271896 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni A. et al. L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium 42, 397–408, 10.1016/j.ceca.2007.04.015 (2007). [DOI] [PubMed] [Google Scholar]
- Xu W. & Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A. et al. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106, 10.1074/jbc.M101469200 (2001). [DOI] [PubMed] [Google Scholar]
- Lipscombe D., Andrade A. & Allen S. E. Alternative splicing: functional diversity among voltage-gated calcium channels and behavioral consequences. Biochim. Biophys. Acta 1828, 1522–1529, 10.1016/j.bbamem.2012.09.018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb A., Ortner N. & Striessnig J. C-terminal modulatory domain controls coupling of voltage-sensing to pore opening in Cav1.3 L-type Ca(2+) channels. Biophys. J. 106, 1467–1475, 10.1016/j.bpj.2014.02.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholze A., Plant T. D., Dolphin A. C. & Nurnberg B. Functional expression and characterization of a voltage-gated Cav1.3 (alpha1D) calcium channel subunit from an insulin-secreting cell line. Mol. Endocrinol. 15, 1211–1221, 10.1210/mend.15.7.0666 (2001). [DOI] [PubMed] [Google Scholar]
- Satin J., Schroder E. A. & Crump S. M. L-type calcium channel auto-regulation of transcription. Cell Calcium 49, 306–313, 10.1016/j.ceca.2011.01.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross M. R., Ben Johny M. & Yue D. T. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J. Gen. Physiol. 135, 197–215, 10.1085/jgp.200910308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. J., Ben-Johny M., Dick I. E., Inoue T. & Yue D. T. Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell 159, 608–622, 10.1016/j.cell.2014.09.047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Yang P. S., Yang W. & Yue D. T. Enzyme-inhibitor-like tuning of Ca(2+) channel connectivity with calmodulin. Nature 463, 968–972, 10.1038/nature08766 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B. Z. et al. Functional characterization of alternative splicing in the C terminus of L-type Cav1.3 channels. J. Biol. Chem. 286, 42725–42735, 10.1074/jbc.M111.265207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzazi H., Ben Johny M., Adams P. J., Soong T. W. & Yue D. T. Continuously tunable Ca(2+) regulation of RNA-edited Cav1.3 channels. Cell Rep 5, 367–377, 10.1016/j.celrep.2013.09.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. et al. RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca(2)(+)-dependent inactivation. Neuron 73, 304–316, 10.1016/j.neuron.2011.11.022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Crowley M. L. & Rittenhouse A. R. Arachidonic acid inhibition of L-type calcium (Cav1.3b) channels varies with accessory Cavbeta subunits. J. Gen. Physiol. 133, 387–403, 10.1085/jgp.200810047 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin A. C. Calcium channel diversity: multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 19, 237–244, 10.1016/j.conb.2009.06.006 (2009). [DOI] [PubMed] [Google Scholar]
- Shen Y. et al. Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J. Neurosci. 26, 10690–10699, 10.1523/JNEUROSCI.2093-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock G. et al. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 286, 42736–42748, 10.1074/jbc.M111.269951 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. et al. Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744, 10.1074/jbc.M802254200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin A. C. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–555, 10.1038/nrn3311 (2012). [DOI] [PubMed] [Google Scholar]
- Buraei Z. & Yang J. Structure and function of the beta subunit of voltage-gated Ca(2)(+) channels. Biochim. Biophys. Acta 1828, 1530–1540, 10.1016/j.bbamem.2012.08.028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin A. C. Beta subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 35, 599–620 (2003). [DOI] [PubMed] [Google Scholar]
- Colecraft H. M. et al. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J. Physiol. 541, 435–452 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenkamp A. F., Matthes J. & Herzig S. Voltage-gated Calcium Channels and Autism Spectrum Disorders. Curr. Mol. Pharmacol. 8, 123–132 (2015). [DOI] [PubMed] [Google Scholar]
- O’Roak B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250, 10.1038/nature10989 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinggera A. et al. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol. Psychiatry 77, 816–822, 10.1016/j.biopsych.2014.11.020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig S. M. et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84, 10.1038/nn.2694 (2011). [DOI] [PubMed] [Google Scholar]
- Platzer J. et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97 (2000). [DOI] [PubMed] [Google Scholar]
- Azizan E. A. et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 45, 1055–1060, 10.1038/ng.2716 (2013). [DOI] [PubMed] [Google Scholar]
- Scholl U. I. et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45, 1050–1054, 10.1038/ng.2695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H. et al. Null mutation of alpha1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J Assoc Res Otolaryngol 5, 215–226, 10.1007/s10162-003-4020-3 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299, 10.1016/j.neuron.2012.04.009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick I. E., Joshi-Mukherjee, R., Yang W & Yue D. T. Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca2+ dependent inactivation. Nat Commun 7, 10370, 10.1038/ncomms10370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I. et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 102, 8089–8096; discussion 8086-8088, 10.1073/pnas.0502506102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I. et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31, 10.1016/j.cell.2004.09.011 (2004). [DOI] [PubMed] [Google Scholar]
