

Abstract
Salivary gland adenoid cystic carcinoma (ACC) is a rare head and neck malignancy without molecular biomarkers that can be used to predict the chemotherapeutic response or prognosis of ACC. The regulation of gene expression of oncogenes and tumor suppressor genes (TSGs) through DNA promoter methylation may play a role in the carcinogenesis of ACC. To identify differentially methylated genes in ACC, a global demethylating agent, 5-aza-2′-deoxycytidine (5-AZA) was utilized to unmask putative TSG silencing in ACC xenograft models in mice. Fresh xenografts were passaged, implanted in triplicate in mice that were treated with 5-AZA daily for 28 days. These xenografts were then evaluated for genome-wide DNA methylation patterns using the Illumina Infinium HumanMethylation27 BeadChip array. Validation of the 32 candidate genes was performed by bisulfite sequencing (BS-seq) in a separate cohort of 6 ACC primary tumors and 6 normal control salivary gland tissues. Hypermethylation was identified in the HCN2 gene promoter in all 6 control tissues, but hypomethylation was found in all 6 ACC tumor tissues. Quantitative validation of HCN2 promoter methylation level in the region detected by BS-seq was performed in a larger cohort of primary tumors (n=32) confirming significant HCN2 hypomethylation in ACCs compared with normal samples (n=10; P=0.04). HCN2 immunohistochemical staining was performed on an ACC tissue microarray. HCN2 staining intensity and H-score, but not percentage of the positively stained cells, were significantly stronger in normal tissues than those of ACC tissues. With our novel screening and sequencing methods, we identified several gene candidates that were methylated. The most significant of these genes, HCN2, was actually hypomethylated in tumors. However, promoter methylation status does not appear to be a major determinant of HCN2 expression in normal and ACC tissues. HCN2 hypomethylation is a biomarker of ACC and may play an important role in the carcinogenesis of ACC.
Keywords: hyperpolarization-activated cation channel 2, DNA methylation, oncogene, salivary gland, adenoid cystic carcinoma, bisulfite sequencing, methylation-specific PCR, beadchip array
Introduction
Although salivary gland adenoid cystic carcinoma (ACC) is a rare disease with roughly 500 cases annually in the USA (1), the prognosis is grave due to the clinical behavior of ACC, such as indolent growth, but frequent perineural invasion and distant metastasis (2,3). ACCs are currently managed by surgery, with postoperative radiotherapy utilized for the more aggressive cases. The effectiveness of adjuvant chemotherapy or targeted therapy to date has been limited. No molecular biomarkers are yet available that can either be used to predict the chemotherapeutic response or prognosis of ACC, or serve as chemotherapeutic targets, and is due to the fact that the molecular pathogenesis remains poorly understood (2,4).
Recent whole-genome and exome sequencing studies (5,6) have confirmed some known mutations and uncovered a handful of new genomic alterations in the ACC mutational landscape, such as somatic driver mutations in PIK3CA, FGFR2, NOTCH1/2 and the negative NOTCH signaling regulator, SPEN (6). Ho et al (5) found somatic mutations in genes belonging to the DNA damage response and protein kinase A signaling pathways. Both Ho et al (5) and Stephens et al (6) identified a high percentage of mutations in chromatin regulating genes that are epigenetic modifiers of gene activity (5,6). Some of the genetic alterations uncovered by sequencing studies corroborated the previous findings from molecular studies of ACC, such as KIT overexpression (7–10). Notably, several oncogenes and tumor suppressor genes that are altered at high frequency in other types of solid tumors, such as CDKN2A, TP53, EGFR, ERBB2 and PTEN (11), appear unaffected or rarely altered in ACC (5,6,11). The FGF-IGF-PI3K pathway is among these; the Stephen et al found no genetic mutations in this pathway (6), while Ho et al (5) found recurrent mutations in the FGF-IGF-PI3K pathway in only 30% of ACCs. Furthermore, similarly to that found previously by next-generation sequencing in 24 ACCs (6), Stephens et al recently found similar, low frequency of genomic alterations in 28 cases of the relapsed and metastatic ACCs by the same sequencing technique (12). Again, like in the 24 primary ACCs (6), these genetic alterations found in the relapsed and metastatic ACCs were also low frequency events, compared with these same genetic alterations seen in the more common solid tumors (12). This suggests that the low frequency of genomic alterations may not account for the relapse and metastasis of ACCs. Taken together, although some novel and known genetic alterations have been identified in ACCs and these genomic alterations may contribute to the molecular pathogenesis of ACC, the relatively low frequency of any genetic mutation uncovered in primary, relapsed, and metastatic ACCs suggests that epigenetic alterations may also contribute in an important way to the pathogenesis of ACC (11).
The molecular pathogenesis of ACC still remains unclear. The most common molecular alterations found in ACC are the t(6;9)(q22-23;p23-24) translocation resulting in the MYB-NFIB fusion gene, which occurs in 29 to 86% of ACCs (3,5,6,13–16), and overexpression of the MYB protein, observed in 89–97% of ACCs (15,16). The role of these two molecular alterations in ACC pathogenesis is not well understood. MYB overexpression is often (15,17), but not always (13–16,18), associated with the MYB-NFIB fusion, multiple MYB-NFIB fusion variants due to the differential breakpoints have also been reported (13), and NFIB has been found to fuse with non-MYB partners in ACC (19), so that the relationship between these two molecular events is also unclear. Neither MYB-NFIB fusion nor MYB overexpression has consistently been found to be associated with prognostic features. Therefore, while improved understanding of these alterations is imperative for elucidating the pathogenesis of ACC, it is also necessary to explore additional aspects of the unique pathology of ACC.
In the present study, we utilized a global demethylating agent, 5-aza-2′-deoxycytidine (5-AZA), to unmask silencing of putative TSGs in ACC xenograft models and a DNA methylation array to identify oncogene and TSG candidates under the control of promoter methylation in ACC. Our approach was to circumvent the lack of viable ACC cell lines (20) by using primary xenograft tumor models, in an attempt to identify relevant genes exhibiting differential promoter methylation.
