

Abstract
Anaplastic thyroid carcinoma (ATC) is an extremely aggressive thyroid cancer sub-type, refractory to current medical treatment. Among various epigenetic anticancer drugs, BET inhibitors (BETi) are considered an appealing novel class of compounds. BETi target the Bromodomain and Extra-Terminal (BET) proteins that act as regulators of gene transcription, interacting with histone acetyl groups. The goal of this study is to delineate which pathway underlie the biological effects derived from BET inhibition, in order to find new potential therapeutic targets in ATC. We investigated effects of BET inhibition on two human anaplastic thyroid cancer-derived cell lines (FRO and SW1736). The treatment with two BETi, JQ1 and I-BET762, decreased cell viability, reduced cell cycle S-phase and determined cell death. In order to find BETi effectors, FRO and SW1736 were subjected to a global transcriptome analysis after JQ1 treatment. A significant portion of deregulated genes belongs to cell cycle regulators. Among them, MCM5 was decreased at both mRNA and protein levels in both tested cell lines. ChIP experiments indicate that MCM5 is directly bound by the BET protein BRD4. MCM5 silencing reduced cell proliferation, thus underlining its involvement in the block of proliferation induced by BETi. Furthermore, MCM5 immunohistochemical evaluation in human thyroid tumor tissues demonstrated its over-expression in several papillary thyroid carcinomas and in all ATCs. MCM5 was also over-expressed in a murine model of ATC, and JQ1 treatment reduced Mcm5 mRNA expression in two murine ATC cell lines. Thus, MCM5 could represent a new target in the therapeutic approach against ATC.
Keywords: MCM5, BET, epigenetics, RNA-seq
Introduction
Thyroid cancers are the most widespread malignancies of the endocrine system and represent approximately 1–1.5% of all tumor-related diseases. The overall five-year survival of these tumors is approximately 85–90%, with the highest mortality rate reported for poor differentiated histotypes (Patel and Shaha 2014; Durante et al. 2006). Anaplastic thyroid carcinomas (ATC), which are wholly or partially composed of undifferentiated cells arising from thyroid follicular epithelium, represent the least common but most lethal thyroid cancer sub-type. Indeed, they are characterized by genetic and epigenetic aberrations that make them unresponsive to treatments based on radioiodine administration (Arturi et al. 2000; Schlumberger et al. 2007; Perri et al. 2011; Elisei 2012). Thus, innovative approaches for ATC treatment are required.
The term “epigenetics” is commonly used to describe the study of hereditable changes in gene expression that are independent from alteration in the underlying DNA sequence. Since aberrant gene expression is an essential characteristic that distinguishes neoplastic cells from normal ones, compounds able to target epigenetic alterations are becoming increasingly appealing for cancer treatment. Most of these molecules act on epigenetic “writers” or “erasers”, i.e. enzymes regulating DNA or histones covalent modifications. Notably, compounds targeting enzymes involved in DNA methylation and histone acetylation have been greatly investigated in several experimental models of solid tumors, including thyroid cancer (Puppin et al. 2005; Russo et al. 2013). Several other chromatin regulators, instead, act as “readers”: they possess peculiar domains able to interact with nucleosome covalent modifications and, responding to upstream signal cascades, to control gene expression (Dawson et al. 2012). A rising class of compounds able to target chromatin readers is called BET inhibitors. BET is the acronym of bromodomain and extra-terminal family of proteins (Nicodeme et al. 2010). The mammalian BET protein family includes four members: BRD2, BRD3, and BRD4, which are ubiquitously expressed, and BRDT, whose expression is confined to germ cells. These proteins possess a tandem bromodomain at the N-terminal region, by which they interact with histone acetyl-lysines in order to control gene transcription and cell proliferation (Chiang 2009). Due to the relevance of BET proteins, several inhibitors have been recently developed (Nicodeme et al. 2010; Filippakopoulos et al. 2010). BET inhibitors hinder proliferation of a wide range of hematopoietic malignant cell lines and of NUT midline carcinoma-derived cells (Filippakopoulos et al. 2010; Zuber et al. 2011). The use of BET inhibitors, as well as the use of other epigenetic compounds, might be a complicate issue in cancer treatment since these drugs act on a large spectrum of genes (Falahi et al. 2015). However, the anti-proliferative outcome could be reasonably due to the deregulation of only a small subset of genes. For instance, repression of the c-MYC oncogene seems to play a major role in mediating the effects of BET inhibitors in leukemic cells (Ott et al. 2012). Thus, an attractive approach to study epigenetic compounds is to define their critical effector genes. To this purpose, in this study we investigated the effects of BET inhibition in two human ATC cell lines, analyzing the modulation of their global gene expression profiles. By using this approach, we propose MCM5 as a potential new target for ATC treatment.
Materials and Methods
Human and murine cell lines
FRO (purchased from the European Collection of Cell Cultures, Salisbury, United Kingdom) and SW1736 (obtained from Cell Lines Service GmbH, Eppelheim, Germany) are human cell lines derived from ATC, both harboring a BRAF V600E mutation (Pilli et al. 2009; Schweppe et al. 2008). Both cell lines have been tested for being mycoplasma-free and authenticated by STR analysis to be appropriate cell lines of thyroid cancer origin. FRO and SW1736 were grown in DMEM medium (EuroClone, Milan, Italy) and RPMI 1640 medium (EuroClone) respectively, supplemented with 10% fetal bovine serum (Gibco Invitrogen, Milan, Italy), 2 mM L-glutamine (EuroClone) and 50 mg/ml gentamicin (Gibco Invitrogen), in a humidified incubator (5% CO2 in air at 37°C). Cultured cells were treated with vehicle (DMSO, Sigma Aldrich, Saint Louis, MO, USA) or the following agents: JQ1 (50 nM–10 μM in DMSO) (Cayman Chemical, Ann Arbor, MI, USA) and I-BET762 (50 nM –10 μM in DMSO) (Merck Millipore, Darmstadt, Germany). T4888M and T3531L are ATC cell lines derived from tumors developed by [Pten, Tp53]thyr−/− mice (Antico Arciuch et al. 2011). Cells were grown in DMEM supplemented with 10% FBS.
Animals
[Pten, Tp53]thyr−/− and Ptenthyr−/− mice have been previously described (Antico Arciuch et al. 2011; Yeager et al. 2007; Antico Arciuch et al. 2010) and were housed in the Albert Einstein College of Medicine animal facility under specific-pathogen-free conditions.
