

Abstract
Mycobacterium tuberculosis has an unusual natural history in that the vast majority of its human hosts enter a latent state that is both non-infectious and devoid of any symptoms of disease. From the pathogen perspective, it seems counterproductive to relinquish reproductive opportunities to achieve a détente with the host immune response. However, a small fraction of latent infections reactivate to the disease state. Thus, latency has been argued to provide a safe harbour for future infections which optimizes the persistence of M. tuberculosis in human populations. Yet, if a pathogen begins interactions with humans as an active disease without latency, how could it begin to evolve latency properties without incurring an immediate reproductive disadvantage? We address this question with a mathematical model. Results suggest that the emergence of tuberculosis latency may have been enabled by a mechanism akin to cryptic genetic variation in that detrimental latency properties were hidden from natural selection until their expression became evolutionarily favoured.
Keywords: latency, virulence, adaptive dynamics, cryptic genetic variation, niche construction
1. Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), has been infecting human hosts for thousands of years [1]. Evidence suggests that M. tuberculosis originated in Africa—when small human population sizes, characteristic of the hunter–gatherer stage of human evolution, was a bottleneck for highly active and virulent human pathogens [2]—before accompanying the Out-of-Africa migrations of modern humans over 50 000 years ago [3]. While the more ‘ancient’ lineages of M. tuberculosis seem to be restricted to West Africa, evolutionarily ‘modern’ lineages are spread throughout the world and most likely expanded during periods of strong population growth over the last few centuries [3,4].
Given their long association, it is believed that M. tuberculosis and humans coevolved strategies to overcome the negative effects of their antagonistic interactions [2,5]. Long-term latency is proposed to be one such strategy that favours the persistence of M. tuberculosis in humans [2,5,6]. This hypothesis is supported by the observation that latent TB is by far the most common form of M. tuberculosis infection with a staggering one-third of the world's population estimated to be latently infected [7].
From the pathogen perspective, it is not obvious why latency should be favoured, since it apparently forgoes reproductive opportunities to achieve a détente with the host immune response. Recent theoretical work has shed some light onto why latency might be an optimal persistence strategy for modern-day M. tuberculosis. Zheng et al. [6] asked what rate of disease reactivation maximized the epidemiological persistence of TB and found that intermediate rates—and therefore prolonged latency—are optimal for the persistence of M. tuberculosis. Similarly, Blaser & Kirschner [5] modelled multiple scales of nested interactions between hosts and pathogens and concluded that latency is an optimal evolutionary strategy for pathogens.
An important advantage of latency is that if active TB cases disappear in a local population, they can reappear later through reactivation from the reservoir of latent infections [6]. Latent TB also appears to balance the selective pressures experienced by the bacteria within the host (i.e. the immune response) with those experienced between hosts (i.e. transmission) [2,5,6]. This arrangement is enabled by a complex set of host–pathogen interactions. For example, in the latent state, M. tuberculosis bacteria persist inside the lungs within granulomas, which are immunopathologies composed of immune cells surrounded by peripheral fibrosis [8]. Granulomas act to control infection and are thus beneficial for the host. However, they also provide a niche for the M. tuberculosis bacteria to survive, and facilitate transmission upon reactivation to the disease state. These sources of both positive and negative feedback may set conditions for latency to become an evolutionary stable strategy [5].
Yet, if early forms of M. tuberculosis caused only primary progressive disease rather than latent infection [9], it is still unclear how a new strain that is able to enter latency could outcompete active bacteria that immediately cause disease and thereby enhance their own transmission. That is, while the modern form of latency may be evolutionarily optimal [5,6], prototypic bacteria that lack latency properties face a reproductive disadvantage by starting to evolve latency. There is a need to explain how they can access optimal latency.
We address this problem by introducing a model of M. tuberculosis infection that captures epidemiological characteristics of TB and allows these characteristics to evolve over time. We use the model to show how the relationships among pathogen characteristics—transmissibility, virulence, recovery and latency—determine the shape of the reproductive fitness landscape of the pathogen which in turn influences the evolutionary trajectory towards latency. We thus illuminate the conditions under which latency can emerge as an evolutionary stable strategy for M. tuberculosis. Understanding these evolutionary trajectories then allows us to propose a scenario for the history of interaction between M. tuberculosis and human hosts.