Materials and methods
Genomic DNA extraction from mouse xenografts of ACC tumors
Freshly resected ACC tumors from three different patients were transplanted in nude mice to establish ACC xenografts. The establishment of mouse xenografts with ACC tumor has been reported (21). When the xenografts reached 125–250 mm3, mice were randomly assigned into two groups, control and treatment. The control group had 2 mice; the treatment group had 3 mice. The control group of 2 mice received no injection. In contrast, the mice in the treatment group received a daily subcutaneous injection of 5-aza-2′-deoxycytidine (5-Aza) at a dose of 1 mg/kg of body weight for 28 days by the South Texas Accelerated Research Therapeutics Co. (San Antonio, TX, USA). 5-Aza is a nucleoside analog that has been shown to cause hypomethylation of genes at a low dose (22–24) by depleting DNA methyltransferase 1 (25,26) and this demethylation at CpG islands in the gene promoter can often re-activate gene expression of the once methylation-silenced gene (27).
After the termination of the treatment, these xenografts of ACC tumors were harvested, snap-frozen in liquid nitrogen, and stored at −80°C before genomic DNA extraction. Two samples were harvested from each xenograft from each mouse.
The DNA was extracted from the same xenograft using AllPrep DNA/RNA Mini kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions.
Methylation array and expression array of mouse xenografts of ACC tumors
The extracted DNAs from mouse xenografts of ACC tumors were submitted to the Johns Hopkins University core facility for genome-wide DNA methylation screening using the Illumina Infinium HumanMethylation27 BeadChip array (Illumina, Inc., San Diego, CA, USA), according to the manufacturer's instructions. The quality of all DNA samples was subjected to quality control by Agilent's Bioanalyzer before methylation array.
Human tissue samples
Formalin-fixed, paraffin-embedded (FFPE) samples were obtained from 32 patients treated surgically by the Department of Otolaryngology-Head and Neck Surgery of the Johns Hopkins Medical Institutions between the years 1999 and 2009. Tissue blocks with high tumor yield were selected after all blocks had been reviewed by an experienced head and neck pathologist (J.A.B.) to confirm the diagnosis of ACC. Four sections of 20 μm thickness were cut from each block and manually micro-dissected to yield ~80% tumor purity. In addition, 20 FFPE parotid or submandibular glands that were resected either for benign disease or as part of other surgical procedures from 20 patients treated surgically by the Department of Otolaryngology-Head and Neck Surgery of the Johns Hopkins Medical Institutions between the years 1999 and 2004 were included in the present study after histologic confirmation that the tissues to be used were distant from any benign or inflammatory lesion. All tissues were obtained with the approval of the Johns Hopkins Institutional Review Board.
DNA extraction from frozen and FFPE tissues
Genomic DNA was extracted from the frozen samples of 6 normal control salivary gland tissues and 6 ACC primary tumor tissues and from microdissected FFPE tissue sections on glass slides (normal and ACC tumor) using the SDS-PK method as described previously (28,29). Briefly, a small piece of each frozen tissue (normal and ACC tumor) was cut-off using a disposable single-edge razor blade, and then collected in a 1.5-ml Eppendorf tube.
Each FFPE tissue section on a glass slide was scraped off by a disposable single-edge razor blade and then collected in a 1.5-ml Eppendorf tube to undergo deparaffinization in xylenes on a heat block in the fume hood. Xylenes were removed when deparaffinization was complete. Residual amount of xylene in each sample was washed 3 times by 100% ethanol.
Each frozen or FFPE sample was then digested in 0.02% (50 μg/ml) proteinase K (Roche, Indianapolis, IN, USA) reconstituted in 1% sodium dodecyl sulfate (SDS) at 48°C for up to 72 h until no visible tissue was seen. DNA was then purified by phenol-chloroform extraction and ethanol precipitation. DNA was subsequently resuspended in LoTE buffer (10 mM Tris-HCl and 2.5 mM EDTA), and the DNA concentration was quantified using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA was stored at −20°C until further use.
Bisulfite treatment and bisulfite genomic sequencing (BS-seq) of ACC and control genomic DNAs
The EpiTect Bisulfite kit (Qiagen) was used to convert unmethylated cytosines in genomic DNA extracted from frozen samples and FFPE samples (normal and ACC tumor) to uracils, according to the manufacturer's instructions (29). Bisulfite converted xenograft genomic DNA samples were stored at −80°C until use.
The criteria used to select genes and a region in a given gene for validation by BS-seq from the list of methylated genes detected by methylation array (data not shown) were that: i) the region selected for validation by BS-seq had to be in the promoter region (5 kb) or 5′-UTR exons. We did not necessarily validate the region corresponding to the probe set ID in methylation array. ii) There was at least one CpG island in the promoter region (5 kb) and the 5′-UTR exons were detected by MethPrimer (30) in a given gene. iii) Twenty genes selected for validation were among top 100 candidates in the gene list; the rest (12 genes) were not among top candidates, but also included due to general interest.
Validation of the 32 selected candidate genes from the methylation array was performed by BS-seq in a separate cohort of 6 ACC primary tumors and 6 normal control parotid salivary gland tissues. In brief, bisulfite-treated DNA was amplified with their primers designed using MethPrimer (30) to span areas of CpG island(s) in their promoters or with β-actin primers.
Specifically for HCN2, there are several CpG islands in its proximal promoter and 5′-UTR exon (53 bp), but bisulfite sequencing primer sets that span areas of the CpG island(s) were not available due to the high density of CpGs. The amplified area evaluated by BS-seq for the HCN2 promoter is illustrated in Figs. 1 and 2. HCN2 and β-actin primer sequences were specifically designed to contain no CG dinucleotides. The HCN2 primer sequences were: forward, 5′-GGA GGT ATT GGG GGT ATA GTT GTA T-3′, which is located at -901 bases to -877 bases upstream of ATG (the start codon) and reverse, 5′-CCA ACC AAA CAA AAA AAA CTA AAA A-3′, which is located at -656 bases to -632 bases upstream of ATG. β-actin primers were: forward, 5′-TGG TGA TGG AGG AGG TTT AGT AAG T-3′ and reverse, 5′-AAC CAA TAA AAC CTA CTC CTC CCT TAA-3′. Touch-down PCR was performed for all converted DNA samples. All PCR products were purified using the QIAquick Gel Extract PCR Purification kit (Qiagen), according to the manufacturer's instructions (29). Purified PCR products were then submitted to GeneWiz Inc. (Frederick, MD, USA) for sequencing with one of the touch-down PCR primers. The criteria to determine methylation in the electropherograms of the purified PCR products were used as previously reported (31).
