

Abstract
Single molecule studies in recent decades have elucidated the full chemo-mechanical cycle of F1-ATPase, mostly based on F1 from thermophilic bacteria. In contrast, high-resolution crystal structures are only available for mitochondrial F1. Here we present high resolution single molecule rotational data on F1 from Saccharomyces cerevisiae, obtained using new high throughput detection and analysis tools. Rotational data are presented for the wild type mitochondrial enzyme, a “liver” isoform, and six mutant forms of yeast F1 that have previously been demonstrated to be less efficient or partially uncoupled. The wild-type and “liver” isoforms show the same qualitative features as F1 from Escherichia coli and thermophilic bacteria. The analysis of the mutant forms revealed a delay at the catalytic dwell and associated decrease in Vmax, with magnitudes consistent with the level of disruption seen in the crystal structures. At least one of the mutant forms shows a previously un-observed dwell at the ATP binding angle, potentially attributable to slowed release of ADP. We discuss the correlation between crystal structures and single molecule results.
F1Fo-ATP synthase is a rotary molecular complex responsible for the synthesis of ATP in bacteria and eukaryotes under aerobic conditions. ATP synthase couples energy from the electrochemical gradient across the cytoplasmic, mitochondrial, or chloroplast membrane, to the phosphorylation of ADP to ATP. The enzyme is comprised of two separable coaxial motors: F1 and Fo. F1 is water-soluble and contains the catalytic sites used for the synthesis of ATP. Fo is membrane bound and acts as a proton turbine, using the electrochemical gradient to generate rotation that is physically coupled to F1 rotation. When isolated from Fo, F1 behaves as an ATP-hydrolysing enzyme.
Figure 1A shows the atomic structure of yeast F1, and Figure 1B the mechanochemical cycle that couples ATP hydrolysis to rotation of the γ subunit in F1 from the thermophilic Bacillus PS3 (TF1)1 (for reviews, see Refs. 2,3,4,5). This enzyme's high functional stability and slow kinetics at room temperature facilitate extended single-molecule observation and resolution of mechano-chemical transitions. F1 from E. coli rotates 4–5 times faster at room temperature, with a similar mechanism6.
Figure 1. X-ray crystal structure and proposed mechanochemical cycle.

(A) Locations of the mgi residues in the X-ray crystal structure of yeast F1 (pdb: 2HLD, Complex I, Ref. 22). The αTP, βE, γ, δ, and ε subunits are shown along with the residues that were mutated for this study. Also shown are the Catch 1 and 2 regions which interact with the γ subunit and help define the structure of the catalytic sites. The P-loop, which forms part of the Pi and ATP binding site, is coloured in red and labelled. (B) A proposed mechanochemical coupling scheme of thermophilic F1. The catalytic sites of the three β subunits are shown in orange (β1), green (β2), and purple (β3). We define the 0° binding dwell to correspond to ATP binding at a particular site labelled β1 and ADP release at β241,54. The subunit shown in orange binds ATP at the 0° mark, and each subunit follows the processes and states listed in the table at the angles shown. The angular position of the γ subunit is represented by the central yellow arrow. Once a catalytic site binds an ATP molecule, it remains bound for 200° rotation of the γ subunit, whereupon it is hydrolysed. ADP is released at 240° and Pi at 320° from the binding site. Each β subunit completes one hydrolysis of an ATP molecule per 360° rotation of the γ subunit, and the three β subunits are staggered in their catalytic state by 120°. This means that at each ATP waiting dwell, ADP will release from one subunit and ATP will bind to the previous subunit. At each catalytic dwell, the ATP bound to one subunit will hydrolyze and the Pi from the previous subunit will be released. Refer to Refs. 2,3,4,5 for detailed reviews of the mechanochemical cycle of F1. Fig. redrawn from Ref. 2 with modifications.
The TP, E and DP sites of the ground state crystal structure are believed to correspond to the states of subunits β1–β3 respectively at the 80° catalytic dwell, as illustrated in Fig. 1B7,8,9. All crystal structures to date have γ subunits within 20° of the ground state angle, most within 5°, indicating that all are variations on this ground state10. About half of these structures contain phosphate or an analogue at the E site, consistent with phosphate release at 80°. TP, E and DP states are best defined by their position with respect to the asymmetric γ subunit, as their nucleotide occupancy varies across different crystal structures. We label sites at the 0° dwell E*, TP*, and DP*, corresponding to the ground state they were most recently in if rotating in the ATP hydrolysis direction. A mechanochemical cycle can then be defined whereby a single β subunit in the E* conformation binds ATP at 0°, then moves through the TP and TP* conformations to hydrolyse ATP in the DP conformation at 200° and release ADP at 240° (DP*). Following rotation to 320° (E) it releases phosphate and is ready to bind ATP again at 360°7,11,12 The other two β subunits follow the same cycle delayed by 120° and 240°. TF1 and EF1 rotate unidirectionally but not continuously, stochastically entering a paused state due to Mg2+ ADP inhibition13.
Importantly, while most single molecule studies have been performed in TF1, the majority of high resolution crystal structures are for mitochondrial F1, with high resolution structures of bacterial F1 proving much harder to obtain14,15. Recent single-molecule studies of EF16 have demonstrated the ability to resolve the kinetics of mesophilic F1 at room temperature. A very recent study of human F1 (HF1) reported that phosphate release and hydrolysis occur at 65° and 90° respectively after the previous ATP binding dwell, rather than both at 80° as in TF1 and EF116. Single-molecule experiments on F1 from yeast will allow us to compare the mechanochemical cycles of different mitochondrial enzymes and to determine to what extent the extensive knowledge of TF1 can be applied to mitochondrial F1. It also allows the first comparison between single-molecule rotation studies and multiple high resolution crystal structures of F1 from the same organism. High-resolution X-ray crystal structures have been determined for bovine F1 in a number of different states, including a transition state with AlF4:ADP17,18,19,20,21 and for wild type yeast F1 in both the presence and absence of bound nucleotides22,23. For the yeast enzyme, the crystal lattice contained 3 complexes, with a well resolved phosphate bound to the E site in Complex II but absent in Complex I and III22.
