

Abstract
Context and objective
Growth hormone insensitivity with immune dysfunction caused by signal transducer and activator of transcription 5B (STAT5B) mutations is an autosomal recessive condition. Heterozygous mutations in other genes involved in growth regulation were previously associated with a mild height reduction. Our objective was to assess for the first time the phenotype of heterozygous STAT5B mutations.
Methods
We genotyped and performed clinical and laboratorial evaluations in 52 relatives of 2 previously described Brazilian brothers with homozygous STAT5B c.424_427del mutation (21 heterozygous). Additionally, we obtained height data and genotype from 1,104 adult control individuals from the same region in Brazil and identified 5 additional families harboring the same mutation (18 individuals, 11 heterozygous). Furthermore, we gathered the available height data from first-degree relatives of patients with homozygous STAT5B mutations (17 individuals from 7 families). Data from heterozygous individuals and non-carriers were compared.
Results
Individuals carrying heterozygous STAT5B c.424_427del mutation were 0.6 SDS shorter than their non-carrier relatives (p= 0.009). Heterozygous subjects also had significantly lower SDS for serum concentrations of IGF-1 (p=0.028) and IGFBP-3 (p=0.02) than their non-carrier relatives. The 17 heterozygous first-degree relatives of patients carrying homozygous STAT5B mutations had an average height SDS of −1.4 ± 0.8 when compared with population-matched controls (p < 0.001).
Conclusions
STAT5B mutations in heterozygous state have a significant negative impact on height (approximately 3.9 cm). This effect is milder than the effect seen in the homozygous state, with height usually within the normal range. Our results support the hypothesis that heterozygosity of rare pathogenic variants contributes to normal height heritability.
Keywords: genetic, height, STAT5B, growth hormone insensitivity, height heritability, IGF-I
Introduction
Previous studies have demonstrated that while homozygous mutations in genes involved in growth regulation are causal of severe syndromic short stature, heterozygosity of the same variants can be associated with a milder height reduction 1–4. For instance, in the growth hormone (GH) - insulin-like growth factor 1 (IGF-1) axis, heterozygous carriers of mutations in acid-labile subunit gene (IGFALS) 3 and IGF1 gene 1 were shown to be significantly shorter than non-carriers, although generally still within the normal height range. These data support the concept that rare mono-allelic variants with moderate effects on phenotype can be associated with height variability 5 and, as such, can be an etiology for non-syndromic short stature 6, 7.
Signal transducer and activator of transcription 5B (STAT5B) is a key mediator of GH signaling, as well as of other signaling pathways, including those of prolactin and interleukin 2 (IL2) 8. Since 2003, ten patients have been reported harboring seven different homozygous STAT5B mutations 9–16. These rare homozygous mutations in STAT5B cause growth hormone insensitivity (GHI) and manifestations of immune dysregulation, such as increased susceptibility for opportunistic infections, lymphoid interstitial pneumonia and eczema. GHI syndrome, classically associated with homozygous mutations in the growth hormone receptor gene (GHR), is characterized by severe postnatal growth failure, normal to elevated GH levels and low serum concentrations of ALS, IGF1 and insulin-like growth factor binding protein 3 (IGFBP-3). Unlike most GHI patients carrying defects in GHR, however, serum concentrations of growth hormone binding protein (GHBP), the proteolytically-cleaved extracellular domain of GHR, were normal and prolactin levels were increased in patients carrying homozygous STAT5B mutations (reviewed in 8).
To date, STAT5B deficiency is considered an autosomal recessive condition. The impact of heterozygous STAT5B mutations on growth and the GH-IGF axis, however, has not been carefully evaluated, due in part to the rarity of described cases and families. To address this issue, we evaluated a large community, in which multiple members carry a previously described STAT5B frameshift mutation 15. By comparing their data with data from other families harboring other mutations in STAT5B, we provide evidence that heterozygous STAT5B mutations can influence stature.
Subjects and methods
Subjects
We evaluated 52 relatives of two Brazilian brothers with characterized GHI due to homozygous STAT5B c.424_427del mutation. Furthermore, an active search was done to investigate the prevalence of this mutation in the region where the index cases were born, identifying five unrelated heterozygous individuals among 1,104 evaluated adult control subjects. Relatives of these five individuals were subsequently evaluated, totaling 18 subjects. Height data gathered from the remaining 1,099 adult control individuals (non-carriers of STAT5B c.424_427del mutation) in the same region were used to assess the local population height.
