

Abstract
Rhodococcus equi pneumonia is a major cause of morbidity and mortality in neonatal foals. Much effort has been made to identify preventative measures and new treatments for R. equi with limited success. With a growing focus in the medical community on understanding the genetic basis of disease susceptibility, investigators have begun to evaluate the interaction of the genetics of the foal with R. equi. This review describes past efforts to understand the genetic basis underlying R. equi susceptibility and tolerance. It also highlights the genetic technology available to study horses and describes the use of this technology in investigating R. equi. This review provides readers with a foundational understanding of candidate gene approaches, single nucleotide polymorphism‐based, and copy number variant‐based genome‐wide association studies, and next generation sequencing (both DNA and RNA).
Keywords: Copy number variants, Genome‐wide association study, Horses, Pneumonia, Rhodococcus equi
Abbreviations
- aCGH
array comparative genomic hybridization
- bp
base‐pairs
- cDNA
complementary DNA
- CFU
colony forming units
- CNV
copy number variant
- FoSTeS
fork stalling and template switching
- GWAS
genome‐wide association study
- IL
interleukin
- Kb
kilobases
- LD
linkage disequilibrium
- MMEJ
microhomology‐mediated end‐joining
- NAHR
non‐allelic homologous recombination
- NGS
next‐generation sequencing
- NHEJ
non‐homologous end joining
- SLC11A1
solute carrier family 11 member 1
- SNP
single nucleotide polymorphism
- TBA
tracheobronchial aspirate
- TRPM2
transient receptor protein potential cation channel, subfamily M, member 2
- Tf
transferrin gene
- VapA
virulence‐associated protein A
Rhodococcus equi pneumonia is an important disease of foals most commonly characterized by chronic progression associated with development of large pulmonary abscesses.1 Treatment of R. equi pneumonia is prolonged and expensive, and prevention is limited because transfusion of hyperimmune plasma is incompletely effective,2 chemoprophylaxis is inconsistently effective3, 4 and may promote antimicrobial resistance,5 and no effective vaccine is currently available.6 Isolates of R. equi that are virulent in foals express the virulence‐associated protein A (VapA), which is encoded by a gene, located within a pathogenicity island, on an approximately 85‐ to 90‐kilobase (kb) plasmid. Expression of VapA alone, however, is not sufficient to cause disease.7, 8 Many different strains of virulent (and avirulent) R. equi have been shown to be present in a common environment (ie, the same farm), and multiple genotypic virulent strains may exist even in an individual foal with R. equi pneumonia.5, 9, 10, 11 Although exposure to R. equi is widespread in farms where foals are affected, only a variable proportion of the foals will develop clinical disease at a given farm, whereas other foals at the same location will not develop disease.12, 13 In addition, anecdotal reports by veterinarians indicate that some mares recurrently have affected foals, whereas other mares from the same environment consistently have foals that do not develop R. equi pneumonia. Taken together, these findings support the possibility of an important role for a genetic predisposition (ie, susceptibility, resistance, or tolerance) to the development of R. equi pneumonia and have prompted investigations of the genetic basis for this disease. Pneumonia caused by R. equi is a complex trait. Thus, it is unlikely that it will have a monogenic basis. Nevertheless, studying the genetic basis of R. equi pneumonia is important because it could reveal information about crucial biological processes and pathways that influence the outcome of infection in foals, and identifying these pathways and processes might consequently lead to novel approaches for diagnostic testing, treatment, control, and prevention. The purpose of this report is to review what is known about the genetic predisposition to pneumonia caused by R. equi in foals and to describe the genetic techniques currently available to study the genetic determinants for the development of R. equi pneumonia or other diseases in horses. We begin by summarizing the current literature regarding genetic associations with R. equi pneumonia, and then discuss some more advanced genetic tools available for future studies to further investigate the genetic basis of R. equi pneumonia.
Literature Search
A literature search was conducted to identify English language studies from any year that focused on foals, R. equi, and genetics. Databases were searched in April 2014 through Ovid including CAB Abstracts, MEDLINE, Embase, and BIOSIS. Search words included (foals or equus or equine) and (r equi or Rhodococcus equi*) and gene*, where the asterisk indicates truncation. Within each database, appropriate subject headings or index terms also were added. A total of 744 articles were retrieved and deduplicated, with 5 articles selected for inclusion. This search was updated in September 2014.
Candidate Genes
We identified 5 studies that attempted to identify genes associated with R. equi pneumonia. Four of these 5 studies have utilized a candidate gene approach (Table 1). The candidate gene approach involves either use of prior knowledge pertaining to known gene functions that might predispose to the disease of interest (eg, the interferon‐gamma pathway and R. equi pneumonia)14, 15 or use of genes implicated in other but similar diseases that could be potential candidates for involvement(eg, genes important for Mycobacterium tuberculosis which, like R. equi, is a gram‐positive, facultative intracellular organism that replicates primarily within macrophages and causes pneumonia and could be potential candidates for R. equi pneumonia).16, 17, 18 To the authors' knowledge, the first candidate gene association study of R. equi pneumonia compared the frequencies of single nucleotide polymorphisms (SNPs) in the transferrin gene (Tf) among Thoroughbred foals from Kentucky that died of R. equi pneumonia with those of control Thoroughbred mares.19 In one study, the Tf gene was selected on the basis of its product's ability to bind iron because iron sequestration is a known host defense mechanism against bacterial replication.20, 21 The authors postulated that polymorphisms in the Tf gene might result in enhanced (or decreased) iron binding, which then could confer a selective advantage (or disadvantage) to survive infections with bacteria such as R. equi.19 The authors used SNP frequencies to infer Tf alleles present within the study population, and allele frequencies were subsequently compared between the case and control groups. The authors documented a significant (P < 0.05) abundance of the Tf F allele and a deficiency of the D 1 allele among the cases (diseased foals) when compared with controls. Limitations of this study included the fact that the sample size was relatively small, it was restricted to a single breed, a separate population for validation was not included, and no mechanistic studies (ie, documentation that the F allele was associated with decreased iron‐binding) were incorporated or cited. Nonetheless, a significant association of SNPs in the Tf gene with R. equi pneumonia was demonstrated, and this finding represented an important advance in knowledge.
Table 1.