In this paper, we investigate two classes of mutations to yeast F1: mitochondrial genome integrity mutations and the “liver” isoform. Mitochondrial genome integrity (mgi) is a class of mutations in the genes encoding the α, β, and γ subunits that allow petite negative yeast K. lactis to lose mitochondrial DNA24,25,26,27. The mgi mutations have been shown in yeast S. cerevisiae to uncouple proton flow from the synthesis of ATP28. While these mutations have been mapped exclusively to the α, β, and γ subunits of ATP synthase (Fig. 1A), the relationship between their effect on ATP synthase and the resulting yeast phenotypes is unclear. X-ray crystal structures have been determined for four mgi mutations29. These structures suggest two mechanisms for uncoupling and identify crucial regions involved. Mutations were grouped into two classes according to which mechanism is postulated: Class 1 mutations alter the structure of the E site and apparently disrupt phosphate binding, while Class 2 may disrupt interaction of the central stalk with the αβ pair29.
There are two isoforms of the γ subunit in humans and other mammals: a heart form and a liver form30. The heart form is expressed in heart, skeletal muscle, and intercostal muscle diaphragm, tissues of rapid and high-energy demand. The liver form is expressed in liver, cerebellar cortex, cerebrum, thyroid, spleen, pancreas, kidney and testis, tissues of relatively low or steady energy demand30. The isoforms are the result of alternative splicing. Isoform switching is regulated in a tissue specific manner and the alternative splicing can be reversibly induced by acid stimulation in muscle cells, but not in non-muscle cells31,32, suggesting that isoforms of the γ subunit in humans are a result of a carefully regulated alternate splicing process. This regulation is relevant to the physiology of the cell, yet the resulting effect on the biochemistry of the ATP synthase is unknown. The γ subunit expressed in the liver differs from that expressed in the heart only by the presence of an additional residue, Asp, at the C-terminus30. The tertiary structure of the yeast F1 is highly conserved with that of the bovine heart F1. Further, the primary sequence of the α and β subunit of the F1 ATPase is highly conserved from yeast to human. Based on the crystal structure of yeast and bovine F1 ATPase, it was predicted that the Asp at the C-terminus of the γ subunit could form a salt bridge with αArg288 in αE (Pagadala V. and Mueller D.M., unpublished results, see SI for further details). If formed, the salt bridge is predicted to slow the catalytic cycle without decreasing the efficiency of the enzyme.
In this study, we use new high throughput detection and analysis tools to investigate the behaviour of wild type mitochondrial F1 from S. cerevisiae at varying ATP concentrations. We quantify the lifetimes of the ATP-binding and catalytic states, and present evidence that mitochondrial F1 rotation is governed by the same mechanism as that of the well-studied TF1 and the more recently studied EF1. We also use these tools to investigate the behaviour of seven forms of yeast F1 containing a point mutation, including six mgi mutations and a mutation that mimics the heart-to-liver isoform in humans. For four of these mutants, we discuss structure-function relationships with reference to high-resolution crystal structures.
Results
Yield of rotating gold beads
The rotation of yeast F1-ATPase was monitored by darkfield microscopy after attachment of gold particles to the γ subunit, as described in Methods. The position of all gold beads in the field of view was calculated and examined. A small fraction of beads showed clear circular trajectories (Fig. 2A), similar to those presented in previous work on F1. Many beads rotated unidirectionally as revealed by asymmetry in the power spectrum of their motion, but with signal-to-noise ratios too low for clear circular trajectories (Fig. 2B).
Figure 2. Example traces for F1 conjugated to neutravidin beads.

(A–C) Bead trajectory (inset, scale bar 40 nm) and rotational power spectrum (counterclockwise, CCW, in green, clockwise, CW, in blue), for three representative molecules at 100 μM ATP. Trajectories show rotation clearly in (A) only, power spectra show clear evidence for a unidirectional rotation peak in both (A) and (B). (D–G) Power spectral bias against mean apparent angular speed for many molecules at different ATP concentrations. Without ATP, data are centred on (0, 0). With ATP, CCW rotating appear at positive speed and bias. The beads shown in (A–C) are marked with red circles. A 4-sigma ellipse is drawn based on the no-ATP data, and the external region split into CW rotation (lower left) and CCW rotation (upper right). 99.5% of the 0 μM ATP beads lie within this circle (955/960). When ATP is added, 21%, 28% and 27% of beads are on the counter-clockwise side of these thresholds at 10, 30 and 100 μM ATP respectively, with 0%, 0.3% and 0.2% on the clockwise side. The fraction of beads with bias above zero is 50.2%, 79%, 83% and 77% in (D–G) respectively.
To assess the fraction of beads which were rotating, the directional ‘bias’ in rotation was examined (Methods). Rotation rate was assessed independently by fitting an ellipse to the bead trajectory and tracking the bead's angular speed around the ellipse - this accurately measures speed in high signal to noise conditions but underestimates speed when noise is significant. Fig. 2D-G shows bias vs. angular speed for F1-bead complexes at various ATP concentrations under identical imaging conditions. F1 is not expected to rotate in the absence of ATP, and under such conditions the plot is symmetric around the origin. When ATP is present, a ‘tail’ is visible in the direction corresponding to counter-clockwise rotation. Significantly, there is no corresponding tail in the direction that corresponds to clockwise rotation. This ATP dependent asymmetry indicates that beads with significant values of bias are rotating due to F1, even though many traces are too noisy to detect clear circular trajectories. In the ‘best samples,’ like those shown in Fig. 2, a conservative estimate is that at least 25% of the beads are rotating, and this is discussed further in SI.
The bias of a bead is related to the signal-to-noise value, which suggests that beads with a high bias should have high quality data. Qualitatively, this is observed. In work on bacterial F1, rotating molecules have typically been identified by eye, a laborious procedure subject to human selection. By measuring the bias of all recorded beads (potentially greater than 1000 per sample), the rotating molecules providing the highest quality data were quickly and automatically identified. In the SI, we discuss why a spread in bias/quality exists, and whether high quality spinners are representative of the broader population. In summary, data quality is principally determined by the orientation of the gold bead bound to F1 but is unlikely to significantly affect the kinetics. The spread in bias is not unique to F1 from yeast and is likely a common feature of all F1 single molecule spinning bead assays.
Characterisation of wild type F1
Fig. 3 shows speeds measured for wild type yeast F1 as a function of ATP concentration using power spectra, and the more conventional angular rotation speed (Methods). Both fit well to Michaelis-Menten kinetics, with fit parameters of Vmax = 586 ± 23 rev/s and Km = 62 ± 9 μM, and 471 ± 79 rev/s and 59 ± 18 μM, respectively. Quoted errors correspond to the 95% confidence range of the fit. The difference between the two fits is primarily a result of selection bias in the smaller number of molecules used for the angular rotation speeds (see SI). Rotation speeds tended to be steady for individual molecules, but varied between molecules at the same ATP concentration, with a spread (sample deviation as fraction of mean) of 16% and 13% for the two methods when averaged over all ATP concentrations. The fit parameters are similar to those determined by biochemical methods (see SI). Biotinylation of the γ subunit caused no significant changes in the steady-state ATPase kinetic values.
