

Mutations in CSF1R, which encodes colony stimulating factor 1 receptor, cause hereditary diffuse leukoencephalopathy with axonal spheroids. Eichler et al. describe a family in which the mother is unaffected despite carrying a CSF1R mutation in ~20% of blood cells. Haematopoietic stem cell transplantation halted disease progression in her affected daughter.
Keywords: leukodystrophy, whole exome sequencing, dementia, neuroinflammation, neurodegeneration
Mutations in CSF1R, which encodes colony stimulating factor 1 receptor, cause hereditary diffuse leukoencephalopathy with axonal spheroids. Eichler et al. describe a family in which the mother is unaffected despite carrying a CSF1R mutation in ~20% of blood cells. Haematopoietic stem cell transplantation halted disease progression in her affected daughter.
Abstract
Mutations in the colony stimulating factor 1 receptor (CSF1R) have recently been discovered as causal for hereditary diffuse leukoencephalopathy with axonal spheroids. We identified a novel, heterozygous missense mutation in CSF1R [c.1990G > A p.(E664K)] by exome sequencing in five members of a family with hereditary diffuse leukoencephalopathy with axonal spheroids. Three affected siblings had characteristic white matter abnormalities and presented with progressive neurological decline. In the fourth affected sibling, early progression halted after allogeneic haematopoietic stem cell transplantation from a related donor. Blood spot DNA from this subject displayed chimerism in CSF1R acquired after haematopoietic stem cell transplantation. Interestingly, both parents were unaffected but the mother’s blood and saliva were mosaic for the CSF1R mutation. Our findings suggest that expression of wild-type CSF1R in some cells, whether achieved by mosaicism or chimerism, may confer benefit in hereditary diffuse leukoencephalopathy with axonal spheroids and suggest that haematopoietic stem cell transplantation might have a therapeutic role for this disorder.
Introduction
Leukodystrophies are inherited disorders affecting the white matter of the CNS. While most disorders have predominantly glial cell or myelin sheath abnormalities, some disorders include significant axonal pathology. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) is a progressive autosomal dominant leukodystrophy with an onset typically occurring in the fourth or fifth decade of life. Recently, Rademakers and colleagues (2012) identified mutations in CSF1R, encoding colony stimulating factor 1, as the cause of HDLS.
We performed exome sequencing in a family with four siblings affected by an unknown adult-onset leukoencephalopathy and found a novel heterozygous mutation in CSF1R. This was particularly surprising as the parents were unaffected, suggesting an autosomal recessive disorder. One of the four siblings carried the presumed diagnosis of metachromatic leukodystrophy and had stabilized after haematopoietic stem cell transplantation 15 years earlier.
Materials and methods
Human subjects
Ethical approval was obtained from Massachusetts General Hospital (protocol #2007P002248). Informed consent was obtained from all subjects. Epstein-Barr virus transformed lymphoblast cell lines were established in all subjects. DNA was subsequently extracted from whole blood and/or lymphoblast cell lines and from saliva and buccal swabs from suspected family members with mosaicism or chimerism of CSF1R.
The brain MRI in all affected siblings was consistent with a leukodystrophy, i.e. symmetrical, confluent, hyperintense signal on T2-weighted images and prominent T1 hypointensity of the affected white matter relative to grey matter structures. Brain tissue was not available.
Case histories
Patient II-1
This female patient developed behavioural and memory difficulties at 35 years of age. She was described as impulsive and was noted to frequently yell and shout insults. Once, she was found disoriented outside her home. Around the same time, she developed gait difficulties, described as shuffling gait with balance problems. By 36 years of age, she was wheelchair-bound and no longer able to ambulate. She was diagnosed with presumed adult-onset metachromatic leukodystrophy and a year later received an allogeneic haematopoietic stem cell transplantation (HSCT); her unaffected brother (Patient II-5) was the donor. She successfully engrafted and her disease stabilized within 6 months from the transplant. For the next 15 years she maintained stable language function and personality and remained non-ambulatory.