Figure 1.

The schematic illustration by MethPrimer of the CpG islands and CpG dinucleotides in the HCN2 proximal promoter (2650 bp), 5′-UTR exon (53 bp to the start codon, ATG), and first CDS exon (632 bp including ATG). The total sequence length: 4685 bp. The island 1 (far left) is 140 bp (1615-1754); island 2 is 162 bp (2081-2242); the island 3 is 112 bp (2466-2577), the island 4 is 167 bp (2647-2813), and the island 5 (far right) that is composed of 5′-UTR exon, first CDS exon, and the proximal part of the first intron is 2290 bp (2202-4491). F4 and R4 are the primers used for bisulfite sequencing (BS-seq).
Figure 2.
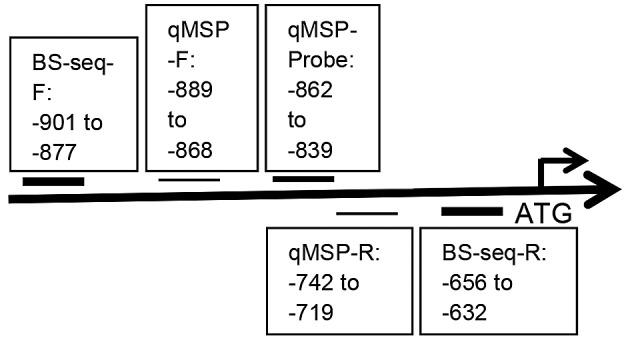
The schematic illustration of the regions in the proximal HCN2 promoter that are validated by bisulfite sequencing (BS-seq) and qMSP. BS-seq-F, bisulfite sequencing forward primer; BS-seq-R, bisulfite sequencing reverse primer; qMSP-F, qMSP forward primer; qMSP-R, qMSP reverse primer; qMSP-Probe, qMSP probe. All numberings are in reference to the start codon, ATG.
Quantitative methylation-specific PCR (qMSP) by TaqMan assay
qMSP conditions and data interpretation were previously described (28,32). Leukocyte DNA from a healthy individual was first methylated in vitro with excess SssI methyltransferase (New England Biolabs, Ipswich, MA, USA) to generate universally methylated genomic DNA according to the manufacturer's instruction, and then this universally methylated genomic DNA was treated with the EpiTect Bisulfite kit (Qiagen) to convert unmethylated cytosines in leukocyte DNA to uracil, according to the manufacturer's instructions (29). These universally methylated, bisulfite converted leukocyte DNA was made in a serial of 10-fold dilutions (90–0.009 ng) to construct two calibration curves (HCN2 and β-actin) for each 384-well microtiter plate on each run. All samples were within the range of sensitivity and reproducibility of the TaqMan assay based on the amplification of the internal reference standard, β-actin (threshold cycle value for β-actin of ≤40), as previously reported (31). The relative level of methylated DNA in each sample was determined as a ratio of qMSP-amplified HCN2 gene to β-actin (reference gene) and then multiplied by 100 for easier tabulation (average value of triplicates of the gene of interest divided by the average value of triplicates of β-actin x 100). The HCN2 qMSP primer sequences were designed by MethPrimer and their sequences were: forward, 5′-GTA TAG TTG TAT TCG GAG TTC G-3′, which is located at -889 bases to -868 bases upstream of ATG and reverse, 5′-AAC AAT ACC CTA AAA AAC CGT ACG-3′, which is located at -742 bases to -719 bases upstream of ATG. The HCN2 probe (sense) was manually designed and its sequence was 5′-/56-FAM/TCG GGG AAA GGA GGT AAT TTC GGG/36-TAMSp/-3′, which is located at -862 bases to -839 bases upstream of ATG (Figs. 1 and 2).
qMSP was carried out using the following conditions: 95°C for 5 min, followed by 45 cycles at 95°C for 15 sec and 60°C for 1 min.
ACC tissue microarray (TMA), detection of MYB-NFIB fusion by fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) staining
An ACC TMA was constructed in house. This TMA comprises of the cores of 93 ACC tumors and 10 normal salivary gland tissues. As previously reported (3), FISH was performed on formalin-fixed paraffin-embedded sections in this ACC TMA to detect MYB-NFIB fusion. For IHC, a mouse monoclonal HCN2 antibody (S71-37) was obtained from Novus Biologicals LLC (Littleton, CO, USA). HCN2 antibody was validated in house using positive control tissue sections (heart and brain) before it was used for IHC staining on ACC TMA sections. IHC staining was evaluated by a pathologist with expertise in the area of head and neck (J.A.B.) As previously reported (33), each sample was assigned an H-score, which is the product of percentage of cells in each sample with positive staining (range, 0–100%) multiplied by the intensity of staining (range, 0–3).
Statisticalanalysis, methods, and bioinformatics
Retrospective medical record abstraction was carried out to ascertain clinical and pathologic variables of interest.
Methylation level for HCN2 and IHC H-scores were considered as continuous variables and reported as median and interquartile range (IQR), or as binary variables categorized as <median or ≥median. Median methylation level and H-score were compared in ACC cases vs. controls, and then across clinicopathological variables of interest among cases, using the non-parametric Wilcoxon rank-sum test for binary variables and the non-parametric Kruskall Wallis test for categorical variables. Overall survival (OS) was calculated from the date of diagnosis to the date of death from any cause. Disease-free survival (DFS) was calculated from the date of diagnosis to the date of any (local, regional or distant) recurrence. Patients with distant metastases at the time of diagnosis, or who died of incident disease without recurrence, were excluded from DFS analysis. OS and DFS by binary methylation status of HCN2 gene (categorized as < median or ≥ median) were reported as median and standard error (SE) and estimated using the Kaplan-Meier method and compared using the log-rank test for equality of survival functions. Pairwise correlation analysis was used to determine correlation between methylation level and H-score.
