

Glycine is required for various cellular functions, including cell wall synthesis, protein synthesis, and the biosynthesis of several important metabolites. Regulating levels of glycine metabolism allows P. aeruginosa to maintain the metabolic flux of glycine through several pathways, including the metabolism of glycine to produce other amino acids, entry into the trichloroacetic acid cycle, and the production of virulence factors such as hydrogen cyanide. In this study, we characterized GcsR, a transcriptional regulator that activates the expression of genes involved in P. aeruginosa PAO1 glycine metabolism. Our work reveals that GcsR is the founding member of a novel class of TyrR-like EBPs that likely regulate glycine metabolism in Pseudomonadales.
KEYWORDS: Glycine metabolism, Pseudomonas aeruginosa PAO1, TyrR, enhancer-binding proteins, transcription factors
ABSTRACT
Glycine serves as a major source of single carbon units for biochemical reactions within bacterial cells. Utilization of glycine is tightly regulated and revolves around a key group of proteins known as the glycine cleavage system (GCS). Our lab previously identified the transcriptional regulator GcsR (PA2449) as being required for catabolism of glycine in the opportunistic pathogen Pseudomonas aeruginosa PAO1. In an effort to clarify and have an overall better understanding of the role of GcsR in glycine metabolism, a combination of transcriptome sequencing and electrophoretic mobility shift assays was used to identify target genes of this transcriptional regulator. It was found that GcsR binds to an 18-bp consensus sequence (TGTAACG-N4-CGTTCCG) upstream of the gcs2 operon, consisting of the gcvH2, gcvP2, glyA2, sdaA, and gcvT2 genes. The proteins encoded by these genes, namely, the GCS (GcvH2-GcvP2-GcvT2), serine hydroxymethyltransferase (GlyA2), and serine dehydratase (SdaA), form a metabolic pathway for the conversion of glycine into pyruvate, which can enter the central metabolism. GcsR activates transcription of the gcs2 operon in response to glycine. Interestingly, GcsR belongs to a family of transcriptional regulators known as TyrR-like enhancer-binding proteins (EBPs). Until this study, TyrR-like EBPs were only known to function in regulating aromatic amino acid metabolism. GcsR is the founding member of a new class of TyrR-like EBPs that function in the regulation of glycine metabolism. Indeed, homologs of GcsR and its target genes are present in almost all sequenced genomes of the Pseudomonadales order, suggesting that this genetic regulatory mechanism is a common theme for pseudomonads.
IMPORTANCE Glycine is required for various cellular functions, including cell wall synthesis, protein synthesis, and the biosynthesis of several important metabolites. Regulating levels of glycine metabolism allows P. aeruginosa to maintain the metabolic flux of glycine through several pathways, including the metabolism of glycine to produce other amino acids, entry into the trichloroacetic acid cycle, and the production of virulence factors such as hydrogen cyanide. In this study, we characterized GcsR, a transcriptional regulator that activates the expression of genes involved in P. aeruginosa PAO1 glycine metabolism. Our work reveals that GcsR is the founding member of a novel class of TyrR-like EBPs that likely regulate glycine metabolism in Pseudomonadales.
INTRODUCTION
Amino acids are a preferred nutrient source for the Gram-negative opportunistic pathogen Pseudomonas aeruginosa (1, 2). This bacterium can use almost any amino acid as either a sole carbon or a sole nitrogen source (2). The amino acid glycine is a major source of single carbon units for biochemical reactions in the cell and is necessary for the biosynthesis of several important metabolites such as purines, thymidine, methionine, threonine, and lipids (3–5). Aside from basic metabolism, glycine is also a precursor of hydrogen cyanide (HCN), which P. aeruginosa uses as a virulence factor (6).
The metabolism of glycine is dependent on a multienzyme complex known as the glycine cleavage system (GCS), which is composed of three proteins, GcvH, GcvP, and GcvT (7). GcvP is a dehydrogenase that catalyzes the decarboxylation of glycine and transfers the aminomethyl moiety to the lipoyl prosthetic group of GcvH. GcvT is an aminomethyltransferase that mediates the release of ammonia from the intermediate attached to GcvH and the synthesis of 5,10-methylene tetrahydrofolate (5,10-methylene-THF) in the presence of tetrahydrofolate (THF). Overall, the GCS catalyzes the conversion of glycine into carbon dioxide, ammonia, and 5,10-methylene-THF. In addition to the GCS, there are two other proteins that play important roles in glycine metabolism. First, the serine hydroxymethyltransferase (GlyA) transfers a methylene group from 5,10-methylene-THF to glycine, thus forming serine and THF as products. The GlyA reaction is reversible and is necessary for the biosynthesis of glycine from serine (8, 9). The other key protein for glycine metabolism is serine dehydratase (SdaA), which catalyzes the deamination of serine into pyruvate, an intermediate of central metabolism (10, 11). The GCS-GlyA-SdaA pathway provides a route for the utilization of glycine as a carbon and nitrogen source by bacteria.
In the case of P. aeruginosa PAO1, two sets of genes coding for GCS proteins are found on the chromosome. There are also three genes coding for GlyA and two serine dehydratase genes in the P. aeruginosa PAO1 genome. Among these, one set of glycine metabolism genes, the gcvH2-gcvP2-glyA2-sdaA-gcvT2 genes, are located together in the chromosome and are collectively known as the gcs2 cluster. This gene cluster was previously found to be downregulated in the absence of the gene PA2449 (here gcsR [for glycine cleavage system regulator]) (12). Indeed, the gcsR gene was also essential for the utilization of glycine as a sole carbon source by P. aeruginosa PAO1 (12). The gcsR gene encodes a transcriptional regulator belonging to the enhancer-binding protein (EBP) family. EBPs are an interesting family of regulators because they possess domains that enable them to specifically interact with the alternative sigma factor σ54 (RpoN) to initiate transcription from their target promoters (13). Upstream of the gcs2 cluster is a putative RpoN promoter, supporting a model in which GcsR regulates the transcription of gcs2 genes in response to glycine availability.
In addition to being an EBP, GcsR also shows >40% identity to the TyrR transcriptional regulator of Escherichia coli. This further classifies GcsR as a TyrR-like EBP. The members of the TyrR family of regulators are known for their roles in aromatic amino acid biosynthesis and metabolism (14–17). The P. aeruginosa PAO1 genome has two genes encoding the TyrR-like EBPs PhhR and GcsR (18). PhhR was previously identified as a regulator of aromatic amino acid metabolism in P. aeruginosa PAO1 (16, 19). GcsR shows 44% sequence homology to PhhR (12).
In this study, we show a novel mechanism for the regulation of the P. aeruginosa PAO1 gcs2 cluster by the TyrR-like EBP GcsR. We demonstrate that, unlike other TyrR regulators that respond to aromatic amino acids, GcsR activates the transcription of the gcs2 cluster in response to glycine. In addition, we show that the five gcs2 genes are transcribed as an operon and GcsR binds to an 18-bp tandem repeat sequence in the promoter region of the gcs2 operon to activate transcription. Although the P. aeruginosa PAO1 genome contains multiple homologs of each of the gcvH, gcvP, gcvT, glyA, and sdaA genes, our work indicates that only the gcs2 operon is essential for the metabolism of glycine as a sole carbon source. GcsR also appears to link glycine metabolism to virulence since the paralytic killing of the nematode Caenorhabditis elegans is significantly enhanced in the absence of gcsR.
Our work also reveals that the gcsR gene is conserved in nearly all of the sequenced genomes of the members of the order Pseudomonadales and is found adjacent to genes encoding a GCS. The results presented in this report indicate that GcsR is the prototype of a new family of TyrR-like EBPs that regulate glycine metabolism.
RESULTS
GcsR is essential for expression of glycine metabolism genes.
Our previous work had shown that the gcsR gene is essential for the metabolism of glycine as a sole carbon source and for pyocyanin production by P. aeruginosa PAO1 (12). We also found that more than 300 genes were differentially expressed in the gcsR transposon mutant strain PW5126 (12). Curiously, except for the gcs2 genes, most of the genes that were affected by the disruption in gcsR were genes involved in or regulated by quorum signaling. This previous result may be explained by a recent study that showed that many mutants of the P. aeruginosa PAO1 transposon mutant library had acquired unrelated mutations that led to altered pyocyanin production and quorum signaling phenotypes (20). It has also been observed in other pathogenic bacteria that transposon mutations can affect the quorum signaling system (21). Therefore, we constructed an in-frame deletion of the gcsR gene in P. aeruginosa PAO1 (ΔgcsR PAO1) to verify if it did indeed affect quorum signaling and pyocyanin production.
Compared to our original transcriptomic study using the gcsR transposon mutant strain PW5126, transcriptome sequencing (RNA-Seq) analysis of the ΔgcsR PAO1 strain showed that only 20 genes were differentially expressed (Table 1). Interestingly, these did not include any of the quorum-sensing or quorum-regulated genes that had been affected by the gcsR transposon mutation. The genes differentially expressed in ΔgcsR PAO1 were mostly involved in metabolism. These include the gcs2 genes that were also downregulated in the gcsR transposon mutant. The gcs2 genes gcvH2, gcvP2, glyaA2, sdaA, and gcvT2 were downregulated 190-, 112-, 28-, 2-, and 3-fold, respectively, in ΔgcsR PAO1. Other genes affected in the ΔgcsR PAO1 strain include the narK1, narK2, narG, and narH genes belonging to a putative operon involved in nitrogen metabolism, which were upregulated ~2.8-fold; an operon containing genes involved in formaldehyde metabolism (PA3628-adhC) that was downregulated ~2.5-fold; and an operon encoding small RNAs that regulate iron homeostasis that was downregulated ~2.7-fold. The genes encoding tRNAs for Lys, Met, Ala, and Leu were also downregulated at least 2-fold in the ΔgcsR PAO1 strain.
TABLE 1 .
Genes with >2-fold changes in transcript levels in the P. aeruginosa ΔgcsR PAO1 compared to wild-type P. aeruginosa PAO1 grown in PB
| Gene ID | Gene name | Mean fold change | Biological function of product |
|---|---|---|---|
| PA0976.1 | −2.2 | tRNA-Lys | |
| PA1183 | dctA | −2.08 | C4-dicarboxylate transport |
| PA2442 | gcvT2 | −2.7 | Glycine metabolism |
| PA2443 | sdaA | −2.19 | Glycine metabolism |
| PA2444 | glyA2 | −38 | Glycine metabolism |
| PA2445 | gcvP2 | −111.58 | Glycine metabolism |
| PA2446 | gcvH2 | −188.91 | Glycine metabolism |
| PA3516 | 2.53 | Purine metabolism | |
| PA3628 | −2.3 | Formaldehyde metabolism | |
| PA3629 | adhC | −2.46 | Formaldehyde metabolism |
| PA3874 | narH | 3 | Nitrogen metabolism |
| PA3875 | narG | 2.8 | Nitrogen metabolism |
| PA3876 | narK2 | 2.85 | Nitrogen metabolism |
| PA3877 | narK1 | 2.53 | Nitrogen metabolism |
| PA4153 | 2.58 | Butanediol catabolic process | |
| PA4280.3 | −7.5 | tRNA-Ala | |
| PA4704.1 | prrF1 | −2.2 | Iron homeostasis |
| PA4704.3 | prrF2 | −3.22 | Iron homeostasis |
| PA4746.1 | −2.04 | tRNA-Met | |
| PA4937.1 | −2.49 | tRNA-Leu |
The five glycine metabolism genes gcvH2, gcvP2, glyA2, sdaA, and gcvT2 were the only genes that were differentially expressed in both the gcsR transposon mutant and the ΔgcsR mutant. The transcription levels of these genes were also the most affected in ΔgcsR PAO1. Additionally, we found that, like the transposon mutant, the ΔgcsR mutant was unable to grow on glycine as a sole carbon source (Fig. 1), providing evidence that the glycine metabolism genes are likely to be an important target of GcsR.