Patient II-2
A 56-year-old male developed confusion and word-finding difficulties. Over the next 2 years, he experienced gait difficulties and required a walker. Around the same time, he developed a seizure disorder, with seizures occurring several times per month. At the present time, expressive language has deteriorated, but he understands simple commands. He can get quite aggressive and, on occasion, punch and spit at his caregivers. He does not verbalize spontaneously and is no longer able to ambulate. Testing for arylsulphatase A enzyme abnormalities was negative. No sulphatides in urine were present.
Patient II-3
A 54-year-old female with a history of psychological problems began having gait difficulties. Her parents noted that she was shuffling and had difficulties walking up stairs. She also became more depressed and tearful. She had a seizure-like event and was placed on gabapentin. After being unable to walk, she was placed in a long-term care facility. Testing for arylsulphatase A enzyme abnormalities was negative. No sulphatides in urine were present.
Patient II-4
A 55-year-old female began having behavioural difficulties, depression, and psychotic episodes. Soon after these first symptoms she developed gait difficulties and needed full-time assistance. Due to swallowing problems, she required placement of a gastric tube. At 58 years of age, she was admitted to a nursing home following a hospital admission for status epilepticus. On assessment 2 years later, she was non-verbal and non-ambulatory. She grunted and grimaced to pain, but showed no interactions with her surroundings otherwise. She passed away at 60 years of age.
Parents and unaffected siblings
The mother (Patient I-2) is an 83-year-old homemaker with intact cognition, speech and motor abilities. The father (Patient I-1) is an 85-year-old retired tool maker and machinist. Neurological examination on parents and unaffected siblings is normal.
Exome sequencing and Sanger sequencing
Exome sequencing of blood DNA was performed in five family members: two affected siblings (Patients II-2 and II-3), one unaffected sibling (Patient II-6) and both parents (Patients I-1 and I-2). A recessive mode of inheritance was assumed, as both parents were unaffected.
A solution hybrid method was used to enrich for DNA targeting the exome (Agilent Sure-Select Human All exon 50 Mb), and sequencing was performed on the Illumina HiSeq2000 using 90-bp paired-end reads.
Unaligned Illumina reads were processed through the Broad Institute Picard suite pipeline and mapped to the hg19 reference genome using the Burrows-Wheeler alignment (BWA) algorithm (Li and Durbin, 2009). High quality indel and single nucleotide variant calling and annotation were performed using the Broad GATK2 joint analysis pipeline using standard filtering criteria (read depth ≥10%, genotype quality score ≥30). Details of solution hybrid selection and target sequence coverage are presented in Supplementary Table 1. We prioritized novel variants by excluding single nucleotide polymorphisms (SNPs) present in whole genome sequencing data from 1092 individuals (1000 Genomes Project Consortium et al., 2012) or in dbSNP (Sherry et al., 2001). Any known pathogenic mutations found in dbSNP were filtered out and were included in further analysis.
Sanger sequencing of a 407-bp amplicon encompassing the mutation was performed to confirm the CSF1R mutation in all exome-sequenced subjects and to test for segregation of the variant with disease in the family. Polymerase chain reaction (PCR) was performed in a 25 µl volume with 40–50 ng DNA, 1 µl of 10 mM forward primer 5’ TTCATGAGCCATCCAACC3’and reverse primer 5’AGGCACAAGGAAACTTGCTC3’. Amplification conditions were 95 °C for 5 min followed by 35 cycles at 95 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min and final extension at 72 °C for 10 min. PCR amplification was followed by Sanger sequencing on an ABI 3170 sequencer. PCR amplification and Sanger sequencing was also performed using DNA extracted from saliva and lymphoblastoid cell lines for two of the subjects. Mosaicism for the CSF1R c.1990G=/>A, p.(E664K) mutation in DNA from the mother’s blood was confirmed by cloning of a 2481-bp PCR product (amplified using forward primer 5’CAAGCAGGAACATGCTCTCA 3’ and reverse primer 5’TGGCTACTTCCCATGACACA3’) encompassing the mutation into vector pGL3 (Promega) in 5’-3’ orientation using a naturally occurring BamHI site. Sanger sequencing of six clones was performed using the 5’TTCATGAGCCATCCAACC3’ primer.