Additionally, we gathered the available height data from first-degree heterozygous relatives of previously reported patients with homozygous STAT5B mutations. We also included in this group two recently diagnosed individuals heterozygous for STAT5B c.424_427del mutation, who lost two children with the same phenotype seen in patients with homozygous STAT5B mutations. In total, height data from 17 first-degree relatives from 7 families were analyzed.
These studies were approved by the local ethics committees, and the patients or guardians gave their written informed consent.
Genotyping in families with STAT5B c.424_427del mutation
Genomic DNA was isolated from peripheral blood leukocytes using standard techniques. Genotyping for STAT5B c.424_427del mutation was done by fragment analysis technique. The primers were designed to amplify the region around this mutation (primer sequences and amplification protocols are available upon request). Genotyping was performed after the clinical evaluation.
Clinical and laboratory assessment in families with STAT5B c.424_427del mutation
Individuals from families with STAT5B c.424–427del mutation were evaluated by an investigator blinded for STAT5B genotype. They were questioned about pneumopathies, eczema and other immune dysfunctions. Height and weight were assessed in all individuals. Total blood count, fasting glucose and insulin, immunoglobulins G, A and E, basal GH, IGF-1, IGFBP-3 and prolactin were tested in 91% of the evaluated individuals. Serum GH, IGF-1, IGFBP-3, prolactin and immunoglobulin E were measured through chemiluminescence assays and immunoglobulins A and G through turbidimetry. IGF-1 and IGFBP-3 were transformed to SD scores (SDS) 17.
Whole-exome sequencing
Whole-exome sequencing of genomic DNA, obtained from the peripheral blood of one individual heterozygous for STAT5B c.424_427del mutation and with pneumopathy of unknown etiology, was performed with Illumina’s Nextera Exome Enrichment kits (Illumina, San Diego, CA) for library preparation and exome capture, and the Illumina HiSeq sequencer. Alignments and variant annotation were made as previouly described 18.
Statistical methods
Because the patients came from many ethnic groups, height data were expressed as SDS for the appropriate country/ethnic group. The effect of one mutant allele vs. wild type was determined in the whole group.
Groups were compared by unpaired t-test or ANOVA followed by Tukey test for numerical variables with normal distribution. Numerical variables without parametric distribution were analyzed by Mann-Whitney Rank Sum Test or Kruskal-Wallis ANOVA on Ranks. Categorical data were compared between groups through chi-square test or Fisher’s exact test as appropriate. Statistical significance was assumed for p < 0.05. Statistical analysis were made with SigmaStat 3.5 (Systat Software Inc. Chicago, USA) and MedCalc version 11.1.1.0 (MedCalc Software, Mariakerke, Belgium).
Results
Families harboring STAT5B c.424_427del mutation
The largest Brazilian family consisted of two patients with homozygous STAT5B c.424_427del mutation, 21 heterozygous carriers (including their non-consanguineous parents) and 31 non-carrier relatives (Supplemental Figure 1). The other five families identified consisted of eleven heterozygous carriers and seven non-carrier relatives. When polymorphic markers around this mutation were studied, the same haplotype was found in these six families, which was consistent with the presence of a founder effect (data not shown) 19. Consequently, we analyzed all individuals from the six families together (Table 1).
Table 1.
Clinical and biochemical characteristics of heterozygous carriers of STAT5B c.424_427del mutation vs. non-carriers
| WT/Mut | WT/WT | p | |
|---|---|---|---|
| n | 32 | 38 | |
| Dermopathy | 9:32 | 1:38 | 0.004 |
| Severe pneumopathy | 1:32 | 0:38 | n.s. |
| Height SDS | −0.8 ± 0.9* | −0.2 ± 1.0 | 0.009 |
| Basal GH (ng/mL) | 1.4 ± 2.2 | 1.0 ± 1.6 | n.s. |
| IGF-1 SDS | −0.4 ± 1.2 | 0.3 ± 1.2 | 0.028 |
| IGFBP-3 SDS | −0.9 ± 1.4 | 0.0 ± 1.5 | 0.02 |
| Prolactin (ng/mL) | 12.5 ± 7.2 | 13.0 ± 10.6 | n.s. |
| Glucose (mg/dL) | 90 ± 14 | 83 ± 13 | n.s. |
| Insulin (μUI/mL) | 6.9 ± 7.3 | 6.4 ± 3.9 | n.s. |
| Hemoglobin (g/dL) | 13.3 ± 1.3 | 13.4 ± 1.5 | n.s. |
| Leucocytes (cells/mm3) | 6893 ± 2008 | 6935 ± 2695 | n.s. |
| Lymphocytes (cells/mm3) | 3998 ± 1292 | 3598 ± 1329 | n.s. |
| IgG (mg/dL) | 1022 ± 205 | 1054 ± 249 | n.s. |
| IgA (mg/dL) | 244 ± 141 | 225 ± 123 | n.s. |
| IgE (UI/mL) | 220 ± 325 | 143 ± 191 | n.s. |
SDS – standard deviation score; GH – growth hormone; IGF-1 – insulin-like growth factor 1; IGFBP-3 – insulin-like growth factor binding protein 3; IgG/IgA/IgE – immunoglobulins G, A and E; n.s. – non-significant.