FIG 1 .
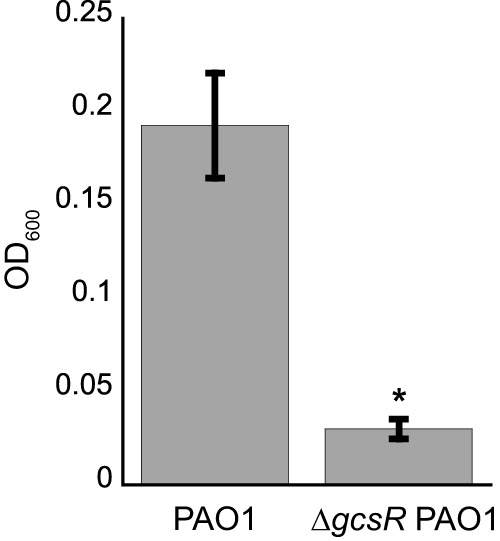
ΔgcsR PAO1 is unable to grow in glycine as a sole carbon source. P. aeruginosa PAO1 and ΔgcsR PAO1 were grown with glycine as the sole carbon source for 48 h at 37°C. Data points represent mean values ± the standard deviations (n = 3). Student’s t test was performed to identify significant differences (P < 0.0001; marked with an asterisk).
The gcs2 cluster genes are cotranscribed in P. aeruginosa PAO1.
The glycine metabolism genes downregulated in the ΔgcsR PAO1 strain are arranged together in the gcs2 gene cluster, with the gcvH2 gene being the first gene, followed by the gcvP2, glyA2, sdaA, and gcvT2 genes (Fig. 2A). The gcvH2, gcvP2, and gcvT2 gene products together make up the GCS (4), the glyA2 gene encodes a serine hydroxymethyltransferase that catalyzes the reversible conversion of glycine to serine (9), and sdaA encodes a serine dehydratase that catalyzes the deamination of serine to a pyruvate (11). Because of their organization in the genome, their function in glycine metabolism, and the fact that they were all downregulated in the ΔgcsR PAO1 strain, we proposed that these genes are cotranscribed and hence regulated by GcsR. In order to test this hypothesis, we determined the operon structure by reverse transcriptase (RT) PCR analysis of RNA isolated from P. aeruginosa PAO1 grown for 48 h to an optical density at 600 nm (OD600) of 0.25 in minimal medium with glycine as the sole carbon source (Fig. 2B). To check whether adjacent genes were cotranscribed, the cDNA was obtained from the isolated RNA and used as a template for PCR amplification with primers that amplified 500- to 600-bp fragments that spanned intergenic regions from the 3′ end of the upstream gene to the 5′ end of the adjacent downstream gene. PCR products were observed for all four primer sets, indicating that these five genes are transcribed together as a single operon (Fig. 2B).
FIG 2 .
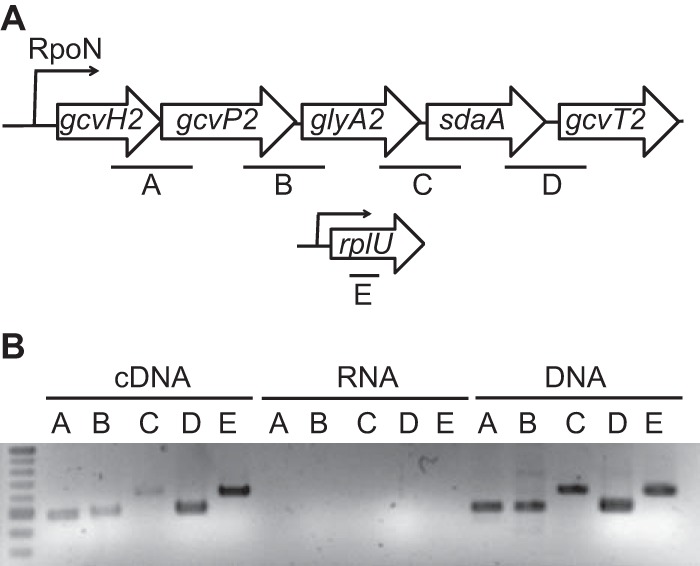
The five gcs2 genes are transcribed as an operon. (A) At the top is a schematic of the gcs2 gene cluster. Intergenic regions that were amplified are designated A to D. At the bottom is a schematic of the rplU gene, encoding the 50S ribosomal protein (L21), which was was used as a control for the RT-PCR analysis (amplified region designated E). (B) RT-PCR analysis of the gcs2 gene cluster. The regions designated A to E were amplified by PCR with cDNA, RNA, or genomic DNA obtained from P. aeruginosa PAO1 as the template.
Expression of a PgcvH2-lacZ fusion in E. coli is dependent on GcsR and is regulated by glycine availability.
GcsR was expected to regulate the expression of the gcs2 operon in response to glycine. To test this hypothesis, the 5′ regulatory region (~500 bp) upstream of gcvH2 (PgcvH2) was fused to the β-galactosidase (lacZ) open reading frame (ORF) of E. coli. The PgcvH2-lacZ fusion was then cotransformed with a plasmid harboring the gcsR gene into nonnative E. coli. As shown in Fig. 3A, β-galactosidase (LacZ) levels increased 3-fold with the addition of 10 mM glycine to our recombinant E. coli strain. The addition of serine or glutamate had no effect on LacZ levels, suggesting that induction of the PgcvH2-lacZ fusion was specific to glycine.
FIG 3 .

Expression of PgcvH2::lacZ is induced by glycine. (A) Wild-type or ΔgcvA or ΔgcvR E. coli cells harboring the PgcvH2::lacZ reporter construct and the pBRL620-5 plasmid for expression of GcsR were grown in LB to an OD600 of ~0.3 and then challenged with 10 mM glycine, 10 mM glutamate, 10 mM serine, or no substrate. (B) E. coli ΔgcvA mutant cells harboring empty plasmids pTrc99a and ΔPlac-pBBR1MCS-5, plasmids pBRL456 containing the PgcvH2::lacZ reporter and pBRL620-5 for the expression of GcsR, plasmid pBRL456 and plasmid pBRL620-3 harboring gcsR without a promoter, plasmids ΔPlac-pBBR1MCS-5 and pBRL620-5, or plasmids pBRL456 and pTrc99a were grown in LB to an OD600 of ~0.3 and then challenged with 10 mM glycine or no substrate. Data points represent mean values ± the standard deviations (n = 3). Analysis of variance was performed by using Dunnett’s post hoc test (α value of 0.05) to identify significant differences (P < 0.0001; marked with an asterisk).
Since wild-type E. coli can metabolize glycine, the internal glycine concentrations were too low to induce lacZ expression, as evidenced by the relatively minor differences in LacZ activity levels in the absence of glycine (20 Miller units [MU]) and in the presence of glycine (70 MU) (Fig. 3A). GcvA is an essential activator of the GCS in E. coli (22). Because an E. coli ΔgcvA mutant is unable to assimilate glycine, the LacZ activity of the PgcvH2-lacZ fusion was 12-fold higher in this strain than in wild-type E. coli (Fig. 3A). In contrast, GcvR is a negative regulator of glycine metabolism in E. coli (23), so its absence was not expected to alter the regulation of the PgcvH2-lacZ fusion by GcsR. Accordingly, in the absence of exogenous glycine, the LacZ level was 259 MU in the ΔgcvA mutant compared to 20 and 19 MU in the wild-type and ΔgcvR mutant strains, respectively (Fig. 3A). The addition of glycine increased the LacZ level by 2-fold in ΔgcvA mutant E. coli, which was similar to the fold change observed in wild-type and ΔgcvR mutant E. coli (Fig. 3A). Lastly, the presence of the gcsR gene was required for glycine induction of PgcvH2-lacZ in E. coli (Fig. 3B). Replacement of the plasmid harboring the gcsR gene with either an empty plasmid equivalent or a plasmid having the gcsR gene in the opposite orientation did not generate significant levels of LacZ activity.
Since GcsR is predicted to be an EBP and since there is a putative RpoN binding site in the PgcvH2 promoter, we wanted to check whether RpoN is required for transcription from this promoter. We found that an E. coli ΔrpoN mutant expressing GcsR and harboring the reporter plasmid showed relatively low LacZ levels and no significant difference in LacZ levels in the presence or absence of glycine (17.9 MU in the absence of substrate and 27.7 MU in the presence of glycine) (Fig. 3A). This indicates that RpoN is required for initiation of transcription from the PgcvH2 promoter.
In order to verify that GcsR regulates the PgcvH2 promoter in vivo, we measured the activity of the PgcvH2-lacZ fusion in the wild-type PAO1, ΔgcsR PAO1, and ΔrpoN PAO1 strains (Fig. 4). We found that the LacZ levels in PAO1 increased 3-fold in the presence of exogenous glycine compared to that in cells grown with no substrate or in the presence of serine or glutamate. Additionally, there was no difference in LacZ levels in the presence or absence of glycine in the ΔgcsR PAO1 strain or in the ΔrpoN PAO1 strain, further indicating that both GcsR and RpoN are required for transcription from the PgcvH2 promoter.
FIG 4 .
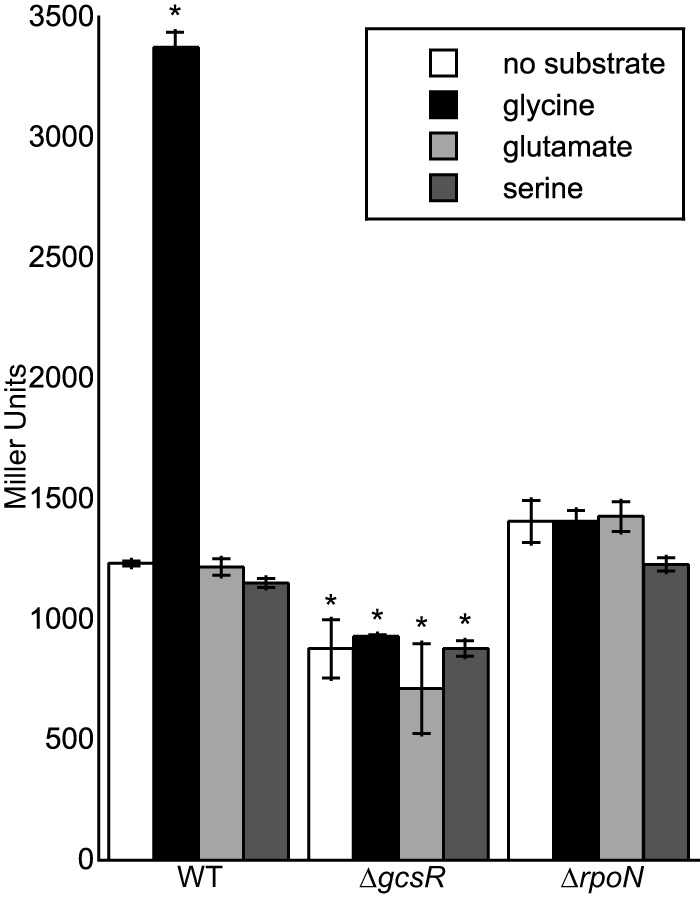
Expression of PgcvH2::lacZ is induced with glycine in P. aeruginosa PAO1. P. aeruginosa wild-type (WT) PAO1, ΔgcsR PAO1, or ΔrpoN PAO1 cells harboring the PgcvH2::lacZ reporter construct was grown in M9 minimal medium to an OD600 of ~0.3 and then challenged with 10 mM glycine, 10 mM glutamate, 10 mM serine, or no substrate. Data points represent mean values ± the standard deviations (n = 3). Analysis of variance was performed by using Dunnett’s post hoc test (α value of 0.05) to identify significant differences (P < 0.0001; marked with an asterisk).