For chimerism analyses, DNA isolated from donor blood, recipient (post-transplant saliva and buccal swab), and post-transplant blood samples were PCR amplified for 15 microsatellite markers and amelogenin and analysed by fluorescent capillary electrophoresis (University of North Carolina).
Results
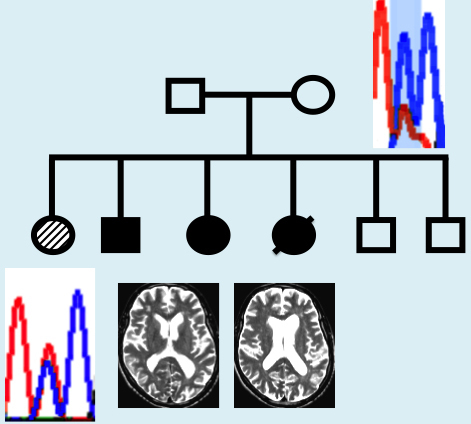
Affected siblings had characteristic white matter abnormalities and presented with progressive neurologic decline. Clinical characteristics are listed in Table 1. In one affected sibling (Patient II-1), early progression halted after allogeneic HSCT from her unaffected brother (Patient II-5) 15 years ago. Representative MRI findings are shown in Fig. 1.
Table 1.
Clinical characteristics and CSF1R mutation in family with HDLS
| ID | CSF1R p.E664K | Sex | Onset age | Death age | Initial symptom |
Clinical features during course of disease development |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Behavioural | Dementia | Depression | Parkinsonism | Seizures | ||||||
| I-2 | Mosaic | F | . | Alive | Healthy | None | None | None | None | None |
| II-1 | Chimeric | F | 35 | Alive | Impulsivity | + | + | Not described | Not described | Not described |
| II-2 | Heterozygous | M | 56 | Alive | Confusion | +++ | ++ | + | Rigidity | + |
| II-3 | Heterozygous | F | 54 | 60 | OCD | ++ | ++ | +++ | Not described | + |
| II-4 | Heterozygous | F | 55 | 60 | Depression | ++ | + | +++ | + | ++ (status) |
OCD = obsessive compulsive disorder.
Figure 1.

MRI findings in HDLS. Affected sibling Patient II-2: 60-year-old male with progressive cognitive deterioration and motor problems. His MRI shows enlarged ventricles with diffuse patchy white matter lesions more prominent in the frontal (A, white arrows) than in the posterior regions (B, black arrows). There is no contrast enhancement present.
To identify the underlying causal gene, we targeted >45 million base pairs in 157 523 exons from 15 994 genes for exome sequencing in five family members (parents, affected siblings Patients II-2 and II-3 and unaffected sibling Patient II-6), with each targeted base covered on average 51 times per individual (Supplementary Table 1). For each participant, ∼872 novel protein-altering variants were found, of which ∼837 were missense mutations. To find causal mutations inherited in an autosomal recessive manner, we searched for genes with novel, deleterious mutations or known recessive pathogenic mutations from dbSNP present in both alleles in both affected siblings (either homozygous or compound-heterozygous mutations) but with only one of the two deleterious mutations in the father and the other in the mother, and at most one deleterious mutation in the unaffected sibling. No such genes or variants were found.
