

Abstract
Nonalcoholic fatty liver disease is a spectrum that ranges from benign steatosis to steatohepatitis. It has become the most common cause of chronic liver disease, and yet there continues to be a lack of effective therapeutic options. This article reviews current concepts underlying the pathophysiologic basis of nonalcoholic steatohepatitis from development of insulin resistance to the establishment of fibrosis. Then using a physiology-based approach, specific targeted therapeutics are reviewed along with their drawbacks. The evidence behind current therapies are based predominantly on small trials and as such, no recommendations can be made until larger randomized trials are conducted.
Keywords: nonalcoholic fatty liver, nonalcoholic steatohepatitis, cirrhosis, vitamin E, thiazolidinediones, betaine, pentoxyfylline, insulin resistance, fatty acids, pathophysiology
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in North America.1 It is a spectrum that ranges from benign hepatic steatosis to nonalcoholic steatohepatitis (NASH). NASH can subsequently progress to cirrhosis and predispose patients to hepatocellular carcinoma (HCC) and to an increased risk of cardiovascular mortality. What was once referred to as “cryptogenic” cirrhosis is now thought to be sequelae of NASH. Recent advances in deciphering NASH pathogenesis have led to numerous clinical trials aimed at halting progression to cirrhosis. To date, there is no optimal treatment, underscoring the need for further efforts in delineating causality from correlative inferences. This article focuses on some proposed mechanisms of NASH, the corresponding therapeutics and their drawbacks, and future potential targets.
Pathophysiologic basis for therapeutics in NASH
The histologic hallmark of nonalcoholic fatty liver disease is the development of predominantly macrovesicular steatosis. In NASH, there is additional hepatic inflammation, hepatocyte injury manifesting as cytologic ballooning with or without Mallory’s hyaline, and varying stages of fibrosis. To understand the pathophysiologic rationale for the treatment of NASH, it is useful to consider the development of hepatic steatosis, then hepatic injury-apoptosis, and lastly inflammation and fibrosis.
A: Development of hepatic steatosis
It has been known for at least two decades that NAFLD is associated with obesity, hypertension, type 2 diabetes mellitus, and dyslipidemia, the clinical hallmarks of the metabolic syndrome. It is therefore not surprising that both non-alcoholic fatty liver (NAFL) and NASH are strongly associated with insulin resistance (IR). IR develops from macrophage infiltration into mainly visceral adipose tissue where it incites an inflammatory response and secretion of adipokines with a predominantly pro-inflammatory, pro-fibrotic profile. These are further augmented by the acute phase reaction of the liver. Thus, both systemically and in the hepatic milieu, there is an excess of pro-inflammatory cytokines such as TNF-α and IL-6. The metabolic consequence of this state is recognized as IR and is operationally defined by the ability to clear glucose from circulation at a given level of insulin. IR is thus not a categorical state but rather a continuous variable. Several factors have been implicated in the initial genesis of adipose tissue inflammation including relative ischemia and production of the hypoxia inducible factor-1, specific gut microflora and microflora-dependent inflammatory responses and hormones such as leptin.2,3
A key consequence of IR is resistance to insulin-mediated suppression of lipolysis; this results in a net increase in lipolytic activity and release of free fatty acids (FA) into the circulation. Free FA are derived from diet and by de novo lipogenesis (DNL) in the liver. DNL is driven by hyperinsulinemia, the initial pancreatic response to peripheral IR, and retained sensitivity to the lipogenic effects of insulin in the liver. The accumulation of triacylglycerol (TAG) is a function of the dynamic balance between TAG formation and turnover. This is modulated by the host genetic background, cytokine milieu of the liver, and cellular elements other than hepatocytes in the liver. The endocannabinoid system has been shown to be an important driver of DNL. Recently, autophagy has been shown to be a key mediator of turnover of cellular components and to minimize lipid accumulation in the liver. Decreased autophagy is associated with hepatic steatosis.