GcsR binds to the gcvH2 promoter region with high specificity and affinity.
On the basis of homology, the gcsR gene encodes a TyrR family EBP (12). Since all of the characterized TyrR family EBPs regulate the metabolism of aromatic amino acids (14, 15), this makes GcsR a unique member of this group. To understand how GcsR regulates glycine metabolism, we first identified its target promoters. The σ54 Promoter Database (http://www.sigma54.ca/promoterdata/Web/data.aspx) predicts a putative strong RpoN binding site (score of 92) 79 bp upstream of the predicted gcvH2 ORF, the first gene of the gcs2 operon. Furthermore, the gcs2 operon is located adjacent to the gcsR gene in the PAO1 genome (18). This made the gcvH2 promoter the most likely target for GcsR. To verify this, we used electrophoretic mobility shift assays (EMSAs) to monitor the binding of His6-GcsR to a Cy5-labeled DNA probe containing the 200-bp region immediately upstream of the putative RpoN binding site of the gcvH2 promoter (PgcvH2). EMSAs showed that His6-GcsR was indeed able to bind to PgcvH2 (Fig. 5A). The affinity of GcsR for this piece of DNA is very high, judging from the shift produced by as little as 6.25 nM His6-GcsR, and the intensity of the shift increased with increasing concentrations of His6-GcsR (Fig. 5B). We also found that His6-GcsR bound to this region with high specificity. To test the specificity of His6-GcsR binding, we compared its binding of PgcvH2 with its binding to the nonspecific probe PPA5530, which contains a 200-bp region upstream of the RpoN binding site on the PA5530 promoter (24). Figure 5A shows that while 200 nM His6-GcsR completely shifts the PgcvH2 probe (lane 4), there is no shift in the nonspecific PPA5530 probe (lane 2). Also, Fig. 5C shows that addition of the nonlabeled PgcvH2 probe led to depletion of the shift (lanes 3 to 5), but addition of the nonlabeled PPA5530 probe did not have any effect on the shift (lane 6). Taken together, these data suggest that the gcvH2 promoter region is indeed a target for GcsR.
FIG 5 .
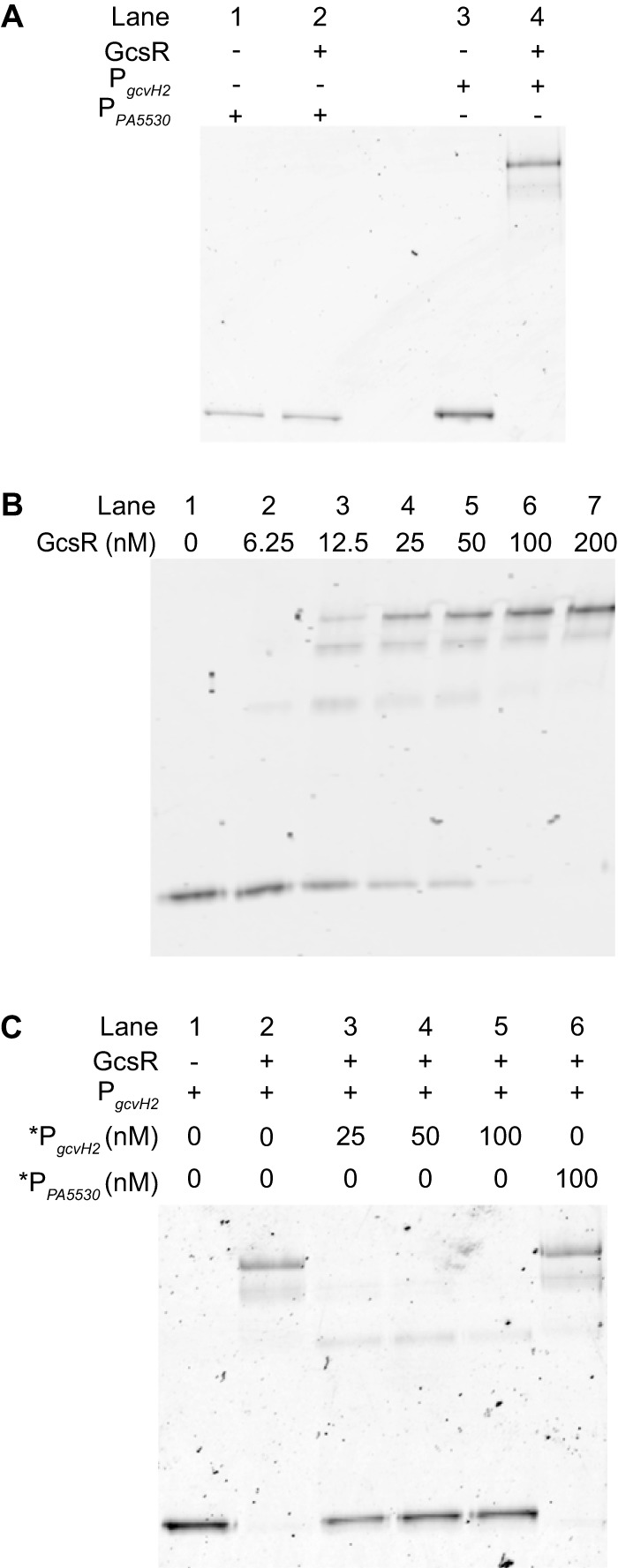
GcsR binds the gcvH2 promoter region. EMSAs were performed with His6-GcsR and 2 nM Cy5-labeled probe DNA unless specified otherwise. (A) PPA5530 (nonspecific) or PgcvH2 (specific) was incubated in the absence (lanes 1 and 3, respectively) or presence (lanes 2 and 4, respectively) of 200 nM His6-GcsR. (B) His6-GcsR (0 to 200 nM) was incubated with PgcvH2. (C) A 200 nM concentration of His6-GcsR was incubated with PgcvH2 and increasing concentrations of unlabeled specific competitor *PgcvH2 (lane 3 to 5) or 100 nM unlabeled nonspecific competitor *PPA5530 (lane 6).
The EMSAs revealed that the binding of His6-GcsR with the PgcvH2 probe resulted in shifts to three distinct positions (Fig. 5). This suggests that there are at least three binding sites for GcsR in the gcvH2 promoter region. This is consistent with the binding patterns of known EBPs. EBPs bind as dimers to tandem repeat sequences known as enhancer elements upstream of the RpoN binding site in the target promoter region (13). Typically, three sets of EBP dimers bind to the promoter region. Upon activation, EBPs form a hexameric ring that goes on to interact with RpoN to initiate transcription with the help of ATP hydrolysis. Since we observed three distinct shifts, we expected there to be three GcsR binding sites in the gcvH2 promoter. Sequence analysis revealed three 18-bp tandem repeat sequences in the 200 bp PgcvH2 probe (Fig. 6A). In order to investigate the effects of these sequences on GcsR binding, we mutated the first tandem repeat sequence. We found that the binding pattern changed from three shifts with the wild-type sequence to two with the mutated sequence (Fig. 6B). We then inserted mutations into each of the other two tandem repeat sequences separately. We found that GcsR was able to bind each site on its own but the binding was abolished when the individual sites were mutated (Fig. 6C). Taken together, these data indicate that this 18-bp tandem repeat sequence is indeed the binding site for GcsR.
FIG 6 .

GcsR binds to three 18-bp tandem repeats in the gcvH2 promoter region. EMSAs were performed with His6-GcsR and 2 nM Cy5-labeled probe DNA. (A) Sequence of the 217-bp region upstream of the gcvH2 gene. The three 18-bp GcsR binding sites are in bold type. The underlined nucleotides in the GcsR binding sites were mutated to G in this study. The RpoN binding site is in bold italic type. (B) A 200 nM concentration of His6-GcsR was incubated with PgcvH2 (lane 2) or PgcvH2-mut1 (lane 4). (C) PgcvH2-2, PgcvH2-2-mut, PgcvH2-3, and PgcvH2-3-mut were incubated in the absence (lanes 1, 3, 5, and 7, respectively) or presence (lanes 2, 3, 6, and 8, respectively) of 200 nM His6-GcsR. (D) Consensus sequence of the GcsR binding site derived from the three 18-bp tandem repeats in the PgcvH2 sequence constructed with WebLogo 3.4.
Using these three binding sites, we were able to identify the following 18-bp consensus site for GcsR binding: TGTAACG-N4-CGTTCCG (Fig. 6D). The binding site is composed of two 7-bp palindromic arms separated by four nucleotides. Upon comparison with the TyrR binding site TGTAAA-N6-TTTACA, we find that although the GcsR and TyrR binding sites are very similar, the GcsR binding site has an invariable CG pair at positions 6 and 7 and a CG or GG pair at positions 12 and 13 that is not present at the TyrR binding site.
In addition to the glycine metabolism genes, the RNA-Seq analysis revealed that 15 other genes were differentially expressed in ΔgcsR PAO1 (Table 1). However, none of these had a putative RpoN binding site or a putative GcsR binding site and therefore they are probably not direct targets of GcsR.
GcsR shows TyrR-like phosphatase activity.
The TyrR protein of E. coli exhibits phosphatase activity that is dependent on the presence of divalent cations (25). The role of this phosphatase activity in vivo has yet to be determined for TyrR. Nonetheless, we decided to determine if GcsR also possesses phosphatase activity in vitro. His6-GcsR was incubated with or without different divalent cations at 2 mM for 3 h at 37°C. Among the divalent cations tested, Zn2+ and Co2+ had the most significant effect on GcsR phosphatase activity (Fig. 7). The presence of glycine in the assay mixture did not have any effect on the phosphatase activity of GcsR.
FIG 7 .