2. Material and methods
The mathematical model, shown schematically in figure 1, allows us to track the evolution of host–pathogen interactions [6,10–12]. Two classes of M. tuberculosis infections are considered: active and latent. A variable spectrum of latent infection likely exists [8]. Infections are thus further described by two characters:
— The degree of latency x of the infecting strain in cases where it enters a latent state (i.e. its ‘potential degree of latency’). This variable ranges from zero latency at x = 0 up to complete latency at x = 1.
— The probability y that the infecting strain enters a latent state upon infection, which ranges from y = 0, whereby all new infections progress immediately to disease, up to latency for all new infections without fail at y = 1.
Thus, at the start of a new infection a strain with a potential degree of latency x will realize this potential with probability y, but it will otherwise cause active infection (i.e. primary progressive disease, which gives the same behaviour as x = 0) with probability 1−y. The dynamics of the latent and active infection populations are described by the density functions L(x, y, t) ≥ 0 and A(x, y, t) ≥ 0, where t is time and 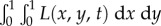 and
and 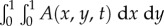 are the respective numbers of latent and active infections at time t.
are the respective numbers of latent and active infections at time t.
Figure 1.

Flow diagram of the M. tuberculosis evolution model. Mycobacterium tuberculosis infections are either latent (with density L) or active (with density A) and are further characterized by the potential latency of the infecting strain x, and the probability y that the infecting strain enters a latent state upon infection. Infection strains undergo stochastic incremental changes in phenotype at rate μ. Active infections regress to latency at rate ω, while latent infections reactivate at a rate a. Infections are lost if the host dies or recovers from the infection. Current infections can cause new infections that inherit the latency traits (x, y) of the infecting strain and undergo fast progression to the active state with probability 1−y. Otherwise, new infections will be initially latent (slow progression). Here, the grey colouring indicates a mutant strain with latency trait values (x*, y*).
We assume that new infections inherit the latency traits (x, y) of the infecting M. tuberculosis strain regardless of whether latency actually occurs. Moreover, bacteria may undergo mutation (or stable epigenetic change) in these characters which results in incremental changes to a strain's trait of a size Δ at a rate μ. We also assume that active infections regress into the latent state at a rate ω, while latent infections reactivate to the disease state at a rate a.
Within-host competition is not modelled explicitly since our understanding of the relationship between latency potential or the speed of onset of disease and within-host competition is limited for M. tuberculosis. For instance, while mixed-strain M. tuberculosis infections have been reported [13], there is limited understanding of how strains interact within infected hosts and of their interactions with the host immune system [13,14]. Instead, we assume that mutants always outcompete resident bacteria and that hosts cannot be infected with multiple strains.
The mixed effectiveness of the BCG vaccine against TB [15,16] and studies reporting relapse and reinfection rates in cured patients [17,18] suggest that immune memory is not completely effective for M. tuberculosis infection [19,20], although the interpretation of this evidence in terms of host susceptibility remains unresolved. Therefore, we make the simplifying assumption that infection does not offer any immunity or cross-immunity and do not include an immune class of hosts in our model formulation. Instead, recovered hosts return to the susceptible subpopulation and their infection history does not impact their susceptibility.
We denote the actual phenotype of a given infection (i.e. the ‘realized infection phenotype’) xr = x for latent infections and xr = 0 for active infections. This variable influences the transmission rate β(xr), the rate γ(xr) at which hosts recover from an infection and the host death rate θ(xr) which includes virulence, defined here as the additional death rate due to the disease. These pathogen characteristics are therefore interrelated through a mutual dependence on the realized infection phenotype xr of a strain.
For most of the analysis, we do not commit to any particular form of host–pathogen interaction and our analytical results, summarized below, apply to host–pathogen systems where the relationship between the transmission rate, virulence and recovery rate can be defined. However, the simulations we present are specific to M. tuberculosis and for these we specified the transmission rate, virulence and recovery rate functions for two different periods in the history of TB in human populations: (i) when M. tuberculosis was an emerging infectious disease with a short history of association with human hosts and (ii) when M. tuberculosis became a specialized human pathogen with a long history of association with human hosts. For both simulation scenarios, we chose a decreasing β(·) and decreasing θ(·) since we expect that more latent infections will always be less transmissible and less virulent for M. tuberculosis. This set-up conforms to the virulence–transmission trade-off hypothesis [21,22], whereby a positive correlation exists between virulence and transmission rate [β′(·) ≤ 0, θ′(·) ≤ 0]. Our choice of γ(·), on the other hand, varied between the two cases. The M. tuberculosis precursor that first encountered humans likely resembled opportunistic environmental mycobacteria [23] which tend to mostly infect immunocompromised patients [24]. Therefore, for emerging M. tuberculosis we assume that clearance of the infection was possible and that latent infections were cleared more easily than active infections. Hence, for these simulations we chose an increasing and non-zero host-recovery rate function [γ ′(·) ≥ 0, γ(·) > 0] and in this case, xr can be related to the strength of within-host pathogen growth, with maximal growth occurring when xr = 0. Alternatively, for simulations of M. tuberculosis as a specialized human pathogen, we specified a decreasing host-recovery rate function [γ′(·) ≤ 0] with very low values of γ(xr) for high xr, since we expect that latent infections will be more protected from the host's immune defences, through, for instance, the protective effects of granuloma formation. In this case, xr measures the extent of pathogen manipulation of the host immune response, with maximal manipulation occurring when xr = 1.