B: Development of steatohepatitis
The major differences between hepatic steatosis and steatohepatitis are the greater apoptosis and inflammation in NASH along with cytologic ballooning. NASH is associated with an increased propensity to progress to cirrhosis. Several mechanisms have been implicated in the pathogenesis of cell injury in NASH. These include free FA induced cell toxicity (lipotoxicity), oxidative stress (OS), endoplasmic reticulum (ER) stress and activation of the innate immune system, and cytokine-mediated cellular changes (Figure 1). Free FA can cause cellular injury in several ways including direct activation of inflammatory pathways, ER stress and activation of the innate immune system via toll like receptors.
Figure 1.
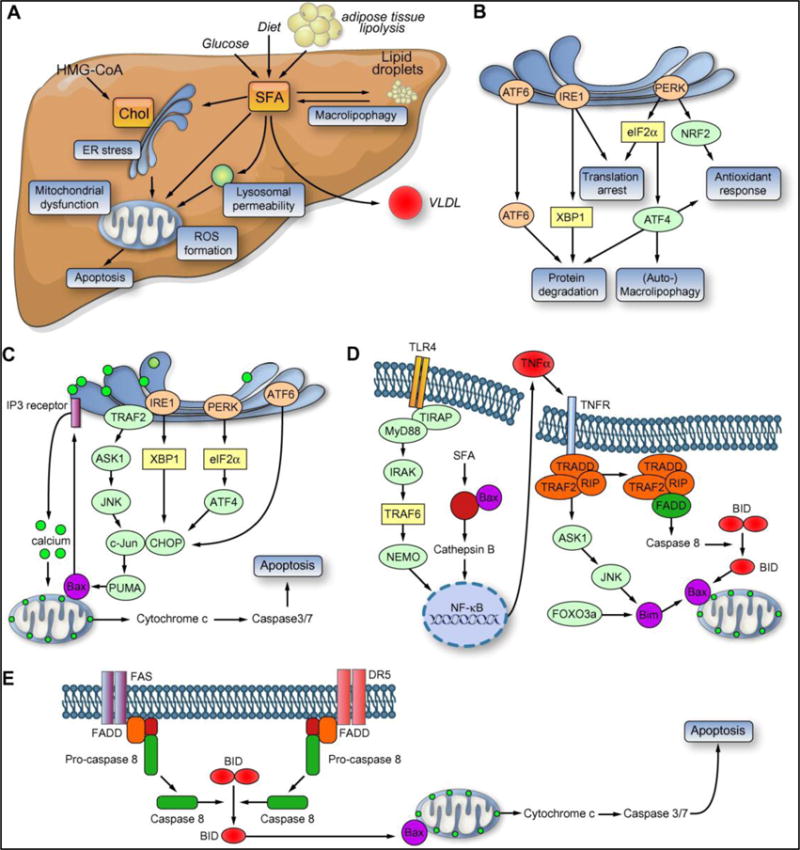
Chol, cholesterol; SFA, saturated fatty acid; VLDL, very low density lipoprotein; ER, endoplasmic reticulum; ROS, reactive oxygen species; IRE1, inositol-requiring enzyme 1; JNK, c-Jun N-terminal kinase; PUMA, p53 upregulated modulator of apoptosis; PERK, PKR-like endoplasmic reticulum kinase; eIF2a, alpha subunit of the eukaryotic initiation factor 2; ATF4/6, activating transcription factor 4/6; CHOP, CCAAT/enhancer-binding homologous protein; Bim, Bcl-2 protein family member; DR5, death receptor 5. NASH, non-alcoholic steatohepatitis; XBP1, X-box protein 1; IP3, inositol triphosphate activated calcium channel; TRAF2, tumor necrosis factor receptor-associated factor 2; ASK1, apoptosis signalregulating kinase 1; CHOP, C/EBP homologous protein; Bax, B-cell lymphoma 2-associated X protein; PUMA, p53 upregulated modulator of apoptosis; TIRAP, Toll/IL-1 receptor domain containing adaptor protein; MyD88, myeloid differentiation factor 88; IRAK, interleukin 1 receptor associated kinase; TRAF2/6, TNF receptor associated factor 2/6; NEMO, NFjB essential modulator; TRADD, TNF receptor associated death domain protein; RIP, receptor interacting protein; FADD, Fas-associated protein with death domain; BID, pro-apoptotic BCL-2 interacting domain; FoxO3a, forkhead box-containing protein, class O member 3a; TNFa, tumor necrosis factor a; NFjB, nuclear factor j B; NRF2, NF-E2 related factor 2.