Figure 3. Rotation speed for wild type yeast F1 as a function of ATP concentration.

Black points and fit line shows data from fitting the power spectra of 2177 beads. Error bars mark the sample deviation of speeds at each ATP concentration, and grey bars mark the 95% confidence interval on the mean. Red points and fit line show speeds of 139 beads selected by eye for trajectories such as that of Fig. 2A, determined from bead angles, points offset horizontally for clarity. The discrepancy between fits is due to selection bias for slow beads in angle speed data; see SI for comparisons. Michaelis-Menten fits were calculated using Origin software.
Fig. 4B-D shows a single molecule displaying typical stepping patterns for low, intermediate and high ATP concentrations. At low and high ATP concentrations, three dwell positions are observed, while at intermediate ATP concentrations six dwells are visible. This is consistent with the model of F1 rotation developed using TF1, in which the ATP binding dwell, observed at low ATP concentrations, is located approximately 40° after the catalytic dwell, observed at high ATP concentrations33,34. By contrast, in HF1 six dwell positions were reported at high ATP concentrations16, indicating that HF1 is different from YF1 and TF1.
Figure 4. Dwells at low, intermediate, and saturating levels of ATP.

(A–D) One F1 molecule observed at low, intermediate and saturating levels of ATP. (A) Unwrapped angle vs time for the full record of this molecule: after recording for 0.6 s at a low ATP concentration (~5 μM), 3 mM ATP was introduced to the flow slide and a further 1.5 s record collected. All data were collected at 9 kHz. (B–D) Angle vs time for a few revolutions at the locations marked in (A), and angle histograms (red) and trajectories (right, scale bar 40 nm) collected (B) prior to ATP flow, (C) in the first 0.3 s recorded after flow began, (D) in the remainder of the trace (once speed was stable). The precise ATP concentration at any moment at time is not known. This molecule was tagged with a gold duplex, giving a very clear rotation signal. Angular separation between the dwells was measured using k-means clustering to determine the location of dwells at low and high ATP, and measuring the angle change between the vectors connecting dwell locations (blue triangle on x-y plots). (E) Histogram of 54 dwell angle separations observed from 18 molecules and least squares Gaussian fit to the histogram. The molecule shown in (A–D) produced atypically large angular deviations (48, 49, 64°), compared to population median of 31°.
In contrast to reports from TF1, significant variation was observed in the measured angles between the binding and hydrolysis dwells. At intermediate ATP concentrations, typically either three broad peaks– likely due to unresolved closely separated dwells – or six peaks with separations above 40° were observed. To resolve closely separated peaks, flow cells were used to change the ATP concentration from 5 μM to 3 mM, and bead trajectories were analysed to compare the position of the dwells at low and high ATP concentration. Fig. 4A–D shows one such molecule captured before, during and after the flow of 3 mM ATP. Typically, dwell kinetics were assessed before and after the addition of 3 mM ATP (4B and 4D), see SI for details of the other molecules observed. Fig. 4E shows a histogram of the measured angles between the binding (5 μM) and hydrolysis (3 mM) dwells, as analysed using k-means clustering to identify the dwell locations at low and saturating ATP (Methods). A least squares Gaussian fit to the results from 18 molecules (54 dwell separations) gave a peak at 33° with a width of 19° (Fig. 4E). The median and mean separations were 31° and 34°. These measurements indicate that the separation between the binding and hydrolysis dwells in wild type yeast F1 is similar to the separation previously measured in TF134 and EF16, albeit the variation is larger.
The number of frames that rotating molecules spent in each dwell state for saturating (≥1 mM) and low (≤10 μM) concentrations of ATP were counted. A Bayesian information criterion (BIC)35 was used across models with 1, 2 or 3 sequential irreversible Poisson steps, both with and without a delay for bead rotation, to judge the quality of fit to each individual trace, as in previous work6. At saturating ATP, the data fit best using a short delay (0.13 ± 0.06 ms) and two ATP-independent steps (rate constants 2.5 ± 1.2 (ms)−1, 4.3 ± 2.3 (ms)−1). At low ATP (3–10 μM), the data fit best using one ATP-dependent step (18 ± 6 μM−1s−1) and one ATP-independent step with a rate (1.8 ± 1.2 (ms)−1) consistent with being an unresolved combination of the two steps fit at saturating ATP. Dwell distributions of a composite of 15 molecules at low and saturating ATP, and the best kinetic fits that result, are shown in Fig. 5. These results suggest that the rotation of wild type yeast F1 is governed by one ATP-dependent process located at the ATP binding dwell and two ATP-independent processes at the catalytic dwell.
Figure 5.

Dwell distributions for a composite of 15 wild type F1 molecules at (A) low (10 μM) ATP and (B) saturating (3 mM) ATP. The low ATP data are fit by two sequential Poisson processes, saturating ATP by two sequential Poisson processes with a time offset. The values for the kinetic rates and time offset from the fits of the composite dwell distributions agree with the kinetic rates and time offset calculated from the median values of single molecule fits within the 95 percent confidence intervals.
Taken together, these results suggest that the fundamental kinetics of yeast F1 are similar to those of F1 from E. coli and the thermophilic Bacillus PS3, but different to those of human F1. That is, there is one ATP sensitive process located at the ATP dwell angle, then two ATP insensitive processes which occur following a 30–40° rotation of the central stalk. In addition, we observe a short delay at high ATP which is consistent with the expected time required for rotation of the gold bead following a mechanochemical step in F1.
Analysis of “liver isoform” and six mutant forms
We used the methods developed here to screen seven forms of yeast F1 containing a single point mutation relative to the wild type enzyme. One mutant form corresponds to the “liver isoform”, while six forms contain mitochondrial genome integrity (mgi) mutations.