We studied the model numerically with two approaches. First, we used an agent-based model where individual agents in a simulation represent a human host infected by a particular strain of M. tuberculosis. This type of implementation is able to capture emergent evolutionary phenomena as well as stochastic effects such as extinction, and unusual evolutionary trajectories and events. Second, the model was formulated mathematically as a system of integrodifferential equations that describe the average dynamics of the density of hosts in the active and latent classes of infection, A(x, y, t) and L(x, y, t), with phenotype (x, y) at time t:
 |
2.1 |
Here, 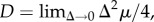
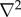 is the Laplacian operator with respect to the phenotypic variables (x, y), and
is the Laplacian operator with respect to the phenotypic variables (x, y), and 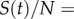
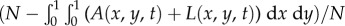 is the susceptible fraction of the population at time t. This type of mathematical formalization has been used in the past to study adaptive and evolutionary processes in population dynamics [25,26], and it has the advantages of being computationally less expensive than an agent-based model and providing population-level descriptions of the expected behaviour of large populations. The electronic supplementary material provides details of the agent-based computational algorithm and shows its relationship to the integrodifferential equation model (2.1) through an analysis of its continuum limit (electronic supplementary material, appendices A, B and figure S1).
is the susceptible fraction of the population at time t. This type of mathematical formalization has been used in the past to study adaptive and evolutionary processes in population dynamics [25,26], and it has the advantages of being computationally less expensive than an agent-based model and providing population-level descriptions of the expected behaviour of large populations. The electronic supplementary material provides details of the agent-based computational algorithm and shows its relationship to the integrodifferential equation model (2.1) through an analysis of its continuum limit (electronic supplementary material, appendices A, B and figure S1).
3. Results
To explain how M. tuberculosis might have evolved from its original state in ancient human populations to its present-day form as a well-adapted species engaged in complex interactions with its human host, we use the model to predict which traits are evolutionarily favoured and the conditions under which such traits are accessible.
(a). Fitness landscape as a heuristic for predicting pathogen evolution
The evolution of pathogen traits can be viewed as an adaptive walk on a phenotype fitness landscape which maps the phenotype of an organism to its reproductive fitness [27,28]. A pathogen strain can survive at a point on the landscape if its absolute fitness is larger than unity and if it avoids stochastic loss. Pathogens can evolve towards a local maximum on the landscape (an evolutionary stable state) if the local peak is accessible through viable states. If the global maximum is viable and accessible it will act as an attractor for evolutionary trajectories (the mean phenotype of a pathogen population tracked through time).
The fitness of a pathogen can be quantified by its basic reproductive number R0 which is the expected number of secondary infections produced by a typical infection in a completely susceptible population. We used the next generation method [29] to derive the following expression for R0 when the phenotype-space length scale of the discrete stochastic model approaches zero, i.e. Δ → 0+ (details are provided in the electronic supplementary material, appendix C):
 |
3.1 |
Contributions to R0 can be separated into new active infections (top line of equation (3.1)) and new latent infections (bottom line of equation (3.1)).
Figure 2a,c,e illustrates evolutionary trajectories from the agent-based model on the fitness landscape given by R0 (equation (3.1)), while the corresponding expected evolutionary trajectories computed from the integrodifferential equation model are provided in the electronic supplementary material, figure S2, alongside the expected equilibrium phenotype distributions. The evolutionary trajectories of surviving strains are clearly attracted to the maximum points of R0. Furthermore, an invasion analysis (electronic supplementary material, appendix D) reveals that R0 is an important threshold parameter in our model. These results confirm the use of R0 to predict pathogen evolution for our model.
Figure 2.