Genetic studies of Rhodococcus equi pneumonia in foals
| Author | Study design | Country | Breed(s) | Number of horses | Markers investigated | Observed outcome | Findings |
|---|---|---|---|---|---|---|---|
| Mousel et al.19 | Candidate gene | United States | Thoroughbred | N = 84 | Tf SNPs | Clinical pneumonia or control | Allelic association of Tf with disease |
| Horin et al.22 | Candidate gene | Czech Republic | Thoroughbred | N = 51 | SNPs, Microsatellites | Burden of R. equi in TBA fluid | Association of IL1RN and IL1β with burden of R. equi |
| Halbert et al.29 | Candidate gene | United States | Arabian and Thoroughbred | N = 103 | SLC11A1 SNPs | Clinical pneumonia or control | Variation in SLC11A1 associated with disease |
| Horin et al.30 | Candidate gene | Czech Republic | Thoroughbred | N = 51 | SNPs | Burden of R. equi in TBA fluid | Association of IL7R with burden of R. equi |
| McQueen et al.52 | GWAS | United States | Quarter Horse |
N = 72 N = 248 |
Genome‐wide SNPs | Clinical and Subclinical pneumonia, or control | Associated SNP in TRPM2 with disease |
A later study seeking to identify a genetic predisposition to R. equi pneumonia utilized the candidate gene approach by comparing frequencies of 22 genetic markers among 51 Thoroughbred foals from the Czech Republic.22 These markers were either SNPs or polymorphic microsatellites in or near immune‐related genes that had been previously identified (except for 5 markers that were first identified in this study). No genetic variants were significantly associated with R. equi pneumonia, but some genetic variants were significantly associated with a higher burden of R. equi in tracheobronchial aspirate (TBA) fluid from foals. Specifically, loci on chromosome 10 and 15 were associated with R. equi infection when comparing the subset of foals with extreme phenotypes (ie, foals with the highest numbers of R. equi in TBA fluid) to those with no R. equi. The strongest association with TBA fluid phenotype was for the microsatellite HMS01 located on chromosome 15 which encodes the genes for interleukin (IL)‐1β (IL1β) and the IL‐1 receptor antagonist (IL1RN). Although the associations were relatively weak and the phenotype was for burden of R. equi in TBA fluid (rather than for pneumonia caused by R. equi), these results offer further evidence of a genetic basis for host response to infection with R. equi.
A third study from our laboratory utilized previous findings indicating association between the solute carrier family 11 member 1 gene (SLC11A1) and susceptibility to intracellular bacterial infections in other species of animals.23, 24, 25, 26 The SLC11A1 gene encodes a protein relevant to innate immune responses to intracellular bacteria.27, 28 Direct sequencing of the beginning of the gene transcript (ie, the 5′ end of the gene) was used to identify SNPs that were compared between cases of R. equi and unaffected foals (controls) among Arabian horses at 2 farms (1 farm in Texas and 1 farm in Arizona). A novel SNP, ‐57T, in the 5′ untranslated region (UTR) was significantly associated with R. equi pneumonia.29 The authors further screened for this polymorphism in 5 domestic horse breeds, donkeys, and zebras, and found that it was represented in 4 of the 5 horse breeds. The observation that this SNP was represented across multiple breeds strengthened the study's findings because if a marker were present in only 1 breed, it would be possible (if not probable) that the identified marker was more likely associated with breed differences than disease. Limitations of this study included the fact that association between the candidate gene and disease was only assessed within a single breed at 2 farms, and no validation of the association in another population was conducted. In addition, inconsistencies in diagnostic practices for R. equi pneumonia among farms in the study existed, which might have impacted the results.29
A fourth candidate gene study investigated the frequency of SNPs in selected immune response genes from DNA samples collected from 31 Thoroughbred foals from the Czech Republic30 that had been used in a previous candidate gene study (described above).22 The candidate markers were used to asses allele frequencies between the groups of foals classified on the basis of a binary outcome using a cut‐point of >5,000 colony forming units (CFU)/mL of vapA‐positive R. equi in TBA fluid. Twenty‐five foals were categorized as below the cut‐point because they had no bacteria cultured from them, and 6 were categorized as above the cut‐point.30 An association was identified between a SNP in the IL‐7 receptor (IL7R) gene and the presence of >5,000 CFU of R. equi in TBA fluid. Limitations of this study included lack of a validation population in which the association could be replicated, and, similar to the earlier study using these foals, the association was not made between the marker and disease but rather between the marker and the concentrations of bacteria present in TBA fluid. Regardless of these limitations, this study was scientifically important in that it implicated the IL7R gene in particular, and innate immunity in general, as having a role in host response to infection with R. equi.
The candidate gene approach is a valid method for genetic investigation and yielded positive associations in the aforementioned studies, strengthening the plausibility of a genetic contribution to susceptibility or resistance to R. equi pneumonia in foals. Moreover, the commonality of identifying innate immune responses as playing a role in host defense against infection with R. equi in these various candidate gene association studies is important to our understanding of the pathogenesis of R. equi pneumonia.
Despite these positive results, the candidate gene approach has important limitations for making genetic associations. Bias is introduced into the study design by selecting a small number of genes for evaluation, on the basis of either function of the gene product(s) or prior association of the gene with disease. This selection process effectively eliminates the ability of the investigators to examine both the enormous amount of genetic information in the remainder of the genome or the relationship and interaction of other genes with the genes of interest.17 Other genetic elements present in the genome (eg, sites critical to gene regulation) are missed by restricting analysis to candidate genes, because in most cases probes used to detect variation are not near each other and offer no information about neighboring genetic variation. Assessing variation across the genome circumvents these limitations of the candidate gene approach. Genome‐wide studies in horses are now feasible because of recent technological developments.
Genome‐Wide Association Studies
Genome‐wide association studies (GWASs) rapidly gained popularity after the sequencing of the genomes of several animal species, including human beings.31, 32, 33, 34 The completion of the sequencing and assembly of these reference genomes (an assembly of the DNA sequence and its chromosomal locations representing the genetic baseline of a species) provided a tool that could be used as a map indicating where elements of the genome reside. Substantial efforts were then made to catalog the locations of the genes and genetic variation identified within these species.35, 36 Single nucleotide polymorphisms proved useful for characterizing the genetic variation among individuals of a given species, and the development of SNP array technology made it possible to perform >1 million association tests simultaneously of markers across the entire genome without the expense or labor of genome sequencing.
Single nucleotide polymorphism arrays are glass slides with genomic probes (sequences of DNA) that capture SNPs present within a given species. Through a hybridization process, the probes bind DNA of samples to identify which polymorphisms are present in that sample.37 These SNP arrays enabled clinical researchers to compare clinically affected horses with unaffected controls so as to examine the association of various health disorders with markers on a genome‐wide basis, and the interplay among different genetic variants associated with disease.38 Results from a GWAS are readily identifiable because they typically are visualized by plotting the negative logarithm of the P value for the association of a given SNP with the outcome of interest as the ordinate (vertical axis) and the chromosome number as the abscissa (horizontal axis). The resultant scatter plots are known as Manhattan plots because they resemble the skyline of a major city with some points that tower over the majority of others. Determining the genome sequence of the domestic horse led to the development of 2 equine SNP arrays that could be used for GWAS by researchers.32, 39
Currently, a single SNP array has been developed, well characterized, made commercially available, and utilized in numerous GWAS in horses. For example, the EquineSNP70 BeadChip Array1 contains approximately 74,000 SNPs positioned across the equine genome that can be simultaneously tested to identify their associations with a phenotype of interest.39, 40 Recently, a higher density SNP array with approximately 770,000 SNPs across the equine genome has been developed but has not been characterized in peer‐reviewed literature to date. Several GWAS in horses using SNP arrays and yielding positive associations have been reported.41, 42, 43, 44, 45, 46 Genome‐wide association studies rely on observing different frequencies of alleles (identified by SNPs) that segregate with a phenotype of interest. These associations have identified regions of interest (Fig 1A), which are further investigated to understand what elements (eg, genes, promoters, other variants),pathways or processes are associated with the phenotype.