Fig. 6 shows speed-ATP curves for these mutant forms, and for comparison the wild type control from Fig. 3. The values associated with Michaelis Menten fits to each mutant are summarised in Table 1. Data obtained from the “liver isoform” mutant enzyme, γG278D, are indistinguishable from those of the wild type enzyme. This indicates that despite the tight regulation between the liver and heart isoforms, the Asp residue at the terminal end of the γ subunit has no measurable effect on kinetics of rotation under ATP hydrolysis. In contrast, all the data from the enzyme forms with mgi mutations show a significant reduction in the maximum rotation rate, by a factor of 2–8. However, the rotation rates of these mutant enzymes are only slightly affected at low ATP concentrations. There was a significant (at 95% confidence) decrease in the observed rate of ATP binding for the mutants βV279F, αN67I and αF405S, which had a binding rate approximately two thirds that of the wild type enzyme. Data of the mutant enzymes generally fit well to Michaelis Menten kinetics, with the exception of βR408I.
Figure 6. Rotation rate as a function of [ATP], for seven mutant forms of yeast F1, and associated Michaelis Menten fits (solid lines) for contrast to the wild type fit (grey dashed line) from Fig. 3.

Class 1 mutations are shown in green, Class 2 mutations in red, with classes assigned according to crystal structures. Thin error bars mark the standard deviation of observed speeds; fat bars mark the 95% CI on the mean speed (in some cases these are too small to observe). Michaelis-Menten fits were calculated in Origin.
Table 1. Dwell state kinetics.
| Form | Michaelis Menten fits | Dwell analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| vmax (rev s−1) | Km (μM) | kATP (s−1 μM−1) | n | kATP (s−1 μM−1) | k2 (ms)−1 | k3 (ms)−1 | n | |
| Wild type | 586 ± 24 | 62 ± 9 | 28.3 ± 3.6 | 2177 | 18 ± 6 | 2.5 ± 1.2 | 4.3 ± 2.3 | 18, 28 |
| γG278D | 582 ± 137 | 63 ± 33 | 27.6 ± 10.2 | 89 | 26 ± 2 | 2.6 ± 0.6 | 5.0 ± 1.7 | 4, 5 |
| βV279F | 144 ± 13 | 24 ± 5 | 17.9 ± 2.8 | 537 | 15 ± 2 | 0.36 ± 0.08 | 2.3 ± 2.7 | 4, 8 |
| αN67I | 248 ± 47 | 41 ± 12 | 18.0 ± 2.6 | 432 | 14 ± 3 | 0.46 ± 0.09 | 2.3 ± 0.7 | 7, 3 |
| βR408G | 80 ± 11 | 10.0 ± 2.6 | 24.1 ± 4.0 | 439 | 19 ± 5 | 0.21 ± 0.08 | 1.3 ± 0.5 | 10, 17 |
| αF405S | 336 ± 40 | 49 ± 11 | 20.4 ± 3.2 | 1044 | 16 ± 6 | 0.8 ± 0.6 | 2.0 ± 0.9 | 62, 40 |
| γI270T | 377 ± 92 | 44 ± 15 | 25.7 ± 4.4 | 2190 | 22 ± 7 | 0.8 ± 0.5 | 2.5 ± 2.0 | 109, 27 |
| βR408I | 69 ± 13 | 7.9 ± 2.5 | 26.4 ± 4.7 | 6519 | 23 ± 6 | 0.20 ± 0.07 | 3.3 ± 2.3 | 384, 51 |
| Bacillus PS333 | 129 ± 22 | 15 ± 5 | 25.8 | |||||
| E. coli6 | 449 ± 39 | 21.1 ± 1.0 | 43 ± 4 | |||||
The results of the analysis from the Michaelis-Menten fits and the dwell time histogram analysis for wild type and the seven mutants screened. All values are quoted with 95% confidence intervals, with the exception of Bacillus PS3 and E. coli, where the values were sourced from previous studies as indicated in the table. The errors in these studies were typically quoted as standard deviations; for comparison to the 95% CIs listed here they have been multiplied by 1.96 for data where the number of degrees of freedom were unknown, or by the appropriate t-statistic where the number of degrees of freedom was known (2.45 to 1.96 for df = 6 to ∞). In some cases the error could not be calculated from the available data and has been left blank. The number of traces used is denoted by n. For the dwell analysis, the first value of n corresponds to the number of traces used to calculate kATP and the second value for the number of traces used to calculate k2 and k3.
Kinetic analyses were performed on the dwell states for the six mgi mutant forms of the enzyme, as for the wild type, in order to determine the kinetics at low and saturating ATP. In all mgi mutant forms, the ATP binding dwell was best fit by a single exponential with an ATP-dependent rate constant assigned as kATP. The catalytic dwell was best fit with two ATP-independent steps and an offset, similar to the wild type. The results are summarised in Table 1. Consistent with the Michaelis Menten fits (Fig. 6), the kinetics of the ATP binding dwell were changed relatively little, and those of the catalytic dwell much more, for all mgi mutants with respect to wild type. The rate of one of the processes occurring at the catalytic dwell dropped by as much as 10-fold, while the rate of the other process remained relatively unchanged. By comparison to TF1, we expect that these rates correspond to hydrolysis of ATP and release of phosphate, but this kinetic analysis does not determine which rate is altered, nor whether the same rate is altered in all mutants.
Data from the βR408I mutant showed an additional dwell at high ATP. At 3 mM, the rate of ATP binding is approximately 60 (ms)−1, which is too fast to be resolved in this study, and the expectation was this dwell would not be observed. Surprisingly, six dwells were clearly visible in the majority of high quality traces at both intermediate and saturating levels of ATP, as shown in Fig. 7A, despite the fact that kATP was not measurably different from wild type. Although these dwells were not typically observed at saturating ATP in the other forms of F1 investigated, a similar weak signal was occasionally present in traces from the wild type enzyme, as well as βR408G, αF405S and γI270T (Fig. 7B–E). However, due to the short duration in forms other than βR408I, and variability in its observation, we were unable to isolate and characterise this state. It is clear, however, that some process at the ATP binding angle was significantly slowed in βR408I even at saturating ATP concentrations. Accordingly, it is possible that a short, poorly resolvable ATP-insensitive dwell at the ATP binding angle may be a general feature of yeast mitochondrial F1 but amplified in the βR408I mutant form.
Figure 7. Six dwells at saturating ATP.

(A) Six dwells are visible at saturating ATP for mutant βR408I. (B–E) Examples showing occasional dwells between major steps for (B) βR408G, (C) αF405S, (D) wild type and (E) γI270T. Note that while (A) is typical of data for βR408I, (substeps were detected by eye in only 1/21, 6/160, 3/45 and 5/360 high quality data traces for (B) βR408G, (C) αF405S, (D) wild type and (E) γI270T respectively. All traces shown were recorded at 10 kHz and 3 mM ATP, except (C) which was at 1 mM ATP. The expected ATP-dependent dwell is ~1/6 frame at 3 mM ATP, and would not be resolved. Scale bar marks 40 nm.