The basic reproductive number R0 of a pathogen acts as a heuristic for predicting evolutionary trajectories. R0(x, y) varies according to a strain's potential latency x (horizontal axis) and probability y that an infecting strain enters a latent state upon infection (vertical axis). Under our model, either active disease is favoured and the speed of progression is not under selection (a); an intermediate level of latency is favoured with slow progression (c); or latency is favoured with slow progression (e). (b,d,f) Display the functional forms of the transmission rate β(x) (dotted line), mortality rate θ(x) (dashed line) and recovery rate γ(x) (solid line) which lead to the respective fitness landscapes shown in (a,c,e). (b) The recovery rate γ(x) (grey dashed-dotted line) which, in combination with the β(x) and θ(x) displayed here, leads to the fitness landscapes shown in figure 3a,b. Evolutionary trajectories (the population mean trait values tracked through time) computed from separate simulations of evolving pathogen populations are shown as coloured lines superimposed over the associated density plot of R0, with the initial population mean trait values indicated by their respective coloured ‘s’. The black lines indicate the contour R0 = 1. The forms of parameters as functions of potential latency x and other parameter values are given in the electronic supplementary material, appendix G. (Online version in colour.)
(b). Evolutionarily stable strategies for pathogen persistence
Only three types of global optima of R0 are possible in our model: either R0 is maximized at x = 0 or (x, y) = (x*, 1) where 0 < x* < 1 or (x, y) = (1, 1). For this result and other results presented in this section, we assume that the rates of transmission, host mortality, recovery, reactivation and regression are all positive/non-negative, i.e. β(·) ≥ 0, θ(·) ≥ 0, γ(·) > 0, a > 0, ω > 0 (see the electronic supplementary material, appendix E, for details of this and other results in this section). In other words, the three types of evolutionary optimum are (i) active disease with no selection on the probability of new infections entering latency, (ii) an intermediate degree of latency with all new infections entering latency, and (iii) full latency with all new infections entering latency. Figure 2 displays an example of each of the three types of fitness landscapes possible alongside the transmission, virulence and recovery rate functions that result in the pictured landscapes.
Rugged fitness surfaces with multiple global maxima are also possible if the transmission, virulence and recovery functions are non-monotonic, or if active disease is favoured (x = 0) and other rather stringent criteria on the transmission, virulence and recovery functions are met. Given that these conditions are unlikely to occur, especially simultaneously, we only consider cases that produce a fitness surface with a single global maximum.
A necessary (but not sufficient) condition for latency to emerge as the optimal persistence strategy is
| 3.2 |
The conditions that support a favoured intermediate latency of degree x = x* with new infections mostly entering latency include constraints on the concavity and rate of change of the transmission and infection loss functions at x = x* which requires these functions to be twice differentiable at this point. A necessary (but not sufficient) condition for this to occur is that
| 3.3 |
If the transmission rate is a decreasing function of latency, i.e. β′(·) < 0, then the above-listed necessary condition for latency, condition (3.2), can only be satisfied when
| 3.4 |
while the necessary condition for partial latency of degree x=x*, condition (3.3), requires
| 3.5 |
Therefore, latency or partial latency can only emerge as the optimal persistence strategy in our model if latency offers a survival advantage over activity.
Inequalities (3.4) and (3.5) are always satisfied when both the recovery rate and virulence decrease with increasing latency which holds for modern-day M. tuberculosis. The assumption that host recovery rate decreases as a pathogen adopts more latent properties is less reasonable for a very early form of M. tuberculosis before it established a history of interaction with humans.
Finally, persistence (or sustained transmission) of the evolutionarily favoured strain is only possible if its R0 is greater than unity. This requires β(1) > γ(1) + θ(1) if latency is favoured, β(x*) > γ(x*) + θ(x*) if partial latency of degree x = x* is favoured, while persistent activity requires β(0) > γ(0) + θ(0). Thus, persistent latency in our model requires that the loss of transmission potential due to latency be outweighed by the gain in infection lifetime from reducing the recovery and host death rates.
(c). Evolutionary accessibility of the latent phenotype
The accessibility of latent regions of the fitness landscape is found by exploring the behaviour of the landscape's gradient vector 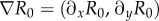 . It is straightforward to show that the gradient of R0 in the x-direction
. It is straightforward to show that the gradient of R0 in the x-direction 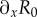 is always steepest when y = 1 since
is always steepest when y = 1 since
| 3.6 |
In other words, the fitness loss or gain from adopting latent properties is always greatest for pathogen strains that enter a latent state upon infection (y = 1). In an evolutionary sense, this result means that strains that are heavily invested in latency have the most to gain and lose from changes to the fitness value of latency traits. Thus, active strains (x = 0) initially faced with incurring a fitness cost for acquiring a more latent phenotype pay the minimal cost if they always enter an active state upon infection (y = 0). This cost is further curtailed for those strains with a small probability of regression (ω) compared with host death or recovery [θ(0) + γ(0)], which are the least invested in latency.