excluding one heterozygous carrier with severe short stature (height SDS −3.5)
Values are expressed as mean ± SD
In total, we analyzed data from 32 heterozygous carriers of STAT5B c.424_427del mutation (17 males) and 38 non-carrier family members (12 males). Unrelated spouses were not included. One heterozygous carrier was excluded from the height analysis because of severe short stature (height SDS −3.5) of unknown cause. Among the seventy evaluated individuals (aged 32.7 ± 18.5 years old), sixteen were children (7 heterozygous for STAT5B mutation).
Non-carrier subjects in these families had a similar height SDS to individuals from the local population (height SDS −0.2 ± 1.0 vs. −0.4 ± 0.8 respectively, p = 0.63). Heterozygous STAT5B c.424_427del individuals were significantly shorter (height SDS −0.8 ± 0.9) than their non-carrier relatives (height SDS difference of −0.6, p = 0.009, confidence interval 95% −1.1 to −0.2), although all were within the normal height range (Table 1). When the analysis was done excluding the children, the same results were obtained (height SDS −0.8 ± 0.9 vs. −0.2 ± 1.0 for heterozygous and non-carrier relatives respectively, p = 0.02, Figure 1). Furthermore, heterozygous carriers had significantly lower IGF-1 and IGFBP-3 SDS than their non-carrier relatives (Table 1). Other parameters, such as basal GH and prolactin concentrations, were not different between these groups.
Figure 1.
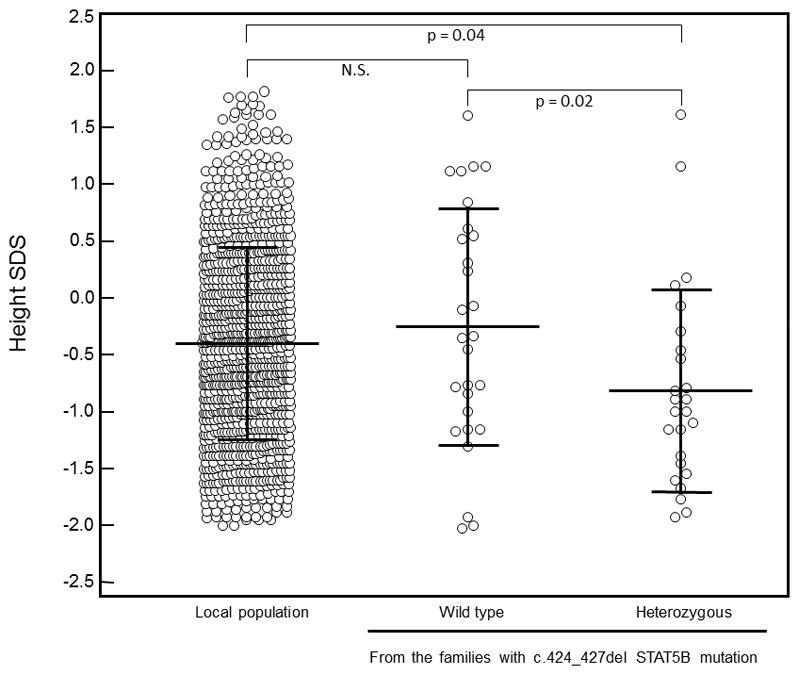
Comparison of height SDS distribution among adult heterozygous carriers of STAT5B c.424_427del mutation (n = 25), their non-carrier relatives (n = 28) and a population sample from the same region in the south of Brazil (n=1,099).
Present or past history of dermopathies was reported in 9 out of 32 individuals heterozygous for STAT5B c.424_427del mutation and in 1 out of 38 individuals who were non-carriers (p = 0.004). We clinically diagnosed eczema in four carriers. No differences in total blood count and immunoglobulin levels between carriers and non-carriers were observed (Table 1).