Discussion
This study provides the first single molecule rotation analysis of F1 from a species where multiple x-ray crystal structures are also available, and the first comparison between high-resolution single molecule results from different mitochondrial F1s. In S. cerevisiae F1 as in bacterial F1, three ATP-independent processes occur approximately 33° before ATP binding. Given the similarity of our results to those with TF1 and EF1, we ascribe these to bead transit, ATP hydrolysis and phosphate release. This is identical to TF1, except that the equivalent work on TF1 lacked the time resolution necessary to observe bead transit33,34. By contrast, ATP hydrolysis and phosphate release in mitochondrial HF1 occur 30° and 55° before ATP binding, respectively16. It is possible that small differences between the angles of hydrolysis and phosphate release in YF1, TF1 and EF1 will be resolved by future improvements in temporal and spatial resolution36, but at present it appears that HF1 is different from the others in this respect. It remains to be seen whether these differences between different forms of F1 will show any discernible pattern; our results indicate that neither bacterial/mitochondrial nor thermophilic/mesophilc sources uniquely determine the pattern of catalytic dwell angles.
This study also demonstrates that the power spectral bias of gold bead position is a powerful method for quick, systematic detection of rotating molecules, facilitating the screening of several mutant forms. The power spectra can also be used to obtain speeds from noisy data traces, giving results comparable to those obtained using methods which require higher signal-to-noise.
The heart and liver isoforms of the γ subunit differ only by the addition of a single Asp residue at the C-terminus in the liver form, predicted to form a salt bridge with αE that would slow the catalytic cycle. However, the results of the rotational studies here suggest this is not the mechanism of regulation, as the kinetics are unchanged. Instead, if the Asp serves in a regulatory mechanism, it likely occurs in conjunction with another molecule. The lack of observable effect of the C-terminal Asp is consistent with observations that crosslinking the equivalent residue to the α-subunit changes neither the catalytic activity nor the torque generated by ATP hydrolysis37,38.
Six mgi mutant forms of S. cerevisiae F1-ATPase were studied to investigate molecular defects which uncouple the yeast ATP synthase28. Mutations that largely or completely uncouple the enzyme are technically very difficult to work with; all of the mgi mutations studied here are “leaky”, meaning that significant coupling is still present. The most consistent and significant change across all mutants was a substantial slowing of one of the processes, presumably ATP hydrolysis or phosphate release, at the catalytic dwell39. Another clear feature of our results is that the ATP binding rates observed at limiting [ATP] were 70–100% of the wild type rate, while the catalytic rates at saturating [ATP] were 15–50% of the wild type rate. This indicates that these mutations strongly affect either the rate of ATP hydrolysis or the rate of phosphate release, but have negligible effect on ATP binding.
mgi mutations have been categorized into two classes based upon their effect and location in the high-resolution structure of yeast F1 ATPase29. Class 1 mutations are located at the top of F1 (Fig. 1a) and form interactions with the E site at the Catch 1 region. The three Class 1 mutants studied here, γI270T, αN67I, and βV279F, showed no, minor, and significant conformational changes in the high-resolution crystal structure, respectively. Consistent with this pattern, the decrease in rotation speed was greatest for βV279F and smallest for γI270T. The changes to the high-resolution crystal structures were primarily around the E site, and the structure of the mutant forms αN67I and βV279F indicated that, unlike the wild type enzyme29, phosphate was not bound in the E site, suggesting that the affinity for phosphate has been reduced. Given the minimal disruption to the DP site in the crystal structure, it is probable that the slow rate observed for Class 1 mutants corresponds to phosphate release. The combination of structural and kinetic data therefore suggests that both the rate of phosphate release and the affinity for phosphate are reduced in the mutant. This implies that the binding rate of phosphate may be greatly reduced, consistent with impaired ATP synthesis.
The Class 2 mutations (αF405S, βR408I and βR408G) also show a significant reduction in the rate of one of the processes occurring at the catalytic dwell. Given the structural similarity of the E site to wild type in structural data, it seems likely that ATP hydrolysis at the DP site, rather than phosphate release, is slowed in Class 2 mutants.
αF405S, βR408I and βR408G are located at the bottom of F1, and while they are near the α/β interface, they do not change the conformation of the DP, TP or E active sites, but do alter the structure near the Catch 2 region. mgi residues αF405 and βR408 share a cation/π interaction40 and the crystal structure of αF405S shows a disruption in this interaction29. Both βR408G and βR408I also likely disrupt this interaction. The kinetic data for βR408I (and occasionally for βR408G and αF405S) showed an unexpected pattern of slow dwells at the ATP binding angle at saturating ATP. This slower dwell indicates a structural change in the conformation of F1 at the ATP binding dwell (in TP*, DP* or E*) and indicates a role for the cation/π interaction in one of the three reactions associated with the ATP binding dwell in bacterial F1; ATP binding, ADP release, or an unidentified temperature-sensitive step7,33,41. To discriminate between these three we note that the principal change in the crystal structure of αF405S is a disruption of the interactions between the γ and β subunits. Release of ADP occurs in the DP* state at or just after 240°, with rotation of the γ subunit greatly increasing the off-rate for ADP42. While ADP is being released from the DP* site, ATP is binding at the TP* site, and the two processes are co-ordinated by the γ subunit. We hypothesize that Class 2 mutations, as exemplified by αF405S, disrupt the αF405/βR408 cation/π interaction and interactions between the β and γ subunits that co-ordinate ATP binding and ADP release, and that the slowing of the binding dwell is due to the resulting decrease in the rate of ADP release.
Current crystal structures of F1 correspond to the catalytic dwell, and hence provide no direct structural information into the impact of Class 2 mutations on ADP release and the binding dwell. The speed of βR408I reduces with increasing [ATP] past Km, suggesting that conformational changes in the disrupted DP* site may be slowed and that ATP may weakly interact with the disrupted site. Alternatively, it is plausible that disruptions to coordination via the gamma subunit could delay the hydrolysis of ATP and allow rotation of F1 after phosphate release, but before ATP hydrolysis. The release of ADP would then be delayed until hydrolysis was complete. These predictions may be testable by computationally regenerating the ATP binding intermediate for Class 2 mutants.