Data analysis was performed using STATA 11.2 (StataCorp LP, College Station, TX, USA). P-values <0.05 were considered statistically significant.
Results
Differentially methylated genes identified by methylation array
The genome-wide methylation array identified 3,481 genes (data not shown) that were significantly differentially methylated in ACC tumors that were treated with 5-Aza, compared with control ACC tumors that received no treatment. Among these genes, 32 candidate genes (Table I) were selected for validation by BS-seq in a small set composed of 6 ACC and 6 normal frozen tissues.
Table I.
Top 32 genes detected by methylation array.
| Gene symbol | Rank in the gene list | Probe Set ID | Normal | ACC |
|---|---|---|---|---|
| RGPD3 | 1 | cg06148997 | 6/6 | 6/6 |
| ABLIM2 | 9 | cg18665513 | 0/6 | 0/6 |
| ZNF653 | 11 | cg13798986 | 0/6 | 0/6 |
| ATF4 | 12 | cg13462160 | 0/6 | 0/6 |
| BCL2 | 16 | cg06881186 | 0/6 | 0/6 |
| PCMT1 | 20 | cg07671221 | 0/6 | 0/6 |
| TNFRSF11A | 23 | cg19524723 | 0/6 | 0/6 |
| ZNF527 | 28 | cg09011348 | 0/6 | 0/6 |
| SIM2 | 31 | cg21697851 | 0/6 | 0/6 |
| PINX1 | 39 | cg26027776 | 0/6 | 0/6 |
| ZNF703 | 45 | cg25487404 | 0/6 | 0/6 |
| HCN2 | 47 | cg25367758 | 6/6a | 0/6 |
| POU4F3 | 49 | cg04701505 | 0/6 | 0/6 |
| EXOSC2 | 52 | cg14638609 | 0/6 | 0/6 |
| ZNF749 | 53 | cg23077461 | 0/6 | 0/6 |
| DUSP4 | 55 | cg05972070 | 0/6 | 0/6 |
| LGI3 | 62 | cg18317494 | 0/6 | 0/6 |
| PEX5 | 68 | cg07748017 | 0/6 | 0/6 |
| FBXO41 | 87 | cg02063488 | 0/6 | 0/6 |
| GPR39 | 97 | cg19309079 | 0/6 | 0/6 |
| RUNX3 | 128 | cg27360282 | 0/6 | 0/6 |
| BHLHE41 | 130 | cg19243777 | 0/6 | 0/6 |
| GPR123 | 196 | cg20559403 | 0/6 | 0/6 |
| SOX21 | 287 | cg18368297 | 0/6 | 0/6 |
| TBX2 | 292 | cg27470066 | 0/6 | 0/6 |
| EHD1 | 652 | cg21739289 | 0/6 | 0/6 |
| TOLLIP | 2012 | cg12308164 | 0/6 | 0/6 |
| FRMD6 | 4423 | cg09410986 | 6/6 | 6/6 |
| BSG | 7360 | cg10362365 | 0/6 | N.D. |
| MALT1 | 9548 | cg21074092 | 0/6 | 0/6 |
| MORN1 | 10003 | cg22045975 | 0/6 | 0/6 |
| CT62 | 26514 | cg13125884 | 0/6 | 0/6 |
N.D., no data;
5 CpG sites were methylated in all 6 normal samples, whereas other 5 CpG sites were methylated in most of 6 normal samples.
Among these 32 genes, FRMD6 and RGPD3, were methylated in both normal and ACC tumor samples by BS-seq (Table I). Using BS-seq, 10 CpG dinucleotides were reliably sequenced in HCN2 (NM_001194), as illustrated in Figs. 1 and 2. The differential methylation status was confirmed between normal and ACC (Table II). Among these 10 CpGs, 5 CpG sites were methylated in all normal samples, whereas other 5 CpG sites were methylated in most of the normal samples. No methylation, except 1 CpG (CpG #4) in 2 samples, was found in all 6 ACC tumor tissues (Table II). The differential methylation status detected by the methylation array for the rest of the top 32 genes was found to be without differential methylation between normal and ACC tumor samples on BS-seq analysis (Table I). These data indicate that these specific CpGs on the promoter region of HCN2 could be demethylated during the carcinogenesis of ACC.
Table II.
The schematic illustration of 10 CpG dinucleotides in the proximal region of HCN2 promoter and their methylation status validated by bisulfite sequencing in a small validation cohort of 6 normal and 6 ACC tumor samples.
| CpG 1 | CpG 2 | CpG 3 | CpG 4 | CpG 5 | CpG 6 | CpG 7 | CpG 8 | CpG 9 | CpG 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| N1 | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● |
| N2 | ● | ● | ● | ● | ● | ○ | ● | ● | ○ | ○ |
| N3 | ○ | ● | ● | ● | ● | ○ | ● | ○ | ○ | ● |
| N4 | ● | ● | ● | ● | ● | ○ | ● | ● | ● | ● |
| N5 | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● |
| N6 | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● |
| T1 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| T2 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| T4 | ○ | ○ | ○ | ● | ○ | ○ | ○ | ○ | ○ | ○ |
| T4 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| T5 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| T6 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
●, methylation; ○, unmethylation. ACC, adenoid cystic carcinoma. N1-N6, 6 DNA samples from 6 normal salivary gland tissues; T1-T6, 6 DNA samples from 6 ACC tissues. CpG 1–10 are 10 CpG dinucleotides that were reliably sequenced by bisulfite sequencing in each PCR product of these 12 DNA samples.
Validation of the differential methylation of HCN2 by quantitative methylation-specific PCR (qMSP)
A separate cohort of normal (n=20) and ACC tumor (n=32) FFPE samples were used to further validate by qMSP the differential methylation status of HCN2 in the same proximal promoter region detected by BS-seq. Further qMSP validation of HCN2 promoter methylation levels confirmed the hypomethylation of the same region in this cohort of ACC tumor FFPE samples (Table III and Fig. 3; P=0.04). These data indicate that promoter demethylation of HCN2 could be a frequent event in the carcinogenesis of ACC.
Table III.