These results are illustrated in figure 3 and in the electronic supplementary material, movie S1, where we simulate the evolution of a population of active infections previously adapted to the fitness landscape shown in figure 2a following changes to host–pathogen interactions (more details of landscape choice are provided in the electronic supplementary material, appendix F and figure S3). Two sets of changes to host–pathogen interactions are considered; in both cases, strains with an intermediate degree of latency that mostly enter a latent state upon infection are newly favoured and the population of active strains has to pay a fitness cost to access the newly optimal trait values.
Figure 3.

Active pathogens can cryptically accumulate costly latency properties. (a) A deep fitness valley separates active strains from the latent region of the fitness landscape when the regression rate ω = 0.3. (b) The fitness valley is less deep when the regression rate is reduced to ω = 0.05, particularly for those strains where all cases progress quickly to disease (y = 0). This allows active strains to traverse the fitness landscape. (a,b) The population mean phenotype of the respective infection populations are shown here as the trajectories starting at ‘s’, and the contours indicate R0 = 1. The transmission rate β(x), virulence θ(x) and recovery rate γ(x) which lead to the fitness landscapes shown in (a,b) are displayed as the dotted, dashed and dashed-dotted lines in figure 2b. (c) A density plot of the equilibrium realized infection phenotype distribution for the case when ω = 0.05 and latency evolves (i.e. 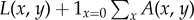 at t = 5000). The forms of parameters as functions of potential latency x and other parameter values are given in the electronic supplementary material, appendix G and table S1. (Online version in colour.)
at t = 5000). The forms of parameters as functions of potential latency x and other parameter values are given in the electronic supplementary material, appendix G and table S1. (Online version in colour.)
Our simulations demonstrate that active strains of infection are able to overcome the fitness cost of adopting latency properties and achieve the optimal persistence strategy when the probability of regression as opposed to host death or recovery is sufficiently small (compare the trajectories, starting at ‘s’, in figure 3a,b which show the evolutionary trajectories of the population mean trait values). When this occurs, the evolutionary trajectory of the population follows a neutral corridor (a flat region) along the fitness landscape where the probability y of causing latent infections remains low and where strains are effectively hidden from the negative selective effects that adopting latent properties carries. Once the population traverses far enough along the neutral corridor, investment in latency (y → 1) begins to be rewarded, and the final (equilibrium) realized infection phenotype distribution (figure 3c; see the electronic supplementary material, movie S1) reveals the emergence of latency.
(d). Dynamic fitness landscape
So far we have set the functional relationships among the infection parameters. However, if these relationships themselves evolve due to niche construction effects [30], whereby a pathogen alters its environment and therefore the selective pressures it experiences within the host, then the landscape can evolve along with the pathogen. Depending on the relative time-scale of these niche construction effects compared with the timescale of phenotypic change for the pathogen (μ), it may be possible for organisms to avoid fitness valleys altogether by accompanying fitness peaks as they move around the landscape. We provide an example of a dynamic fitness landscape model in the electronic supplementary material, appendix F and figure S3. In this case, small alterations in the host recovery rate function γ(·) progressively change the fitness landscape until a single peak at active infection transforms into two peaks: one at latency and the other at active infection with a fitness valley in between.
4. Discussion
(a). The evolutionary emergence of latency
Our analysis shows how latency can appear as a peak on the fitness landscape of pathogen evolution. Latency is an optimal evolutionary strategy for pathogens when the gain from reducing the recovery rate outweighs the disadvantage of losing transmission opportunities. If latency does not reduce recovery sufficiently, then an alternative fitness peak occurs at phenotypes conferring active infections. A long association between pathogens and their hosts would promote conditions for latency leading to persistence (low recovery rates) within hosts, thereby allowing a fitness peak at high degrees of latency. These findings agree with previous analyses of TB latency [5,6]. It also accords with the notion that latency is a bet-hedging strategy that enables a latent pool of infections to act as a safe harbour for future active infections [31].