One cousin of the probands, who was heterozygous for STAT5B c.424_427del mutation, presented with a moderate to severe restrictive pneumopathy of unknown etiology. Her disease was milder than the pneumopathy observed in patients homozygous for STAT5B mutations, since she was in her thirties and still not oxygen-dependent. Exome sequencing excluded other STAT5B mutations and mutations in genes normally associated with pneumopathies (data not shown). A lung biopsy of this patient showed areas of interstitial thickening near respiratory bronchioles, inflammatory interstitial infiltrate with lymphocytes, plasmocytes and histiocytes and mild interstitial fibrosis, which is compatible with lymphoid interstitial pneumonia. Her father, an obligatory heterozygous carrier for the same mutation, died of respiratory failure secondary to an uninvestigated chronic pneumopathy, but he had confounding factors such as smoking and working as a miner.
First-degree relatives of index cases carrying STAT5B mutations
Height data were obtained in seventeen first-degree relatives of ten patients homozygous for STAT5B mutations (Table 2). Two Argentinian patients were adopted soon after birth and, consequently, data from their biological relatives were not available. Parents were consanguineous in four families and not consanguineous in three families. All these relatives were heterozygous for STAT5B mutations with an average height SDS of −1.4 ± 0.8 when compared with appropriate population-matched controls (p < 0.001).
Table 2.
Data of index patients homozygous for STAT5B mutations and their first-degree relatives.
| Family n° | Ref | Consanguinity | cDNA mutation | Origin | Local height SDS
|
|||
|---|---|---|---|---|---|---|---|---|
| Patient | Fathers | Mothers | Siblings | |||||
|
|
|
|
||||||
| 1 | 9 | Yes | c.1888G>C | Argentina | −7.5 | −0.3 | −1.2 | |
| 2 | 10 | Yes | c.1191insG | Turkey | −7.8 | −0.9 | −0.6 | |
| 3 | 11 | No | c.1102insC | Caribbean | −5.9 | −2.8 | −0.8 | −2.3 / −0.8 |
| 4 | 12 | No | c.454C>T | Argentina | −9.9 | −2.2 | −3.3 | −2.0 |
| 5 | 13 | Yes | c.1680delG | Kuwait | −5.6 / −5.8 | −1.3 | −0.6 | |
| 6 | 14 | Adopted | c.454C>T | Argentina | −5.3 | NA | NA | |
| 7 | 15 | No | c.424_427del | Brazil | −5.6 / −3.0 | −1.5 | −1.0 | |
| 8 | 16 | Adopted | c.1937T>C | Argentina | −5.95 | NA | NA | |
| 9 | 19 | Yes | c.424_427del | Brazil | NA* | −0.9 | −1.9 | |
|
|
|
|
||||||
| Mean ± SDS | −6.2 ± 1.8 | −1.4 ± 0.8 | ||||||
| Median (range) | −5.9 (−9.9; −3.0) | −1.2 (−3.3; −0.3) | ||||||
NA: not available
patients died before anthropometric assessment.
Discussion
In adequate health and nutritional conditions, genetic variation is the main determinant of stature, accounting for approximately 80% of height variability 20. Recent genome-wide association studies (GWAS) identified 697 variants in 423 loci that, together, accounted for only one-fifth of adult height heritability 21. The individual effect of single nucleotide polymorphisms (SNPs) found in these studies, furthermore, is very small (less than 0.5 cm) 22. The inability of GWAS to explain all height heritability, despite the increasing number of evaluated individuals, suggests that numerous rare variants with large to moderate effect have a role in height variability 23. However, it is difficult to evaluate the importance of rare variants in height variability through the current available methods, since each private allele is restricted to a few families or small populations.
In the present study, the analysis of a large family with many heterozygous carriers of the STAT5B c.424_427del mutation showed that these individuals are significantly shorter than their non-carrier relatives and local population controls (mean height SDS difference of 0.6). Assuming that the mean SD for adult height distribution is 6.5cm, the mean height loss seen in these individuals can be estimated at 3.9 cm, which is a much larger individual effect than the 0.5 cm attributed to SNPs identified in GWAS. The significant reduction in IGF-1 SDS and IGFBP-3 SDS seen with STAT5B c.424_427del heterozygous carriers, furthermore, suggests that a decreased responsiveness to GH action could explain, at least in part, the observed height reduction.