The correlation of the clinical variables and patient outcomes with the median HCN2 promoter methylation status in a proximal region detected by the methylation array.
| Variables | n | Median methylation level of HCN2 (IQR) | P-value |
|---|---|---|---|
| Tumor vs. control (n=52) | 0.04 | ||
| Control | 20 | 14.1 (0–28.3) | |
| Case | 32 | 0.4 (0–9.5) | |
| Tumor only (n=31) | |||
| Gender | 0.25 | ||
| Male | 11 | 0.7 (0–25.9) | |
| Female | 20 | 0.09 (0–8.1) | |
| Age (years) | 0.68 | ||
| <50 | 17 | 0.7 (0–9.5) | |
| 50+ | 14 | 0.3 (0–7.1) | |
| Smoking history | 0.89 | ||
| No | 16 | 1.0 (0–9.5) | |
| Yes | 13 | 0.2 (0–10.5) | |
| Site | 0.48 | ||
| Major | 14 | 0.6 (0–9.4) | |
| Minor | 17 | 0.6 (0–20.0) | |
| T-stage | 0.76 | ||
| I–II | 11 | 4.8 (0–9.5) | |
| III–IV | 16 | 1.0 (0–15.3) | |
| N-stage | 0.44 | ||
| 0 | 21 | 4.8 (0–16) | |
| 1+ | 6 | 0.4 (0–7.1) | |
| M-stage | 0.53 | ||
| 0 | 24 | 1.0 (0–10.0) | |
| 1 | 3 | 7.1 (0–96.4) | |
| Overall stage | 0.79 | ||
| I–II | 9 | 5.4 (0–9.5) | |
| III–IV | 18 | 0.6 (0–10.5) | |
| Margin | 0.34 | ||
| Negative/close | 6 | 0.10 (0–9.5) | |
| Positive | 21 | 4.8 (0–10.5) | |
| Pattern | 0.10 | ||
| Cribriform or tubular | 26 | 3.0 (0–10.5) | |
| Solid | 2 | 0 (0–0) | |
| Nodal metastases on neck dissection | 0.63 | ||
| No | 9 | 4.8 (0–9.4) | |
| Yes | 6 | 0.4 (0–7.1) | |
| Perineural invasion | 0.09 | ||
| No | 1 | 39.7 (n/a) | |
| Yes | 18 | 0.6 (0–5.4) | |
| MYB-NFIB translocation | 0.22 | ||
| Negative | 11 | 6.7 (0–16.0) | |
| Positive | 14 | 0.4 (0–7.1) | |
| Any recurrencea | 0.02 | ||
| No | 14 | 5.4 (0.2–16.0) | |
| Yes | 14 | 0 (0–4.8) | |
| Local recurrence | 0.03 | ||
| No | 22 | 3.0 (0–10.5) | |
| Yes | 9 | 0 (0–0) | |
| Regional recurrence | 0.30 | ||
| No | 30 | 0.6 (0–9.5) | |
| Yes | 1 | 0 (n/a) | |
| New distant metastases | 0.05 | ||
| No | 20 | 4.0 (0–13.3) | |
| Yes | 11 | 0 (0–4.8) | |
| Vital status at last follow-up | 0.10 | ||
| Alive | 14 | 7.1 (0–16.0) | |
| Expired | 17 | 0 (0–5.4) |
IQR, interquartile range;
any recurrence denotes a regional, local, or distant recurrence after primary treatment, excluding cases with distant metastases at presentation.
n/a, not applicable.
Figure 3.

qMSP validation of the differential methylation status in the proximal region of HCN2 promoter detected by methylation array. qMSP validation was done between control (n=20) and ACC (n=32) cases in a separate, large FFPE cohort. P=0.04 between control and ACC cases.
Comparison of HCN2 promoter methylation with clinico-pathological parameters
After confirming that the promoter region of HCN2 is significantly hypomethylated in ACC compared to normal tissue, we investigated whether the quantitative methylation status of the HCN2 promoter in ACC specimens was associated with clinicopathological characteristics (Table III). Patients who developed disease recurrence had significantly lower HCN2 promoter methylation levels than those who did not (0 vs. 5.4, P=0.02; Table III and Fig. 4). The same was true when considering only local recurrence (0 vs. 3.0, P=0.03) and only new distant metastases (0 vs. 4.0, P=0.05). Thus, hypomethylation of the HCN2 promoter region may increase the risk of local and distant tumor recurrence. In addition, as previously reported (3), the local (9 out of 31 cases) and distant (11 out of 31 cases) recurrences were the predominant recurrences and the regional (1 out of 31 cases) recurrence was rare in this cohort of ACCs.
Figure 4.

HCN2 methylation status is correlated with any recurrence (regional, local or distant) after primary treatment in ACC cases. ‘No’ means ACC cases that had no recurrence after primary treatment (n=16). ‘Yes’ means ACC cases that had any recurrence after primary treatment (n=15). P=0.02 by the non-parametric Wilcoxon rank-sum test between ‘No’ and ‘Yes’.
Since the MYB-NFIB fusion gene and MYB over-expression are found in most ACC patients (13,19,34), the presence of the MYB-NFIB fusion gene was examined. There were 11 MYB-NFIB translocation-negative patients and 14 MYB-NFIB translocation-positive patients (Table III). No correlation was found between MYB overexpression and HCN2 promoter hypomethylation (Table III, P=0.22), indicating that HCN2 promoter demethylation occurred independently of MYB-NFIB translocation in this cohort.
The differential methylation status in the proximal region of HCN2 promoter was not correlated with DFS (P=0.78, log-rank test; Table IV and Fig. 5) or OS (P=0.56, log-rank test; Table IV and Fig. 5).
Table IV.
Survival by promoter methylation level of HCN2 gene among ACC cases.
| Disease-free survival | Overall survival | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Promoter methylation level | n | Median months (SE) | Log-rank P-value | Median months (SE) | Log-rank P-value |
| HCN2, also see Fig. 5 | 0.78 | 0.56 | |||
| < median | 16 | 124.9 (20.5) | 180.0 (62.5) | ||
| ≥ median | 16 | 93.5 (33.6) | 163.7 (13.5) | ||
ACC, adenoid cystic carcinoma; SE, standard error.