Figure 1.
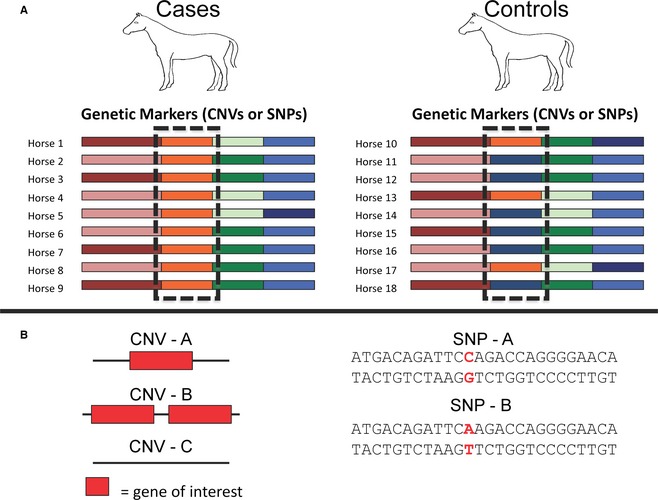
Association studies, CNVs, and SNPs. (A) The colored blocks indicate different alleles or haplotypes present in the horse genome. These have been identified by either a CNV or a SNP but any type of genetic variation can be used to identify alleles. The boxed regions show a greater frequency of the orange allele in the cases compared to the controls. The increased frequency of this allele in the cases suggests that it may harbor a variant(s) causing or contributing to the associated phenotype. (B) CNV – A represents a single copy of a gene; CNV – B represents a duplication of the gene; and, CNV – C represents a deletion of the gene. These examples demonstrate how CNVs can affect a single gene and can be used to identify different alleles in a population. SNPs, represented as red bases, offer the ability to identify alleles because of their polymorphic nature. Either type of genetic variation can be used in a GWAS to identify disease‐associated alleles.
The reason marker associations require region investigation is because of linkage disequilibrium (LD), which is defined as the nonrandom association of genetic information.47 Linkage disequilibrium is a phenomenon that allows for the prediction of the nongenotyped genetic information around a genotyped marker because of an assumption that the genetic material around a marker differs, and thus can be based on the allele represented by the marker (ie, SNP). The use of LD to make disease associations leverages inheritance patterns, selection, and evolution and is a fundamental concept underlying GWAS. The association of a marker, whether it is located in a gene or in a noncoding region of the genome, should only be treated as an indicator of the need to further investigate the area. An association of an SNP with disease neither indicates that the SNP is causally associated with the disease nor that the specific gene in which the SNP lies is the gene of interest. An SNP only indicates that there might be genetic variation in the area of the genome where the SNP is located. The size of the area of interest is largely described by the length of the LD (ie, the number of bases for which another gene or genetic element can be expected to be in LD with the marker). Using LD to assist in making associations is a powerful tool that is genome‐wide and efficient because not all markers across a genome must be tested to find an associated region, should one exist. The power of LD allows fewer markers to be present on an array, and hence decreases the number of necessary test corrections. Furthermore, the longer the LD of the species the fewer SNPs are necessary to identify significant associations. Estimates of LD for breeds of horses are markedly longer than those for humans.48, 49 Thus, one might expect to need fewer SNPs on an equine array to have the same discriminatory power as a human array or to have greater power in a GWAS for horses than humans for an array of the same size or density of SNPs.
Although SNP arrays are proving to be a powerful tool for investigating the relationship of genetic and phenotypic variation, challenges exist with validating and reproducing results generated by SNP‐based GWAS. There are likely many contributing risk alleles for all complex traits and complex diseases such that no single allele can explain all of the phenotypic variation.50 This becomes problematic during replication using different breeds and populations because the markers identified might merely reflect breed differences, or the markers might represent different alleles conferring different levels of risk across breeds or populations. The number of association studies in equine genetics will only continue to increase and the equine research community will continue to face these challenges. Appropriate study designs, accurately defined and categorized phenotypes, and functional follow‐up assays will be essential to maximize the utility of GWAS results in future studies.51
The first report of a GWAS with R. equi pneumonia recently was published by our laboratory.52 The study53 population included 248 foals born in 2011 at a large Quarter Horse breeding farm. For a separate study characterizing the accuracy of screening tests for R. equi pneumonia, foals at the farm were examined by thoracic ultrasonography every 2 weeks beginning at 3 weeks of age and continuing through 19 weeks of age (or until weaned) to identify foals with areas of pulmonary consolidation or abscess formation attributed to R. equi infection. Farm personnel were blinded to the ultrasonographic findings and a separate team of individuals performed thoracic ultrasonography. Foals at the farm were classified as having R. equi pneumonia (N = 43; on the basis of clinical signs of pneumonia, isolation of virulent R. equi from the TBA fluid, cytologic evidence of sepsis in TBA fluid, and ultrasonographic evidence of pulmonary consolidation or abscess formation >1 cm in maximal diameter), no pneumonia (N = 49; on the basis of the absence of clinical signs of pneumonia and no ultrasonographic evidence of pulmonary consolidation or abscess formation >1 cm diameter), and subclinical pneumonia (N = 156; on the basis of absence of clinical signs of pneumonia with ultrasonographic evidence of pulmonary consolidation or abscess formation >1 cm diameter). From each of these 3 subpopulations of foals, a sample of 24 foals was randomly selected for genotyping using the EquineSNP70 BeadChip Array. Comparisons among the 3 groups identified a significant association of a region on chromosome 26 that included the gene for the transient receptor potential cation channel, subfamily M, member 2 (TRPM2).
These results are notable because the TRPM2 gene is known to play a role in neutrophil function and recruitment. In a study using TRPM2 knock‐out mice and a model of ulcerative colitis, TRPM2‐deficient mice had less tissue damage at sites of inflammation than did wild‐type mice.54 The association of the TRPM2 was validated using polymerase chain reaction (PCR)‐based genotyping of the locus in the remaining 176 study foals that were not tested using the SNP array. The principal limitations of this study were that only a single breed at a single farm was studied, and that no association of the genotype with function of the TRPM2 gene‐product or associated signaling pathways was identified. Nonetheless, this study is interesting in that, consistent with previous candidate gene studies, a gene related to innate immunity was associated with R. equi pneumonia, and the study provides further evidence of the underlying genetic basis for R. equi pneumonia.