Moreover, the analysis of the available height data from carriers of the different STAT5B mutations also displayed a significant reduction in height when compared to their population controls, and was equivalent to a height decrease of 9.1 cm, an even greater difference than that observed for carriers of STAT5B c.424_427del mutation. This difference could be due to the relatively smaller number of first-degree relatives available for study, and/or to variable effects on height dependent on the individual STAT5B mutation itself. For the STAT5B c.424_427del mutation, the lack of expression of the mutant protein in reconstitution experiments (Hwa V, unpublished data) suggests that partial haploinsufficiency could explain the modest height reduction seen in heterozygous carriers. No dominant-negative STAT5B mutations have been reported to date, although, interestingly, a heterozygous STAT5B p.Gln177Pro variant was recently described in two GHI patients with severe short stature but no immunological dysfunction 24.
Heterozygous mutations in other genes along the GH-IGF1 axis similarly show larger individual effects on height than SNPs, supporting our finding. For example, in a family carrying IGF1 p.V44M mutation, individuals heterozygous for this mutation were 0.6 SDS shorter (equivalent to 3.9 cm) than their non-carrier relatives 1. Moreover, heterozygous carriers of IGFALS mutations were 0.9 SDS shorter (equivalent to 5.8 cm) than their non-carrier relatives 3. Heterozygous mutations in genes associated with bone growth regulation similarly impacted height: in a large family with many individuals heterozygous for a NPR2 mutation, carriers were 1.4 SDS shorter than non-carriers (equivalent to 9.1 cm) 2. In all of these studies, the clinical presentation of heterozygous individuals was much milder than the disorder seen in patients homozygous for the same mutations. Altogether, the presence of these rare pathogenic mutations in heterozygous state suggests that loss of one functional allele may result in low-normal height and borderline short stature.
Finally, we observed that individuals with heterozygous STAT5B c.424_427del mutation reported more dermopathies and skin allergies when compared to their non-carrier relatives (p = 0.004). There was no difference in pneumopathies or other allergies reported by both groups, although two heterozygous carriers (a cousin of the probands and her father) had severe pneumopathy of unknown etiology. Further investigations are necessary to better characterize the potential effects of heterozygous STAT5B mutations in immunologic alterations.
In conclusion, we demonstrated that STAT5B mutations in heterozygous state exert a significant negative impact on height. This effect is milder than the effect seen in homozygous state, with height usually within the low normal range. Our results support the hypothesis that heterozygosity of rare pathogenic variants contributes to normal height heritability. Whether the cumulative effect of such variants could be responsible for a proportion of the missing height heritability posed by GWAS studies remains to be determined.
Supplementary Material
The largest Brazilian family with STAT5B c.424_427del mutation. Subjects homozygous for STAT5B c.424_427del mutation are indicated by black symbols; heterozygous carriers by gray symbols and non-carriers by white symbols. The symbol * refers to two individuals with severe pneumopathy of unknown etiology.
Acknowledgments
Funding: This work was supported by Grants 2013/03236–5, 2010/19809-6 (to A.A.L.J.) and 2011/15078-0 (to R.C.S) from the Sao Paulo Research Foundation (FAPESP); Grant 304678/2012–0 (to A.A.L.J.) from the National Council for Scientific and Technological Development (CNPq) and NIH R01HD078592 (to V.H.).
The authors thank Dr. Julio Cezar Cechinel from Laboratorio Pasteur – Criciúma, SC, Brazil for performing laboratory tests in individuals from families with STAT5B c.424_427del mutation.
Footnotes
Declaration of interest: The authors have nothing to disclose.