- Limpitikul W. B. et al. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol. 74, 115–124, 10.1016/j.yjmcc.2014.04.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoda J. C., Zaghetto F., Koschak A. & Striessnig J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels. J. Neurosci. 25, 252–259, 10.1523/JNEUROSCI.3054-04.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohaus A. et al. Structural determinants of L-type channel activation in segment IIS6 revealed by a retinal disorder. J. Biol. Chem. 280, 38471–38477, 10.1074/jbc.M507013200 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus R. L. et al. Three new familial hemiplegic migraine mutants affect P/Q-type Ca(2+) channel kinetics. J. Biol. Chem. 275, 9239–9243 (2000). [DOI] [PubMed] [Google Scholar]
- Raybaud A. et al. The role of distal S6 hydrophobic residues in the voltage-dependent gating of Cav2.3 channels. J. Biol. Chem. 282, 27944–27952, 10.1074/jbc.M703895200 (2007). [DOI] [PubMed] [Google Scholar]
- Raybaud A. et al. The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J. Biol. Chem. 281, 39424–39436, 10.1074/jbc.M607405200 (2006). [DOI] [PubMed] [Google Scholar]
- Barrett C. F. & Tsien R. W. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of Cav1.2 L-type calcium channels. Proc. Natl. Acad. Sci. USA 105, 2157–2162, 10.1073/pnas.0710501105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz S. C., Hamid J., Spaetgens R. L., Jarvis S. E. & Zamponi G. W. Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? J. Biol. Chem. 275, 24575–24582, 10.1074/jbc.M000399200 (2000). [DOI] [PubMed] [Google Scholar]
- Stotz S. C., Jarvis S. E. & Zamponi G. W. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J. Physiol. 554, 263–273, 10.1113/jphysiol.2003.047068 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz S. C. & Zamponi G. W. Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J. Biol. Chem. 276, 33001–33010, 10.1074/jbc.M104387200 (2001). [DOI] [PubMed] [Google Scholar]
- Dafi O. et al. Negatively charged residues in the N-terminal of the AID helix confer slow voltage dependent inactivation gating to Cav1.2. Biophys. J. 87, 3181–3192, 10.1529/biophysj.104.045559 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stea A. et al. Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc. Natl. Acad. Sci. USA 91, 10576–10580 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick I. E. et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of Cav channels. Nature 451, 830–834, 10.1038/nature06529 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross M. R., Dick I. E. & Yue D. T. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240, 10.1016/j.cell.2008.05.025 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P. & Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science 202, 1203–1206 (1978). [DOI] [PubMed] [Google Scholar]
- Brehm P., Eckert R. & Tillotson D. Calcium-mediated inactivation of calcium current in Paramecium. J. Physiol. 306, 193–203 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imredy J. P. & Yue D. T. Mechanism of Ca(2+)-sensitive inactivation of L-type Ca2+ channels. Neuron 12, 1301–1318 (1994). [DOI] [PubMed] [Google Scholar]
- Yang P. S. et al. Switching of Ca2+-dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J. Neurosci. 26, 10677–10689, 10.1523/JNEUROSCI.3236-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Yu D. & Soong T. W. C-terminal alternative splicing of Cav1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol. Pharmacol. 84, 643–653, 10.1124/mol.113.087155 (2013). [DOI] [PubMed] [Google Scholar]
- Schulz J. B. Mechanisms of neurodegeneration in idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 13 Suppl 3, S306–308, 10.1016/S1353-8020(08)70021-X (2007). [DOI] [PubMed] [Google Scholar]
- Zhang L., Yang H., Zhao H. & Zhao C. Calcium-related signaling pathways contributed to dopamine-induced cortical neuron apoptosis. Neurochem. Int. 58, 281–294, 10.1016/j.neuint.2010.11.021 (2011). [DOI] [PubMed] [Google Scholar]
- Mizuseki K. & Buzsaki G. Preconfigured, skewed distribution of firing rates in the hippocampus and entorhinal cortex. Cell Rep 4, 1010–1021, 10.1016/j.celrep.2013.07.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase H., Leinekugel X., Czurko A., Csicsvari J. & Buzsaki G. Firing rates of hippocampal neurons are preserved during subsequent sleep episodes and modified by novel awake experience. Proc. Natl. Acad. Sci. USA 98, 9386–9390, 10.1073/pnas.161274398 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A., Shmigol A., Kirischuk S., Pronchuk N. & Kostyuk P. Age-dependent changes in calcium currents and calcium homeostasis in mammalian neurons. Ann. N. Y. Acad. Sci. 747, 365–381 (1994). [DOI] [PubMed] [Google Scholar]
- Hemara-Wahanui A. et al. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc. Natl. Acad. Sci. USA 102, 7553–7558, 10.1073/pnas.0501907102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A., Flockerzi V. & Hofmann F. Regional expression and cellular localization of the alpha1 and beta subunit of high voltage-activated calcium channels in rat brain. J. Neurosci. 17, 1339–1349 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnby G. et al. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966, 10.1086/430454 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. G. et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889, 10.1126/science.1086446 (2003). [DOI] [PubMed] [Google Scholar]
- Zhou Z. et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269, 10.1016/j.neuron.2006.09.037 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey J. F. & Dolmetsch R. E. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr. Opin. Neurobiol. 17, 112–119, 10.1016/j.conb.2007.01.010 (2007). [DOI] [PubMed] [Google Scholar]
- Peterson B. Z., DeMaria C. D., Adelman J. P. & Yue D. T. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22, 549–558 (1999). [DOI] [PubMed] [Google Scholar]
- Lee S. R., Adams P. J. & Yue D. T. Large Ca2+-dependent facilitation of Cav 2.1 channels revealed by Ca2+ photo-uncaging. J. Physiol. 593, 2753–2778, 10.1113/JP270091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A. & Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815, 10.1006/jmbi.1993.1626 (1993). [DOI] [PubMed] [Google Scholar]
- Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N. & Catterall W. A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139, 10.1038/nature11077 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. A. et al. Clockwise domain arrangement of the sodium channel revealed by (mu)-conotoxin (GIIIA) docking orientation. J. Biol. Chem. 276, 11072–11077, 10.1074/jbc.M010862200 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.