Figure 5.

Disease-free survival curve and overall survival curve by the methylation level in the proximal region of HCN2 promoter. No statistical significance was found for each curve by the log-rank test (P=0.78 or P=0.56).
HCN2 IHC staining was performed on a tissue microarray (TMA) sections comprised of ACC tumors (n=93) and normal salivary gland tissue cores as controls (n=10). The HCN2 staining intensity (P<0.001) and H-score (P=0.01), but not the percentage of the positively stained cells (P=0.10), were significantly stronger in normal control tissues than those of ACC tissues (Table V and Figs. 6–8). However, IHC parameters were not significantly correlated with HCN2 promoter methylation level (P=0.67), clinicopathological characteristics (P>0.09 for all), or survival (DFS P=0.74, OS P=0.53; log-rank test).
Table V.
HCN2 IHC analysis between normal control and ACC cases.
| Score | ACC cases median (IQR) (n=93) | Controls median (IQR) (n=10) | P-value |
|---|---|---|---|
| HCN2 H-score also see Fig. 6 | 27 (10–77) | 60 (60–90) | 0.01 |
| HCN2 intensity also see Fig. 7 | 2 (1–2) | 3 (3–3) | <0.001 |
| HCN2 percent staining also see Fig. 8 | 13 (5–33) | 20 (20–30) | 0.10 |
IHC, immunohistochemical staining; ACC, adenoid cystic carcinoma; IQR, interquartile range.
Figure 6.
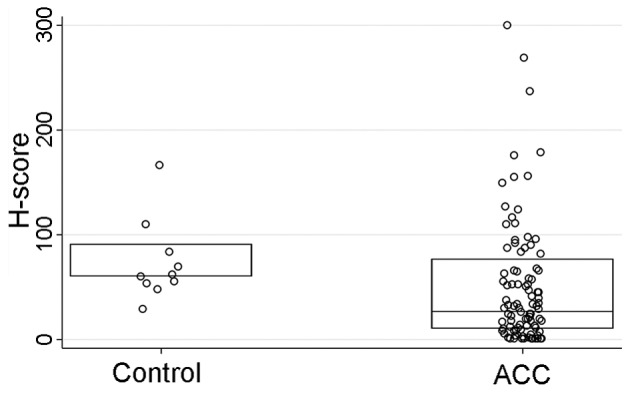
The comparisons of H-scores between control and ACC cases. H-scores (P=0.01) were statistically significant between control and ACC cases.
Figure 7.
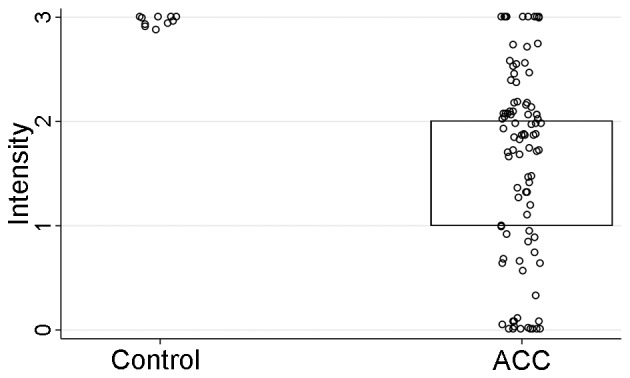
The comparisons of staining intensity between control and ACC cases. Staining intensity (P<0.001) was statistically significant between control and ACC cases.
Figure 8.
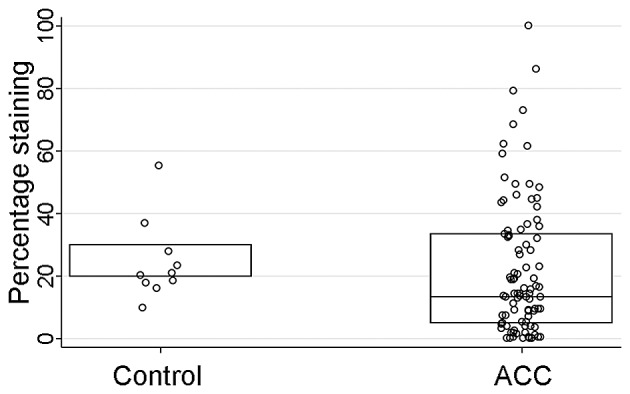
The comparisons of percentage of the positively stained cells between control and ACC cases. The percentage of the positively stained cells (P=0.1) was not statistically significant between control and ACC cases.
Discussion
While there is increasing insight being gained into the genetic alterations in ACC, the epigenetic landscape remains somewhat unknown. Our objective was to use a novel method to identify differentially methylated genes in ACC in an attempt to broaden our understanding of its pathogenesis. Currently, there are no FDA-approved chemotherapeutic drugs available for the treatment of ACC, and there is a need to find targetable molecular alterations to develop effective adjuvant treatment options.
The regulation of gene expression of oncogenes and tumor suppressor genes (TSGs) through DNA promoter methylation plays an important role in the carcinogenesis of many types of human cancers (27) and may also play a role in the carcinogenesis of ACC. In fact, the methylome of ACC has been profiled (35) with four genes validated (35).
We used a xenograft-based pharmacological demethylation and genome-wide methylation array approach, as there is a lack of viable cell lines (20) in which to perform these studies. Through this non-biased screening for genes whose promoters were under the control of methylation in ACC patients, we have identified HCN2 as a putative oncogene whose promoter was hypomethylated in ACC patients. The hypomethylation of HCN2 promoter suggests that HCN2 may act as an oncogene in the pathogenesis of ACC. Furthermore, the hypomethylation in the HCN2 promoter is correlated with the any recurrence, local recurrence, and distant metastasis of ACC primary tumors.
Hyperpolarization-activated cyclic nucleotide-gated (HCN) 2 (HCN2) belongs to a non-selective cation channel family that has 4 members, HCN1-4. The molecular identity of HCN channels remained unknown until the first member of HCN was cloned from mouse brain (36,37) and the other members were later cloned (38–43). HCN isoforms were not originally named as HCNs, but the HCN nomenclature was proposed and adopted later (44,45). HCN channels were originally named If (f for ‘funny’), Ih (h for hyperpolarization activated), or Iq (q for queer in neurons), based on the unique electrophysiological properties of the current that HCN channels carry (44,46). Furthermore, HCN1-4 share ~60% sequence identity of each other.