Our single molecule data show no direct evidence of F1 moving into an uncoupled kinetic cycle where more than one ATP is consumed in a 120° rotation. However, the biochemical assays that show uncoupling involve situations where F1 experiences high load or is operated in the synthesis direction, and it is likely that these mutants remain tightly coupled when no load is present. Two technologies demonstrated on thermophilic F1 might be useful for investigating coupling – fluorescent ATP has been used to track ATP binding and ADP release from F143, and magnetic handles have been used with magnetic tweezers to rotate F1 in the synthesis direction44, with one suggestion that uncoupling (“slips”) are observable in the wild type thermophilic enzyme45. Furthermore, we have not measured torque output from these mutants, which might correlate with coupling. We expect further work along these lines will help elucidate the mechanism of uncoupling in these mutants. Further, a crystallographic goal is to obtain the structure of F1 in a number of additional intermediate conformations including the ATP binding dwell conformation. The results in this study illustrate the power of single molecule analysis, the limitations of x-ray crystallographic data, and the complementarity of the approaches.
Methods
Covalent Modification of F1
Yeast F1 was genetically engineered using the QuikChange method (Agilent Tech.) to include two surface cysteine residues, γD101C and γE189C and a His6-tag on the amino end of the β-subunit, and was purified as described46. The cysteine residues were biotinylated by incubating: 10 mM (3.5 mg/ml) F1, 20 mM Biotin-PEAC5-maleimide (6-{N′-[2-(N-Maleimido)ethyl]-N-piperazinylamido}hexyl D-biotinamide, hydrochloride, Dojindo Laboratories), 50 mM HEPES, pH 8.0, 2 mM ATP, 2 mM MgSO4; for 30 min at 23°C, quenching with 100 mM DTT, and desalting on a centrifuge column containing Biogel P6 resin (Biorad). The protein concentration was adjusted to 0.4 mg/ml in buffer containing 50% glycerol, flash frozen and stored in liquid N2. This modification does not alter the activity of the enzyme.
Gold bead preparation
60 nm gold beads (BBInternational) were functionalised with either Neutravidin or Streptavidin. Neutravidin beads were prepared by combining gold beads with 8 mg/mL Neutravidin in 10 mM Tris pH 8.0, and storing at 4°C for a minimum of 12 hours47. Streptavidin beads were prepared based on Ref. 48 by combining 5 μL each of 0.5 mM DSP, 10 mM TCEP and 100 μM streptavidin, each in 10 mM MOPS pH 7.0 with 50 mM KCl. This was incubated for five minutes, then 1 mL of gold beads was added as supplied, incubated for 2 hours at room temperature, and stored at 4°C. Beads were washed before use with excess 10 mM HEPES pH 7.0 (Neutravidin) or Tris pH 8.0 (streptavidin), followed by 10 mM MOPS pH 7.65, 50 mM KCl, 2 mM MgCl2.
Rotation assays
Glass coverslips were functionalised with Ni2+ NTA as described previously49. Tunnel slides used double-sided tape to define a channel between a microscope slide and a Ni2+ NTA functionalised coverslip. Where the buffer was changed during microscopy, flow slides were used as described in SI, with two short tubes and the input tube connected to a syringe. F1 (1–10 nM in buffer A - 10 mM MOPS, 50 mM KCl, 2 mM MgCl2, 10 μM ATP, pH 7.65) and then gold beads (in buffer B = buffer A + 1 mg/mL BSA for streptavidin beads, or buffer A + 10 mg/mL BSA for neutravidin beads) were each added for ten minutes and washed with at least 5 volumes of buffer B. Rotation was observed in assay buffer (buffer B + ATP 1 μM–3 mM, 2 mM phosphoenolpyruvate and 20 units/mL pyruvate kinase). Where the concentration of ATP in the assay buffer was less than 10 μM, the ATP concentration of buffers A and B was reduced to the same level.
Tunnel slides were viewed under a high-speed backscattering darkfield microscope50 and images of multiple gold beads recorded at frame rates between 60 Hz and 30 kHz using a CMOS camera (Fastcam 1024 PCI, Photron) with 85 nm per pixel. For flow slides a custom stage with high mechanical stiffness replaced the piezo mount described in Ref. 50.
Temperature
Microscope temperature was measured using a K-type thermocouple, and was 23.0 ± 0.5°C for all experiments used for speed or kinetic rate measurements. For flow slide experiments, used to measure the angle separation of substeps, the temperature was 22.5 ± 1.0°C.
Video Analysis
Analysis of bead motion was performed using custom-written MATLAB software, as follows (see SI for details). Bead positions and intensities in each frame were calculated using a Gaussian mask algorithm51. Spinning beads were identified from directional bias in bead rotation,
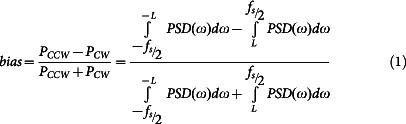 |
where PSD(ω) is the power spectral density at angular frequency ω of the bead's position (x(t), y(t)) expressed as the complex number z = x + iy, fS is the sampling rate and L is a low frequency cutoff used to remove noise associated with microscope drift, typically set to the lower of 5% of the typical rotation speed under the assay conditions used or 10 Hz. Beads identified as duplexes based on either radius of motion or intensity, were removed from the data set for all speed measurements and for kinetics measurements at high ATP (where the extra drag is significant). The software allows screening a sample to reliably detect rotating probes prior to acquisition of long videos.
Rotation speed
Ellipses were fit to the x-y data using an algebraic algorithm52, from which an unwrapped angular position was derived. Pauses, during which a bead stopped rotating, were identified as described below in the sub-section “kinetic fits”. Angular speed was estimated as the total angular displacement during the video divided by the total time, having first removed any pauses. Beads were excluded from speed measurements if more than 2% of the data points were closer to the centre of the fitted ellipse than one-third of the radius of the ellipse. Traces rejected by this criterion tend to have multiple false centre-crossings, and produce measured speeds that are lower than the actual rotation speed, as shown in Fig. 2.
Speed was also estimated as the peak in a Gaussian fit to the ‘subtracted’ power spectrum (PSD(−ω) - PSD(ω)), which isolates rotation in the CCW direction from non-rotational noise in the bead trajectory. If the trace contained pauses, data were restricted to the longest period for which an L153 fit to the angle data indicated that the bead was rotating above one-third the maximum speed in the trace. Beads were excluded if they failed any of the following criteria: bias < 0.15 over the video, bias < 0.2 over the spinning period, the integral of the fit Gaussian <80% of the integral of the subtracted spectrum. A further symmetry criterion excluded data where the fitting mask moved between separate beads during the video (see SI for details).