Copy Number Variants
Although the genetic determinants of phenotypic variation are largely dependent on the gene or genes and the manner in which they exert their effect (eg, altering the biochemical properties of a protein, changing the expression patterns or levels of messenger RNA), recent studies have implicated copy number variants (CNVs) as major determinants of phenotypic variation in humans and animals.55, 56, 57 As the name implies, CNVs are characterized by changes in the number of copies of DNA between at least 2 individuals (Fig 1B).58 Their sizes can range from hundreds to millions of base‐pairs (bps). Although they often are enriched in certain regions of the genome that predispose to their formation, CNVs have been detected throughout the genome, with many CNVs involving multiple genes, individual genes, or components of a single gene.
Several mechanisms have been shown to cause the formation of CNVs. During meiosis in the germ cells, homologous chromosomes align with each other to exchange genetic information between the parental genomes. This process, called homologous recombination or crossing over, plays an instrumental role in expanding the genetic diversity of a population. In rare instances, however, the exchange of genetic material can occur between 2 different sites (non‐allelic homologous recombination [NAHR]), resulting in an unequal exchange of genetic material.59 Although NAHR often is the source of many disease‐causing CNVs, this process plays a key role in the formation of gene families and the birth of new genes. Naturallyoccurring DNA repair mechanisms also can delete or duplicate DNA. For example, the non‐homologous end joining (NHEJ) and microhomology‐mediated end‐joining (MMEJ) pathways are used to repair double‐stranded DNA breaks that occur in the genome. During the repair of the breaks, the NHEJ and MMEJ pathways either add or remove DNA to ligate the broken strands back together.60, 61, 62 Fork stalling and template switching (FoSTeS) is a mechanism used to circumvent a stalled replication complex during DNA synthesis. When this happens, the FoSTeS machinery identifies a similar sequence at a nearby replication site to re‐engage the stalled complex, leading either to a deletion or duplication of the circumvented segment of DNA.63 Microhomology‐mediated break‐induced repair is another mechanism believed to give rise to CNVs under stressed cellular conditions in which traditional break‐induced repair does not occur and therefore homologous sequences are identified to continue replication.57, 64 Overall, there are numerous pathways and processes that can lead to the formation of a CNV.
Identification of CNVs across the genome has proven to be challenging because of the dependency on probe density to increase resolution and the physical limitation of the number of probes that can be placed on a single array. Array design technology continues to advance and undoubtedly will increase our ability to identify CNVs by enhancing genome resolution via probe density. Two studies in horses have used the EquineSNP70 BeadChip Array to search for the equine genome for CNVs.65, 66 The use of the equine SNP array to identify CNVs highlights the usefulness of the SNP array, but, there are limitations when SNP arrays are used solely for identifying CNVs. The probes present on SNP arrays are often evenly distributed across the genome, thus spanning large distances and allowing only for the identification of large CNVs. The SNP arrays also are not well suited for identifying CNVs in structurally complex regions (eg, gene families, segmentally duplicated regions). Probe design often is difficult in these regions, thus they are excluded from the array.67, 68, 69
Several studies have used technologies other than SNP arrays to identify CNVs in horses, such as next generation sequencing (NGS) and comparative genomic hybridization arrays (aCGH).52, 70, 71, 72, 73, 74 Arrays for CGH are designed by tiling oligonucleotide probes across the genome to which DNA of interest then can be hybridized for the identification of CNVs (Fig 2). Use of aCGH also has limitations for the identification of CNVs, principally related to probe placement and density. The currently published equine arrays are a whole genome tiling array (ie, probes tiled across the whole genome) and an exon tiling array (ie, probes tiled across noncoding and coding exons of genes).71, 72 Thus, these arrays only permit evaluation of CNVs within specific regions of the genome. The results of studies identifying CNVs by NGS are limited by variation in read‐depth (ie, the number of copies of sequences aligned to a specific area) across the genome and the size of the CNVs identified. Specifically, CNVs of lengths ranging from 197 bp to 3.5 Mb have been identified and confirmed using a CGH array designed to identify CNVs in genes of the equine genome.71 In a subsequent study using NGS, CNVs ranging in length from 3.74 kb to 4.84 Mb were identified.70 There is, however, a trade‐off when using either approach. Targeted arrays can identify smaller CNVs, but they are only able to identify CNVs within regions targeted on the array. Conversely, NGS can be used to identify CNVs throughout the entire genome, but NGS approaches to identifying CNVs are limited because of their bias toward larger CNV size. Although there are discrepancies among the approaches used to identify CNVs, the studies to date have identified CNVs in genes involved in similar pathways, such as sensory perception, signal transduction, and immune‐related pathways. Results from some CNV studies of horses also have found concordant results between aCGH and NGS whole genome sequencing in which CNVs of horses have been shown to be enriched in genes relating to sensory perception, signal transduction, and immune‐related functions.71, 72
Figure 2.
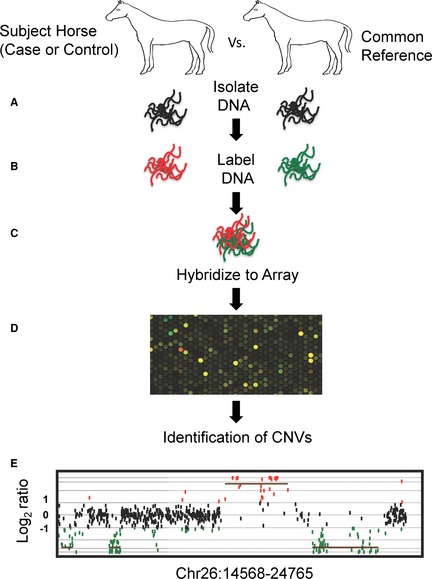
Comparative genomic hybridization method to identify CNVs in horses. (A) Genomic DNA is isolated from subject horses (cases and controls) and a single reference horse. (B) Genomic DNA from the subject horses are independently labeled with a red dye and genomic DNA from a single reference horse is labeled with a green dye. (C) Labeled DNA from a single subject horse and the reference horse are mixed together at equal ratios and competitively hybridized onto a comparative genomic hybridization array. (D) Fluorescent image of array after hybridization of subject and reference DNA. The spots on the array represent individual oligonucleotides. Yellow spots reflect regions with equal DNA content, and red and green spots reflect unequal ratios of DNA between the subject and reference horse, respectively. (E) Plot of normalized log2 ratios of oligonucleotides on the array. The Y‐axis represents normalized log2 ratios of fluorescent signals for each spot on the array. The X‐axis represents the relative genomic coordinates of each oligonucleotide. For example, a log 2 ratio <1 and >‐1 (black dots) indicates equal DNA content between the subject and reference horses. A log2 ratio >1 and <‐1 indicates unequal DNA content between the subject and reference horses.