There are several potential sources of OS in NASH. NASH has been shown to be associated with mitochondrial injury and impairment of the electron transport system.4,5 There is also activation of the cytochrome P450 2E1 with futile cycling which leads to production of reactive oxygen species. There is also evidence for peroxisomal dysfunction in NASH.6,7 A consequence of OS is activation of transcriptional factors such as Nrf-1 which turn on genetic pathways to limit injury due to OS. OS leads to activation of inflammatory pathways such as the JNK and NFκB pathways. Glutathione is a major hepatic anti-oxidant and its turnover is increased under conditions of OS. Glutathione store repletion requires extracellular release of glutamyl-cysteine, which is taken up and converted to glutathione. Cystathionine serves as another source of cysteine and is linked to s-adenosyl homocysteine and s-adenosyl methionine (sAME). OS can deplete sAME and also affect gene expression via epigenetic modulation of the methylation status of DNA.
Apoptosis occurs due to both lipotoxicity and OS. ER stress contributes to apoptosis especially because the proteosomal degradation arm of ER stress response is impaired in NASH.8
Ultimately, disease progression is a function of cell injury and tissue restitution versus fibrosis and progressive architectural destruction of the liver. It has been shown that bone marrow derived macrophages play an important role in the development of steatohepatitis. The role of resident and circulating stem cells in disease progression remain to be defined. The activation of the sonic hedgehog pathway and its downstream target osteopontin appear to be important in this process.9
All of these provide treatment targets for NASH. The statuses of current therapies are discussed below in their pathophysiology-based context.
Dietary Excess and Obesity
The mainstay of NASH therapy has been to promote diet and lifestyle modifications as these have been shown to improve insulin sensitivity. A high fat diet increases hepatic levels of anandamide, cannabinoid receptor 1 (CB1) density, and basal rates of FA synthesis in the liver. Anandamide is an endogenous cannabinoid that activates CB1 receptors on hepatocytes that subsequently leads to over expression of transcription factors (TF) and lipogenic genes, contributing to de novo FA synthesis in the liver.
Currently, there is no consensus on a specific diet for NASH. A 2003 review of a variety of diets and formal guidelines from the American Heart Association and the American Diabetes Association comparing the relative composition of macronutrients may be helpful in guiding clinicians in counseling patients with concomitant cardiovascular disease or renal dysfunction as certain diets are higher in protein versus essential FA.10 Because compliance is a major limiting factor, clinicians should individualize diet plans and avoid being too aggressive. Weight loss of 7–10% has been shown to lead to histologic improvement in NASH.11 Weight loss should be gradual over 6 months as sudden rapid weight loss has been associated with further progression of liver disease.12 Bariatric surgery has yielded conflicting results according to a 2010 Cochrane review, and there is no general recommendation for this procedure due to a lack of randomized control trials.13
Insulin Resistance and Pharmacologic Treatment (Table 1)
Table 1.
Drugs Tested for Use in NASH
Insulin sensitizers:
|
| Angiotensin Converting Enzyme Inhibitors and receptor blockers |
Anti-oxidants and hepatoprotectants
|
Bile Acids
|
Cytokine mediated pathways
|
Lipid Lowering Agents
|
Endocannabinoid System
|
Insulin Sensitizers
Metformin is a biguanide that inhibits hepatic gluconeogenesis and enhances insulin sensitivity in muscle and fat. It improves hepatic IR by enhancing 5′AMP-activated protein kinase signaling. This reduces lipid accumulation, glucose output and TNF-α signaling. At this time, metformin use remains in clinical trials, many of which are small and lack histologic evidence of improvement.14,15 A recent large clinical trial demonstrated that metformin had no impact on any of the histologic features of NASH.16 The use of this drug for NASH is therefore discouraged.