The topography of the fitness landscape raises the problem of how an early form of a pathogen that causes active disease can begin to evolve latency properties that are initially disadvantageous. In other words, how can active pathogens cross a fitness valley to reach a higher peak at latency? In our model a shallow passage appears in the fitness valley under some conditions. Namely, if the probability of a new infection entering latency is low and if the rate of regression to latency is relatively low, then most infections enter and stay in the active form. Under these conditions, latency properties are occasionally expressed but not enough to heavily disadvantage the pathogen. This creates a neutral corridor along which pathogens can randomly drift in their latency properties until latency is refined enough to become advantageous. Selection can then drive the probability of entering latency to high levels.
Because latency properties are hidden from selection while pathogens drift along the neutral corridor, these dynamics are similar to the evolution of cryptic genetic variation [32] under which genetic variation accumulates in a population by being selectively neutral. If conditions change, for example, through a change in the environment or additional mutations, this variation is ‘revealed’ and becomes a significant substrate on which natural selection can act [32,33]. Stochasticity is an important feature of these dynamics, because it allows the hidden latency property to sometimes wander across the landscape to a region where it can be selected.
(b). A scenario for the early evolution of Mycobacterium tuberculosis
Mycobacterium tuberculosis is proposed to have evolved from an ancestral mycobacterial population similar to the smooth tubercle bacilli M. canettii [34] by gaining persistence and virulence mechanisms [9,35]. Presumably then, prototypic M. tuberculosis was similar to present-day opportunistic mycobacteria, which have poor human-to-human transmission and invade immunocompromised individuals but are otherwise controlled by host immunity [24,36]. Accordingly, we propose the following scenario for the early evolution of M. tuberculosis.
The prototypic forms of M. tuberculosis first encountered by humans did not possess the ability to manipulate its host's immune response, and likewise host immune responses were unlikely to have been appropriately regulated to limit inflammation, particularly at the level that is observed in modern-day TB infections. However, as prototypic M. tuberculosis established itself in the human population it acquired the ability to transmit between hosts and replicate within hosts sufficiently well to continue chains of transmission. With these characteristics, our model predicts a fitness landscape for prototypic M. tuberculosis similar to that shown in figure 2a. In this setting, host–pathogen interactions favour pathogen strains with low levels of latency, and there is little selective pressure on whether new transmissions are active or latent since the latency trait of an adapted pathogen would be very close to its active state. In addition, early forms of TB may have evolved as low a level of virulence as is compatible with the interdependence of parameters [35], as well as the small average susceptible host population size typical of early hunter–gatherer populations [2,5].
As the association between host and pathogen continued, M. tuberculosis increased its ability to avoid the immune response (perhaps due to niche construction effects) and natural selection enriched for host populations that downregulate inflammation [37]. It is therefore conceivable that one of the first significant shifts in the fitness landscape was due to recovery slowing down in latently infected hosts. Such a change in the relationship between latency and recovery (while keeping the same relationships among latency, transmission and mortality) would have altered the fitness landscape to one similar to that shown in figure 3b. Under these new conditions, latency is the new optimal strategy for M. tuberculosis. Yet, as outlined above, latency might also have been initially disadvantageous. In this case, the valley in the fitness landscape might have been crossed through the cryptic accumulation of latency properties such as the interference with phagosome–lysosome fusion in M. tuberculosis-infected macrophages [38], the inhibition of phagosomal maturation and acidification [39], and/or the suppression of macrophage apoptosis [40].
(c). Limitations and future work
Our modelling framework focuses on the evolution of a pathogen with constant host immune response and thus constant fitness landscapes. While this is generally reasonable for modelling the evolution of pathogens with much shorter generation times than their hosts, M. tuberculosis and humans have had a long history of association and coevolution is likely to have occurred [2,3]. We have considered alternative possible landscapes for different periods in the history of human TB but a more general approach would be to explicitly model fitness as a dynamic landscape. Specifically, we have not considered any structure in the host population. If an extended model were to include variation in host susceptibility for instance, the latency properties could be a function of not just the pathogen but also the host. In such a model, it would be possible to study more explicitly the role of bet-hedging in latency evolution, because the environment would be less predictable from the point of view of the pathogen [31].
Finally, our model considers the evolution of a pathogen where only single-strain infections are possible. Of interest is to consider whether the evolutionary trajectories predicted under our model change significantly when hosts are allowed to be reinfected and/or sustain multiple infections at once.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank Adelle Coster and James Wood for helpful discussions, and Tommaso Lorenzi who provided feedback on the manuscript.
Data accessibility
The data supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
Both authors contributed to the study design, analyses and the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was supported in part by a grant from the Australian Research Council (FT140100398).