Substep angle measurements
Data were collected using a flow slide, observing the same field of molecules at low followed by saturating ATP concentrations. Comparison of videos collected before, during, and after buffer exchange, showed that there was typically translation of the field of view associated with flowing buffer through the slide, but no detectable rotation of the field of view.
For each molecule rotating before and after the buffer exchange, the last spinning episode before, and the first spinning episode after buffer exchange were selected. Episodes were discarded where low ATP speed was greater than 50 Hz or the final speed was less than 400 Hz (to ensure full buffer exchange), or the ratio of the long to short axis of the fit ellipse was greater than 1.5 (when angle recovery is less accurate). Measuring angles via ellipse fits before and after buffer exchange was unreliable, as a misplacement of the ellipse fit by a few nm significantly distorted the recovered angles. Instead, k-means clustering with 3 clusters was used to identify the dwell locations (x, y) at low and high ATP. The vectors between successive dwell states were determined, and the rotation of these vectors was used as the measurement of the angular displacement from the ATP hydrolysis to the ATP waiting state. This measures angle changes directly without requiring ellipse fitting or drift correction between the two sets of data.
Kinetic fits
Kinetics were fit to angle-time traces in a manner similar to previously6. For fitting three dwells per revolution, the angle trace was unwrapped and broken into 120° segments, and the number of data points in each segment was used to generate a histogram of dwell times, pooling dwells from all 3 states. Pauses were removed by ignoring dwells longer than 7 times the mean dwell length. For single exponential kinetics this corresponds to removing the longest 0.1% of real (i.e., non-pause) dwells and a 0.6% (i.e. negligible) systematic underfit of the rate constants; for every other kinetic model the fraction of real events removed and effect on the fit is smaller. The dwells immediately preceding and following pauses were also removed.
A histogram of the dwell times was constructed for each enzyme. Six kinetic models were considered as potential fits to the histogram, comprised of 1, 2 or 3 kinetic rates per dwell both with and without a rotation-delay time offset. The dwell time histograms were fit to each model and rate constants were derived with a maximum likelihood estimation (MLE) algorithm. The likelihood of these six models and thus the quality of fit was compared using the Bayesian information criterion (BIC).
Supplementary Material
Supplementary Information
Acknowledgments
Thanks to Yoshiyuki Sowa for building the first version of the darkfield microscope on which this data was collected, to Tom Bilyard for demonstrating the practical aspects of working with F1, and to Richard Wang for the bulk ATPase assays and analysis. This work was supported in part, by the National Institute of Health Grant, R01GM66223 (to DMM).
Footnotes
Author Contributions B.C.S. collected all experimental data. Y.W. and V.P. performed all genetic transformations, enzyme purifications, and biotinylation modifications. B.C.S. and A.L.N. were responsible for software, algorithms and analysis. A.L.N., B.C.S., D.M.M. and R.M.B. wrote the manuscript.
References
- Noji H., Yasuda R., Yoshida M. & Kinosita K. Jr Direct observation of the rotation of F1-ATPase. Nature 386, 299–302 (1997). [DOI] [PubMed] [Google Scholar]
- Iino R. & Noji H. Operation mechanism of F(o) F(1)-adenosine triphosphate synthase revealed by its structure and dynamics. IUBMB Life 65, 238–246 (2013). [DOI] [PubMed] [Google Scholar]
- Junge W., Sielaff H. & Engelbrecht S. Torque generation and elastic power transmission in the rotary F(O)F(1)-ATPase. Nature 459, 364–370 (2009). [DOI] [PubMed] [Google Scholar]
- Pilizota T., Sowa Y. & Berry R. M. Single-Molecule Studies of Rotary Molecular Motors. Handbook of Single-Molecule Biophysics. Hinterdorfer, P. & van Oijen, A. (eds.) (Springer, Verlag, 2009). [Google Scholar]
- von Ballmoos C., Wiedenmann A. & Dimroth P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Review. Biochem. 78, 649–672 (2009). [DOI] [PubMed] [Google Scholar]
- Bilyard T. et al. High-resolution single-molecule characterization of the enzymatic states in Escherichia coli F1-ATPase. Phi.l Trans. R. Soc. Lond. B. Biol. Sci. 368, 20120023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaike T., Koyama-Horibe F., Oiwa K., Yoshida M. & Nishizaka T. Cooperative three-step motions in catalytic subunits of F(1)-ATPase correlate with 80 degrees and 40 degrees substep rotations. Nat. Struct. Mol. Biol. 15, 1326–1333 (2008). [DOI] [PubMed] [Google Scholar]
- Sielaff H., Rennekamp H., Engelbrecht S. & Junge W. Functional halt positions of rotary FOF1-ATPase correlated with crystal structures. Biophysical Journal 95, 4979–4987 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno D. et al. Correlation between the conformational states of F1-ATPase as determined from its crystal structure and single-molecule rotation. Proc. Natl. Acad. Sci. U.S.A. 105, 20722–20727 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K. & Takada S. Structural comparison of F1-ATPase, interplay among enzyme structures, catalysis, and rotations. Structure (London, England, 1993) 19, 588–598 (2011). [DOI] [PubMed] [Google Scholar]
- Shimo-Kon R. et al. Chemo-mechanical coupling in F(1)-ATPase revealed by catalytic site occupancy during catalysis. Biophys. J. 98, 1227–1236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R. et al. The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. 100, 9314–9318 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono-Hara Y. et al. Pause and rotation of F(1)-ATPase during catalysis. Proc. Natl. Acad. Sci. 98, 13649–13654 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani G. & Duncan T. M. Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nature structural & molecular biology 18, 701–707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Hutcheon M. L., Duncan T. M. & Cingolani G. Improved crystallization of Escherichia coli ATP synthase catalytic complex (F1) by introducing a phosphomimetic mutation in subunit epsilon. Acta crystallographica. Section F, Structural biology and crystallization communications 68, 1229–1233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Tanaka K., Wakabayashi C., Saita E. & Yoshida M. Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat Chem Bio. 10, 930–936 (2014). [DOI] [PubMed] [Google Scholar]