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor (PPAR)γ agonists. PPARγ is a nuclear hormone receptor that is expressed predominantly in adipose tissue, and regulates adipocyte uptake of FA as well as glucose metabolism. The exact mechanisms of TZDs in NAFLD remain unclear. Recent data suggest that hepatic overexpression of PPARγ, specifically the isoform PPARγ1, promotes hepatic steatosis via inducing adipogenic transformation of hepatocytes.
To date, there are 5 published trials comparing rosiglitazone or pioglitazone to placebo, metformin and lifestyle modification. These trials vary in size, duration and endpoints, making a generalizable conclusion difficult. A 2010 meta-analysis of these 5 trials found that TZDs improved steatosis, hepatocyte ballooning and serum ALT levels.17 Of interest is the lack of improvement in fibrosis for diabetic patients; however, in non-diabetics, fibrosis was improved. Liver enzymes worsened after discontinuation of pioglitazone, suggesting that long-term use may be necessary.18 Due to significant side effects of TZDs (i.e. weight gain, cardiovascular events, fractures), long-term use of TZDs have not been studied. Given these limitations, TZDs should be reserved as second line treatment.
Incretin Mimetics
Incretins are endogenous hormones that are secreted from the gastrointestinal tract such as glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). These hormones increase β-cell proliferation, delay gastric emptying, and regulate appetite. GLP-1 inhibits glucose dependent pancreatic glucagon secretion, decrease gluconeogenesis and glycogenolysis, and decrease liver fat. In diabetics, GLP-1 secretion after meals is reduced. These hormones are rapidly deactivated by dipeptidyl peptidase (DPP)-4.
The drugs exanatide and liraglutide are GLP-1 receptor agonists that are resistant to DPP-4 breakdown. Preliminary data from a 3 year study demonstrated reduction in aminotransferase levels, weight loss, and better glycemic control.19 A newly published trial has shown that exanitide in combination with pioglitazone compared to pioglitazone alone reduced hepatic steatosis and plasma fibroblast growth factor 21 levels.20 Studies at this time are limited and are not histologically proven. Given the cost of exanatide, the need for parenteral route of administration, the risks benefits of GLP-1 agonists remain unclear and therefore no recommendations can be made at this time.
Angiotensin converting enzyme inhibitors and receptor blockers
The renin-angiotensin system (RAS) is an enzymatic cascade with a wide array of systemic effects. Some proposed mechanisms include: 1) interactions with insulin receptors and intracellular signaling pathways; 2) modulation of adipogenesis; 3) influences on cytokine and adipokine production; 4) interference with pancreatic β-cell insulin secretion; 5) local hepatic effects that interfere with hepatocellular regulatory mechanisms.21 Small trials have demonstrated improvement in biomarkers and histology.22,23 A 2009 randomized trial compared the effects of telmisartan and valsartan in patients with the metabolic syndrome and showed that telmisartan improved IR, necrosis, and fibrosis. Valsartan did not improve histology apart from steatosis and did not improve plasma lipids.24 Due to the lack of large randomized controlled trials, no recommendations can be made at this time.