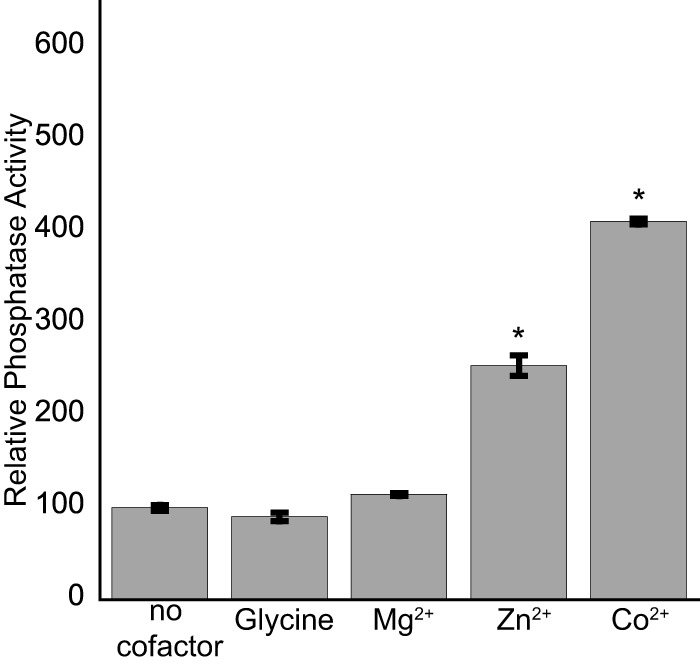
GcsR has divalent-cation-dependent phosphatase activity. A 5 µM sample of His6-GcsR was incubated with the substrate p-nitrophenylphosphate in the absence of any cofactor or in the presence of 2 mM glycine, 2 mM Mg2+, 2 mM Zn2+, or 2 mM Co2+. Data points represent mean values ± the standard deviations (n = 3). Analysis of variance was performed by using Dunnett’s post hoc test (α value of 0.05) to identify significant differences (P < 0.0001; marked with an asterisk).
GcsR is not essential for pyocyanin biosynthesis in P. aeruginosa PAO1.
Our previous study indicated that GcsR was crucial for the production of pyocyanin in P. aeruginosa PAO1 (12). Specifically, a gcsR transposon mutant (PW5126) was observed to be deficient in pyocyanin production. Plasmid-derived expression of the gcsR gene did restore pyocyanin production in PW5126, suggesting that pyocyanin biosynthesis was dependent on gcsR. However, the gcsR mutant generated in this study exhibited wild-type levels of pyocyanin production (Fig. 8A), contradicting these previous findings.
FIG 8 .
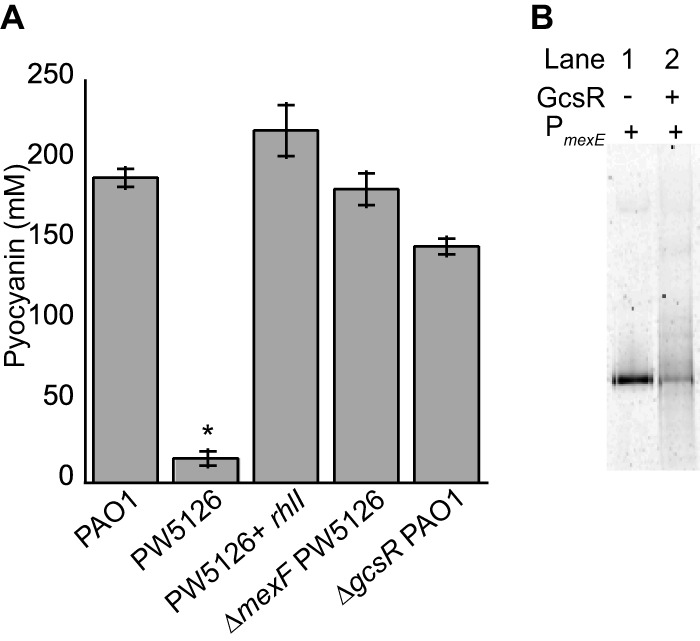
GcsR affects pyocyanin production through the MexEF-OprN efflux pump. (A) Overexpression of rhlI or deletion of the mexF gene inactivating the MexEF-OprN pump in the gcsR::Tn mutant strain PW5126 restores pyocyanin production in PW5126. The ΔgcsR PAO1 strain produces pyocyanin levels similar to those of PAO1. Data points represent mean values ± the standard deviations (n = 3). Analysis of variance was performed by using Dunnett’s post hoc test (α value of 0.05) to identify significant differences (P < 0.0001; marked with an asterisk). (B) The PmexE probe was incubated in the absence (lane 1) or presence (lane 2) of 200 nM His6-GcsR.
A reasonable explanation that might account for the discrepancies in pyocyanin production between the two gcsR mutants is the deregulation of quorum sensing-related genes in PW5126. Microarray analysis revealed that numerous quorum sensing genes were significantly downregulated in PW5126 (12). One of these genes was rhII, which encodes the N-butyryl homoserine lactone (C4-HSL) synthase (26, 27). Because C4-HSL positively regulates pyocyanin biosynthesis (28), the downregulation of rhlI and, consequently, the absence of C4-HSL would explain why the PW5126 strain was unable to produce pyocyanin. Consistent with this hypothesis, plasmid-derived expression of rhlI did rescue pyocyanin production in PW5126 (Fig. 8A).
In addition to downregulation of rhlI, the previous transcriptome analysis indicated that there was >100-fold upregulation of the mexEF and oprN genes in PW5126. The MexEF-OprN pump has been shown to be responsible for the efflux of biosynthetic precursors of 2-alkyl-4-quinolone, the Pseudomonas quinolone signal, which affects the expression of rhlI (29–31). Indeed, deletion of the mexF gene from PW5126 did restore pyocyanin production (Fig. 8A). This result suggests that upregulation of the mexEF and oprN genes from PW5126 was the likely cause of its pyocyanin deficiency. Unexpectedly, we found that GcsR did bind with low affinity to the promoter region of mexEF-oprN (Fig. 8B). This low-affinity binding might explain the previous observation that plasmid-derived expression of gcsR rescued pyocyanin biosynthesis in PW5126; i.e., low-affinity binding of GcsR to the PmexE promoter region antagonized the transcription of the mexEF and oprN genes.
HCN-induced killing of C. elegans by ΔgcsR PAO1.
One of the metabolic fates of glycine in P. aeruginosa is its conversion into HCN through the actions of an HCN synthase (HcnABC). HCN is a virulence factor and causes paralytic killing (HCN-mediated lethal paralysis) of C. elegans (32, 33). Since GcsR was necessary for the assimilation of glycine, we wanted to determine if GcsR has any impact on the paralytic-killing capabilities of P. aeruginosa PAO1.
After 2 h of exposure to ΔgcsR PAO1, ~60% of the worms were dead (Fig. 9A). Figure 9B and C show live, healthy, motile C. elegans on a lawn of PAO1 after 2 h of exposure. In contrast, Fig. 9D and E show worms on a lawn of ΔgcsR PAO1 after 2 h. The worms in Fig. 9 D and E are dead, as indicated by a lack of pharyngeal pumping. The worm have kinked or curved bodies, as dead C. elegans worms commonly do. In some worms, the nose was hypercontracted (data not shown), as previously described (32). C. elegans exposed to control strains P. aeruginosa lecA::luxΔlasR PAO1 and E. coli OP50 had motility and appearance similar to those of worms on wild-type P. aeruginosa PAO1 lawns (data not shown).
FIG 9 .
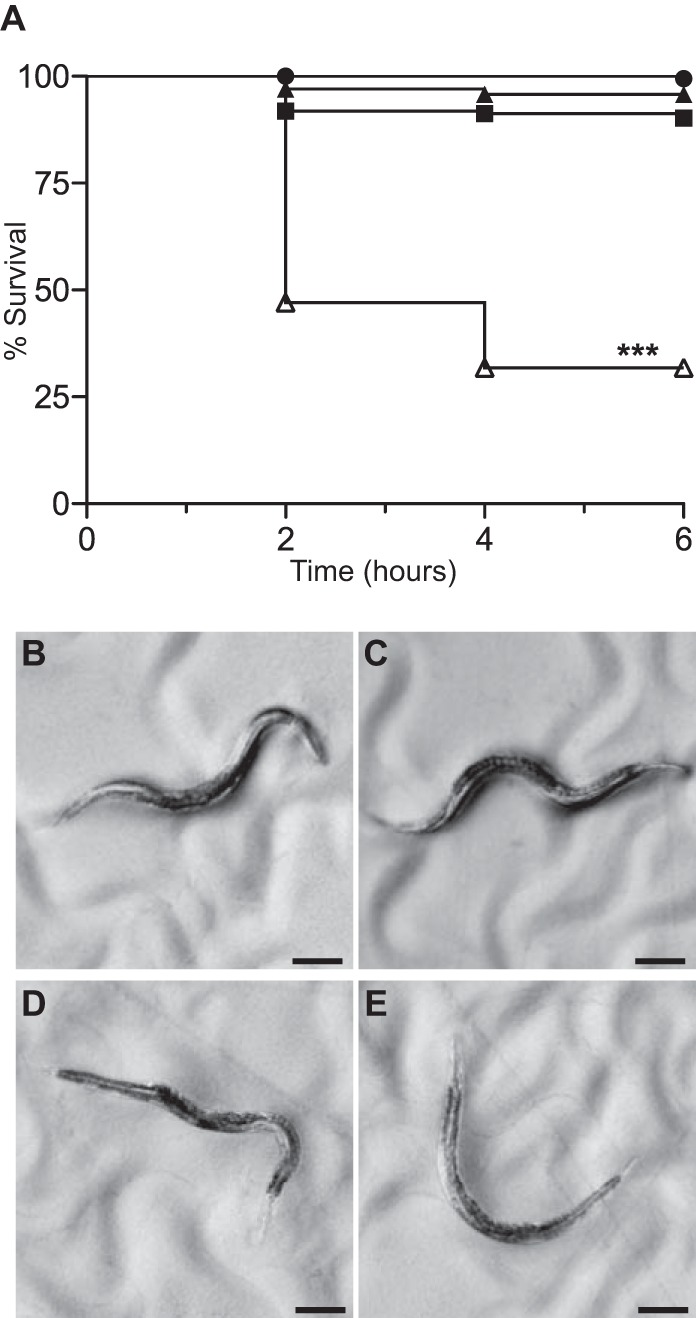
P. aeruginosa ΔgcsR PAO1 is lethal to C. elegans in a paralytic-killing model. (A) Course of C. elegans survival in a paralytic-killing assay with E. coli OP50 (closed circles; n = 195), P. aeruginosa lecA::luxΔlasR PAO1 (closed squares; n = 197), P. aeruginosa PAO1 (closed triangles; n = 273), and P. aeruginosa ΔgcsR PAO1 (open triangles; n = 351). C. elegans survival was significantly reduced when it was exposed to P. aeruginosa ΔgcsR PAO1. The log rank test was performed to identify significant differences (P < 0.0001; marked with asterisks). (B, C) Appearance of C. elegans after a 2-h exposure to P. aeruginosa PAO1. Scale bars are 100 µm. (D, E) Appearance of C. elegans after a 2-h exposure to P. aeruginosa ΔgcsR PAO1. Scale bars are 100 µm.
Despite the results of the paralytic-killing assays, RNA-Seq data (see Materials and Methods) showed no significant differences in the expression of the hcnABC genes in ΔgcsR PAO1 and EMSAs indicated that there was no binding of GcsR to the 5′ upstream regulatory region of hcnA (data not shown). This suggests that GcsR does not directly regulate HCN production. Since glycine catabolism is abolished in the GcsR mutant, we hypothesize that more glycine flux into the HCN production pathway is occurring, leading to increased HCN production and lethality.
DISCUSSION
TyrR family EBPs regulate the expression of genes involved in aromatic amino acid metabolism (14, 15, 19, 34). Many members of this family have been well characterized, including PhhR from P. aeruginosa PAO1 (16, 19). Interestingly, in addition to PhhR, P. aeruginosa PAO1 has another TyrR homolog (18). The second one is GcsR, and we have found that, unlike other previously characterized TyrR homologs, it regulates the expression of genes required for glycine metabolism.