References
- 1.Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, van Thiel SW, Sluimers CA, Bax JJ, de Laat JA, Breuning MB, Romijn JA, Wit JM. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–2864. doi: 10.1210/jc.2004-1254. [DOI] [PubMed] [Google Scholar]
- 2.Olney RC, Bukulmez H, Bartels CF, Prickett TC, Espiner EA, Potter LR, Warman ML. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J Clin Endocrinol Metab. 2006;91:1229–1232. doi: 10.1210/jc.2005-1949. [DOI] [PubMed] [Google Scholar]
- 3.Fofanova-Gambetti OV, Hwa V, Wit JM, Domene HM, Argente J, Bang P, Hogler W, Kirsch S, Pihoker C, Chiu HK, Cohen L, Jacobsen C, Jasper HG, Haeusler G, Campos-Barros A, Gallego-Gomez E, Gracia-Bouthelier R, van Duyvenvoorde HA, Pozo J, Rosenfeld RG. Impact of heterozygosity for acid-labile subunit (IGFALS) gene mutations on stature: results from the international acid-labile subunit consortium. J Clin Endocrinol Metab. 2010;95:4184–4191. doi: 10.1210/jc.2010-0489. [DOI] [PubMed] [Google Scholar]
- 4.Jorge AA, Funari MF, Nishi MY, Mendonca BB. Short stature caused by isolated SHOX gene haploinsufficiency: update on the diagnosis and treatment. Pediatr Endocrinol Rev. 2010;8:79–85. [PubMed] [Google Scholar]
- 5.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malaquias AC, Scalco RC, Fontenele EG, Costalonga EF, Baldin AD, Braz AF, Funari MF, Nishi MY, Guerra-Junior G, Mendonca BB, Arnhold IJ, Jorge AA. The sitting height/height ratio for age in healthy and short individuals and its potential role in selecting short children for SHOX analysis. Horm Res Paediatr. 2013;80:449–456. doi: 10.1159/000355411. [DOI] [PubMed] [Google Scholar]
- 7.Vasques GA, Arnhold IJ, Jorge AA. Role of the natriuretic peptide system in normal growth and growth disorders. Horm Res Paediatr. 2014;82:222–229. doi: 10.1159/000365049. [DOI] [PubMed] [Google Scholar]
- 8.Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab. 2011;25:61–75. doi: 10.1016/j.beem.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, Heinrich JJ, Rosenfeld RG. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- 10.Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005;90:4260–4266. doi: 10.1210/jc.2005-0515. [DOI] [PubMed] [Google Scholar]
- 11.Vidarsdottir S, Walenkamp MJ, Pereira AM, Karperien M, van Doorn J, van Duyvenvoorde HA, White S, Breuning MH, Roelfsema F, Kruithof MF, van Dissel J, Janssen R, Wit JM, Romijn JA. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol Metab. 2006;91:3482–3485. doi: 10.1210/jc.2006-0368. [DOI] [PubMed] [Google Scholar]
- 12.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, Ornani A, Paz R, Rivarola MA, Zelazko M, Belgorosky A. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118:e1584–1592. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 13.Hwa V, Camacho-Hubner C, Little BM, David A, Metherell LA, El-Khatib N, Savage MO, Rosenfeld RG. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene. Horm Res. 2007;68:218–224. doi: 10.1159/000101334. [DOI] [PubMed] [Google Scholar]
- 14.Boyanovsky A, Lozano A, Testa G. Growth hormone insensitivity and immunodeficiency: mutation in the STAT5B gene. 8th Joint Meeting of the Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology; 2009. pp. P01–P67. [Google Scholar]
- 15.Pugliese-Pires PN, Tonelli CA, Dora JM, Silva PC, Czepielewski M, Simoni G, Arnhold IJ, Jorge AA. A novel STAT5B mutation causing GH insensitivity syndrome associated with hyperprolactinemia and immune dysfunction in two male siblings. Eur J Endocrinol. 2010;163:349–355. doi: 10.1530/EJE-10-0272. [DOI] [PubMed] [Google Scholar]
- 16.Scaglia PA, Martinez AS, Feigerlova E, Bezrodnik L, Gaillard MI, Di Giovanni D, Ballerini MG, Jasper HG, Heinrich JJ, Fang P, Domene HM, Rosenfeld RG, Hwa V. A Novel Missense Mutation in the SH2 Domain of the STAT5B Gene Results in a Transcriptionally Inactive STAT5b Associated with Severe IGF-I Deficiency, Immune Dysfunction, and Lack of Pulmonary Disease. J Clin Endocrinol Metab. 2012;97:E830–E839. doi: 10.1210/jc.2011-2554. [DOI] [PubMed] [Google Scholar]