Treatments directed at mechanisms of hepatocyte injury
Anti-oxidants and hepatoprotectants
Vitamin E consists of eight tocopherols, of which α-tocopherol is the most active. Its primary function is to prevent the non-enzymatic oxidation of cell components by molecular oxygen and free radicals. There are few randomized controlled trials with overall small numbers (Table 2).15,16,25–28 Improvements in ALT, and histology were noted, but were not statistically significant in some smaller trials. The PIVENS trial29 is the largest randomized controlled trial to date comparing pioglitazone and vitamin E at 800 IU per day to placebo. Vitamin E improved hepatic steatosis, lobular inflammation, and serum ALT and AST levels, but no improvement was found in fibrosis.18 It is important to note that these patients were non-diabetics, and that not all patients would benefit from high doses of Vitamin E. Other studies have implicated Vitamin E with an increased risk of hemorrhagic strokes, and at high doses to increased all cause mortality.30,31 It must be remembered that in making decisions to use vitamin E, one has to consider the competing risks of the drug versus the disease it is being used for. In patients with active NASH (NAS ≥ 4) without diabetes, Vitamin E at 800 IU/day is recommended. The cardiovascular risk profile should be optimized as standard of care in such patients. Also, in light of recent data implicating vitamin E in prostrate cancer risk, men over the age of 50 should undergo close monitoring and these risks should be discussed with the patients before starting treatment.32
Table 2.
Clinical Trials of Vitamin E for NASH
| Authors | n | Dose | Comparators | Histology |
|---|---|---|---|---|
| Arendt | 80 | 1000 IU/day | Placebo | Improvedˆ |
| Sanyal | 247 | 800 IU/day | Pioglitazone, placebo | Improved* |
| Lavine | 173 | 800 IU/day | Metformin, placebo | Improved*** |
| Harrison | 45 | 1000 IU/day | Placebo | Improved** |
| Sanyal | 10 | 400 IU/day | Vit E + pioglitazone | Improved* |
| Dufour | 48 | 800 IU/day | UDCA+placebo, placebo | Improved* |
CT scan assessment of steatosis only
All histologic parameters excluding fibrosis
fibrosis improvement
steatohepatitis and ballooning
Betaine (trimethylglycine), a metabolite of choline, has been demonstrated in animal models to prevent development of steatosis via increasing activation of hepatic AMP-activated protein kinase. It also reduces levels of S-adenosylhomocysteine and homocysteine via increasing levels of S-adenosylmethionine (sAME). It improves IR, reduces abnormal adipokine production and attenuates of ER stress response induced by a high-fat diet. Adipokines induce lipolysis of TAG stores in macrophages, stimulate adipocyte triglyceride lipolysis, and ultimately increase cellular levels of free FA. However, clinical trials have yet to prove efficacy. One trial demonstrated that betaine did not improve steatosis.33 At this time, betaine is not recommended as further clinical trials are warranted.
Silymarin (milk thistle) is a flavolignan extract from the plant Silybum marianum. It has demonstrated antioxidant and hepatoprotectant properties, but clinical trials are lacking. One trial showed improvement in steatosis with combination Vitamin E, silymarin, and phospholipids.34 It is difficult to distinguish if this improvement was due to silymarin itself or Vitamin E or both. No recommendations can be made until further clinical trials have dealt specifically with silymarin supplementation.
Bile Acids
Bile acids, apart from aiding in dietary lipid absorption and cholesterol homeostasis also act as metabolic signaling molecules. They are reabsorbed into the enterohepatic circulation and coordinate hepatic triglyceride and glucose metabolism by activating protein kinase pathways. They activate nuclear receptor farsenoid X receptor (FXR), which is involved in hepatic DNL, VLDL-TG export, plasma triglyceride turnover, hepatic gluconeogenesis, glycogen synthesis, and insulin sensitivity. Bile acids bind TGR5, with subsequent stimulation of GLP-1 secretion in the small intestine and increased metabolism within adipose and skeletal tissue.
Ursodeoxycholic acid (UDCA) has been shown in animal models to improve hepatic endoplasmic stress and insulin sensitivy. NorUDCA is a side chain shortened homologue of UDCA and has been shown to attenuate progression to NASH in murine models. In a 2007 Cochrane review of 4 randomized clinical trials involving UDCA, there was no significant improvement in liver function tests, no association with mortality, and histological data was lacking.35 No recommendations could be drawn from the review.
FXR agonists, such as 6-ethyl-chedeoxycholic acid (6E-CDCA), is currently under investigation for primary biliary cirrhosis and NAFLD. Preliminary data show reduced ALP levels and amelioration of insulin.36 Further human studies are warranted.