Cell viability and apoptosis
To test cell viability, an MTT assay was performed as previously described (Baldan et al. 2014). Briefly, 5000 cells were seeded onto 96-well plates. On the next day, the growth medium was replaced with vehicle-treated medium (NT, untreated cultures) or with medium containing JQ1 or I-BET762; plates were incubated for 0, 24, 48 and 72 h. All experiments were run in quadruplicate and cell viability was expressed as a percentage relative to baseline samples (T0). The percentage of cell viability assessed by MTT assay was used to determine EC50 concentrations from dose–response curves after 72 h treatment.
Caspase 3/7 activity was measured as a marker of apoptosis using the Apo-ONE Homogeneous Caspase-3/7 Assay kit (Promega, Milan, Italy) according to the manufacturer’s protocol. 5000 cells were seeded onto 96-well plates. On the next day, the growth medium was replaced with vehicle-treated medium (NT, untreated cultures) or medium containing JQ1 5 μM or I-BET762 5 μM; plates were incubated for 48 h. All experimental points were run in triplicate and apoptosis levels were expressed as a percentage relative to baseline samples (NT).
Cell cycle analysis
Cell cycle was determined by fluorescence-activated cell-sorting (FACS) analysis of DNA content. Briefly, FRO and SW1736 cells were collected after a 24, 48 and 72 h treatment either with JQ1 or vehicle (NT); cells were fixed in cold ethanol 70% and stained with a propidium iodide solution containing RNase and Triton-X100. Flow cytometric analysis was performed on a FACSCalibur (Becton Dickinson) using Modfit LT analysis software (Verity Software House, Inc., Topsham, ME, USA). A minimum of 20,000 cells was analyzed for each sample. All experiments were run in triplicate.
High throughput RNA-sequencing and analysis
1.5 μg of good quality RNA (RNA Integrity Number >7) was used as starting material for library preparation using the Illumina mRNASeq Sample Prep kit v2.0, following the manufacturer’s instructions. The poly-A mRNA was fragmented for 1.5 minutes at 94°C and all purification steps were performed by using 1X Agencourt AMPure XP beads. Quality and quantity of each library were assessed by using an Agilent Bioanalyzer 2100 High Sensitivity and Qubit DNA High Sensitivity (Invitrogen). Libraries were pooled together and the obtained pool was checked on an Agilent Bioanalyzer 2100 in order to determine the molarity. Single read sequencing was performed on the HiSeq2000 (Illumina, San Diego, CA, USA) generating 50-base reads. The CASAVA 1.8.2 version of the Illumina pipeline was used for image analysis, base calling, and demultiplexing. Reads were first trimmed in order to remove lower base quality data with ERNE (Vezzi et al. 2012) and adapter sequences were removed with Cutadapt (https://pypi.python.org/pypi/cutadapt/1.3). TopHat (Trapnell et al. 2012) was used for mapping and annotation on the hg19 reference sequence. Differential gene expression analysis was performed according to the experimental design using Cufflinks (Trapnell et al. 2012).
Gene expression assays
Total RNA from human cell lines, treated either with JQ1 5 μM or vehicle, was extracted with RNeasy mini kit according with manufacturer’s instructions (Qiagen, Hilden, Germany). 500 ng of total RNA were reverse transcribed to cDNA using random exaprimers and MMLV reverse transcriptase (Life Technologies, Carlsbad, CA, USA). Real-time PCRs were performed using Platinum Sybr Green QPCR supermix (Life Technologies) with the ABI Prism 7300 Sequence Detection Systems (Applied Biosystems). The ΔΔCT method, by means of the SDS software (Applied Biosystems), was used to calculate mRNA levels. Oligonucleotide primers for MCM5 (forward, 5′-GATCCTGGCATTTTCTACAG-3′; reverse, 5′-CCCTGTATTTGAAGGTGAAG-3′) and β-actin (forward, 5′-TTGTTACAGGAAGTCCCTTGCC-3′; reverse, 5′-ATGCTATCACCTCCCCTGTGTG-3′) were purchased from Sigma Aldrich. Total mouse RNA (treated either with 0.5μM or vehicle) was extracted with Trizol and reverse transcribed using the Maxima First Strand cDNA Synthesis Kit kit (Thermo Scientific, Waltham, MA). qRT-PCR was performed on a StepOne Plus apparatus using SYBR Green mix (Applied Biosystems) and custom-designed primers (forward, 5′-CAGAGGCGATTCAAGGAGTTC-3′; reverse, 5′-CGATCCAGTATTCACCCAGGT-3′). Each sample was run in triplicate and 18S was used to control for input RNA. Data analysis was based on the Ct method, and experiments were repeated at least three times using at least two independent organ and cell pools.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as previously described (Baldan et al., 2015). For immunoprecipitation, samples were incubated with 10 μg of rabbit polyclonal anti-BRD4 (Active Motif) or Rabbit IgG, (Millipore), as negative control. After incubation with Dynabeads Protein A, washing and elution, the DNA was extracted with phenol/chloroform, the aqueous phase recovered and precipitated. The extracted DNA was used as a template in qPCR with the following primers: MCM5 Forward Primer (CCTGTTCTGGCCGTTTGTTC), MCM5 Reverse Primer (GATCGTCGAATCCCGACATG); NCR Forward Primer (TGCTGTTACTTTTTACAGGGAGTT), NCR Reverse Primer (TTTGAGCAAAATGTTGAAAACAA). Fold enrichment was, then, calculated, as signal over background (IgG). Data are representative of three different experiments.
RNA silencing
For transient silencing of endogenous MCM5, TriFECTa RNAi Kit (Integrated DNA Technologies Inc, Coralville, IA, USA) was used. A ‘universal’ negative control duplex (NC) that targets a site that is absent from human, mouse and rat genomes was used. siRNA oligonucleotides were transfected at a concentration of 5 nM using DharmaFECT Transfection reagent (Thermo Scientific Inc, Waltham, MA, USA), according to manufacturer’s instructions. The day before transfection, cells were plated in antibiotics-free medium and cells were harvested 72 h after transfection. The gene-silencing efficiency was evaluated by mRNA and protein levels analysis.