Our laboratory conducted a CNV‐based GWAS by applying the aforementioned equine exon tilling array71 to the 72 foals studied in our SNP GWAS.48 Although similar lengths and numbers of CNVs were observed in these foals as in the previous report using this array, no CNVs were significantly associated with R. equi pneumonia in these foals. This finding does not preclude the possibility that CNVs contribute to susceptibility to R. equi pneumonia because only CNVs within exons were considered. The CNVs located within other elements such as promoters and silencers that were not detected by the array might influence the odds of foals developing R. equi pneumonia. Moreover, sample size was small, which might have limited our ability to detect anything less than very strong associations. Future efforts should include the design and implementation of adequately powered studies using tiling arrays focused at gene promotors, gene expression enhancers, and other regulatory elements that are both near and within genes. Much remains to be investigated to characterize CNVs in horses and to study the role of CNVs in susceptibility to R. equi foal pneumonia and other diseases of horses. Because CNVs represent a change in genetic content (ie, deletions and duplications), they may have the potential to greatly impact many phenotypes. A 4.6‐kb duplication in an intron has been associated with graying and melanomas in horses.75 A 16.1‐kb duplication has been shown to cause wrinkling of the skin in Shar‐Pei dogs.76 In humans, CNVs are believed to play critical roles in neurodevelopmental disorders, psychiatric disorders, and cancers.77, 78 A number of studies have described CNVs in cattle. Overall, the CNVs identified to date are enriched in genes related to immune function and sensory perception, which also has been observed in horses.79
Next Generation Sequencing Techniques
The invention of next generation sequencing (NGS) technologies has opened a new world of opportunities for understanding genetic variation and its role in disease pathogenesis. The NGS technology has enabled rapid sequencing of the genomes of individuals at a low cost and with maximum genome coverage.40 Before NGS, the gold standard for sequencing technology was automated Sanger sequencing.80 Sanger sequencing technology was developed in the late 1970s and later automated to increase throughput.81 Next generation sequencing technologies differ among companies, but they all share a principal advantage over Sanger sequencing in that they are capable of sequencing multiple DNA fragments (e, an entire genome) in a single sequencing reaction (versus sequencing small fragments piece‐by‐piece in multiple reactions).82 The opportunities provided by NGS technology are accompanied by the challenge of managing and analyzing datasets of enormous size. The ability of NGS to generate data has out‐paced the ability of the scientists to interpret it. Developments in bioinformatic and biostatistical software have facilitated our ability to visualize and make inferences from large datasets.
Both DNA and RNA can be sequenced using NGS methods. Sequencing the genome (DNA) and transcriptome (RNA) offers 2 interrelated but distinct biological approaches. Genome sequencing using NGS can characterize all of the variants known to exist in gene sequences, including single base changes (SNPs), insertions and deletions, CNVs, and genetic variation in nongenomic regions. The first application of NGS for genome sequencing in horses yielded the genome sequence of a Quarter Horse mare.70
Sequencing of the genome, however, does not reflect which elements of the DNA are transcribed. Moreover, transcription generally should be considered at the level of a specific tissue or cell type because of intercellular variation in gene expression. Although the DNA sequence is common to all cells in an individual, the genes expressed vary among cells or organs of the same individual. Sequencing RNA yields a snapshot of the expressed genes of the tissue or cell type that cannot be identified by DNA sequencing. The process of sequencing RNA using NGS methods is termed RNA‐Seq; it may be applied either to total RNA (all forms of RNA) or specific types of RNA. Most commonly, RNA‐Seq is applied to messenger RNA (mRNA) to reflect which portions of the genome are being transcribed in the specimen. Arriving at RNA‐sequencing is a multistep process which first requires deciding from which tissue or body‐fluid RNA should be extracted to best answer the biological questions being asked. Briefly, isolated RNA is converted to complementary DNA (cDNA) in order to construct a library that represents all of the RNA isolated and to be sequenced (Fig 3). The representative libraries are then sequenced, generally in a paired‐end fashion. Paired‐end sequencing reads are generated by sequencing from both ends of a cDNA fragment (ie, from the 5′ end of both strands of the cDNA fragment). Paired‐end reads are extremely valuable because 2 complementary pieces of information have been generated about the same cDNA fragment, and this greatly increases the accuracy of mapping these RNA sequences back to their respective genes of the genome.
Figure 3.
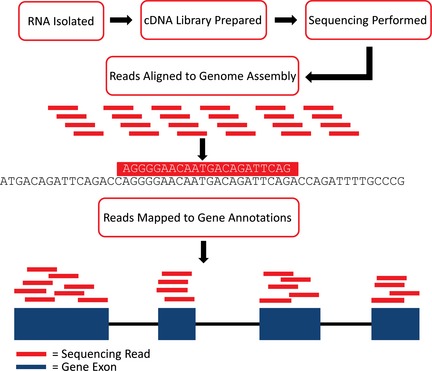
RNA‐Seq flowchart. Isolated RNA is converted to cDNA, a stable molecule, which can then be amplified and sequenced. Sequencing reads are then aligned to the genome assembly (sequence only) to identify their locations based on nucleotide matches. Mapping the reads to a gene annotation list will generate the number of sequencing reads that have aligned with a particular gene and are called counts. These counts at any particular gene are representative of the amount of gene expression in the sample and can be compared across horses to identify differentially expressed genes.
RNA‐Sequencing is an invaluable tool for gaining insight into biologically relevant questions such as differences in gene expression by different alleles and gene expression of a target specimen under different biological or biochemical conditions. Several downstream RNA‐Seq processing and analysis programs can be used to identify differentially expressed genes, novel transcripts, and multiple isoforms of gene transcripts in order to find answers to biological or clinical questions.83, 84, 85 The conclusions inferred from these analyses can lead to identifying potential biological pathways and processes that can be targeted for the development of novel interventions, including treatments and preventative measures.