References
- 1.Donoghue HD, Spigelman M, Greenblatt CL, Lev-Maor G, Bar-Gal GK, Matheson C, Vernon K, Nerlich AG, Zink AR. 2004. Tuberculosis: from prehistory to Robert Koch, as revealed by ancient DNA. Lancet Infect. Dis. 4, 584–592. ( 10.1016/S1473-3099(04)01133-8) [DOI] [PubMed] [Google Scholar]
- 2.Gagneux S. 2012. Host–pathogen coevolution in human tuberculosis. Phil. Trans. R. Soc. B 367, 850–859. ( 10.1098/rstb.2011.0316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comas I, et al. 2013. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182. ( 10.1038/ng.2744) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hershberg R, et al. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6, e311 ( 10.1371/journal.pbio.0060311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaser MJ, Kirschner D. 2007. The equilibria that allow bacterial persistence in human hosts. Nature 449, 843–849. ( 10.1038/nature06198) [DOI] [PubMed] [Google Scholar]
- 6.Zheng N, Whalen CC, Handel A. 2014. Modeling the potential impact of host population survival on the evolution of M. tuberculosis latency. PLoS ONE 9, e105721 ( 10.1371/journal.pone.0105721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. 2014. Global tuberculosis report 2014. Technical report. Geneva, Switzerland: World Health Organization.
- 8.Barry CE, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. 2009. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855. ( 10.1038/nrmicro2236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veyrier FJ, Dufort A, Behr MA. 2011. The rise and fall of the Mycobacterium tuberculosis genome. Trends Microbiol. 19, 156–161. ( 10.1016/j.tim.2010.12.008) [DOI] [PubMed] [Google Scholar]
- 10.Blower SM, Mclean AR, Porco TC, Small PM, Hopewell PC, Sanchez MA, Moss AR. 1995. The intrinsic transmission dynamics of tuberculosis epidemics. Nat. Med. 1, 815–821. ( 10.1038/nm0895-815) [DOI] [PubMed] [Google Scholar]
- 11.Castillo-Chavez C, Song B. 2004. Dynamical models of tuberculosis and their applications. Math. Biosci. Eng. 1, 361–404. ( 10.3934/mbe.2004.1.361) [DOI] [PubMed] [Google Scholar]
- 12.Basu S, Galvani AP. 2009. The evolution of tuberculosis virulence. Bull. Math. Biol. 71, 1073–1088. ( 10.1007/s11538-009-9394-x) [DOI] [PubMed] [Google Scholar]
- 13.Cohen T, van Helden PD, Wilson D, Colijn C, McLaughlin MM, Abubakar I, Warren RM. 2012. Mixed-strain Mycobacterium tuberculosis infections and the implications for tuberculosis treatment and control. Clin. Microbiol. Rev. 25, 708–719. ( 10.1128/CMR.00021-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sergeev R, Colijn C, Cohen T. 2011. Models to understand the population-level impact of mixed strain M. tuberculosis infections. J. Theor. Biol. 280, 88–100. ( 10.1016/j.jtbi.2011.04.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michelsen S, et al. 2014. The effectiveness of BCG vaccination in preventing Mycobacterium tuberculosis infection and disease in Greenland. Thorax 69, 851–856. ( 10.1136/thoraxjnl-2014-205688) [DOI] [PubMed] [Google Scholar]
- 16.Roy A, et al. 2014. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. Br. Med. J. 349, g4643 ( 10.1136/bmj.g4643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verver S, Warren RM, Beyers N, Richardson M, van der Spuy GD, Borgdorff MW, Enarson DA, Behr MA, van Helden PD. 2005. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am. J. Respir. Crit. Care Med. 171, 1430–1435. ( 10.1164/rccm.200409-1200OC) [DOI] [PubMed] [Google Scholar]
- 18.Marx FM, Dunbar R, Enarson DA, Williams BG, Warren RM, van der Spuy GD, van Helden PD, Beyers N. 2014. The temporal dynamics of relapse and reinfection tuberculosis after successful treatment: a retrospective cohort study. Clin. Infect. Dis. 58, 1676–1683. ( 10.1093/cid/ciu186) [DOI] [PubMed] [Google Scholar]
- 19.van Rie A, Warren R, Richardson M, Victor TC, Gie RP, Enarson DA, Beyers N, van Helden PD. 1999. Exogenous reinfection as a cause of recurrent tuberculosis after curative treatment. N. Engl. J. Med. 341, 1174–1179. ( 10.1056/NEJM199910143411602) [DOI] [PubMed] [Google Scholar]