- 17.Elmlinger MW, Kuhnel W, Weber MM, Ranke MB. Reference ranges for two automated chemiluminescent assays for serum insulin-like growth factor I (IGF-I) and IGF-binding protein 3 (IGFBP-3) Clin Chem Lab Med. 2004;42:654–664. doi: 10.1515/CCLM.2004.112. [DOI] [PubMed] [Google Scholar]
- 18.Bertola DR, Yamamoto GL, Almeida TF, Buscarilli M, Jorge AA, Malaquias AC, Kim CA, Takahashi VN, Passos-Bueno MR, Pereira AC. Further evidence of the importance of RIT1 in Noonan syndrome. Am J Med Genet A. 2014;164A:2952–2957. doi: 10.1002/ajmg.a.36722. [DOI] [PubMed] [Google Scholar]
- 19.Scalco RC, Funari MF, Aracava RM, Tonelli CA, Jorge AA. E Society, editor. Endocrine Society’s 96th Annual Meeting and Expo. Chicago: Endocrine Review; 2014. Evidence for a Founder Effect of C.424_427del STAT5B Mutation Causing Growth Hormone Insensitivity in the South of Brazil; p. MON-0161. [Google Scholar]
- 20.Silventoinen K, Sammalisto S, Perola M, Boomsma DI, Cornes BK, Davis C, Dunkel L, De Lange M, Harris JR, Hjelmborg JV, Luciano M, Martin NG, Mortensen J, Nistico L, Pedersen NL, Skytthe A, Spector TD, Stazi MA, Willemsen G, Kaprio J. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. 2003;6:399–408. doi: 10.1375/136905203770326402. [DOI] [PubMed] [Google Scholar]
- 21.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, Croteau-Chonka DC, Day FR, Duan Y, Fall T, Fehrmann R, Ferreira T, Jackson AU, Karjalainen J, Lo KS, Locke AE, Magi R, Mihailov E, Porcu E, Randall JC, Scherag A, Vinkhuyzen AA, Westra HJ, Winkler TW, Workalemahu T, Zhao JH, Absher D, Albrecht E, Anderson D, Baron J, Beekman M, Demirkan A, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Fraser RM, Goel A, Gong J, Justice AE, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Lui JC, Mangino M, Mateo Leach I, Medina-Gomez C, Nalls MA, Nyholt DR, Palmer CD, Pasko D, Pechlivanis S, Prokopenko I, Ried JS, Ripke S, Shungin D, Stancakova A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, van der Laan SW, van Setten J, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Afzal U, Arnlov J, Arscott GM, Bandinelli S, Barrett A, Bellis C, Bennett AJ, Berne C, Bluher M, Bolton JL, Bottcher Y, Boyd HA, Bruinenberg M, Buckley BM, Buyske S, Caspersen IH, Chines PS, Clarke R, Claudi-Boehm S, Cooper M, Daw EW, De Jong PA, Deelen J, Delgado G, Denny JC, Dhonukshe-Rutten R, Dimitriou M, Doney AS, Dorr M, Eklund N, Eury E, Folkersen L, Garcia ME, Geller F, Giedraitis V, Go AS, Grallert H, Grammer TB, Grassler J, Gronberg H, de Groot LC, Groves CJ, Haessler J, Hall P, Haller T, Hallmans G, Hannemann A, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hemani G, Henders AK, Hillege HL, Hlatky MA, Hoffmann W, Hoffmann P, Holmen O, Houwing-Duistermaat JJ, Illig T, Isaacs A, James AL, Jeff J, Johansen B, Johansson A, Jolley J, Juliusdottir T, Junttila J, Kho AN, Kinnunen L, Klopp N, Kocher T, Kratzer W, Lichtner P, Lind L, Lindstrom J, Lobbens S, Lorentzon M, Lu Y, Lyssenko V, Magnusson PK, Mahajan A, Maillard M, McArdle WL, McKenzie CA, McLachlan S, McLaren PJ, Menni C, Merger S, Milani L, Moayyeri A, Monda KL, Morken MA, Muller G, Muller-Nurasyid M, Musk AW, Narisu N, Nauck M, Nolte IM, Nothen MM, Oozageer L, Pilz S, Rayner NW, Renstrom F, Robertson NR, Rose LM, Roussel R, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Schunkert H, Scott RA, Sehmi J, Seufferlein T, Shi J, Silventoinen K, Smit JH, Smith AV, Smolonska J, Stanton AV, Stirrups K, Stott DJ, Stringham HM, Sundstrom J, Swertz MA, Syvanen AC, Tayo BO, Thorleifsson G, Tyrer JP, van Dijk S, van Schoor NM, van der Velde N, van Heemst D, van Oort