Cytokine mediated therapy
Chronic OS leads to the abnormal secretion of inflammatory cytokines TNF-α and IL-6. Adipose tissue can produce adipocytokine. These in turn increase lipolysis and release of FA, which are pro-inflammatory. Free FA activate the lysosomal pathway of cell death and regulate death receptor gene expression.
Pentoxifylline (PTX) is a TNF-α antagonist. Small human studies have found that PTX significantly improved steatosis, lobular inflammation, but not cellular ballooning. Fibrosis was improved, but not statistically significant compared to placebo, and transaminase reduction was also insignificant.37,38 Other small studies showed significant reduction in AST and significant improvement in fibrosis.39,40 Side effects noted were nausea and headache. Prelimary data are promising, but larger trials are needed still. Given its safe profile, PTX can be considered in patients with severe disease and who have not responded to diet and exercise.
Lipid Lowering Agents
N-6-polyunsaturated fatty acids (PUFAs) such as arachidonic acid (AA) can oxidize into eicosanoids and potentiate inflammation. Oxidized PUFAs lead to lipid peroxidation and alter the phospholipid composition of cellular membranes. N-3 polyunsaturated fatty acids (N-3-PUFA) unlike the n-6 PUFAs have anti-inflammatory properties by inhibiting lymphocyte proliferation, antibody and cytokine production, adhesion molecule expression, and natural killer cell activity. Examples of N-3 PUFAs are α-linolenic acid, docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA). These inhibit conversion of AA to thromboxane A2 (TXA2). In a 2008 pilot trial, 23 patients were given EPA 2700mg/day for 12 months. Biopsies demonstrated improvement in steatosis, fibrosis, ballooning, and lobular inflammation in 6 patients. Serum ALT, free FA, TNF receptor 1 and 2, and ferritin levels were reduced.41 There were no significant reported side effects. Larger clinical trials are needed.
Fibrates have been used in mice models, and small pilot studies in humans have shown potential in modulating development of NASH.42 Larger trials are warranted. Ezetimibe is a cholesterol absorption inhibitor. A small trial of 6 patients given ezetimibe for 6 months demonstrated reduction in serum hepatic biomarkers. Liver biopsies showed improvement in steatosis, but not fibrosis.43
Statins are HMG coA reductase inhibitors, antioxidants. They increase adiponectin levels, which is associated with higher insulin sensitivity, enhance PPAR-gamma activation, and lower TNF-α levels. Multiple small studies were conducted with varying results in terms of significant histologic improvement.44–46 Further studies are needed before recommendations can be made for NAFLD therapy.
Endocannabinoid antagonists
The ultimate progression to fibrosis and cirrhosis is a result of chronic inflammation. Recent data implicate the endocannabinoid system with development of fibrosis. Endocannabinoids are endogenous lipid compounds that bind and activate the cannabinoid receptors (CB1 and CB2). They modulate mood, energy balance, immune and inflammatory responses, and have vasoregulatory and lipogenic effects. CB1 receptors are found in the liver and adipose tissue, and their activation within adipose tissue increases lipolysis with subsequent triglyceride influx into the liver. Within the liver, activation of CB1 leads to overexpression of lipogenic transcription factor sterol regulatory element binding proteins (SREBP-1), promoting de novo FA synthesis. In striking contrast, CB2 receptors portend anti-inflammatory effects and prevent steatosis and fibrosis.47 Several large clinical trials with rimonabant, a CB1 receptor antagonist, have shown a significant reduction in weight, triglyceride levels, and increase in HDL levels, however studies were limited by report of severe depression.48,49 No recommendations can be made at this time and further studies are warranted.
Conclusions
The mechanisms underlying NAFLD pathogenesis remain elusive, but many animal studies of late hold promise for future clinical trials. These targeted therapies have been studied in small human trials, which have varied in statistical significance that could not be powered within meta-analyses. At this time, there is no consensus on optimal management of NAFLD and further research is needed in this rapidly evolving field.