We did identify a novel heterozygous G to A transition within exon 15 of CSF1R (c.1990G > A) in both affected siblings (49% of exome sequencing reads in Patient II-2 and 40% of sequencing reads in Patient II-3) that was absent in the sequences from the unaffected sibling and the father (Supplementary Table 1). The A allele is also absent in the dbSNP database (Sherry et al., 2001), in 2184 chromosomes from the 1KGP (1000 Genomes Project Consortium et al., 2012), in 13 006 exome sequences from the National Heart Lung and Blood Institute’s Exome Sequencing Project [Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (URL:http://evs.gs.washington.edu/EVS/) (8 June 2013)] and in the ExAC database of 60 706 unrelated individuals [Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) (accessed 9 July 2015)]. The resulting non-synonymous amino acid substitution, p.(E664K), is predicted to be deleterious by prediction algorithms PolyPhen-2 (Adzhubei et al., 2013), SIFT (Kumar et al., 2009), MutationTaster (Schwarz et al., 2010), and CADD (score of 36, ranking in the 0.025 percentile of deleterious mutations) (Kircher et al., 2014) and alters a site conserved across Euteleostomi species (NCBI Resource Coordinators, 2013) and across other CSF1/PDGF receptor family members (Fig. 2). As this residue is predicted to lie within the tyrosine kinase domain of CSF1R (Coussens et al., 1986), encoded by a gene implicated previously in HDLS, a familial neurodegenerative condition with a similar phenotype (Rademakers et al., 2012), we examined this variant more closely in all family members.
Figure 2.

Genomic and protein localization of novel CSF1R mutation. (A) Genomic organization of the 60 kb CSF1R gene with 22 exons (vertical hatches); (B) exon structure of the human CSF1R cDNA, with start codon (ATG) and stop codon (TGA) shown. Arrow shows position of c.1990G > A, p.(E664K) mutation in exon 15. (C) Domain structure of the CSF1R protein showing the immunoglobulin domains (Ig) and the protein tyrosine kinase domain (PTK), interrupted by the kinase insert at amino acid positions 670-740 (shaded). (D) The position of the p.E664K mutation adjacent to the kinase insert within the PTK domain. (E) Alignment for the parts of the PTK domain surrounding p.E664K, including human CSF1/PDGF receptor family members and multiple CSF1R homologues.
Sanger sequencing at the novel CSFR1 mutation site p.(E664K) confirmed exome variant calls in all samples, and revealed that the mutation segregated with disease in affected siblings (Fig. 3). The proportion of mutant A allele for affected siblings was estimated at ∼50% from Sanger sequencing of DNA from saliva (Patients II-1 and II-4) or from blood (Patients II-2 and II-3; Fig. 3). While the father (Patient I-1) did not carry the mutant A allele, DNA from the blood and saliva of the unaffected mother (Patient I-2) displayed mosaicism, with the A allele fraction of DNA estimated at ∼15–20% from Sanger sequencing of saliva or blood DNA and 20% from exome sequencing of blood DNA (Fig. 3 and Supplementary Fig. 1). Based on the Sanger sequence traces, the fraction of cells carrying the mutant A allele appeared to be greater in DNA from saliva (∼20% A allele; mixture of epithelial and haematopoietic cells) than from the blood (∼15% A allele; haematopoietic cells only), providing suggestive evidence that mosaicism may differ by cell type (Fig. 3). Sequencing of six subclones from the PCR product derived from the mother’s blood DNA revealed one clone with the mutant A allele and five clones with the wild-type G allele (Supplementary Fig. 1). Chimerism analysis was performed for affected sibling Patient II-1 using blood spot DNA obtained >15 years after HSCT. The per cent donor chimerism, calculated from the average of 11 informative short tandem repeat markers, was 15% (standard deviation 5%).
Figure 3.

Pedigree of family with HDLS and sequence at the novel CSF1R p.E664K mutation site. Filled circles (black or hashed pattern) indicate affected family members. Transplanted individual (Patient II-1) is labelled with ‘HSCT’ and filled with a hashed pattern to indicate a milder clinical course. Deceased individual is marked by a slash. Sanger sequence traces of DNA surrounding the c.1990G > A, p.(E664K) mutation shaded in blue [reverse strand sequence corresponding to wild-type CC homozygous genotype (wt) or heterozygous CT genotype (het) are shown for each family member]. The source of DNA for each trace is listed below the trace diagram and fraction of mutant allele reads from exome sequencing or estimated by Sanger sequencing is indicated in brackets after source of DNA. Mosaicism is seen in DNA from blood and saliva from Patient I-2 (mother). Sanger trace for transplanted Patient II-1 in blood spot DNA is ∼50% (consistent with 57.5% normal allele expected based on 15% donor chimerism).