All four HCN channels share one unusual property-activation by a hyperpolarized cell membrane potential. HCN channels open upon hyperpolarization of the cell membrane potential and close at a positive cell membrane potential. They all also share another property, a cyclic nucleotide binding domain (CNBD) for cAMP and cGMP in their C-termini. Because they contain a CNBD, their activity can be furthermore modulated by hormones and neurotransmitters that regulate the productions of cAMP and cGMP to control heart rate and rhythm as well as neuronal pacemaking. The binding of cAMP and cGMP make HCNs active even at positive cell membrane potential (44,46). HCNs are slow activating, inward non-selective cation channels. The current of HCNs is carried by K+ and Na+ (ratio 4:1) (44,46). In addition, HCN channels exist as tetramers on cell membrane (44,46).
HCN4 is predominantly expressed in the sinoatrial (SA) node of heart, while HCN2 is predominantly expressed in ventricular cardiomyocytes and also in the sinoatrial node. Overall HCN channels are predominantly expressed in human heart, central and peripheral neurons, and photoreceptors in retina (40). HCN channels account for the rhythmic activity of cells in the sinoatrial node of heart and neurons due to their spontaneous, repetitive depolarization (47,48).
A variety of ion channels have been implicated in malignancies and can function to regulate tumor cell survival, proliferation, growth and metastasis (49), therefore, ion channels may be employed as drug targets. However, it remains challenging to define the role of an individual ion channel in malignancy. There is no inherited HCN2 mutation reported in human diseases, although acquired dysfunction of HCN2 has been reported in epilepsy and neuropathic pain (50).
HCNs including HCN2 have not been studied in salivary gland tumors and any other type of tumor, except that HCN2 was reported to be overexpressed in lung carcinoma cells in which HCN2 acted as an upstream regulator of cell apoptosis induced by Ca2+ overload by protein kinase C inhibitors (51). Although HCN2 normally conducts K+ and Na+ (44,46), it has been reported that HCN2 was permeable to Ca2+, and it was suggested that they may participate in pathological Ca2+ signaling when HCN2 is overexpressed (52).
Since there were no reliable HCN2 antibodies available for immunohistochemical staining until now, it remained unknown whether HCN2 is overexpressed in human ACC tissues and whether these channels exist on the cell membrane. IHC staining was done on our ACC TMA sections. We found that the hypomethylation of the HCN2 promoter did not increase the number of cells with HCN2 expression in ACC tumor samples, though there did appear to be an increase in staining intensity compared with the normal control samples.
In conclusion, through our screening and sequencing, we have identified hypomethylation of HCN2 as a potential biomarker for ACC that may be associated with more aggressive disease. The functional role of HCN2 remains to be seen, and as viable cell lines of ACC are more available, this can be further explored.
Acknowledgements
Dr Patrick K. Ha is supported by the National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research grant (5R03DE022591 and 5R01DE023227).
References
- 1.Renehan A, Gleave EN, Hancock BD, Smith P, McGurk M. Long-term follow-up of over 1000 patients with salivary gland tumours treated in a single centre. Br J Surg. 1996;83:1750–1754. doi: 10.1002/bjs.1800831228. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Shao C, Tan ML, Mu D, Ferris RL, Ha PK. Molecular biology of adenoid cystic carcinoma. Head Neck. 2012;34:1665–1677. doi: 10.1002/hed.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rettig EM, Tan M, Ling S, Yonescu R, Bishop JA, Fakhry C, Ha PK. MYB rearrangement and clinicopathologic characteristics in head and neck adenoid cystic carcinoma. Laryngoscope. 2015;125:E292–E299. doi: 10.1002/lary.25356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskaluk CA. Adenoid cystic carcinoma: Clinical and molecular features. Head Neck Pathol. 2013;7:17–22. doi: 10.1007/s12105-013-0426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holst VA, Marshall CE, Moskaluk CA, Frierson HF., Jr KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol. 1999;12:956–960. [PubMed] [Google Scholar]
- 8.Vila L, Liu H, Al-Quran SZ, Coco DP, Dong HJ, Liu C. Identification of c-kit gene mutations in primary adenoid cystic carcinoma of the salivary gland. Mod Pathol. 2009;22:1296–1302. doi: 10.1038/modpathol.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tetsu O, Phuchareon J, Chou A, Cox DP, Eisele DW, Jordan RCK. Mutations in the c-Kit gene disrupt mitogen-activated protein kinase signaling during tumor development in adenoid cystic carcinoma of the salivary glands. Neoplasia. 2010;12:708–717. doi: 10.1593/neo.10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung JY, Ahn HK, Kwon JE, Jeong H, Baek CH, Son YI, Ahn YC, Park K, Ahn MJ, Ko YH. Reappraisal of KIT mutation in adenoid cystic carcinomas of the salivary gland. J Oral Pathol Med. 2012;41:415–423. doi: 10.1111/j.1600-0714.2011.01105.x. [DOI] [PubMed] [Google Scholar]
- 11.Frierson HF, Jr, Moskaluk CA. Mutation signature of adenoid cystic carcinoma: Evidence for transcriptional and epigenetic reprogramming. J Clin Invest. 2013;123:2783–2785. doi: 10.1172/JCI69070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross JS, Wang K, Rand JV, Sheehan CE, Jennings TA, Al-Rohil RN, Otto GA, Curran JC, Palmer G, Downing SR, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol. 2014;38:235–238. doi: 10.1097/PAS.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 13.Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C, El-Naggar AK. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010;16:4722–4731. doi: 10.1158/1078-0432.CCR-10-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West RB, Kong C, Clarke N, Gilks T, Lipsick JS, Cao H, Kwok S, Montgomery KD, Varma S, Le QT. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brill LB, II, Kanner WA, Fehr A, Andrén Y, Moskaluk CA, Löning T, Stenman G, Frierson HF., Jr Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol. 2011;24:1169–1176. doi: 10.1038/modpathol.2011.86. [DOI] [PubMed] [Google Scholar]
- 16.Persson M, Andrén Y, Moskaluk CA, Frierson HF, Jr, Cooke SL, Futreal PA, Kling T, Nelander S, Nordkvist A, Persson F, et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer. 2012;51:805–817. doi: 10.1002/gcc.21965. [DOI] [PubMed] [Google Scholar]