The primary route of glycine catabolism is catalyzed by the GCS (4). P. aeruginosa PAO1 harbors two GCS gene clusters (18). We have found that the gcs2 gene cluster, comprising the gcvH2P2T2, glyA2, and sdaA genes, forms an operon and is essential for the metabolism of glycine as a sole carbon source. The best-characterized regulation of the expression of a bacterial gcs operon is that of E. coli, where its expression is regulated by the LysR-type transcriptional activator GcvA (22) and the repressor GcvR (23). Our work has revealed that in P. aeruginosa PAO1, a novel TyrR family EBP, GcsR, regulates the expression of this gcs2 operon and hence the metabolism of glycine. GcsR is the first characterized member of the TyrR family that regulates the metabolism of a nonaromatic amino acid.
GcsR has 41% sequence identity with E. coli TyrR. Sequence analysis shows that its domain architecture is identical to that of other TyrR homologs (15, 35): an N-terminal regulatory domain composed of an ACT and PAS domain, a central AAA+ ATPase and RpoN interaction domain, and a C-terminal DNA-binding domain. Like other TyrR family EBPs, GcsR responds to an amino acid effector (glycine). We found that GcsR upregulates the expression of the gcs2 operon in the presence of glycine. Another piece of evidence indicating that GcsR is a TyrR homolog is that it has divalent-cation-dependent phosphatase activity like E. coli TyrR (25). How GcsR binds to metals is unclear since its sequence does not have any recognizable metal-binding motifs such as stretches of multiple cysteine or histidine residues. Although the exact purpose of the phosphatase activity of TyrR or GcsR is not known, one possibility is that GcsR regulates expression from its target promoter by dephosphorylating other phosphorylated proteins. Further work is needed to better characterize the phosphatase activity of GcsR.
P. aeruginosa PAO1 is not the only pseudomonad with a genome that harbors a gcsR gene. We found that there are GcsR orthologs in a number of other sequenced genomes (Table 2). All of the genomes that harbor a GcsR ortholog belong to the order Pseudomonadales. Additionally, all of these GcsR orthologs are located adjacent to a glycine metabolism gene cluster (18) (Fig. 10A). This likely indicates a conserved mechanism for regulating glycine metabolism in members of the order Pseudomonadales.
TABLE 2 .
Members of the order Pseudomonadales harboring GcsR orthologs
| Bacterial strain | GcsR ortholog IDa |
|---|---|
| Azotobacter chroococcum NCIMB 8003 | AChR_RS10790 |
| Azotobacter vinelandii Avop | Avin_25940 |
| Pseudomonas alcaligenes OT 69 | L682_31660 |
| Pseudomonas amygdali 2250 | IC51_RS0115310 |
| Pseudomonas brassicacearum DF41 | CD58_RS22465 |
| Pseudomonas chlororaphis O6 | PchlO6_4751 |
| Pseudomonas cremoricolorata ND07 | LK03_RS10195 |
| Pseudomonas denitrificans 106_PDEN | ADM39_RS28200 |
| Pseudomonas entomophila L48 | PSEEN4432 |
| Pseudomonas fluorescens Pf0-1 | PFL01_4390 |
| Pseudomonas fragi A22 | O5G_RS0105995 |
| Pseudomonas fuscovaginae CB98818 | Y53_RS0126010 |
| Pseudomonas mandelii JR | OU5_RS24435 |
| Pseudomonas mendocina ymp | Pmen_1348 |
| Pseudomonas monteilii SB3078 | X969_03350 |
| Pseudomonas mosselii SJ10 | O165_RS17570 |
| Pseudomonas oleovorans MGY01 | GL31_RS19330 |
| Pseudomonas oryzihabitans RIT370 | UM91_RS16980 |
| Pseudomonas parafulva CRS01-1 | NJ69_RS01535 |
| Pseudomonas plecoglossicida NyZ12 | RK21_RS19015 |
| Pseudomonas protegens Pf-5 | PFL_4639 |
| Pseudomonas pseudoalcaligenes ad 6 | AU05_RS20595 |
| Pseudomonas psychrophila DSM 17535 | TU76_RS13130 |
| Pseudomonas putida KT2440 | PP_0997 |
| Pseudomonas resinovorans NBRC 106553 | PCA10_RS15640 |
| Pseudomonas savastanoi BO76 | PsgB076_20487 |
| Pseudomonas simiae WCS417 | PS417_RS06035 |
| Pseudomonas syringae pv. tomato DC3000 | PSPTO_1280 |
| Pseudomonas taiwanensis DSM 21245 | H620_RS0100750 |
| Pseudomonas thermotolerans DSM 14292 | H165_RS0110300 |
| Pseudomonas tolaasii PMS117 | PTOL117_RS0124240 |
| Pseudomonas umsongensis 20MFCvi1.1 | D470_RS0108450 |
| Pseudomonas veronii 1YB2 | Y055_RS30055 |
| Pseudomonas viridiflava CC1582 | N029_RS0112150 |
GcsR ortholog sequences are >80% identical to the P. aeruginosa PAO1 GcsR protein.
FIG 10 .
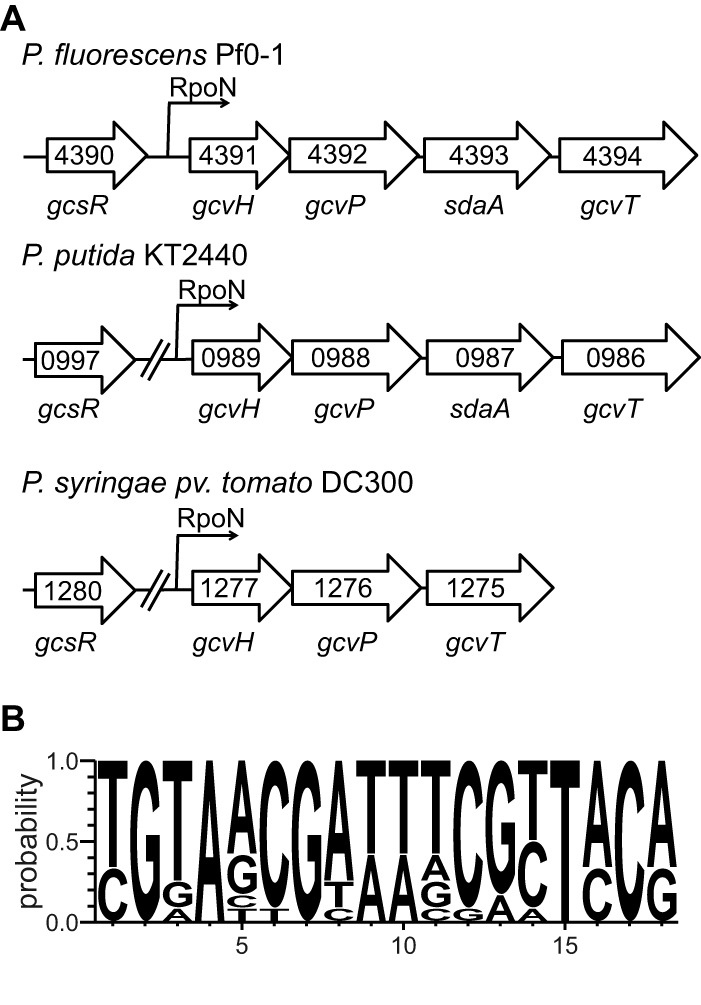
The P. fluorescens Pf0-1, P. putida KT2440, and P. syringae pv. tomato DC3000 genomes harbor gcsR orthologs. (A) Organization of the gcsR gene and the gcs gene cluster in P. fluorescens Pf0-1, P. putida KT2440, and P. syringae pv. tomato DC3000. (B) Consensus sequence of putative binding sites of GcsR orthologs from P. fluorescens Pf0-1, P. putida KT2440, and P. syringae pv. tomato DC3000 constructed with WebLogo 3.4.
The binding site of GcsR is an 18-bp tandem repeat sequence with two palindromic arms that is very similar to that of TyrR binding sites (Fig. 6D). We analyzed the genome sequences of P. putida KT2440, P. fluorescens Pf0-1, and P. syringae pv. tomato DC300 and identified binding sites for GcsR in the 5′ regulatory regions of the gcs gene clusters in these genomes (Fig. 10B). The σ54 Promoter Database (http://www.sigma54.ca/promoterdata/Web/data.aspx) indicates that there are putative RpoN binding sites upstream of the gcs cluster in all three strains. Additionally, RpoN has been shown to be required for glycine metabolism in P. putida KT2440 (36). Taken together, these data suggest that the regulation of glycine metabolism by GcsR and RpoN is likely a common mechanism among members of the order Pseudomonadales.
The P. aeruginosa genome harbors two homologs of the gcvHPT genes, three homologs of the glyA gene, and two homologs of the l-serine dehydratase genes, namely, sdaA and sdaB. However, this does not translate to redundancy in the function of the glycine metabolism genes. The glyA1 and sdaB genes have been shown to function in the glycine betaine catabolism pathway, and their expression is regulated by the AraC-like transcriptional activator GbdR (37, 38). Our work indicated that only the gcs2 operon, gcvH2-gcvP2-glyA2-sdaA-gcvT2, is required for the metabolism of glycine as the sole carbon source. We also found that the growth defect displayed by ΔgcsR PAO1 with glycine as the sole nitrogen source was much less pronounced than the growth defect with glycine as the sole carbon source. This suggests that GcsR and, hence, the GCS2 proteins are not essential for the metabolism of glycine as a nitrogen source. Both the GCS and GlyA have been shown to be important for the metabolism of glycine as a nitrogen source in E. coli (39). However, since GcvH2P2T2 or GlyA2 does not appear to be essential for the metabolism of glycine as a nitrogen source by P. aeruginosa PAO1, it is likely that the other gcv genes and perhaps the glyA3 gene are involved in that process. This is an excellent example of the diverse metabolic capacity of P. aeruginosa PAO1.
Glycine also acts as the precursor for the production of HCN gas by an HCN synthase in P. aeruginosa (6). P. aeruginosa is one of the few bacteria that produce HCN (40). HCN has been detected in patients with P. aeruginosa infection and is thought to contribute to the pathogenesis of P. aeruginosa in cystic fibrosis patients (41). Although how HCN production by P. aeruginosa contributes to its virulence in cystic fibrosis lungs is not yet clear, it has been shown that P. aeruginosa uses HCN for paralytic killing of the nematode C. elegans (33). We found that the level of C. elegans paralytic killing by ΔgcsR PAO1 was much greater than that by the parent P. aeruginosa PAO1 strain. Since the GCS is not functioning in the ΔgcsR PAO1 mutant, more glycine is available for conversion to HCN, which is likely responsible for the increased lethality of the ΔgcsR PAO1 strain.