Acknowledgments
This review was written entirely by the authors without any external assistance. It is partly supported by a grant from the NIH to Dr. Sanyal T32 DK 007150-35.
Footnotes
Conflict of Interest
No conflict of interest has been declared by the authors.
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Gruen ML, Hao M, Piston DW, Hasty AH. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am J Physiol Cell Physiol. 2007;293:C1481–8. doi: 10.1152/ajpcell.00062.2007. [DOI] [PubMed] [Google Scholar]
- 3.Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr. 2008;100:227–35. doi: 10.1017/S0007114508971282. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Carreras M, Del Hoyo P, Martin MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 5.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 6.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–8. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 7.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–90. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 8.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–76. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 9.Syn WK, Jung Y, Omenetti A, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–88 e8. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86:285–300. doi: 10.1093/ajcn/86.2.285. [DOI] [PubMed] [Google Scholar]
- 11.Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotler SJ, Vitello JM, Guzman G, Testa G, Benedetti E, Layden TJ. Hepatic decompensation after gastric bypass surgery for severe obesity. Dig Dis Sci. 2004;49:1563–8. doi: 10.1023/b:ddas.0000043364.75898.c8. [DOI] [PubMed] [Google Scholar]
- 13.Chavez-Tapia NC, Tellez-Avila FI, Barrientos-Gutierrez T, Mendez-Sanchez N, Lizardi-Cervera J, Uribe M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst Rev. 2010:CD007340. doi: 10.1002/14651858.CD007340.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uygun A, Kadayifci A, Isik AT, et al. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2004;19:537–44. doi: 10.1111/j.1365-2036.2004.01888.x. [DOI] [PubMed] [Google Scholar]
- 15.Bugianesi E, Gentilcore E, Manini R, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100:1082–90. doi: 10.1111/j.1572-0241.2005.41583.x. [DOI] [PubMed] [Google Scholar]
- 16.Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305:1659–68. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rakoski MO, Singal AG, Rogers MA, Conjeevaram H. Meta-analysis: insulin sensitizers for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2010;32:1211–21. doi: 10.1111/j.1365-2036.2010.04467.x. [DOI] [PubMed] [Google Scholar]
- 18.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satapathy SK, Sanyal AJ. Novel treatment modalities for nonalcoholic steatohepatitis. Trends Endocrinol Metab. 2010;21:668–75. doi: 10.1016/j.tem.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Samson SL, Sathyanarayana P, Jogi M, et al. Exenatide decreases hepatic fibroblast growth factor 21 resistance in non-alcoholic fatty liver disease in a mouse model of obesity and in a randomised controlled trial. Diabetologia. 2011 doi: 10.1007/s00125-011-2317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Georgescu EF. Angiotensin receptor blockers in the treatment of NASH/NAFLD: could they be a first-class option? Adv Ther. 2008;25:1141–74. doi: 10.1007/s12325-008-0110-2. [DOI] [PubMed] [Google Scholar]
- 22.Enjoji M, Kotoh K, Kato M, et al. Therapeutic effect of ARBs on insulin resistance and liver injury in patients with NAFLD and chronic hepatitis C: a pilot study. Int J Mol Med. 2008;22:521–7. [PubMed] [Google Scholar]
- 23.Yokohama S, Yoneda M, Haneda M, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–5. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 24.Georgescu EF, Ionescu R, Niculescu M, Mogoanta L, Vancica L. Angiotensin-receptor blockers as therapy for mild-to-moderate hypertension-associated non-alcoholic steatohepatitis. World J Gastroenterol. 2009;15:942–54. doi: 10.3748/wjg.15.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dufour JF, Oneta CM, Gonvers JJ, et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4:1537–43. doi: 10.1016/j.cgh.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 26.Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003;98:2485–90. doi: 10.1111/j.1572-0241.2003.08699.x. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther. 2001;15:1667–72. doi: 10.1046/j.1365-2036.2001.01083.x. [DOI] [PubMed] [Google Scholar]