Annexin V assay
In other to test the percentage of viable/apoptotic cells after MCM5 silencing, an Annexin V assay was performed. Briefly, cells were washed with cold PBS, transferred to a polystyrene round-bottomed flow tube (Falcon, Becton Dickinson, Franklin Lakes, NY) and resuspended in 195 μL of 1x binding buffer (BB 10 mM; HEPES/NaOH, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2). 5 μL of Fluorescein-conjugated Annexin V (Annexin V-FITC; Bender Med Systems, Vienna, Austria) was added and samples were incubated for 10 minutes at room temperature. After washing, cells were resuspended in 190 μL of BB in which 10 μL of propidium iodide stock solution (final concentration 1 μg/mL) was added. Flow cytometry analysis was done on CyAN, Dako Cytomation using the Summit software (Flow cytometer, Beckman Coulter). Forward scatter (FSC) and side scatter (SSC) were acquired in linear mode. FITC and PI fluorescent signals derived from 488 nm excitation were detected in logarithmic mode at FL1/PMT3 and FL2/PMT4, with FITC 530/30 nm filters, and PI 585/42 nm filters, respectively, and a FL1/2 560 nm short pass dichroic filter. Signals for forward and side scatter and fluorescence were collected for 104 cells using the forward light scatter parameter as the master signal. Data are expressed as mean fluorescence intensity (FI) values. All experiments were run in triplicate.
MCM5 over-expression
To over-express MCM5 protein, we purchased the TrueClone MCM5 cDNA cloned in pCMV6-AC, which contains a full open reading frame of the human MCM5 gene (Origene, Rockville, MD, USA). The over-expression vector was transfected with Turbofect reagent (Fisher Scientific S.A.S., Illkirch, France) at 2 μL per well. pCMV- empty vector was used as negative control (NC). Once verified the over-expression by immuonoblotting, we performed a rescue experiment, in order to verify the direct involvement of MCM5 in JQ1 mechanism of action. Cells were transfected with pCMV-MCM5 or NC and exposed to 5 μM JQ1 or vehicle for 48 h. Cell viability was evaluated by MTT assay, as previously described.
Protein extraction and Western blot
Total protein extraction was performed as described previously (Passon et al. 2012). Briefly, FRO and SW1736 cells, incubated in vehicle-treated medium (NT, untreated cultures) or with JQ1 5 μM, were harvested by scraping and lysed with total lysis buffer (Tris HCl 50 mM pH8, NaCl 120 mM, EDTA 5 mM, Triton 1%, NP40 1%, protease inhibitors). For subcellular fractionation, pellets were resuspended in buffer A (10 mM Tris-HCl [pH 7.5], 1.5 mM MgCl2, and 10 mM KCl supplemented with 1× protease inhibitor cocktail, 0.5 mM PMSF, 1 mM NaF and 1 mM Na3VO4) and centrifugated at 2,000 × g for 10 min at 4°C. For nuclear extracts collection, pellets were resuspended in buffer B (20 mM Tris-HCl [pH 7.5], 0.42 M KCl, 1.5 mM MgCl2, 20% [vol/vol] glycerol supplemented with 1× protease inhibitor cocktail, 0.5 mM PMSF, 1 mM NaF, and 1 mM Na3VO4) and centrifuged at 15,000 × g for 30 min at 4°C. For Western Blot analysis, proteins were electrophoresed on 10% SDS-PAGE and then transferred to nitrocellulose membranes, which were saturated with 5% non-fat dry milk in PBS/0.1% Tween 20. The membranes were then incubated overnight with rabbit polyclonal anti-BRD4 antibody 1:1000 (Active Motif, Carlsbad, CA, USA), rabbit anti-lamin-β1 antibody 1:1000 (Abcam, Cambridge, UK), rabbit polyclonal anti-MCM5 antibody 1:250 (Sigma Aldrich) or rabbit anti-β-actin antibody 1:1000 (Abcam). The day after, membranes were incubated for 2 h with anti-rabbit immunoglobulin coupled to peroxidase 1:4000 (Sigma-Aldrich). The blots were developed using UVITEC Alliance LD (UVITec Limited, Cambridge, UK) with the SuperSignal Technology (Thermo Scientific Inc).
Human tissues and immunohistochemistry
A series of 12 normal thyroid glands (NTs), 25 follicular adenomas (FAs), 23 follicular thyroid carcinomas (FTCs), 36 papillary thyroid carcinomas (PTCs) and 8 anaplastic thyroid carcinomas (ATCs) were selected from the files of the Institution of Anatomic Pathology of the University of Udine and the most representative block of each lesion was retrieved from the archive. Representative tumor-bearing areas were selected and a tissue microarray was constructed. All samples were diagnosed by referral pathologists of the institution and then reviewed by a single experienced pathologist, thus including only patients with a confirmed diagnosis. Briefly, 5 μM formalin-fixed paraffin tissue sections mounted on SuperFrost Plus slides (Menzel-Gläser, Braunschweig, Germany) were placed in the PT Link pre-treatment module (DAKO A/S, Glostrup, Denmark), which performs automatically the entire pre-treatment process of de-paraffinization, rehydration, and epitope retrieval using the Low pH Target Retrieval Solution (0.001 M citrate buffer pH 6.0) at 98 °C for 40 min from DAKO. Endogenous peroxidase activity was blocked by incubation in the Peroxidase Block solution (DAKO) for 5 minutes. Primary rabbit polyclonal antiserum to MCM5 (Sigma Aldrich) diluted 1:1000 was applied and incubated for 60 minutes at room temperature. After washing slides were incubated with the DAKO EnVision FLEX System (DAKO) according to manufacturer’s guidelines. For reaction visualization, 3-3 diaminobenzidine tetrahydrochloride was used as chromogen. The sections were counterstained with Mayer’s hematoxylin. Using light microscopy, the entire section was scanned at high-power magnification (400×) and nuclear immunostaining was evaluated semi-quantitatively as percentage of positive cells. Staining intensity was scored as light, moderate and strong. A final score was then calculated by multiplying the percent staining by the intensity level.
Statistical analysis
mRNA and protein levels, cell viability and apoptosis levels were expressed as means ± SD, and significances were determined by a one-way ANOVA followed by Dunnett’s test performed with GraphPAD Software for Science (San Diego, CA, USA). *P value <0.05 vas considered statistically significant.