Several studies have reported the application of RNA‐Seq in horses in an attempt to identify differentially expressed genes.86, 87, 88, 89, 90, 91, 92, 93, 94, 95 To the authors' knowledge, the first report of RNA‐Seq in horses was an effort to characterize the transcriptome and tissue‐specific expression profiles from 8 equine tissues.94 A subsequent study focused on characterizing gene expression by RNA‐Seq in immunologically active tissues.95 Investigators have used RNA‐Seq to characterize the expression profile of genes critical to the differentiation and regulation of cells during embryogenesis,90 and to characterize the expression and inferred function of RNAs in the equine sperm transcriptome.89 Several studies also have used RNA‐Seq in horses to identify differentially expressed genes when comparing blood, muscle (obtained by biopsy), or both before and after exercise or racing.86, 87, 92 These studies have successfully identified pathways involved in stress during and while recovering from exercise. Other's studies have sought to answer a more specific question such as identifying expression differences in the cartilage of the metacarpophalangeal joints of young and old horses in an attempt to shed light on genes involved in the development and aging of cartilage.93 Use of RNA‐Seq of hoof lamellar basal epithelial cells has been performed to identify cell‐signaling pathways indicative of the early stages of laminitis.91 Using RNA‐Seq, an association has been demonstrated of a long terminal repeat (a genetic element inserted in the past by conversion of viral RNA to cDNA and subsequently incorporated in the genome of the host) with congenital stationary night blindness and leopard spotting in horses.88
Our laboratory currently is analyzing RNA‐Seq data to identify differentially expressed genes of foals representing the 3 genotypes of the TRPM2 SNP identified in our SNP‐based GWAS to better understand the role of this (and possibly other) gene in susceptibility to R. equi pneumonia. We are also currently applying RNA‐Seq to leukocytes collected from healthy and R. equi‐affected foals to gain insights about gene expression of these immune‐related cells. These studies will further our understanding of R. equi pathogenesis and hopefully identify critical biological pathways and processes involved in disease development.
The genetic basis of a common and complex disease such as R. equi pneumonia is likely polygenic. Gene expression profiling by RNA‐Seq, thus, will be an essential step in understanding the relationships and interactions of multiple genes with this disease. The identification of genes that are up‐ or down‐regulated after pathogen exposure can reveal host responses critical for defense against infection. When evidence of differential gene expression is identified by RNA‐Seq (or other methods), it then becomes necessary to understand the mechanistic cause driving the change in expression(ie, variation within regulatory elements, changes in epigenetic modifications, structural variation, post‐transcriptional and post‐translational modifications).
Conclusions
Research findings regarding genetic relationships with disease continue to substantiate that most common and complex diseases are not monogenic. This likely is true for R. equi pneumonia. The evidence to date, as summarized in this review, indicates that susceptibility to R. equi pneumonia is not controlled by a single gene. It is increasingly clear that both innate and adaptive immune responses as well as their interactions are critical for protecting foals against R. equi infection. Genetic association studies have specifically implicated innate immune responses, but innate immune responses are critical for orchestrating adaptive immune responses and it may be an oversimplification to dichotomize these responses. It is likely that there also are epigenetic factors involved in regulating the gene transcription of critical immune‐related genes, which adds further complexity to the pathogenesis of R. equi pneumonia in foals. Future proteomic studies also will be required to follow‐up on promising genetic findings as protein concentrations, structures, and interactions are critical to disease development.96 Proteomic studies may be able to answer critical questions such as protein concentrations in diseased and nondiseased foals and variable consequences related to protein concentrations and their interactions, which cannot be answered with molecular genetic techniques and sequencing. With more genotypic–phenotypic associations being identified in horses, it will be challenging to investigate the causal implications of genetic variants with functional assays. Mechanistic studies (eg, knock‐out or knock‐in genes) can be become very expensive and would not be feasible in horses. Developing rodent models of important equine diseases and use of mechanistic studies in cell culture assay will be required to understand the functional consequences of identified associations with genetic markers. Moreover, it will be important to remain mindful of the agent‐related and environmental factors that contribute to disease development. No single genetic tool or technique will identify the factors that render some foals susceptible to R. equi, whereas others in the same environment remain clinically unaffected. The future will require a multifaceted approach to integration and analysis of data from multiple sources to successfully identify the critical pathways and processes. We believe that molecular genetic and epigenetic methods will play an important role in solving the complex riddle of susceptibility to R. equi pneumonia in foals.
Acknowledgments
Cole McQueen was supported by a grant from the USDA‐NIFA #2011‐68004‐30367. Noah Cohen was supported in part by the Link Equine Research Endowment, Texas A&M University.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
Footnote
EquineSNP70 BeadChip ArrayIllumina, San Diego, CA
References
- 1. Prescott JF. Rhodococcus equi: an animal and human pathogen. Clin Micro Rev 1991;4:20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hurley JR, Begg AP. Failure of hyperimmune plasma to prevent pneumonia caused by Rhodococcus equi in foals. Aust Vet J 1995;72:418–420. [DOI] [PubMed] [Google Scholar]
- 3. Venner M, Reinhold B, Beyerbach M, et al. Efficacy of azithromycin in preventing pulmonary abscesses in foals. Vet J 2009;179:301–303. [DOI] [PubMed] [Google Scholar]
- 4. Chaffin MK, Cohen ND, Martens RJ, et al. Evaluation of the efficacy of gallium maltolate for chemoprophylaxis against pneumonia caused by Rhodococcus equi infection in foals. Am J Vet Res 2011;72:945–957. [DOI] [PubMed] [Google Scholar]
- 5. Burton AJ, Giguère S, Sturgill TL, et al. Macrolide‐ and rifampin‐resistant Rhodococcus equi on a horse breeding farm, Kentucky, USA. Emerg Infect Dis 2013;19:282–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giguère S, Cohen ND, Chaffin MK, et al. Rhodococcus equi: clinical manifestations, virulence, and immunity. J Vet Intern Med 2011;25:1221–1230. [DOI] [PubMed] [Google Scholar]
- 7. Giguère S, Hondalus MK, Yager JA, et al. Role of the 85‐kilobase plasmid and plasmid‐encoded virulence‐associated protein A in intracellular survival and virulence of Rhodococcus equi . Infect Immun 1999;67:3548–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jain S, Bloom BR, Hondalus MK. Deletion of vapA encoding Virulence Associated Protein A attenuates the intracellular actinomycete Rhodococcus equi . Mol Microbiol 2003;50:115–128. [DOI] [PubMed] [Google Scholar]
- 9. Cohen ND, Smith KE, Ficht TA, et al. Epidemiologic study of results of pulsed‐field gel electrophoresis of isolates of Rhodococcus equi obtained from horses and horse farms. Am J Vet Res 2003;64:153–161. [DOI] [PubMed] [Google Scholar]