We had previously found that, in addition to regulating glycine metabolism, GcsR also plays a role in pyocyanin production (12). Pyocyanin is one of several redox-active phenazines produced by P. aeruginosa that are used as virulence factors (42–47). Pyocyanin causes virulence by affecting gene expression, electron transport, cellular respiration, and innate immune mechanisms (42, 46, 47). We had previously found that pyocyanin production is reduced in the gcsR transposon mutant strain PW5126 (12). In this study, we found that this decrease in production of pyocyanin in PW5126 is due not to the direct regulation by GcsR of biosynthetic genes but to overexpression of the MexEF-OprN efflux pump. However, we found that this was not reproducible in the ΔgcsR PAO1 strain. When we tested the pyocyanin production of this strain, we found that it was not significantly reduced. Furthermore, in contrast to PW5126, RNA-Seq analysis of ΔgcsR PAO1 showed no significant change in mexEF-oprN expression. The parent strain of the gcsR transposon mutant strain PW5126 is the PAO1 strain from Colin Manoil’s lab at the University of Washington (48). Our ΔgcsR PAO1 strain is a derivative of a different PAO1 strain from the Dieter Haas lab at the University of Lausanne in Switzerland. It has been shown that in many PAO1 strains originating from different laboratories, mexEF-oprN is quiescent and uninducible because of mutations in several regulatory genes (49, 50). Recent studies have also shown differences in the genome sequences of different PAO1 strains originating from different laboratories (51). Additionally, unexpected mutations affecting the expression of mexEF-oprN have also been shown to alter the pyocyanin production of several PAO1 strains and a number of mutants from the PAO1 transposon library (20). Thus, one explanation for this discrepancy could be that the PW5126 strain had an inducible mexEF-oprN operon, whereas ΔgcsR PAO1 did not. The other possibility is that an adventitious mutation that allows the overexpression of mexEF-oprN could have occurred in PW5126 because of the transposon mutation. Further studies must be conducted with known mexEF-oprN-inducible strains to fully understand the effect of GcsR on mexEF-oprN.
Our RNA-Seq analysis of the ΔgcsR PAO1 strain revealed that 20 genes were differentially regulated (Table 1). In addition to the five gcs2 operon genes, these genes include genes for several tRNAs, an operon encoding genes for formaldehyde metabolism, and another operon involved in nitrogen metabolism. However, none of the latter 15 genes are likely to be direct targets of GcsR. There are two lines of evidence to suggest this. First, we could not identify a putative GcsR binding site in the 5′ regulatory region of any of these genes. Second, 14 out of these 15 genes do not have an RpoN binding site in their promoter region. The only gene with an RpoN binding site in its promoter region is dctA, which has been previously shown to be regulated by the DctD EBP (52). This, along with the fact that the dctA promoter region lacks a GcsR binding site, indicates that dctA is likely not a direct target of GcsR. All of these genes function in pathways that directly or indirectly depend on products from the glycine metabolism pathway. This could possibly explain why disruption of glycine metabolism likely causes changes in their expression patterns.
In conclusion, our data suggest that GcsR is the first characterized member of a novel family of TyrR-like EBPs that regulate genes required for glycine metabolism. Unlike E. coli, which uses a LysR-type regulator to regulate glycine metabolism, GcsR also presents a novel mechanism of regulation of glycine metabolism involving the alternative σ factor RpoN. Future work will determine how GcsR interacts with glycine to regulate the expression of its target genes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains, plasmids, and oligonucleotides used in this study are listed in Table 3. Bacteria were grown in Difco Lennox broth (LB), peptone broth (PB) (12), modified M9 minimal medium supplemented with 5 µM FeSO4 (12), 2×YT medium (53), or brain heart infusion (BHI) broth (RPI). Solid bacteriological medium was prepared with the addition of BD agar at 15 g liter−1, unless otherwise specified. Liquid cultures were grown at 37°C with shaking at 200 rpm, and cultures on solid plates were grown by incubation at 37°C. When required, the antibiotics carbenicillin (Cb; 100 µg·ml−1 for E. coli or 200 µg·ml−1 for P. aeruginosa), kanamycin (Km; 50 µg·ml−1 for E. coli) or gentamicin (Gm; 10 µg·ml−1 for E. coli or 30 µg·ml−1 for P. aeruginosa) were added to culture medium to maintain plasmids.
TABLE 3 .
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide |
Features | Source |
|---|---|---|
| P. aeruginosa | ||
| PAO1 | Wild type | 61 |
| ΔgcsR PAO1 | ΔgcsR derivative of PAO1 | This study |
| PW5126 | gcsR-E03::ISphoA/hah | 48 |
| ΔmexF PW5126 | ΔmexF derivative of PW5126 | This study |
| lecA::luxΔlasR PAO1 | ΔlasR derivative of lecA::lux PAO1 | 62 |
| E. coli | ||
| JW2779-5 | ΔgcvA736::Kmr | 63 |
| JW2464-1 | ΔgcvR727::Kmr | 63 |
| BL21 | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS | EMD Millipore |
| Top10 | F−
mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15ΔlacX74 nupG recA1araD139
Δ(ara-leu)7697 galE15 galK16 rpsL (Strr) endA1 λ− |
Invitrogen |
| OP50 | Strain used for C. elegans growth | 59 |
| Plasmids | ||
| pBBR1MCS-5 | Broad-host-range plasmid; Gmr | 64 |
| ΔPlac-pBBR1MCS-5 | pBBR1MCS-5 minus lac promoter; Gmr | 12 |
| pCR-Blunt | Cloning plasmid; Kmr | Invitrogen |
| pET28b | Expression plasmid; Kmr | EMD Millipore |
| pTrc99a | Expression plasmid; Cbr | Pharmacia |
| pDONR221 | Cloning plasmid; Kmr | Invitrogen |
| pEX18ApGW | Plasmid for gene deletions in P. aeruginosa; Cbr | 54 |
| pBRL408 | gcsR in pCR-Blunt; Kmr | This study |
| pBRL417 | gcsR in pET28b; Kmr | 12 |
| pBRL620-3 | gcsR cloned in backwards in pTrc99a; Cbr | This study |
| pBRL620-5 | gcsR cloned in forwards in pTrc99a; Cbr | This study |
| pBRL456 | PgcvH2-lacZ fusion in ΔPlac-pBBR1MCS-5; Gmr | 12 |
| pBRL518 | mexF::Gmr in pDONR221; Gmr Kmr | This study |
| pBRL521 | mexF::Gmr in pEX18ApGW; Cbr Gmr | This study |
| pBRL527 | gcsR::Gmr in pDONR221; Gmr Kmr | This study |
| pBRL528 | gcsR::Gmr in pEX18ApGW; Cbr Gmr | This study |
| pMTG01 | rhlI in pCR-Blunt; Kmr | This study |
| pMTG02 | rhlI in pBBR1MCS-5; Gmr | This study |
| pZS406 | PgcvH2 probe in pJET1.2; Cbr | 12 |
| pZS416 | PPA5530 probe in pJET1.2; Cbr | This study |
| pZS420 | PgcvH2-mut1 probe in pJET1.2; Cbr | This study |
| Oligonucleotides | ||
| BL439.f | GCATCTAGAAGAAGGAGACATATGATCGAATTGCTCTCTGAATCG | rhlI gene with XbaI at start |
| BL439.r | GCAGAGCTCTTCACACCGCCATCGACAGC | rhII gene with SacI at stop |
| BL452.f | TACAAAAAAGCAGGCTGCATCCACGTCTCCTTCATC | gcsR UpF-GWL |
| BL452.r | TCAGAGCGCTTTTGAAGCTAATTCGCAACGCATCGAGCTGCAAG | gcsR UpR-Gm |
| BL453.f | AGGAACTTCAAGATCCCCAATTCGCACTTCATGCAGCAGGCCTG | gcsR DnF-Gm |
| BL453.r | TACAAGAAAGCTGGGTCTGACGTAGAGCTTCTCCAG | gcsR DnR-GWR |
| BL454.f | TACAAAAAAGCAGGCTGAATTTCTCCCAATTCTTCATCC | mexF UpF-GWL primer |
| BL454.r | TCAGAGCGCTTTTGAAGCTAATTCGGAAGGTGATGGTCAGGGTC | mexF UpR-Gm |
| BL455.f | AGGAACTTCAAGATCCCCAATTCGGCAAGAACGCGATCCTGATC | mexF DnF-Gm |
| BL455.r | TACAAGAAAGCTGGGTCATGCATGCACCTCTGGCAG | mexF DnR-GWR |
| BL456.f | GGCTCGAGTTTTTCAGCAAGAT | pJET1.2 |
| BL456.r | GAATATTGTAGGAGATCTTCTAGAAAG | pJET1.2 |
| JRH05.f | GGCTCGAGTTTTTCAGCAAGAT | 5′ Cy5-labeled pJET1.2 |
| JRH05.r | GAATATTGTAGGAGATCTTCTAGAAAG | 5′ Cy5-labeled pJET1.2 |
| ZS420F | CGTTCCACTCGTCCCCGAAGCCCCACAGCCGATGGCACATCTTGCTG | PgcvH2-mut1 |
| ZS420R | CAGCAAGATGTGCCATCGGCTGTGGGGCTTCGGGGACGAGTGGAACG | PgcvH2-mut1 |
| ZS426F | GGCTCGAGTTTTTCAGCAAGATGAGTGGAACGAATTCGTTCCGATTGCAGGAAAAACCTT | PgcvH2-2 |
| ZS426R | TTGTAGGAGATCTTCTAGAAAGAAGGTTTTTCCTGCAATCGGAACGAATTCGTTCCACTC | PgcvH2-2 |
| ZS422F | GGCTCGAGTTTTTCAGCAAGATGAGTGGGGCGAATTCGGGCCGATTGCAGGAAAAACCTT | PgcvH2-2-mut |
| ZS422R | TTGTAGGAGATCTTCTAGAAAGAAGGTTTTTCCTGCAATCGGCCCGAATTCGCCCCACTC | PgcvH2-2-mut |
| ZS431F | GGCTCGAGTTTTTCAGCAAGATGGCGGGTCTGTAACGAAAGCGTTCCATGGCGGCCCTG | PgcvH2-3 |
| ZS431R | TTGTAGGAGATCTTCTAGAAAGCAGGGCCGCCATGGAACGCTTTCGTTACAGACCCGCC | PgcvH2-3 |
| ZS425F | GGCTCGAGTTTTTCAGCAAGATGGCGGGTCTGTGGCGAAAGCGCCCCATGGCGGCCCTG | PgcvH2-3-mut |
| ZS425R | TTGTAGGAGATCTTCTAGAAAGCAGGGCCGCCATGGGGCGCTTTCGCCACAGACCCGCC | PgcvH2-3-mut |
| ZS416F | CACTGAGAGCCAAGCGATC | PPA5530 |
| ZS416R | CGCGGGAAAGCCGCGTGGGGAC | PPA5530 |
| ZS448F | ACGCCGCCGGCAGTGAAGTG | gcvH2-gcvP2 RT probe |
| ZS448R | TTGCGCGCGGCGATCGCC | gcvH2-gcvP2 RT probe |
| ZS449F | CCGCCGCCGAACTGCTCG | gcvP2-glyA2 RT probe |
| ZS449R | GGATAGCCCTCGGCATACTTG | gcvP2-glyA2 RT probe |
| ZS454F | CAGGGGCTGACCGGCAAG | glyA2-sdaA RT probe |
| ZS454R | GCTGGAACGGATGGCATCG | glyA2-sdaA RT probe |
| ZS451F | GTCCAGGTGCCCTGCATCG | sdaA-gcvT2 RT probe |
| ZS451R | GCCACATCGGCGCCCACC | sdaA-gcvT2 RT probe |
| ZS452F | CAAGGTCGAGAAACTCGACGTCG | rplU RT probe |
| ZS452R | GACGCTTCATGTGGTGCTTACGAC | rplU RT probe |
Construction of P. aeruginosa PAO1 deletion mutant strains.