Results
Biological effects of BET inhibitors on ATC cells
In a first set of experiments, we evaluated the sensitivity of two human anaplastic thyroid cancer cell lines (FRO and SW1736) to BET inhibitors. As shown in Figure 1, incubation either with JQ1 (Panel A) or I-BET762 (Panel B) significantly reduced FRO and SW1736 cell viability at different time points. Relying on data obtained, we decided to use for further experiments the median effective dose of 5 μM, i.e. the dose required to achieve 50% of the desired response in 50% of the population. To assess whether the decrease in cell viability was due to an increase of apoptotic cell death, caspase 3/7 levels were measured after 48 h treatment. Administration of JQ1 increased caspase levels in both cell lines in a quite striking manner while I-BET762 treatment showed a less pronounced effect (Fig. 1, Panel C). In order to verify whether a 48 h I-BET762 treatment displayed a caspase-independent cell death, we perform a western blot analysis of PARP. PARP is known to be a substrate for several proteins as caspases, calpains and cathepsins; it produces several specific cleavage fragments that resemble signature patterns of different cell death programs (Gobeil et al. 2001; Chaitanya et al., 2010). As shown in Supplementary Figure 2 (Panels A and B) ATC cells showed a marked increase of both apoptotic- and necrotic-specific cleaved-PARP. We, then, corroborated the BET inhibition effects on cell viability and cell death in 8505c cells, a third anaplastic thyroid cell line (Supplementary Fig. 3). Moreover, we used a third BET inhibitor: I-BET151. This latter is a pan-BET inhibitor having a slight different chemical structure compared to I-BET762. I-BET151 reduced cell viability similarly to I-BET762 (Supplementary Fig. 1, Panels A and B, and Supplementary Fig. 3, Panels C). Hereinafter, we decided to proceed in our investigation with JQ1-treatment only, as it revealed to be the most effective drug, in terms of biological effects, in both cell lines. A FACS analysis of the cell cycle was then performed on both cell lines treated with 5 μM JQ1 for 24, 48 and 72 h. JQ1-treatment significantly increased the proportion of cells arrested in G0/G1 at each analyzed time point (Fig. 1, Panel D).
Figure 1. BET inhibitors administration decreases proliferation and survival of anaplastic thyroid carcinoma cell lines.

Panels A and B; FRO and SW1736 were exposed to JQ1 or I-BET762 different doses (rising from 50 nM to 10 μM). Cell viability was determined by MTT assay after 0, 24, 48 and 72 h and expressed as a percentage of baseline samples (T0). All samples were run in quadruplicate. Panel C; apoptosis levels were evaluated 48 h after treatment either with 5 μM BET inhibitors or vehicle (NT). Caspase 3/7 levels were normalized to the vehicle-treated group. Temozolomide (500 μM), a chemotherapy drug, was used as a positive control for apoptosis. Each sample was run in triplicate. * p < 0.05, *** p < 0.001, **** p < 0.0001 by ANOVA test. Panel D; FRO and SW1736 cell cycle distribution was determined after 5 μM JQ1 or vehicle administration. Cells were collected after 24, 48 and 72 hours treatments. Data are representative of 3 independent experiments.
Differential gene expression after BET inhibition
To identify potential molecular targets connected to BET inhibition, we performed a high-throughput RNA sequencing analysis on both FRO and SW1736 cells treated with 5 μM JQ1 or vehicle for 24 h. To assess the transcriptional changes induced by BET inhibition, a comparison between cells treated with JQ1 or vehicle was performed. Heat maps representing each analyzed condition are shown in Figure 2 (Panel A). Expression of 14199 and 13013 genes was detected in FRO and SW1736 cell lines, respectively. Vehicle-treated (NT) FRO and SW1736 cells showed a quite different mRNA profile. After filtering low quantity reads, results showed that approximately 2000 genes were differentially expressed after JQ1 treatment either in FRO or SW1736 cells (at a log2 fold change >2) (Supplementary Tables 1 and 2). Among them, 768 genes were significantly up-regulated and 1229 were down-regulated in response to JQ1 treatment (Fig. 2, Panel B). Venn diagrams showed that the majority of altered genes was specific for each cell line, which likely underlies the differential mRNA profile observed with vehicle-treatment. However, there was a core set of 288 genes commonly affected by JQ1 treatment in both cell lines, most of which decreased their expression (n=213). RNA-seq data, then, were confirmed by qPCR (Supplementary Figure 4). The shared top 20 differentially expressed genes (Tab.1) were then subjected to Ingenuity Pathway Analysis in order to outline which pathways were mostly affected by JQ1 treatment. Various signaling cascades turned out to be modified by BET inhibitors treatment: regulation of DNA and metabolic processes proved out to be the most deregulated one (Supplementary Table 3). The RNA-seq analysis identified a large number of JQ1 putative targets, primarily in cell cycle-linked pathways. These data are consistent with Da Costa’s findings in leukemia cells (Da Costa et al. 2013). Several studies have proposed a possible role of c-MYC down-regulation as a major mechanism of action of BET inhibitors (Zubet et al 2011; Alderton 2011). Our RNA-seq data show that this oncogene is not a critical factor in JQ1-mediated effects in ATC cells, since JQ1-treatment did not significantly affect c-MYC expression in neither of the two cell lines (Supplementary Figure 4). BET inhibitors, however, modified the expression of several cell cycle regulators such as RB proteins, E2F transcription factors and CDK-inhibitors (Supplementary Table 4).
Figure 2. Gene expression modifications in ATC JQ1-treated cell lines.

Panel A; Heat maps showing the hierarchical clustering of mRNA targets in FRO and SW1736 cell lines (N=1997). Cells were treated either with 5 μM JQ1 or vehicle for 24 hours. Panel B; Venn diagrams representing the comparison of both up-regulated (left) and down-regulated (right) genes between FRO and SW1736 cell lines after RNA-seq data analysis. Purple gene clusters represent shared modified genes between FRO and SW1736 cell lines.
Table 1.