- 20.Chiang C-Y, Riley LW. 2005. Exogenous reinfection in tuberculosis. Lancet Infect. Dis. 5, 629–636. ( 10.1016/S1473-3099(05)70240-1) [DOI] [PubMed] [Google Scholar]
- 21.Anderson RM, May R. 1982. Coevolution of hosts and parasites. Parasitology 85, 411–426. ( 10.1017/S0031182000055360) [DOI] [PubMed] [Google Scholar]
- 22.Alizon S, Hurford A, Mideo N, Van Baalen M. 2009. Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J. Evol. Biol. 22, 245–259. ( 10.1111/j.1420-9101.2008.01658.x) [DOI] [PubMed] [Google Scholar]
- 23.Galagan JE. 2014. Genomic insights into tuberculosis. Nat. Rev. Genet. 15, 307–320. ( 10.1038/nrg3664) [DOI] [PubMed] [Google Scholar]
- 24.Cook JL. 2010. Nontuberculous mycobacteria: opportunistic environmental pathogens for predisposed hosts. Br. Med. Bull. 96, 45–59. ( 10.1093/bmb/ldq035) [DOI] [PubMed] [Google Scholar]
- 25.Dieckmann U. 2002. Adaptive dynamics of pathogen–host interactions. In Adaptive dynamics of infectious diseases: in pursuit of virulence management (eds Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K), ch. 4, pp. 39–59. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 26.Lorz A, Mirrahimi S, Perthame B. 2011. Dirac mass dynamics in multidimensional nonlocal parabolic equations. Commun. Part. Diff. Eq. 36, 1071–1098. ( 10.1080/03605302.2010.538784) [DOI] [Google Scholar]
- 27.Wright S. 1932. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. In Proc. 6th Int. Congress Genet., vol. 1, pp. 356–366.
- 28.de Visser JAG, Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. ( 10.1038/nrg3744) [DOI] [PubMed] [Google Scholar]
- 29.Heffernan JM, Smith RJ, Wahl LM. 2005. Perspectives on the basic reproductive ratio. J. R. Soc. Interface 2, 281–293. ( 10.1098/rsif.2005.0042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odling-Smee FJ, Laland KN, Feldman MW. 2003. Niche construction: the neglected process in evolution. Number 37 in Monographs in Population Biology. Princeton, NJ: Princeton University Press.
- 31.Stumpf MP, Laidlaw Z, Jansen VA. 2002. Herpes viruses hedge their bets. Proc. Natl Acad. Sci. USA 99, 15 234–15 237. ( 10.1073/pnas.232546899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paaby AB, Rockman MV. 2014. Cryptic genetic variation: evolution's hidden substrate. Nat. Rev. Genet. 15, 247–258. ( 10.1038/nrg3688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rohner N, Jarosz DF, Kowalko JE, Yoshizawa M, Jeffery WR, Borowsky RL, Lindquist S, Tabin CJ. 2013. Cryptic variation in morphological evolution: HSP90 as a capacitor for loss of eyes in cavefish. Science 342, 1372–1375. ( 10.1126/science.1240276) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez MC, Brisse S, Brosch R, Fabre M, Omas B, Marmiesse M, Supply P, Vincent V et al. 2005. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 1, e5 ( 10.1371/journal.ppat.0010005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Supply P, et al. 2013. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat. Genet. 45, 172–179. ( 10.1038/ng.2517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koeck J-L, et al. 2011. Clinical characteristics of the smooth tubercle bacilli ‘Mycobacterium canettii’ infection suggest the existence of an environmental reservoir. Clin. Microbiol. Infect. 17, 1013–1019. ( 10.1111/j.1469-0691.2010.03347.x) [DOI] [PubMed] [Google Scholar]
- 37.Blaser M. 1993. Malaria and the natural history of Helicobacter pylori infection. Lancet 342, 551 ( 10.1016/0140-6736(93)91674-B) [DOI] [PubMed] [Google Scholar]
- 38.Armstrong J, Hart P. 1975. Phagosome–lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. J. Exp. Med. 142, 1–16. ( 10.1084/jem.142.1.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sturgill-Koszycki S, et al. 1994. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681. ( 10.1126/science.8303277) [DOI] [PubMed] [Google Scholar]
- 40.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. 1998. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-α. J. Immunol. 161, 2636–2641. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been uploaded as part of the electronic supplementary material.