- Abrahams J. P., Leslie A. G., Lutter R. & Walker J. E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 (1994). [DOI] [PubMed] [Google Scholar]
- Bowler M. W., Montgomery M. G., Leslie A. G. & Walker J. E. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 A resolution. J. Biol. Chem. 282, 14238–14242 (2007). [DOI] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A. & Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D. Biol. Crystallog. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- Pu J. & Karplus M. How subunit coupling produces the gamma-subunit rotary motion in F1-ATPase. Proc. Natl. Acad. Sci. 105, 1192–1197 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. & Teplyakov A. Molecular replacement with MOLREP. Acta. Crystallogr. D. Biol. Crystallog. 66, 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- Kabaleeswaran V., Puri N., Walker J. E., Leslie A. G. & Mueller D. M. Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J. 25, 5433–5442 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabaleeswaran V. et al. Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides. J. Biol. Chem. 284, 10546–10551 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. X. & Clark-Walker G. D. Specific mutations in alpha- and gamma-subunits of F-1-ATPase affect mitochondrial genome integrity in the petite-negative yeast Kluyveromyces lactis. EMBO J. 14, 3277–3286 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. J. & Clark-Walker G. D. Mutations in MGI genes convert Kluyveromyces lactis into a petite-positive yeast. Genetics 133, 517–525 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. J. & ClarkWalker G. D. The mitochondrial genome integrity gene, MGI1, of Kluyveromyces lactis encodes the beta-subunit of F-1-ATPase. Genetics 144, 1445–1454 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. J., Hansbro P. M. & Clark-Walker G. D. Suppression of rho0 lethality by mitochondrial ATP synthase F1 mutations in Kluyveromyces lactis occurs in the absence of FO. Mol. Genet. Genomics 259, 457–467 (1998). [DOI] [PubMed] [Google Scholar]
- Wang Y., Singh U. & Mueller D. M. Mitochondrial genome integrity mutations uncouple the yeast Saccharomyces cerevisiae ATP synthase. J. Biol. Chem. 282, 8228–8236 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenieva D., Symersky J., Wang Y., Pagadala V. & Mueller D. M. Crystal structures of mutant forms of the yeast F1 ATPase reveal two modes of uncoupling. J. Biol. Chem. 285, 36561–36569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda C. et al. Tissue-specific isoforms of the bovine mitochondrial ATP synthase gamma-subunit. FEBS Lett. 325, 281–284 (1993). [DOI] [PubMed] [Google Scholar]
- Hayakawa M. et al. Muscle-specific exonic splicing silencer for exon exclusion in human ATP synthase gamma-subunit pre-mRNA. J. Biol. Chem. 277, 6974–6984 (2002). [DOI] [PubMed] [Google Scholar]
- Ichida M. et al. Differential regulation of exonic regulatory elements for muscle-specific alternative splicing during myogenesis and cardiogenesis. J. Biol. Chem. 275, 15992–16001 (1978). [DOI] [PubMed] [Google Scholar]
- Yasuda R., Noji H., Yoshida M., Kinosita K. & Itoh H. Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410, 898–904 (2001). [DOI] [PubMed] [Google Scholar]
- Shimabukuro K. et al. Catalysis and rotation of F-1 motor, Cleavage of ATP at the catalytic site occurs in 1 ms before 40 degrees substep rotation. Proc. Natl. Acad. Sci. 100, 14731–14736 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the Dimension of a Model. Ann. Stat. 6, 461–464 (1978). [Google Scholar]
- Ishmukhametov R., Hornung T., Spetzler D. & Frasch W. D. Direct observation of stepped proteolipid ring rotation in E. coli F0F1-ATP synthase. EMBO Journal 29, 3911–3923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbers F., Junge W. & Sielaff H. The torque of rotary F-ATPase can unfold subunit gamma if rotor and stator are cross-linked. PloS one 8, e53754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiowski K. et al. F-ATPase: forced full rotation of the rotor despite covalent cross-link with the stator. The Journal of biological chemistry 276, 42287–42292 (2001). [DOI] [PubMed] [Google Scholar]
- Okazaki K. & Hummer G. Phosphate release coupled to rotary motion of F1-ATPase. Proc. Natl. Acad. Sci. 110, 16468–16473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H. Z., Abraham C. G., Krishnakumar A. M. & Weber J. A functionally important hydrogen-bonding network at the betaDP/alphaDP interface of ATP synthase. J. Biol. Chem. 283, 24781–24788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R., Iino R., Shimabukuro K., Yoshida M. & Noji H. Temperature-sensitive reaction intermediate of F-1-ATPase. EMBO Rep. 9, 84–90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi K., Oiwa K., Yoshida M., Nishizaka T. & Kinosita K. Controlled rotation of the F1-ATPase reveals differential and continuous binding changes for ATP synthesis. Nat. Commun. 3, 1022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaka T. et al. Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation. Nat. Struct. Mol. Biol. 11, 142–148 (2004). [DOI] [PubMed] [Google Scholar]
- Iino R. & Noji H. F1-ATPase, a highly coupled reversible rotary motor. Biochem. Soc. Trans. 34, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- Palanisami A. & Okamoto T. Torque-Induced Slip of the Rotary Motor F-1-ATPase. Nano Letters 10, 4146–4149 (2010). [DOI] [PubMed] [Google Scholar]
- Mueller D. M. et al. Ni-chelate-affinity purification and crystallization of the yeast mitochondrial F1-ATPase. Protein Expr. Purif. 37, 479–485 (2004). [DOI] [PubMed] [Google Scholar]
- Dunn A. R. & Spudich J. A. Dynamics of the unbound head during myosin V processive translocation. Nat. Struct. Mol. Biol. 14, 246–248 (2007). [DOI] [PubMed] [Google Scholar]
- Furuike S. et al. Temperature dependence of the rotation and hydrolysis activities of F-1-ATPase. Biophys. J. 95, 761–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H. et al. Mechanically driven ATP synthesis by F-1-ATPase. Nature 427, 465–468 (2004). [DOI] [PubMed] [Google Scholar]
- Sowa Y., Steel B. C. & Berry R. M. A simple backscattering microscope for fast tracking of biological molecules. Rev. Sci. Instrum. 81, 113704 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. E., Larson D. R. & Webb W. W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgibbon A., Pilu M. & Fisher R. B. Direct least square fitting of ellipses. IEEE Trans. Pattern Anal. Mach. Intell. 21, 476–480 (1999). [Google Scholar]
- Kim S. J., Koh K., Boyd S. & Gorinevsky D. L(1) Trend Filtering. Siam. Rev. Soc. Ind. Appl. Math 51, 339–360 (2009). [Google Scholar]
- Adachi K. et al. Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information