Top 20 most down-regulated and up-regulated genes
| DOWN-REGULATED GENES | UP-REGULATED GENES | ||||
|---|---|---|---|---|---|
| GENE | Log2 (fold_change) | GENE | Log2 (fold_change) | ||
| FRO | SW1736 | FRO | SW1736 | ||
| E2F8 | −8.18691 | −3.32852 | HIST2H2BE | 5.45681 | 6.36269 |
| IL7R | −7.92256 | −4.28496 | EFR3B | 5.45085 | 2.31549 |
| SCG5 | −7.07872 | −5.36462 | HIST1H2BJ | 5.41635 | 4.29568 |
| MFAP2 | −6.55018 | −4.00946 | HIST1H2BC | 4.91188 | 3.90626 |
| KLF17 | −5.87194 | −2.54046 | AQP3 | 4.82547 | 5.53788 |
| HAS2 | −5.61592 | −2.59963 | KCNJ2-AS1 | 4.77102 | 2.57414 |
| SP140 | −5.51289 | −2.76406 | HIST1H2BD | 4.76918 | 7.37894 |
| SFTA1P | −4.86488 | −2.30007 | HIST1H1C | 4.65229 | 4.36823 |
| APOL3 | −4.76234 | −3.04492 | MSS51 | 4.53001 | 4.03264 |
| UHRF1 | −4.58863 | −2.45864 | PROCA1 | 4.31851 | 2.40037 |
| ELFN2 | −4.56104 | −2.07491 | RNFT2 | 4.20444 | 2.75534 |
| SLC38A5 | −4.50203 | −3.75806 | ARHGAP4 | 4.14859 | 2.29762 |
| WDR76 | −4.50144 | −3.32284 | ALS2CR12 | 4.07061 | 5.36388 |
| SEMA6B | −4.46547 | −2.00721 | CLU | 4.06906 | 3.93869 |
| STAP2 | −4.43699 | −3.34008 | HIST1H2AC | 3.98232 | 4.03878 |
| RFX8 | −4.42127 | −3.66572 | BFSP1 | 3.77102 | 2.63104 |
| FMNL1 | −4.42127 | −2.04374 | ITPR1 | 3.6183 | 7.03299 |
| GINS2 | −4.3919 | −2.49012 | FAM222A | 3.3286 | 2.436 |
| HTR1D | −4.32174 | −3.30238 | TLL2 | 3.24364 | 2.56248 |
| MCM5 | −4.27584 | −3.20364 | TM7SF2 | 3.20111 | 7.24837 |
BET inhibitors effects on MCM5
Considering that cell cycle regulatory genes were the most affected by JQ1 treatment, we focused on MCM5, one of the top 20 down-regulated genes, which is known to be involved in G1/S cell cycle transition, coordinating origin activation and replication fork progression. MCM5 belongs to the minichromosome maintenance complex family, which has been associated with the highly proliferative phenotype of ATC (Guida et al. 2005). Several studies have established that MCM5 protein level is a good diagnostic marker of malignancy for bladder, esophageal, and gastric cancers (Kebebew et al. 2006). After confirming RNA-seq data on mRNA levels (Fig. 3 Panel A), we evaluated whether a 5 μM JQ1 treatment significantly altered MCM5 protein levels in FRO and SW1736 cells (Fig. 3, Panel B). After 72 h incubation with JQ1, a significant reduction in MCM5 protein levels was observed (Fig. 3, Panel C). To extend and further validate this finding, we used two murine ATC cell lines derived from tumors developed by a genetically defined mouse model of ATC. Treatment of both lines with 0.5 μM JQ1 for 72h resulted in a significant downregulation of Mcm5 RNA (Fig. 3, Panel D). To evaluate whether MCM5 is a direct target of BRD4, which is the BET protein whose activity is inhibited by JQ1, we performed a ChIP-qPCR analysis in FRO and SW1736 cells. We first evaluated if the two cell lines expressed comparable BRD4 protein levels (Fig. 4, Panel A). After the ChIP assay, we observed an average 5-fold MCM5 enrichment in the BRD4-immunoprecipitate if compared to the IgG one. To confirm specificity, an intergenic region has been used as negative control for BRD4-binding (Fig. 4, Panel B). In order to test the biological relevance of MCM5 gene repression upon BET inhibition, an RNA interference analysis was performed. MCM5 gene expression was silenced both in FRO and in SW1736 cells, and immunoblot analysis revealed an almost total abolishment of endogenous MCM5 protein level with siRNA#2 treatment (Fig. 4, Panel C). Furthermore, MCM5 silencing, especially with siRNA#2, significantly decreased cell viability (p<0.001), increasing the percentage of apoptotic cells after Annexin V staining compared to negative control (Fig. 4, Panel D). Similarly, to what we observed treating cells with JQ1, this outcome was more pronounced in SW1736 than in FRO cells. To further prove that MCM5 is directly involved in the cell viability decrease after 5 μM JQ1 treatment, we performed a rescue experiment. FRO and SW1736 cells were treated with pCMV-MCM5 or NC and exposed to JQ1 for 48 h. As shown in Fig. 4 Panel E, MCM5 protein over-expression partially rescued its protein levels in JQ1-treated cells. To validate that this phenomenon is associate to a cell viability rescue, an MTT assay was performed. FRO and SW1736 cells over-expressing MCM5 displayed a slight but significative reduction (10% on average) of JQ1 effects when assessing cell viability (Fig. 4, Panel F). This mild rescue could be explained by the multi-target activity of BET inhibitors, whose effects are mediated by a remarkable number of actors, as we showed in our RNA-seq analysis (Fig. 2).
Figure 3. Down-regulation of MCM5 in human and murine ATC cell lines after JQl-treatment.

Panel A; FRO and SW1736 cells were treated either with 5 μM JQ1 or vehicle for 24 hours and MCM5 mRNA expression was evaluated by qPCR. All samples were run in triplicate. Vehicle-treated (NT) FRO cells were arbitrarily set at 1.0 and mRNA levels are expressed as relative expression values. Panel B; FRO and SW1736 were treated either with 5 μM JQ1 or vehicle for 72 h and MCM5 protein levels were detected by western blotting. Panel C; densitometric analysis of MCM5 protein levels in ATC cells after 5 μM JQ1 or vehicle treatment. Panel D; T4888M and T3531L cells were treated either with 0.5 μM JQ1 or vehicle for 72 hours and Mcm5 mRNA expression was evaluated by qPCR. All samples were run in triplicate. Vehicle-treated (NT) T4888M cells were arbitrarily set at 1.0 and mRNA levels are expressed as relative expression values. Results are shown as mean ± SD. *** p < 0.001, **** p < 0.0001 by ANOVA test. Data are representative of 3 independent experiments.
Figure 4. MCM5 is directly involved in BET inhibitors effects in human ATC cell lines.

Panel A; BRD4 protein expression in ATC cell lines. Panel B, chromatin was isolated from ATC cell lines and immunoprecipitated with BRD4 or IgG antibodies. MCM5 DNA was amplified using promoter-specific primers and analyzed by qPCR. All samples were run in triplicate. IgG-immunoprecipitate was arbitrarily set at 1.0 and the enrichment was expressed as relative expression value. An intergenic region (NCR) was used as a negative control for BRD4-binding. Panel C, FRO and SW1736 cells were treated with non-targeting siRNA (NC, negative control) or three different siRNA sequences specific to MCM5 (5 nM). Cells were collected after 72 h treatment and MCM5 protein levels were analyzed. Panel D; FRO and SW1736 were exposed to either siRNA targeting MCM5 or negative control for 72 hours and apoptosis levels was analyzed by Annexin V staining. Panel E, FRO and SW1736 cells were treated with pCMV empty vector (NC, negative control) or pCMV vector specific for MCM5 (1.3 μg). Cells were collected after 48 h treatment and MCM5 protein levels were analyzed. Panel F, FRO and SW1736 were treated with 0.01 μg p-CMV-MCM5 or empty vector and exposed to 5 μM JQ1 or vehicle. Cell viability was determined by MTT assay after 48 h. NT was arbitrarily set at 100 and cell viability was expressed as relative expression value. All samples were run in quadruplicate. Results are shown as mean ± SD. *** p < 0.001, **** p < 0.0001 by ANOVA test. Data are representative of 3 independent experiments.