- 28.Yakaryilmaz F, Guliter S, Savas B, et al. Effects of vitamin E treatment on peroxisome proliferator-activated receptor-alpha expression and insulin resistance in patients with non-alcoholic steatohepatitis: results of a pilot study. Intern Med J. 2007;37:229–35. doi: 10.1111/j.1445-5994.2006.01295.x. [DOI] [PubMed] [Google Scholar]
- 29.Chalasani NP, Sanyal AJ, Kowdley KV, et al. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp Clin Trials. 2009;30:88–96. doi: 10.1016/j.cct.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 31.Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2008;300:2123–33. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein EA, Thompson IM, Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2011;306:1549–56. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdelmalek MF, Sanderson SO, Angulo P, et al. Betaine for nonalcoholic fatty liver disease: results of a randomized placebo-controlled trial. Hepatology. 2009;50:1818–26. doi: 10.1002/hep.23239. [DOI] [PubMed] [Google Scholar]
- 34.Loguercio C, Federico A, Trappoliere M, et al. The effect of a silybin-vitamin e-phospholipid complex on nonalcoholic fatty liver disease: a pilot study. Dig Dis Sci. 2007;52:2387–95. doi: 10.1007/s10620-006-9703-2. [DOI] [PubMed] [Google Scholar]
- 35.Orlando R, Azzalini L, Orando S, Lirussi F. Bile acids for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst Rev. 2007:CD005160. doi: 10.1002/14651858.CD005160.pub2. [DOI] [PubMed] [Google Scholar]
- 36.Fiorucci S, Cipriani S, Mencarelli A, Baldelli F, Bifulco G, Zampella A. Farnesoid X receptor agonist for the treatment of liver and metabolic disorders: focus on 6-ethyl-CDCA. Mini Rev Med Chem. 2011;11:753–62. doi: 10.2174/138955711796355258. [DOI] [PubMed] [Google Scholar]
- 37.Van Wagner LB, Koppe SW, Brunt EM, et al. Pentoxifylline for the treatment of non-alcoholic steatohepatitis: a randomized controlled trial. Ann Hepatol. 2011;10:277–86. [PubMed] [Google Scholar]
- 38.Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology. 2011 doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–8. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 40.Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:634–8. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, Aoyama T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol. 2008;42:413–8. doi: 10.1097/MCG.0b013e31815591aa. [DOI] [PubMed] [Google Scholar]
- 42.Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384. doi: 10.1016/s0168-8278(99)80243-8. [DOI] [PubMed] [Google Scholar]
- 43.Yoneda M, Fujita K, Nozaki Y, et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol Res. 2010;40:613–21. doi: 10.1111/j.1872-034X.2010.00644.x. [DOI] [PubMed] [Google Scholar]
- 44.Hyogo H, Ikegami T, Tokushige K, et al. Efficacy of pitavastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: An open-label, pilot study. Hepatol Res. 2011 doi: 10.1111/j.1872-034X.2011.00849.x. [DOI] [PubMed] [Google Scholar]
- 45.Hyogo H, Tazuma S, Arihiro K, et al. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism. 2008;57:1711–8. doi: 10.1016/j.metabol.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 46.Ekstedt M, Franzen LE, Mathiesen UL, Holmqvist M, Bodemar G, Kechagias S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol. 2007;47:135–41. doi: 10.1016/j.jhep.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Mallat A, Teixeira-Clerc F, Deveaux V, Manin S, Lotersztajn S. The endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openings. Br J Pharmacol. 2011;163:1432–40. doi: 10.1111/j.1476-5381.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Gaal LF, Scheen AJ, Rissanen AM, Rossner S, Hanotin C, Ziegler O. Long-term effect of CB1 blockade with rimonabant on cardiometabolic risk factors: two year results from the RIO-Europe Study. Eur Heart J. 2008;29:1761–71. doi: 10.1093/eurheartj/ehn076. [DOI] [PubMed] [Google Scholar]
- 49.Despres JP, Golay A, Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–34. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]