- 10. Bolton T, Kuskie K, Halbert N, et al. Detection of strain variation in isolates of Rhodococcus equi from an affected foal using repetitive sequence‐based polymerase chain reaction. J Vet Diag Inv 2010;22:611–615. [DOI] [PubMed] [Google Scholar]
- 11. Morton AC, Begg AP, Anderson GA, et al. Epidemiology of Rhodococcus equi strains on Thoroughbred horse farms. Appl Environ Microbiol 2001;67:2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen ND, Carter CN, Scott HM, et al. Association of soil concentrations of Rhodococcus equi and incidence of pneumonia attributable to Rhodococcus equi in foals on farms in central Kentucky. Am J Vet Res 2008;69:385–395. [DOI] [PubMed] [Google Scholar]
- 13. Muscatello G, Anderson GA, Gilkerson JR, et al. Associations between the ecology of virulent Rhodococcus equi and the epidemiology of R. equi pneumonia on Australian thoroughbred farms. Appl Environ Microbiol 2006;72:6152–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boyd NK, Cohen ND, Lim WS, et al. Temporal changes in cytokine expression of foals during the first month of life. Vet Immunol Immunopathol 2003;92:75–85. [DOI] [PubMed] [Google Scholar]
- 15. Breathnach CC, Sturgill‐Wright T, Stiltner JL, et al. Foals are interferon gamma‐deficient at birth. Vet Immunol Immunopathol 2006;112:199–209. [DOI] [PubMed] [Google Scholar]
- 16. Stewart GR, Robertson BD, Young DB. Tuberculosis: a problem with persistence. Nat Rev Microbiol 2003;1:97–105. [DOI] [PubMed] [Google Scholar]
- 17. Tabor HK, Risch NJ, Myers RM. Candidate‐gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet 2002;3:391–397. [DOI] [PubMed] [Google Scholar]
- 18. Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health 2000;24:164–168. [PMC free article] [PubMed] [Google Scholar]
- 19. Mousel MR, Harrison L, Donahue JM, et al. Rhodococcus equi and genetic susceptibility: assessing transferrin genotypes from paraffin‐embedded tissues. J Vet Diag Invest 2003;15:470–472. [DOI] [PubMed] [Google Scholar]
- 20. de Jong G, van Dijk JP, van Eijk HG. The biology of transferrin. Clin Chim Acta 1990;190:1–46. [DOI] [PubMed] [Google Scholar]
- 21. Skaar EP. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog 2010;6:e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horin P, Smola J, Matiasovic J, et al. Polymorphisms in equine immune response genes and their associations with infections. Mamm Genome 2004;15:843–850. [DOI] [PubMed] [Google Scholar]
- 23. Blackwell JM, Goswami T, Evans CA, et al. SLC11A1 (formerly NRAMP1) and disease resistance. Cell Microbiol 2001;3:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blackwell JM, Searle S, Mohamed H, et al. Divalent cation transport and susceptibility to infectious and autoimmune disease: continuation of the Ity/Lsh/Bcg/Nramp1/Slc11a1 gene story. Immunol Lett 2003;85:197–203. [DOI] [PubMed] [Google Scholar]
- 25. Vidal S, Tremblay ML, Govoni G, et al. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J Exp Med 1995;182:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams LG, Templeton JW. Genetic resistance to bacterial diseases of animals. Rev Sci Tec 1998;17:200–219. [DOI] [PubMed] [Google Scholar]
- 27. Forbes JR, Gros P. Divalent‐metal transport by NRAMP proteins at the interface of host‐pathogen interactions. Trends Microbiol 2001;9:397–403. [DOI] [PubMed] [Google Scholar]
- 28. Blackwell JM, Searle S, Goswami T, et al. Understanding the multiple functions of Nramp1. Microbes Infect 2000;2:317–321. [DOI] [PubMed] [Google Scholar]
- 29. Halbert ND, Cohen ND, Slovis NM, et al. Variations in equid SLC11A1 (NRAMP1) genes and associations with Rhodococcus equi pneumonia in horses. J Vet Intern Med 2006;20:974–979. [DOI] [PubMed] [Google Scholar]
- 30. Horin P, Sabakova K, Futas J, et al. Immunity‐related gene single nucleotide polymorphisms associated with Rhodococcus equi infection in foals. Int J Immunogenet 2010;37:67–71. [DOI] [PubMed] [Google Scholar]
- 31. Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 32. Wade CM, Giulotto E, Sigurdsson S, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009;326:865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elsik CG, Tellam RL, Worley KC, et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science 2009;324:522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Waterston RH, Lindblad‐Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002;420:520–562. [DOI] [PubMed] [Google Scholar]
- 35. Consortium TGP . A map of human genome variation from population‐scale sequencing. Nature 2010;467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibbs RA, Taylor JF, Van Tassell CP, et al. Genome‐wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009;324:528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. LaFramboise T. Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Res 2009;37:4181–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barsh GS, Copenhaver GP, Gibson G, et al. Guidelines for genome‐wide association studies. PLoS Genet 2012;8:e1002812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCue ME, Bannasch DL, Petersen JL, et al. A high density SNP array for the domestic horse and extant perissodactyla: utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet 2012;8:e1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finno CJ, Bannasch DL. Applied equine genetics. Equine Vet J 2014;46:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Go YY, Bailey E, Cook DG, et al. Genome‐wide association study among four horse breeds identifies a common haplotype associated with in vitro CD3+ T cell susceptibility/resistance to equine arteritis virus infection. J Virol 2011;85:13174–13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hauswirth R, Haase B, Blatter M, et al. Mutations in MITF and PAX3 cause “splashed white” and other white spotting phenotypes in horses. PLoS Genet 2012;8:e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hill EW, McGivney BA, Gu J, et al. A genome‐wide SNP‐association study confirms a sequence variant (g.66493737C>T) in the equine myostatin (MSTN) gene as the most powerful predictor of optimum racing distance for Thoroughbred racehorses. BMC Genom 2010;11:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lykkjen S, Dolvik NI, McCue ME, et al. Genome‐wide association analysis of osteochondrosis of the tibiotarsal joint in Norwegian Standardbred trotters. Anim Genet 2010;41(Suppl 2):111–120. [DOI] [PubMed] [Google Scholar]
- 45. Raudsepp T, McCue ME, Das PJ, et al. Genome‐wide association study implicates testis‐sperm specific FKBP6 as a susceptibility locus for impaired acrosome reaction in stallions. PLoS Genet 2012;8:e1003139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lykkjen S, Dolvik NI, McCue ME, et al. Equine developmental orthopaedic diseases ‐ a genome‐wide association study of first phalanx plantar osteochondral fragments in Standardbred trotters. Anim Genet 2013;44:766–769. [DOI] [PubMed] [Google Scholar]
- 47. Slatkin M. Linkage disequilibrium–understanding the evolutionary past and mapping the medical future. Nat Rev Genet 2008;9:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Petersen JL, Mickelson JR, Cothran EG, et al. Genetic diversity in the modern horse illustrated from genome‐wide SNP data. PLoS ONE 2013;8:e54997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kruglyak L. Prospects for whole‐genome linkage disequilibrium mapping of common disease genes. Nat Genet 1999;22:139–144. [DOI] [PubMed] [Google Scholar]
- 50. Chanock SJ, Manolio T, Boehnke M, et al. Replicating genotype‐phenotype associations. Nature 2007;447:655–660. [DOI] [PubMed] [Google Scholar]