In vivo MCM5 levels evaluation
Our data underline the relevance of MCM5 in thyroid cancer cell proliferation. Thus, MCM5 expression was evaluated in a panel of 12 normal thyroid samples (NTs), 25 follicular adenomas (FAs), 23 follicular thyroid carcinomas (FTCs), 36 papillary thyroid carcinomas (PTCs) and 8 anaplastic thyroid carcinomas (ATCs). Representative images of MCM5 staining are shown in Figure 5 (Panel A). MCM5 was faintly expressed in normal thyroid tissue and in FAs. PTCs showed a dual behavior: some samples displayed an intense and high MCM5 nuclear staining, while some others a much lesser one, with only a cytoplasmic signal. In FTCs, nuclear staining was lesser intense and frequent, with a predominant cytoplasmic MCM5 staining. ATCs, instead, showed a marked nuclear MCM5 localization. No significant differences were observed in terms of MCM5 nuclear or cytoplasmic staining intensity among different areas of each tumor. Quantification of the nuclear immunohistochemical signal was obtained by computing percentage of cell positivity and intensity of signal (Fig. 5, Panel B). Since cytoplasmic MCM5 protein represents the inactive MCM5 isoform (Abe et al. 2012), the strong nuclear staining observed in all ATCs and several PTCs would indicate its hyper-activation. Finally, we measured the expression of Mcm5 in four primary ATCs developed by [Pten, Tp53]thyr−/− mice (Antico Arciuch et al. 2011). In agreement with the human data, Mcm5 was found significantly overexpressed in mouse ATC, compared to normal thyroids (Fig. 5, Panel C), while its expression was not altered in FTCs developed by Ptenthyr−/− mice (data not shown).
Figure 5. MCM5 expression in thyroid tumors.

Panel A; representative images of MCM5 expression in normal thyroid tissues (NT), FAs, PTCs, FTCs and ATCs. Panel B; quantitation of MCM5 expression in normal and neoplastic thyroid tissues. Quantitation was obtained by using the IHC score, calculated as described in Materials and Methods section. Panel C; Mcm5 expression was evaluated by qPCR in control thyroids from wild type mice and primary ATCs from [Pten, Tp53]thyr−/− mice. All samples were run in triplicate. Wild type (WT) expression was set at 1.0 and mRNA levels are expressed as relative expression values. Results are shown as mean ± SD. * p < 0.05, ** p < 0.01, **** p < 0.0001 by ANOVA test.
Discussion
The cross talk between genomic and epigenomic elements in driving generation and progression of both hematologic malignancies and solid tumors has been extensively highlighted (Martín-Subero et al. 2013). Among epigenetic anticancer drugs, those targeting BET proteins have recently emerged as promising therapeutics in a remarkable range of diseases (Shi and Vakoc 2014; Wee et al. 2014). ATC is the most aggressive thyroid cancer subtype and it does not significantly respond to any therapy. In order to find new strategies to treat this aggressive thyroid cancer sub-type, we investigated BET inhibitors target genes in ATC cell lines. As expected, human ATC cell lines FRO and SW1736, treated with three different BET inhibitors, displayed a decrease in cell viability associated to an increase in cell death phenomena. These data were also been confirmed in a third ATC cell line (Supplementary Fig. 3). In our experimental model, BET inhibition triggers a multi-target cascade resulting in substantial biological effects, as demonstrated by RNA sequencing data. The identification of genes mediating BET inhibitors anti-proliferative effect could provide innovative therapeutic targets. Among hundreds of deregulated genes after BET inhibition, a large fraction belongs to regulators of cell cycle and apoptosis, i.e. E2F family members, CDKs and CDK inhibitors, and other TP53 pathway-related elements (Møller 2003; Smallridge et al. 2009). These data are similar to those previously published in medulloblastoma and metastatic melanoma (Segura et al. 2013; Henssen et al. 2013). All these findings corroborate the hypothesis that BET inhibitors disrupt cell cycle control, driving G0/G1 arrest and inducing cell death, by acting on multiple targets. Ott at al. observed that JQ1 treatment decrease the expression of IL7R in leukaemic cell lines, both at mRNA and protein levels (Ott et al. 2012). In anaplastic thyroid cell lines a 24h JQ1 treatment exhibited a significant down-regulation of IL7R mRNA levels, but this reduction was not confirmed analyzing protein levels (Supplementary Figure 5). We would like to stress that endogenous IL7R protein is not so abundant in these two cell lines; therefore, the misregulation of a protein involved in during lymphocyte development seems not to perform a substantial role in JQ1 effects in thyroid cancer cells. Our data, instead, show an at least 10-fold down-regulation of MCM5 expression in JQ1-treated cells, suggesting a critical role of this protein in the molecular cascade triggered by BET inhibitors. As confirmation, MCM5 proved to be a direct target of BRD4, as highlighted in the ChIP-qPCR assay. Moreover, MCM5 overexpression moderately attenuated FRO and SW1736 JQ1 sensitivity, in terms of cell viability, as shown in the rescue experiment (Fig. 4, Panel F). This mild effect is reasonably due to the multi-target activity of epigenetic drugs, which act simultaneously on a wide range of pathways, as shown in our RNA-seq analysis. Accordingly to our data, poor rescue effects have been obtained for other targets of BET inhibitors (Lockwood et al., 2012; Bandopadhayay et al., 2014). MCM5 belongs to the minichromosome maintenance complex, which includes elements of the pre-replicative complex (pre-RC) that are essential for DNA replication (Noseda and Karsan 2006). MCM proteins are considered licensing components for the S-phase initiation. MCMs oscillate between nuclear and cytoplasmic localizations (Madine et al. 2000). Nuclear MCMs associate with DNA during G1 and early S-phase, allowing the control of replication origin firing, in order to restrict the chromosome replication to only one round per cell cycle (Romanowski and Madine 1997). MCMs are restricted, instead, to cytoplasm during G2/M phase in order to prevent re-replication phenomena (Laskey 2005). Recently, components of MCM family have been described as aberrantly expressed in many malignancies, including thyroid carcinomas (Giaginis et al. 2010). In agreement with these findings and corroborating our in vitro and in vivo results, MCM5 immunostaining demonstrated that ATCs contained more MCM5-positive cells than did normal thyroid gland (Fig. 5).