Discussion
Heterozygous mutations in CSF1R causing HDLS were first identified by Rademakers et al. (2012). CSF1R encodes a tyrosine kinase growth factor receptor for colony stimulating factor 1, the macrophage and monocyte-specific growth factor (Ridge et al., 1990). This cell-surface receptor regulates survival, proliferation, and differentiation of mononuclear phagocytic cells, including microglia of the CNS (Stanley et al., 1997). In the brain, CSF1R protein is predominantly expressed in microglial cells (Ginhoux et al., 2010), although low levels of CSF1R have been reported in cultured neurons (Akiyama et al., 1994; Raivich et al., 1998; Wang et al., 1999). All disease-causing mutations to date—including that of our family—are located within the tyrosine kinase domain of CSF1R.
The phenotype of HDLS is characterized by adult-onset rapidly progressive neurodegenerative disease characterized by behavioural, cognitive and motor changes. Our patients followed this course and—with the exception of the transplanted patient—were all non-verbal or dead within 10 years of symptom onset. Brain imaging showed patchy involvement of white matter, predominantly in the frontal and parietal regions. The disorder is inherited in an autosomal dominant manner, and therefore affected individuals would typically be expected to have an affected parent. In our family, however, the mother was mosaic and unaffected, conveying the false impression of a recessive pedigree and indicating that the proportion of her cells with a mutant allele was insufficient to precipitate a dementia phenotype. Alternatively her asymptomatic state could have represented a lack of penetrance or a late onset of phenotype that so far had not manifested in the 83-year-old female. A limitation of the study is that we did not have access to post-mortem tissue to verify HDLS pathology or mosaic status in various tissues.
Mosaic forms have been reported in other autosomal dominant disorders, such as neurofibromatosis 1 and hereditary haemorrhagic telangiectasia (Ruggieri and Huson, 2001; Lee et al., 2011). Some patients with mosaicism have affected children, while others appear unable to pass the mutation on to offspring, suggesting that the mutation can be limited to a part of the soma and thus absent in the germ line.
No treatment is currently known to halt or alter the course of HDLS. Most patients reported to date succumb to the disease within 10 years of symptom onset (Rademakers et al., 2012). To have a patient retain a high level of communication and survive beyond 15 years after symptom onset is unusual and suggests amelioration following HSCT. An individual who has undergone a successful allogeneic bone marrow transplant is chimeric, i.e. an individual comprised of multiple cell lineages derived from distinct fertilized eggs (Biesecker and Spinner, 2013). Chimerism is thus distinct from the related phenomenon of mosaicism but either chimeric or mosaic status that lowers the overall fraction of haematopoietic cells with mutant CSF1R alleles can be of benefit.
Only a few neurological disorders are known to benefit from HSCT. Among these are other leukodystrophies such as adrenoleukodystrophy, metachromatic leukodystrophy, and globoid cell leukodystrophy. All of these are recessive disorders that require both alleles to be defective for disease to occur. In contrast, HDLS is a dominant disorder where one mutant allele alone can compromise CSF1R function, with haploinsufficiency a likely cause (Konno et al., 2014). HSCT may confer benefit in recessive disorders through non-cell autonomous introduction of wild-type protein, and may similarly enhance CSF1R signalling after partial loss in HDLS. Only a small percentage of cells were found to be corrected in the affected sibling 15 years after HSCT (15%). However, the clinical benefit of HSCT with this small percentage of corrected cells is consistent with per cent correction seen after cell-based correction in the leukodystrophies. In particular, cerebral childhood adrenoleukodystrophy is known to stabilize after correction of only 20% of peripheral blood cells. We were not able to obtain interim assessments of levels of chimerism after transplant to determine the stability of chimerism levels over time. It is possible that stabilization occurred spontaneously in our transplanted patient and is not related to the HSCT. In that case the correction of her peripheral blood cells was coincidental to her clinical course. Late onset phenotypes are known to show variability in course, and we can therefore not exclude this possibility but find it highly unlikely given the immediately preceding precipitous decline.