- 51. Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest 2008;118:1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McQueen CM, Doan R, Dindot SV, et al. Identification of Genomic Loci Associated with Rhodococcus equi Susceptibility in Foals. PLoS ONE 2014;9:e98710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chaffin MK, Cohen ND, Blodgett GP, et al. Evaluation of ultrasonagraphic screening methods for early detection of Rhodococcus equi pneumonia in foals. J Equine Vet Sci 2012;32:S20–S21 (Abstract). [Google Scholar]
- 54. Yamamoto S, Shimizu S, Kiyonaka S, et al. TRPM2‐mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 2008;14:738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hastings PJ, Lupski JR, Rosenberg SM, et al. Mechanisms of change in gene copy number. Nat Rev Genet 2009;10:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hastings PJ, Ira G, Lupski JR. A microhomology‐mediated break‐induced replication model for the origin of human copy number variation. PLoS Genet 2009;5:e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lupski JR. Structural Variation in the Human Genome. N Engl J Med 2007;356:1169–1171. [DOI] [PubMed] [Google Scholar]
- 58. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet 2006;7:85–97. [DOI] [PubMed] [Google Scholar]
- 59. Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet 2002;18:74–82. [DOI] [PubMed] [Google Scholar]
- 60. Weterings E, van Gent DC. The mechanism of non‐homologous end‐joining: a synopsis of synapsis. DNA Repair 2004;3:1425–1435. [DOI] [PubMed] [Google Scholar]
- 61. Haviv‐Chesner A, Kobayashi Y, Gabriel A, et al. Capture of linear fragments at a double‐strand break in yeast. Nucleic Acids Res 2007;35:5192–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McVey M, Lee SE. MMEJ repair of double‐strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 2008;24:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007;131:1235–1247. [DOI] [PubMed] [Google Scholar]
- 64. Sheen CR, Jewell UR, Morris CM, et al. Double complex mutations involving F8 and FUNDC2 caused by distinct break‐induced replication. Hum Mutat 2007;28:1198–1206. [DOI] [PubMed] [Google Scholar]
- 65. Dupuis MC, Zhang Z, Durkin K, et al. Detection of copy number variants in the horse genome and examination of their association with recurrent laryngeal neuropathy. Anim Genet 2013;44:206–208. [DOI] [PubMed] [Google Scholar]
- 66. Metzger J, Philipp U, Lopes MS, et al. Analysis of copy number variants by three detection algorithms and their association with body size in horses. BMC Genom 2013;14:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kato M, Kawaguchi T, Ishikawa S, et al. Population‐genetic nature of copy number variations in the human genome. Hum Mol Gen 2010;19:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population‐scale genome sequencing. Nature 2011;470:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet 2007;39:S16–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Doan R, Cohen ND, Sawyer J, et al. Whole‐genome sequencing and genetic variant analysis of a Quarter Horse mare. BMC Genom 2012;13:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Doan R, Cohen N, Harrington J, et al. Identification of copy number variants in horses. Genome Res 2012;22:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ghosh S, Qu Z, Das PJ, et al. Copy number variation in the horse genome. PLoS Genet 2014;10:e1004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang W, Wang S, Hou C, et al. Genome‐wide detection of copy number variations among diverse horse breeds by array CGH. PLoS ONE 2014;9:e86860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Park KD, Kim H, Hwang JY, et al. Copy number deletion has little impact on gene expression levels in racehorses. Asian‐Aust J Anim Sci 2014;27:1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rosengren Pielberg G, Golovko A, Sundstrom E, et al. A cis‐acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat Genet 2008;40:1004–1009. [DOI] [PubMed] [Google Scholar]
- 76. Olsson M, Meadows JR, Truve K, et al. A novel unstable duplication upstream of HAS2 predisposes to a breed‐defining skin phenotype and a periodic fever syndrome in Chinese Shar‐Pei dogs. PLoS Genet 2011;7:e1001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hehir‐Kwa JY, Pfundt R, Veltman JA, et al. Pathogenic or not? Assessing the clinical relevance of copy number variants. Clin Genet 2013;84:415–421. [DOI] [PubMed] [Google Scholar]
- 78. Krepischi AC, Pearson PL, Rosenberg C. Germline copy number variations and cancer predisposition. Future Oncol 2012;8:441–450. [DOI] [PubMed] [Google Scholar]
- 79. Liu GE, Bickhart DM. Copy number variation in the cattle genome. Funct Integr Genomics 2012;12:609–624. [DOI] [PubMed] [Google Scholar]
- 80. Metzker ML. Emerging technologies in DNA sequencing. Genome Res 2005;15:1767–1776. [DOI] [PubMed] [Google Scholar]
- 81. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain‐terminating inhibitors. Proc Nat Acad Sci USA 1977;74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Metzker ML. Sequencing technologies ‐ the next generation. Nat Rev Genet 2010;11:31–46. [DOI] [PubMed] [Google Scholar]
- 83. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protocols 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Katz Y, Wang ET, Airoldi EM, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Meth 2010;7:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Capomaccio S, Vitulo N, Verini‐Supplizi A, et al. RNA sequencing of the exercise transcriptome in equine athletes. PLoS ONE 2013;8:e83504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Park KD, Park J, Ko J, et al. Whole transcriptome analyses of six thoroughbred horses before and after exercise using RNA‐Seq. BMC Genom 2012;13:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bellone RR, Holl H, Setaluri V, et al. Evidence for a retroviral insertion in TRPM1 as the cause of congenital stationary night blindness and leopard complex spotting in the horse. PLoS ONE 2013;8:e78280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Das PJ, McCarthy F, Vishnoi M, et al. Stallion sperm transcriptome comprises functionally coherent coding and regulatory RNAs as revealed by microarray analysis and RNA‐seq. PLoS ONE 2013;8:e56535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Iqbal K, Chitwood JL, Meyers‐Brown GA, et al. RNA‐seq transcriptome profiling of equine inner cell mass and trophectoderm. Biol Reprod 2014;90:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Leise BS, Watts MR, Roy S, et al. Use of laser capture microdissection for the assessment of equine lamellar basal epithelial cell signalling in the early stages of laminitis. Equine Vet J 2015;47:478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Park W, Kim J, Kim HJ, et al. Investigation of de novo unique differentially expressed genes related to evolution in exercise response during domestication in Thoroughbred race horses. PLoS ONE 2014;9:e91418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Peffers M, Liu X, Clegg P. Transcriptomic signatures in cartilage ageing. Arthritis Res Ther 2013;15:R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Coleman SJ, Zeng Z, Wang K, et al. Structural annotation of equine protein‐coding genes determined by mRNA sequencing. Anim Genet 2010;41(Suppl 2):121–130. [DOI] [PubMed] [Google Scholar]
- 95. Moreton J, Malla S, Aboobaker AA, et al. Characterisation of the horse transcriptome from immunologically active tissues. PeerJ 2014;2:e382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hanash S. Disease proteomics. Nature 2003;422:226–232. [DOI] [PubMed] [Google Scholar]