Several studies have demonstrated that expression of MCM5 is controlled by different pathways (Guida et al. 2005; Abe et al. 2012; Noseda and Karsan 2006; Laskey 2005). Figure 6 summarizes major players controlling MCM5 gene expression, as well as their functional interactions. By comparing Figure 6 and Supplementary Table 4, it is undeniable that many proteins that up-regulate MCM5 expression are also down-regulated by BET inhibitors. Interestingly, JQ1 reduces the expression of some elements involved in the replication fork assembly thus affecting cell cycle mechanisms, in which, in turn, MCM proteins are involved. Altogether, this cascade could lead to G0/G1 arrest (see Fig. 1, Panel D) and cell death. In conclusion, our findings support the hypothesis that MCM5 targeting may be regarded as an effective molecular therapy against ATC.
Figure 6. Biological network related to MCMs protein function.
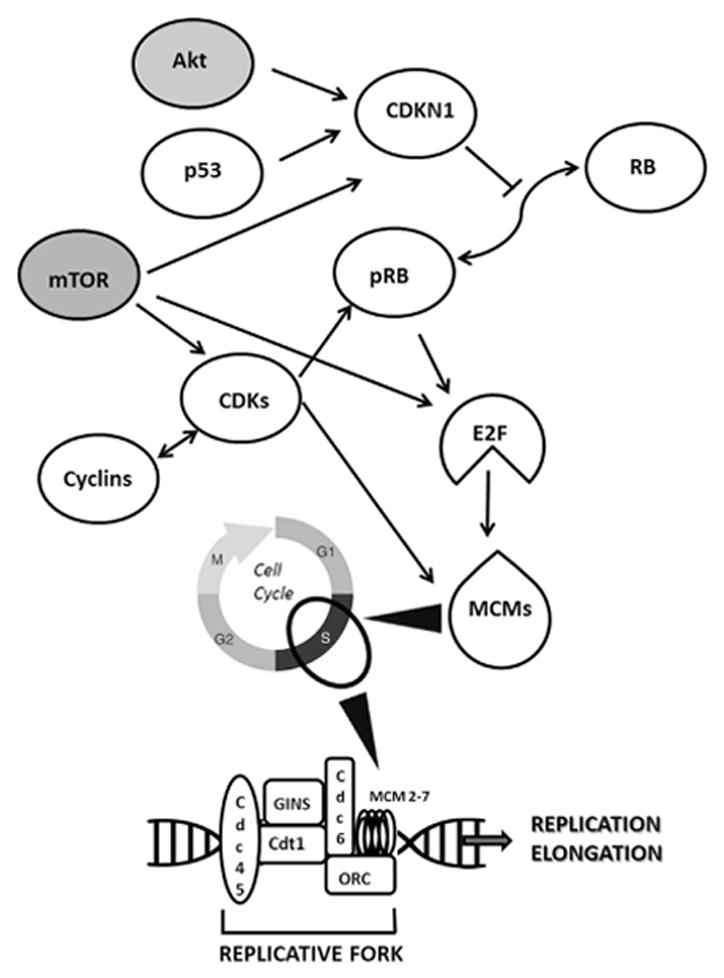
Network diagram (upper part) showing key molecules controlling MCMs protein levels. White symbols represent proteins whose mRNA levels are modulated by BET inhibitors in our transcriptome analysis (see Supplementary Table 4); gray symbols indicate proteins whose mRNA levels are not modulated by BET inhibitors. Arrows indicate functional relationship between proteins (delineated in Guida et al. 2005; Abe et al. 2012; Madine et al. 2000; Laskey 2005). The lower part indicates factors cooperating with MCM proteins in the replicative fork assembly. All of these factors are down-regulated by BET inhibitors (see Supplementary Table 4). Arrowheads show the relationship between MCMs and cell cycle phases.
Supplementary Material
Acknowledgments
Funding
This work was supported by grants to GD from Associazione Italiana per la Ricerca sul Cancro (AIRC) (project n° IG 10296) and from Ministero degli Affari Esteri of Italy (Progetti grande rilevanza 2016, project n° PGR02954), to ADC from the National Institutes of Health (CA128943 and CA172012).
Abbreviations
- ATC
anaplastic thyroid cancer
- BET
Bromodomain and Extra-Terminal proteins
- BETi
BET inhibitors
- FA
follicular adenoma
- FTC
follicular thyroid cancer
- H2O2
hydrogen peroxide
- MCM5
minichromosome manteinance complex 5
- NT
normal thyroid gland
- PTC
papillary thyroid cancer
- TZM
Temozolomide
Footnotes
Declaration of interest
The authors have nothing to disclose.
References
- Abe S, Kurata M, Suzuki S, Yamamoto K, Aisaki K, Kanno J, Kitagawa M. Minichromosome Maintenance 2 Bound with Retroviral Gp70 Is Localized to Cytoplasm and Enhances DNA-Damage-Induced Apoptosis. PLoS One. 2012;7:40129. doi: 10.1371/journal.pone.0040129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton GK. Targeting MYC? You BET. Nat Rev Drug Discov. 2011;10:732–3. doi: 10.1038/nrd3569. [DOI] [PubMed] [Google Scholar]
- Antico Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29:5678–5686. doi: 10.1038/onc.2010.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico Arciuch VG, Russo MA, Dima M, Kang Ks, Dasrath F, Liao XH, Refetoff S, Montagna C, Di Cristofano A. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget. 2011;2:1109–1126. doi: 10.18632/oncotarget.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arturi F, Russo D, Giuffrida D, Schlumberger M, Filetti S. Sodium-Iodide symporter (NIS) gene expression in lymph node metastases of papillary thyroid carcinomas. Eur J Endocrinol. 2000;143:623–627. doi: 10.1530/eje.0.1430623. [DOI] [PubMed] [Google Scholar]
- Baldan F, Lavarone E, Di Loreto C, Filetti S, Russo D, Damante G, Puppin C. Histone post-translational modifications induced by histone deacetylase inhibition in transcriptional control units of NIS gene. Mol Biol Rep. 2014;41:5257–65. doi: 10.1007/s11033-014-3397-x. [DOI] [PubMed] [Google Scholar]
- Baldan F, Mio C, Lavarone E, Di Loreto C, Puglisi F, Damante G, Puppin C. Epigenetic bivalent marking is permissive to the synergy of HDAC and PARP inhibitors on TXNIP expression in breast cancer cells. Oncol Rep. 2015;33:2199–206. doi: 10.3892/or.2015.3873. [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al. BET bromodomain inhibition of M http://www.edmgr.com/ijsem/Default.aspx YC-amplified medulloblastoma. Clin Cancer Res. 2014;20:912–25. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. 2009;1:98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa D, Agathanggelou A, Perry T, Weston V, Petermann E, Zlatanou A, Oldreive C, Wei W, Stewart G, Longman J, et al. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 2013;3:e126. doi: 10.1038/bcj.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–57. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De Vathaire F, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892–9. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- Elisei R. Anaplastic thyroid cancer therapy: dream or reality? Endocrine. 2012;42:468–70. doi: 10.1007/s12020-012-9785-x. [DOI] [PubMed] [Google Scholar]
- Falahi F, Sgro A, Blancafort P. Epigenome Engineering in Cancer: Fairytale or a Realistic Path to the Clinic? Front Oncol. 2015;5:22. doi: 10.3389/fonc.2015.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaginis C, Vgenopoulou S, Vielh P, Theocharis S. MCM proteins as diagnostic and prognostic tumor markers in the clinical setting. Histol Histopathol. 2010;25:351–70. doi: 10.14670/HH-25.351. [DOI] [PubMed] [Google Scholar]
- Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–94. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- Guida T, Salvatore G, Faviana P, Giannini R, Garcia-Rostan G, Provitera L, Basolo F, Fusco A, Carlomagno F, Santoro M. Mitogenic effects of the up-regulation of minichromosome maintenance proteins in anaplastic thyroid carcinoma. J Clin Endocrinol Metab. 2005;90:4703–9. doi: 10.1210/jc.2004-2459. [DOI] [PubMed] [Google Scholar]
- Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schäfers S, Schramm A, et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget. 2013;4:2080–95. doi: 10.18632/oncotarget.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebebew E, Peng M, Reiff E, Duh QY, Clark OH, McMillan A. Diagnostic and prognostic value of cell-cycle regulatory genes in malignant thyroid neoplasms. World J Surg. 2006;30:767–74. doi: 10.1007/s00268-005-0308-2. [DOI] [PubMed] [Google Scholar]
- Laskey R. The Croonian Lecture 2001 hunting the antisocial cancer cell: MCM proteins and their exploitation. Philos Trans R Soc Lond B Biol Sci. 2005;360:1119–32. doi: 10.1098/rstb.2005.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408–13. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madine MA, Swietlik M, Pelizon C, Romanowski P, Mills AD, Laskey RA. The roles of the MCM, ORC, and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells. J Struct Biol. 2000;129:198–210. doi: 10.1006/jsbi.2000.4218. [DOI] [PubMed] [Google Scholar]
- Martín-Subero JI, López-Otín C, Campo E. Genetic and epigenetic basis of chronic lymphocytic leukemia. Curr Opin Hematol. 2013;20:362–8. doi: 10.1097/MOH.0b013e32836235dc. [DOI] [PubMed] [Google Scholar]
- Møller MB. Molecular control of the cell cycle in cancer: biological and clinical aspects. Dan Med Bull. 2003;50:118–38. [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseda M, Karsan A. Notch and minichromosome maintenance (MCM) proteins: integration of two ancestral pathways in cell cycle control. Cell Cycle. 2006;5:2704–9. doi: 10.4161/cc.5.23.3515. [DOI] [PubMed] [Google Scholar]
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–52. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passon N, Gerometta A, Puppin C, Lavarone E, Puglisi F, Tell G, Di Loreto C, Damante G. Expression of Dicer and Drosha in triple-negative breast cancer. J Clin Pathol. 2012;65:320–6. doi: 10.1136/jclinpath-2011-200496. [DOI] [PubMed] [Google Scholar]
- Patel KN, Shaha AR. Poorly differentiated thyroid cancer. Curr Opin Otolaryngol Head Neck Surg. 2014;22:121–6. doi: 10.1097/MOO.0000000000000037. [DOI] [PubMed] [Google Scholar]
- Perri F, Lorenzo GD, Scarpati GD, Buonerba C. Anaplastic thyroid carcinoma: A comprehensive review of current and future therapeutic options. World J Clin Oncol. 2011;2:150–7. doi: 10.5306/wjco.v2.i3.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli T, Prasad KV, Jayarama S, Pacini F, Prabhakar BS. Potential utility and limitations of thyroid cancer cell lines as models for studying thyroid cancer. Thyroid. 2009;19:1333–42. doi: 10.1089/thy.2009.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppin C, D’Aurizio F, D’Elia AV, Cesaratto L, Tell G, Russo D, Filetti S, Ferretti E, Tosi E, Mattei T, et al. Effects of histone acetylation on NIS promoter and expression of thyroid-specific transcription factors. Endocrinology. 2005;146:3967–3974. doi: 10.1210/en.2005-0128. [DOI] [PubMed] [Google Scholar]
- Romanowski P, Madine MA. Mechanisms restricting DNA replication to once per cell cycle: the role of Cdc6p and ORC. Trends Cell Biol. 1997;7:9–10. doi: 10.1016/S0962-8924(97)30077-4. [DOI] [PubMed] [Google Scholar]
- Russo D, Durante C, Bulotta S, Puppin C, Puxeddu E, Filetti S, Damante G. Targeting histone deacetylase in thyroid cancer. Expert Opin Ther Targets. 2013;17:179–193. doi: 10.1517/14728222.2013.740013. [DOI] [PubMed] [Google Scholar]
- Schlumberger M, Lacroix L, Russo D, Filetti S, Bidart JM. Defects in iodide metabolism in thyroid cancer and implications for the follow-up and treatment of patients. Nat Clin Pract Endocrinol Metab. 2007;3:260–269. doi: 10.1038/ncpendmet0449. [DOI] [PubMed] [Google Scholar]
- Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, Fagin JA, Marlow LA, Copland JA, Smallridge RC, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, González-Gomez P, Morante M, Jubierre L, Zhang W, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–76. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–36. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer. 2009;16:17–44. doi: 10.1677/ERC-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzi F, Del Fabbro C, Tomescu AI, Policriti A. rNA: a fast and accurate short reads numerical aligner. Bioinformatics. 2012;28:123–4. doi: 10.1093/bioinformatics/btr617. [DOI] [PubMed] [Google Scholar]
- Wee S, Dhanak D, Li H, Armstrong SA, Copeland RA, Sims R, Baylin SB, Liu XS, Schweizer L. Targeting epigenetic regulators for cancer therapy. Ann N Y Acad Sci. 2014;1309:30–36. doi: 10.1111/nyas.12356. [DOI] [PubMed] [Google Scholar]
- Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007;67:959–66. doi: 10.1158/0008-5472.CAN-06-3524. [DOI] [PubMed] [Google Scholar]
- Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–40. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.