The ΔgcsR PAO1 and ΔmexF PW5126 strains were constructed by previously described methods (24, 54) Briefly, the gcsR::Gm and mexF::Gm cassettes were cloned into pEX18ApGW to give plasmids pBRL528 and pBRL521, respectively. The gcsR gene was deleted from PAO1 with the pBRL528 plasmid, and the mexF gene was deleted from PW5126 with the pBRL521 plasmid. The Gm markers were removed with the pFLP2 plasmid. The ΔgcsR and ΔmexF mutations were verified by PCR.
Preparation of RNA.
RNA of P. aeruginosa PAO1 and ΔgcsR PAO1 were isolated and purified as described previously (12). To isolate RNA for RNA-Seq, each strain was grown in quadruplicate in 50 ml of PB in 500-ml baffled shake flasks at 37°C and 200 rpm to an OD600 of ~0.5. To isolate RNA for RT analysis of operon organization, each strain was grown in quadruplicate in 50 ml of M9 minimal medium supplemented with 20 mM glycine as the sole carbon source in 500-ml baffled shake flasks at 37°C and 200 rpm to an OD600 of ~0.25. Cultures were immediately stabilized by adding 1 ml of RNAprotect Bacteria reagent (Qiagen) to 0.5 ml of culture. Cells were then lysed with lysozyme and proteinase K as described in the manufacturer’s protocol. The total RNA was subsequently purified from the lysed cells with the RNeasy minikit (Qiagen) by using an on-column DNase digestion step. PCR and a Bioanalyzer were used to check the RNA for DNA contamination and quality.
RNA-Seq.
The purified total RNA was sent to the Molecular Analysis Core at SUNY Upstate Medical University for mRNA isolation, cDNA library preparation, and RNA sequencing with the Illumina NextSeq 500 system. CLC Genomics workbench 8.5 (Qiagen) was used to map the sequencing reads to the P. aeruginosa PAO1 genome and to obtain differential gene expression data (a ≥2-fold change in the number of reads per kilobase of transcript per million mapped reads and a Bonferroni-corrected P value of ≤0.05) (55, 56).
Analysis of operon organization of glycine metabolism genes.
In order to map the operon organization of the gcs2 locus, RT analysis of isolated RNA was used. An RT reaction with 500 ng of each RNA sample was set up with the iScript cDNA synthesis kit (Bio-Rad) to obtain cDNA. Primers ZS448F/ZS448R, ZS449F/ZS449R, ZS454F/ZS454R, and ZS451F/ZS451R were designed to amplify 500- to 600-bp regions spanning from the 3′ end of gcvH2 to the 5′ end of gcvP2, from the 3′ end of gcvp2 to the 5′ end of glyA2, from the 3′ end of glyA2 to the 5′ end of sdaA, and from the 3′ end of sdaA to the 5′ end of gcvT2, respectively. Primers ZS452F/ZS452R were designed to amplify the 50S ribosomal protein L21 gene rplU as a positive control (57). PCR analyses of the cDNA samples were subsequently performed with these primers. PCR analyses of the RNA samples and genomic DNA samples were performed as negative and positive controls, respectively. The PCR products obtained were analyzed by agarose gel electrophoresis.
LacZ reporter assays.
Recombinant E. coli strains were grown in M9 minimal medium supplemented with glycine, glutamine, or serine at a 20 mM concentration. Each culture was grown in triplicate. Cells were grown to an OD600 of 0.3. β-Galactosidase (LacZ) activity was measured with the Miller assay as described previously (12).
Heterologous expression and purification of GcsR.
GcsR was expressed and purified as an N-terminally six-histidine-tagged fusion protein (His6-GcsR) from plasmid pBRL417 harboring the gcsR gene using E. coli BL21(DE3) as described previously (53). Cells were grown in 2×YT medium to an OD600 of ~0.6 at 37°C with shaking at 200 rpm. Protein expression was induced by the addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside, and cell cultures were incubated for 12 h at 16°C with shaking at 200 rpm. Cells were harvested, resuspended in lysis buffer (100 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol, 1 mg/ml lysozyme, 5 U/ml DNase I, 1 µg/ml pepstatin, 1 µg/ml leupeptin), and lysed by three 30-s sonication bursts. The lysate was centrifuged at 9,000 × g to remove insoluble material, and the His6-GcsR was then purified with Ni-nitrilotriacetic acid Superflow resin (Qiagen). The protein was eluted off the resin by a step elution method with elution buffer (100 mM Tris [pH 8.0], 300 mM NaCl) containing 20, 100, and 250 mM imidazole. Purified protein was concentrated with Amicon Ultra centrifugal filter units (Millipore). Protein expression and purification were monitored visually by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE). The concentration of purified protein was determined with the Bradford assay.
EMSAs.
DNA fragments containing the promoter region of the gcvH2 gene were amplified by PCR and cloned into the pJET1.2 vector. The mutated promoter regions were obtained with a quick-change site-directed mutagenesis kit (Qiagen). Cy5-labeled primers JRH05.f/JRH05.r were used to PCR amplify the probes from the resulting plasmids, yielding the Cy5-labeled probes used in subsequent EMSAs.
The binding of GcsR to PgcvH2 was investigated by EMSA (58). A 200 nM sample of His6-GcsR was incubated with 2 nM Cy5-labeled PgcvH2 probe (specific probe) or 2 nM Cy5-labeled PPA5530 probe (nonspecific probe) in EMSA buffer (25 mM Tris-acetate [pH 8.0], 8 mM magnesium acetate, 10 mM potassium chloride, 1 mM dithiothreitol) for 30 min at 30°C. For EMSA reactions to examine the binding affinity of His6-GcsR for PgcvH2, 2.0 nM 5′-labeled Cy5 PgcvH2 probe was incubated with 0, 6.25, 12.5, 25, 50, 100, or 200 nM His6-GcsR. For EMSA reactions to determine the binding specificity of His6-GcsR for PgcvH2, a 200 nM sample of His6-GcsR was incubated with 2.0 nM 5′-labeled Cy5 PgcvH2 probe in the presence of 0, 25, 50, or 100 nM unlabeled PgcvH2 probe (specific competitor probe) or 100 nM unlabeled PPA5530 probe (nonspecific competitor probe). For EMSA reactions to determine the binding site of GcsR on the gcvH2 promoter, a 200 nM sample of His6-GcsR was incubated with 2 nM each probe containing either a wild-type or a mutated binding site. For EMSA reactions to determine the binding of GcsR to the mexE promoter region, a 200 nM sample of His6-GcsR was incubated with 2 nM probe PmexE. The samples were then analyzed by PAGE under nondenaturing conditions and imaged with a Typhoon imager.
Phosphatase assay.
Phosphatase activity of His6GcsR was measured as described previously for E. coli TyrR (25). Briefly, a 5 µM sample of His6-GcsR was incubated at 37°C for 3 h in phosphatase buffer (100 mM p-nitrophenylphosphate, 100 mM HEPES [pH 6.5]) in the presence of 2 mM glycine, 2 mM MgCl2, 2 mM ZnCl2, and 2 mM CoCl2 or in the absence of any divalent salts or glycine. Reactions were stopped by the addition of 1 ml of 1 M Na2CO3. The absorbance at 410 nm of the samples was measured with a Genesys 20 spectrophotometer to calculate the amount of substrate dephosphorylated.
Spectroscopic measurement of extracellular pyocyanin
Experiments were done in triplicate. Cells were grown in 2 ml of PB at 37°C and 200 rpm for 24 h. Pyocyanin was measured as described previously (12). Briefly, cells were removed from the culture by centrifugation at 16,000 × g for 5 min and passage through Acrodisc syringe filters with 0.2-µm nylon membranes. The absorbance at 690 nm of the cell-free samples was measured with a Genesys 20 spectrophotometer and converted to the pyocyanin concentration by using a molar extinction coefficient of 4,130 M−1 cm−1.
Paralytic-killing assay.
The C. elegans Bristol (N2) strain was cultivated under standard conditions (59). From synchronized populations of worms, hermaphroditic gravid adults were transferred to fresh nematode growth medium agar on a lawn of E. coli for 2 to 4 h to lay eggs and then removed. Eggs were incubated at 20°C for 3 days to the young adult stage (60).
Bacteria were grown overnight in BHI broth at 37°C with shaking. Cultures were diluted 1:100 in BHI broth, and 170-µl volumes were spread on BHI agar plates (1.7% Bacto agar, 60-mm petri plates) and then incubated at 37°C for 24 h (32). Synchronized worms were transferred to prepared BHI plates and incubated at room temperature for 6 h. Worms were examined for paralysis and scored for survival every 2 h with a stereomicroscope (×4.5 magnification). Worm paralysis was defined as no movement after mechanical stimulation, and death was defined as cessation of pharyngeal pumping. Data were analyzed with GraphPad Prism (GraphPad Software Inc.). Differences were considered significant when the P value was ≤0.05.
Microarray data accession number.
RNA-Seq data were posted to the Gene Expression Omnibus (GEO) under accession number GSE76522.
ACKNOWLEDGMENTS
We acknowledge support from the following for this study: NIH 2R15GM104880, NSF CBET 1263905, the SUNY ESF Honors Program, and the SUNY ESF Center for Applied Microbiology.