- 17.Persson M, Andrén Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stenman G. Fusion oncogenes in salivary gland tumors: Molecular and clinical consequences. Head Neck Pathol. 2013;7(Suppl 1):S12–S19. doi: 10.1007/s12105-013-0462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao YJ, Zhang L, Mitani M, Weber RS, Lippman SM, et al. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: Association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res. 2011;17:7003–7014. doi: 10.1158/1078-0432.CCR-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One. 2009;4:e6040. doi: 10.1371/journal.pone.0006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, Golden WL, Frierson HF., Jr Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91:1480–1490. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17:771–779. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 23.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 24.Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 25.Jones PA, Taylor SM. Hemimethylated duplex DNAs prepared from 5-azacytidine-treated cells. Nucleic Acids Res. 1981;9:2933–2947. doi: 10.1093/nar/9.12.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor SM, Jones PA. Mechanism of action of eukaryotic DNA methyltransferase. Use of 5-azacytosine-containing DNA. J Mol Biol. 1982;162:679–692. doi: 10.1016/0022-2836(82)90395-3. [DOI] [PubMed] [Google Scholar]
- 27.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 28.Shao C, Sun W, Tan M, Glazer CA, Bhan S, Zhong X, Fakhry C, Sharma R, Westra WH, Hoque MO, et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin Cancer Res. 2011;17:4320–4330. doi: 10.1158/1078-0432.CCR-10-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao C, Bai W, Junn JC, Uemura M, Hennessey PT, Zaboli D, Sidransky D, Califano JA, Ha PK. Evaluation of MYB promoter methylation in salivary adenoid cystic carcinoma. Oral Oncol. 2011;47:251–255. doi: 10.1016/j.oraloncology.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li LC, Dahiya R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 31.Kim MS, Louwagie J, Carvalho B, Terhaar Sive Droste JS, Park HL, Chae YK, Yamashita K, Liu J, Ostrow KL, Ling S, et al. Promoter DNA methylation of oncostatin m receptor-β as a novel diagnostic and therapeutic marker in colon cancer. PLoS One. 2009;4:e6555. doi: 10.1371/journal.pone.0006555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durr ML, Mydlarz WK, Shao C, Zahurak ML, Chuang AY, Hoque MO, Westra WH, Liegeois NJ, Califano JA, Sidransky D, et al. Quantitative methylation profiles for multiple tumor suppressor gene promoters in salivary gland tumors. PLoS One. 2010;5:e10828. doi: 10.1371/journal.pone.0010828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling S, Chang X, Schultz L, Lee TK, Chaux A, Marchionni L, Netto GJ, Sidransky D, Berman DM. An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res. 2011;71:3812–3821. doi: 10.1158/0008-5472.CAN-10-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El-Naggar AK. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther. 2011;12:569–573. doi: 10.4161/cbt.12.7.17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell A, Bell D, Weber RS, El-Naggar AK. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer. 2011;117:2898–2909. doi: 10.1002/cncr.25818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santoro B, Grant SGN, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/S0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- 38.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 39.Gauss R, Seifert R, Kaupp UB. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 1998;393:583–587. doi: 10.1038/31248. [DOI] [PubMed] [Google Scholar]
- 40.Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaccari T, Moroni A, Rocchi M, Gorza L, Bianchi ME, Beltrame M, DiFrancesco D. The human gene coding for HCN2, a pacemaker channel of the heart. Biochim Biophys Acta. 1999;1446:419–425. doi: 10.1016/S0167-4781(99)00092-5. [DOI] [PubMed] [Google Scholar]
- 42.Seifert R, Scholten A, Gauss R, Mincheva A, Lichter P, Kaupp UB. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci USA. 1999;96:9391–9396. doi: 10.1073/pnas.96.16.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishii TM, Takano M, Xie LH, Noma A, Ohmori H. Molecular characterization of the hyperpolarization-activated cation channel in rabbit heart sinoatrial node. J Biol Chem. 1999;274:12835–12839. doi: 10.1074/jbc.274.18.12835. [DOI] [PubMed] [Google Scholar]
- 44.Clapham DE. Not so funny anymore: Pacing channels are cloned. Neuron. 1998;21:5–7. doi: 10.1016/S0896-6273(00)80508-5. [DOI] [PubMed] [Google Scholar]
- 45.Hofmann F, Biel M, Kaupp UB. International Union of Pharmacology. LI. Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. Pharmacol Rev. 2005;57:455–462. doi: 10.1124/pr.57.4.8. [DOI] [PubMed] [Google Scholar]
- 46.Pape HC. Queer current and pacemaker: The hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 47.Stieber J, Hofmann F, Ludwig A. Pacemaker channels and sinus node arrhythmia. Trends Cardiovasc Med. 2004;14:23–28. doi: 10.1016/j.tcm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 48.DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434–446. doi: 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- 49.Lang F, Stournaras C. Ion channels in cancer: Future perspectives and clinical potential. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130108. doi: 10.1098/rstb.2013.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benarroch EE. HCN channels: Function and clinical implications. Neurology. 2013;80:304–310. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- 51.Norberg E, Karlsson M, Korenovska O, Szydlowski S, Silberberg G, Uhlén P, Orrenius S, Zhivotovsky B. Critical role for hyperpolarization-activated cyclic nucleotide-gated channel 2 in the AIF-mediated apoptosis. EMBO J. 2010;29:3869–3878. doi: 10.1038/emboj.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michels G, Brandt MC, Zagidullin N, Khan IF, Larbig R, van Aaken S, Wippermann J, Hoppe UC. Direct evidence for calcium conductance of hyperpolarization-activated cyclic nucleotide-gated channels and human native If at physiological calcium concentrations. Cardiovasc Res. 2008;78:466–475. doi: 10.1093/cvr/cvn032. [DOI] [PubMed] [Google Scholar]