FV, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Waldenberger M, Wennauer R, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Arveiler D, Bakker SJ, Beilby J, Bergman RN, Bergmann S, Biffar R, Blangero J, Boomsma DI, Bornstein SR, Bovet P, Brambilla P, Brown MJ, Campbell H, Caulfield MJ, Chakravarti A, Collins R, Collins FS, Crawford DC, Cupples LA, Danesh J, de Faire U, den Ruijter HM, Erbel R, Erdmann J, Eriksson JG, Farrall M, Ferrannini E, Ferrieres J, Ford I, Forouhi NG, Forrester T, Gansevoort RT, Gejman PV, Gieger C, Golay A, Gottesman O, Gudnason V, Gyllensten U, Haas DW, Hall AS, Harris TB, Hattersley AT, Heath AC, Hengstenberg C, Hicks AA, Hindorff LA, Hingorani AD, Hofman A, Hovingh GK, Humphries SE, Hunt SC, Hypponen E, Jacobs KB, Jarvelin MR, Jousilahti P, Jula AM, Kaprio J, Kastelein JJ, Kayser M, Kee F, Keinanen-Kiukaanniemi SM, Kiemeney LA, Kooner JS, Kooperberg C, Koskinen S, Kovacs P, Kraja AT, Kumari M, Kuusisto J, Lakka TA, Langenberg C, Le Marchand L, Lehtimaki T, Lupoli S, Madden PA, Mannisto S, Manunta P, Marette A, Matise TC, McKnight B, Meitinger T, Moll FL, Montgomery GW, Morris AD, Morris AP, Murray JC, Nelis M, Ohlsson C, Oldehinkel AJ, Ong KK, Ouwehand WH, Pasterkamp G, Peters A, Pramstaller PP, Price JF, Qi L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Ritchie M, Rudan I, Salomaa V, Samani NJ, Saramies J, Sarzynski MA, Schwarz PE, Sebert S, Sever P, Shuldiner AR, Sinisalo J, Steinthorsdottir V, Stolk RP, Tardif JC, Tonjes A, Tremblay A, Tremoli E, Virtamo J, Vohl MC, Amouyel P, Asselbergs FW, Assimes TL, Bochud M, Boehm BO, Boerwinkle E, Bottinger EP, Bouchard C, Cauchi S, Chambers JC, Chanock SJ, Cooper RS, de Bakker PI, Dedoussis G, Ferrucci L, Franks PW, Froguel P, Groop LC, Haiman CA, Hamsten A, Hayes MG, Hui J, Hunter DJ, Hveem K, Jukema JW, Kaplan RC, Kivimaki M, Kuh D, Laakso M, Liu Y, Martin NG, Marz W, Melbye M, Moebus S, Munroe PB, Njolstad I, Oostra BA, Palmer CN, Pedersen NL, Perola M, Perusse L, Peters U, Powell JE, Power C, Quertermous T, Rauramaa R, Reinmaa E, Ridker PM, Rivadeneira F, Rotter JI, Saaristo TE, Saleheen D, Schlessinger D, Slagboom PE, Snieder H, Spector TD, Strauch K, Stumvoll M, Tuomilehto J, Uusitupa M, van der Harst P, Volzke H, Walker M, Wareham NJ, Watkins H, Wichmann HE, Wilson JF, Zanen P, Deloukas P, Heid IM, Lindgren CM, Mohlke KL, Speliotes EK, Thorsteinsdottir U, Barroso I, Fox CS, North KE, Strachan DP, Beckmann JS, Berndt SI, Boehnke M, Borecki IB, McCarthy MI, Metspalu A, Stefansson K, Uitterlinden AG, van Duijn CM, Franke L, Willer CJ, Price AL, Lettre G, Loos RJ, Weedon MN, Ingelsson E, O’Connell JR, Abecasis GR, Chasman DI, Goddard ME, Visscher PM, Hirschhorn JN, Frayling TM. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lettre G. Genetic regulation of adult stature. Curr Opin Pediatr. 2009;21:515–522. doi: 10.1097/MOP.0b013e32832c6dce. [DOI] [PubMed] [Google Scholar]
- 23.Johansson A, Jonasson I, Gyllensten U. Extended haplotypes in the growth hormone releasing hormone receptor gene (GHRHR) are associated with normal variation in height. PLoS One. 2009;4:e4464. doi: 10.1371/journal.pone.0004464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klammt J, Neumann D, Andrew SF, Vokurkova D, Stobbe H, Buckham K, Rosenfeld RG, Pfäffle R, Hwa V. Endocrine Society’s 96th Annual Meeting and Expo. Chicago: Endocrine Society; 2014. Severe Short Stature and GH Insensitivity Due to a De Novo Heterozygous STAT5B Missense Mutation; pp. OR24–23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The largest Brazilian family with STAT5B c.424_427del mutation. Subjects homozygous for STAT5B c.424_427del mutation are indicated by black symbols; heterozygous carriers by gray symbols and non-carriers by white symbols. The symbol * refers to two individuals with severe pneumopathy of unknown etiology.