REFERENCES
- 1.Kay WW, Gronlund AF. 1969. Amino acid pool formation in Pseudomonas aeruginosa. J Bacteriol 97:282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kay WW, Gronlund AF. 1969. Influence of carbon or nitrogen starvation on amino acid transport in Pseudomonas aeruginosa. J Bacteriol 100:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutton WB, Schlenk F, Werkman CH. 1951. Glycine as a precursor of bacterial purines. Arch Biochem Biophys 32:85–88. doi: 10.1016/0003-9861(51)90240-8. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi G. 1973. The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem 1:169–187. doi: 10.1007/BF01659328. [DOI] [PubMed] [Google Scholar]
- 5.Kaback HR, Stadtman ER. 1968. Glycine uptake in Escherichia coli. II. Glycine uptake, exchange, and metabolism by an isolated membrane preparation. J Biol Chem 243:1390–1400. [PubMed] [Google Scholar]
- 6.Castric PA. 1977. Glycine metabolism by Pseudomonas aeruginosa: hydrogen cyanide biosynthesis. J Bacteriol 130:826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. 2008. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Phys Biol Sci 84:246–263. doi: 10.2183/pjab.84.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakley RL. 1955. The interconversion of serine and glycine: participation of pyridoxal phosphate. Biochem J 61:315–323. doi: 10.1042/bj0610315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schirch V, Hopkins S, Villar E, Angelaccio S. 1985. Serine hydroxymethyltransferase from Escherichia coli: purification and properties. J Bacteriol 163:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su H, Moniakis J, Newman EB. 1993. Use of gene fusions of the structural gene sdaA to purify l-serine deaminase 1 from Escherichia coli K-12. Eur J Biochem 211:521–527. doi: 10.1111/j.1432-1033.1993.tb17578.x. [DOI] [PubMed] [Google Scholar]
- 11.Su HS, Lang BF, Newman EB. 1989. l-Serine degradation in Escherichia coli K-12: cloning and sequencing of the sdaA gene. J Bacteriol 171:5095–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundgren BR, Thornton W, Dornan MH, Villegas-Peñaranda LR, Boddy CN, Nomura CT. 2013. Gene PA2449 is essential for glycine metabolism and pyocyanin biosynthesis in Pseudomonas aeruginosa PAO1. J Bacteriol 195:2087–2100. doi: 10.1128/JB.02205-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bush M, Dixon R. 2012. The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol Mol Biol Rev 76:497–529. doi: 10.1128/MMBR.00006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panina EM, Vitreschak AG, Mironov AA, Gelfand MS. 2001. Regulation of aromatic amino acid biosynthesis in gamma-proteobacteria. J Mol Microbiol Biotechnol 3:529–543. [PubMed] [Google Scholar]
- 15.Pittard J, Camakaris H, Yang J. 2005. The TyrR regulon. Mol Microbiol 55:16–26. doi: 10.1111/j.1365-2958.2004.04385.x. [DOI] [PubMed] [Google Scholar]
- 16.Song J, Jensen RA. 1996. PhhR, a divergently transcribed activator of the phenylalanine hydroxylase gene cluster of Pseudomonas aeruginosa. Mol Microbiol 22:497–507. doi: 10.1046/j.1365-2958.1996.00131.x. [DOI] [PubMed] [Google Scholar]
- 17.Herrera MC, Duque E, Rodríguez-Herva JJ, Fernández-Escamilla AM, Ramos JL. 2010. Identification and characterization of the PhhR regulon in Pseudomonas putida. Environ Microbiol 12:1427–1438. doi: 10.1111/j.1462-2920.2009.02124.x. [DOI] [PubMed] [Google Scholar]
- 18.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer GC, Palmer KL, Jorth PA, Whiteley M. 2010. Characterization of the Pseudomonas aeruginosa transcriptional response to phenylalanine and tyrosine. J Bacteriol 192:2722–2728. doi: 10.1128/JB.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luong PM, Shogan BD, Zaborin A, Belogortseva N, Shrout JD, Zaborina O, Alverdy JC. 2014. Emergence of the P2 phenotype in Pseudomonas aeruginosa PAO1 strains involves various mutations in mexT or mexF. J Bacteriol 196:504–513. doi: 10.1128/JB.01050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Novick RP. 2007. svrA, a multi-drug exporter, does not control agr. Microbiology 153:1604–1608. doi: 10.1099/mic.0.2007/006247-0. [DOI] [PubMed] [Google Scholar]
- 22.Wilson RL, Urbanowski ML, Stauffer GV. 1995. DNA binding sites of the LysR-type regulator GcvA in the gcv and gcvA control regions of Escherichia coli. J Bacteriol 177:4940–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heil G, Stauffer LT, Stauffer GV. 2002. Glycine binds the transcriptional accessory protein GcvR to disrupt a GcvA/GcvR interaction and allow GcvA-mediated activation of the Escherichia coli gcvTHP operon. Microbiology 148:2203–2214. doi: 10.1099/00221287-148-7-2203. [DOI] [PubMed] [Google Scholar]
- 24.Lundgren BR, Villegas-Peñaranda LR, Harris JR, Mottern AM, Dunn DM, Boddy CN, Nomura CT. 2014. Genetic analysis of the assimilation of C5-dicarboxylic acids in Pseudomonas aeruginosa PAO1. J Bacteriol 196:2543–2551. doi: 10.1128/JB.01615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao S, Zhu Q, Somerville RL. 2000. The σ70 transcription factor TyrR has zinc-stimulated phosphatase activity that is inhibited by ATP and tyrosine. J Bacteriol 182:1053–1061. doi: 10.1128/JB.182.4.1053-1061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parsek MR, Val DL, Hanzelka BL, Cronan JE, Greenberg EP. 1999. Acyl homoserine-lactone quorum-sensing signal generation. Proc Natl Acad Sci U S A 96:4360–4365. doi: 10.1073/pnas.96.8.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson JP, Passador L, Iglewski BH, Greenberg EP. 1995. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 92:1490–1494. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whiteley M, Lee KM, Greenberg EP. 1999. Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:13904–13909. doi: 10.1073/pnas.96.24.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamarche MG, Déziel E. 2011. MexEF-OprN efflux pump exports the Pseudomonas quinolone signal (PQS) precursor HHQ (4-hydroxy-2-heptylquinoline). PLoS One 6:e24310. doi: 10.1371/journal.pone.0024310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Köhler T, van Delden C, Curty LK, Hamzehpour MM, Pechere JC. 2001. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J Bacteriol 183:5213–5222. doi: 10.1128/JB.183.18.5213-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKnight SL, Iglewski BH, Pesci EC, Iglewski BH. 2000. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 182:2702–2708. doi: 10.1128/JB.182.10.2702-2708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darby C, Cosma CL, Thomas JH, Manoil C. 1999. Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:15202–15207. doi: 10.1073/pnas.96.26.15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallagher LA, Manoil C. 2001. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183:6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrera MC, Duque E, Rodríguez-Herva JJ, Fernández-Escamilla AM, Ramos JL. 2010. Identification and characterization of the PhhR regulon in Pseudomonas putida. Environ Microbiol 12:1427–1438. doi: 10.1111/j.1462-2920.2009.02124.x. [DOI] [PubMed] [Google Scholar]
- 35.Verger D, Carr PD, Kwok T, Ollis DL. 2007. Crystal structure of the N-terminal domain of the TyrR transcription factor responsible for gene regulation of aromatic amino acid biosynthesis and transport in Escherichia coli K12. J Mol Biol 367:102–112. doi: 10.1016/j.jmb.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 36.Köhler T, Harayama S, Ramos JL, Timmis KN. 1989. Involvement of Pseudomonas putida RpoN σ factor in regulation of various metabolic functions. J Bacteriol 171:4326–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hampel KJ, LaBauve AE, Meadows JA, Fitzsimmons LF, Nock AM, Wargo MJ. 2014. Characterization of the GbdR regulon in Pseudomonas aeruginosa. J Bacteriol 196:7–15. doi: 10.1128/JB.01055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wargo MJ, Szwergold BS, Hogan DA. 2008. Identification of two gene clusters and a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine catabolism. J Bacteriol 190:2690–2699. doi: 10.1128/JB.01393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newman E, Batist G, Fraser J, Isenberg S, Weyman P, Kapoor V. 1976. The use of glycine as nitrogen source by Escherichia coli K12. Biochim Biophys Acta 421:97–105. doi: 10.1016/0304-4165(76)90173-2. [DOI] [PubMed] [Google Scholar]
- 40.Blumer C, Haas D. 2000. Mechanism, regulation, and ecological role of bacterial cyanide biosynthesis. Arch Microbiol 173:170–177. doi: 10.1007/s002039900127. [DOI] [PubMed] [Google Scholar]
- 41.Anderson RD, Roddam LF, Bettiol S, Sanderson K, Reid DW. 2010. Biosignificance of bacterial cyanogenesis in the CF lung. J Cyst Fibros 9:158–164. doi: 10.1016/j.jcf.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Lau GW, Hassett DJ, Ran H, Kong F. 2004. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med 10:599–606. doi: 10.1016/j.molmed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Jayaseelan S, Ramaswamy D, Dharmaraj S. 2014. Pyocyanin: production, applications, challenges and new insights. World J Microbiol Biotechnol 30:1159–1168. doi: 10.1007/s11274-013-1552-5. [DOI] [PubMed] [Google Scholar]
- 44.Morales DK, Grahl N, Okegbe C, Dietrich LE, Jacobs NJ, Hogan DA. 2013. Control of Candida albicans metabolism and biofilm formation by Pseudomonas aeruginosa phenazines. mBio 4:e00526–e00512. doi: 10.1128/mBio.00526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ortiz-Castro R, Pelagio-Flores R, Méndez-Bravo A, Ruiz-Herrera LF, Campos-García J, López-Bucio J. 2014. Pyocyanin, a virulence factor produced by Pseudomonas aeruginosa, alters root development through reactive oxygen species and ethylene signaling in Arabidopsis. Mol Plant Microbe Interact 27:364–378. doi: 10.1094/MPMI-08-13-0219-R. [DOI] [PubMed] [Google Scholar]
- 46.Caldwell CC, Chen Y, Goetzmann HS, Hao Y, Borchers MT, Hassett DJ, Young LR, Mavrodi D, Thomashow L, Lau GW. 2009. Pseudomonas aeruginosa exotoxin pyocyanin causes cystic fibrosis airway pathogenesis. Am J Pathol 175:2473–2488. doi: 10.2353/ajpath.2009.090166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rada B, Leto TL. 2013. Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol 21:73–81. doi: 10.1016/j.tim.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maseda H, Saito K, Nakajima A, Nakae T. 2000. Variation of the mexT gene, a regulator of the MexEF-OprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol Lett 192:107–112. [DOI] [PubMed] [Google Scholar]
- 50.Sobel ML, Neshat S, Poole K. 2005. Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical strain of Pseudomonas aeruginosa. J Bacteriol 187:1246–1253. doi: 10.1128/JB.187.4.1246-1253.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F, Larbig KD, Heeb S, Schöck U, Pohl TM, Wiehlmann L, Tümmler B. 2010. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol 192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valentini M, Storelli N, Lapouge K. 2011. Identification of C(4)-dicarboxylate transport systems in Pseudomonas aeruginosa PAO1. J Bacteriol 193:4307–4316. doi: 10.1128/JB.05074-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarwar Z, Garza AG. 2012. The Nla28S/Nla28 two-component signal transduction system regulates sporulation in Myxococcus xanthus. J Bacteriol 194:4698–4708. doi: 10.1128/JB.00225-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heussler GE, Cady KC, Koeppen K, Bhuju S, Stanton BA, O’Toole GA. 2015. Clustered regularly interspaced short palindromic repeat-dependent, biofilm-specific death of Pseudomonas aeruginosa mediated by increased expression of phage-related genes. mBio 6:e00129-15. doi: 10.1128/mBio.00129-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Balasubramanian D, Kumari H, Jaric M, Fernandez M, Turner KH, Dove SL, Narasimhan G, Lory S, Mathee K. 2014. Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res 42:979–998. doi: 10.1093/nar/gkt942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacEachran DP, Ye S, Bomberger JM, Hogan DA, Swiatecka-Urban A, Stanton BA, O’Toole GA. 2007. The Pseudomonas aeruginosa secreted protein PA2934 decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect Immun 75:3902–3912. doi: 10.1128/IAI.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lundgren BR, Harris JR, Sarwar Z, Scheel RA, Nomura CT. 2015. The metabolism of (R)-3-hydroxybutyrate is regulated by the enhancer-binding protein PA2005 and the alternative sigma factor RpoN in Pseudomonas aeruginosa PAO1. Microbiology 161:2232–2242. doi: 10.1099/mic.0.000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wood WB. 1988. Introduction to C. elegans biology, p 1–16. In Wood WB (ed), The nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 60.Byerly L, Cassada RC, Russell RL. 1976. The life cycle of the nematode Caenorhabditis elegans. I. Wild-type growth and reproduction. Dev Biol 51:23–33. doi: 10.1016/0012-1606(76)90119-6. [DOI] [PubMed] [Google Scholar]
- 61.Heurlier K, Dénervaud V, Pessi G, Reimmann C, Haas D. 2003. Negative control of quorum sensing by RpoN (σ54) in Pseudomonas aeruginosa PAO1. J Bacteriol 185:2227–2235. doi: 10.1128/JB.185.7.2227-2235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fletcher MP, Diggle SP, Crusz SA, Chhabra SR, Cámara M, Williams P. 2007. A dual biosensor for 2-alkyl-4-quinolone quorum-sensing signal molecules. Environ Microbiol 9:2683–2693. doi: 10.1111/j.1462-2920.2007.01380.x. [DOI] [PubMed] [Google Scholar]
- 63.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]