While introduction of cells with wild-type CSF1R by HSCT may compensate for partial loss of CSF1R function, the alternative of over-expression of CSF1R beyond normal levels in individual cells, by gene therapy for example, may carry risks. An increase in CSF1R copy number and point mutations leading to constitutive activation of the CSF1R receptor has been associated with tumour development, including haematological malignancies and renal cell carcinomas (Ridge et al., 1990; Soares et al., 2009).
In summary, this first report of mosaicism in CSF1R in a family affected by HDLS, involving a mosaic unaffected mother who passed the pathogenic mutation through her germ line to multiple children, one of whom was chimeric and showed a milder course, suggests strongly that HSCT be explored for potential therapeutic benefit in this devastating disorder.
Supplementary Material
Acknowledgements
We thank study participants and the Shenzhen Municipal of Government of China (CXB201108250094A) for support.
Glossary
Abbreviations
- HDLS =
hereditary diffuse leukoencephalopathy with axonal spheroids
- HSCT =
haematopoietic stem cell transplantation
Funding
This work was supported by an Institutional Development Award to the Center for Applied Genomics from The Children’s Hospital of Philadelphia and Massachusetts General Hospital Department of Anaesthesia, Critical Care and Pain Medicine Institutional funds. F.S.E received funding from NIH (R01 NS072446, R01FD004127, R01NS082331) and PCORI; J.M.L received funding from NIH (F32DK102323) and R.S. received funding from NIH (R21HL121728, R01DK102696, R01 HL113338).
Supplementary material
Supplementary material is available at Brain online.
References
- 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; Chapter 7: Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer's disease and amyotrophic lateral sclerosis. Brain Res 1994; 639: 171–4. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet 2013; 14: 307–20. [DOI] [PubMed] [Google Scholar]
- Coussens L, Van Beveren C, Smith D, Chen E, Mitchell RL, Isacke CM, et al. Structural alteration of viral homologue of receptor proto-oncogene fms at carboxyl terminus. Nature 1986; 320: 277–80. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2014; 82: 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–81. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NP, Matevski D, Dumitru D, Piovesan B, Rushlow D, Gallie BL. Identification of clinically relevant mosaicism in type I hereditary haemorrhagic telangiectasia. J Med Genet 2011; 48: 353–7. [DOI] [PubMed] [Google Scholar]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2013; 41: D8–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2012; 44: 200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Haas S, Werner A, Klein MA, Kloss C, Kreutzberg GW. Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous system: a quantitative immunofluorescence study using confocal laser microscopy. J Comp Neurol 1998; 395: 342–58. [DOI] [PubMed] [Google Scholar]
- Ridge SA, Worwood M, Oscier D, Jacobs A, Padua RA. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc Natl Acad Sci USA 1990; 87: 1377–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology 2001; 56: 1433–43. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–6. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29: 308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MJ, Pinto M, Henrique R, Vieira J, Cerveira N, Peixoto A, et al. CSF1R copy number changes, point mutations, and RNA and protein overexpression in renal cell carcinomas. Mod Pathol 2009; 22: 744–52. [DOI] [PubMed] [Google Scholar]
- Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, et al. Biology and action of colony–stimulating factor-1. Mol Reprod Dev 1997; 46: 4–10. [DOI] [PubMed] [Google Scholar]
- Wang Y, Berezovska O, Fedoroff S. Expression of colony stimulating factor-1 receptor (CSF-1R) by CNS neurons in mice. J Neurosci Res 1999; 57: 616–32. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}