

Abstract
Site-directed tryptophan fluorescence has been successfully used to determine the solution structure of tear lipocalin. Here, the technique is extended to measure the binding energy landscape. Single Trp mutants of tear lipocalin are bound to the native ligand and an analogue tagged with a quencher group to both populate and discriminate the excited protein states. Steady-state and time-resolved fluorescence quenching data reveal the intracavitary state of the ligand. The static components of fluorescence quenching identify the residues where nonfluorescence complexes form. An asymmetric distribution of the ligand within the cavity reflects the complex energy landscape of the excited protein states. These findings suggest that the excited protein states are not unique but consist of many substates. The roughness of the binding energy landscape is about 2.5kBT. The excited protein states originate primarily from conformational selections of loops AB and GH, a portal region. In contrast to static quenching, the dynamic components of fluorescence quenching by the ligand are relevant to both local side chain and ligand dynamics. Apparent bimolecular rate constants for collisional quenching of Trp by the nitroxide moiety are ~1 ÷ 5 × 1012M−1 s−1. Estimations made for effective ligand concentrations establish actual rate constants on the order of 12 × 109 M−1 s−1. Prior to exit from the cavity of the protein, ligands explore binding sites in nanoseconds. Although microsecond fluctuations are rate-limiting processes in ligand binding for many proteins, accompanying nanosecond motion may be necessary for propagation of ligand binding.
Implicit to the mechanistic understanding of ligand binding of any protein is the ability to track the conformations of excited protein states because ligands select these states for binding (1). Methods are needed to elicit side chain interactions and loop motion that occur in nanoseconds to milliseconds. By introducing tryptophan at each site of a protein, ligand-induced conformational changes in a nanosecond time scale can be monitored by changes in tryptophan fluorescence parameters. Ligands bearing quenching groups can potentially map intracavitary transitory ligand collisions as well as protein ligand complex formations for an entire protein if dynamic and static quenching are resolved with site-directed tryptophan fluorescence.
In this study, site-directed tryptophan fluorescence (SDTF)1 in concert with the native fatty acid and its analogue tagged with a quencher group (nitroxide) provides a new tool to determine (a) the protein sites that show dynamic and static conformational changes associated with ligand binding, (b) static populations of fatty acids at various sites of the protein, and (c) frequencies of collisions between the ligand (nitroxide moiety) and the side chains of the protein, which relate to ligand as well as side chain dynamics. Finally, because ligand binding selects excited protein states, the intracavitary association constants of the ligand at various sites reveal the binding energy landscape of proteins.
The protein used for SDTF is human tear lipocalin (TL). The gene is called lipocalin 1 (LCN1) because it is a prominent member of the lipocalin superfamily. TL is the principal lipid binding protein in human tears (2) and is found in other human tissues (3–6). The ability to bind to various classes of ligands is critical to the functions of TL (7–14). These features in conjunction with TL’s well-characterized concordant crystal and solution structures make an ideal model to investigate the structure and energetics of ligand binding.
The solution structure of TL was initially determined by site-directed tryptophan fluorescence (15). The typical lipocalin fold consists of eight antiparallel β-strands with a repeated +1 topology (16). Promiscuous ligand binding by TL was attributed to a capacious cavity. These findings were subsequently verified by crystallography (17). Dynamic quenching with nitroxide-labeled ligand unveiled direct collisional contact with specific intracavitary amino acids of the G strand of TL, but to date the crystal structure of the TL-native ligand complex has not been published. Elucidation of the binding site is essential for any mechanistic understanding of TL’s function. Because the ligand orientation in the binding cavity of a protein in solution may differ significantly from that found in crystal form (18), the binding complex was studied in an aqueous environment.
MATERIALS AND METHODS
Materials
Palmitic (PA) and lauric (LA) acids were purchased from Sigma-Aldrich (St. Louis, MO). DAUDA (11-((5-dimethylaminonaphthalene-1-sulfonyl)amino)undecanoic acid) was purchased from Invitrogen (San Diego, CA). C12SL, spin-labeled analogue of PA, was provided by Dr. Wayne Hubbell.
Site-Directed Mutagenesis and Plasmid Construction
The TL cDNA in PCR II (Invitrogen), previously synthesized (19), was used as a template to clone the TL gene spanning bases 115–592 of the previously published sequence (10) into pET 20b (Novagen, Madison, WI). Flanking restriction sites for NdeI and BamHI were added to produce the native protein sequence as found in tears but with an initiating methionine (20). To construct mutant proteins with a single tryptophan, the previously well-characterized TL mutant, W17Y, was prepared with oligonucleotides (Universal DNA Inc., Tigard, OR) by sequential PCR steps (21, 22). Using this mutant as a template, mutant cDNAs were constructed in which selected amino acids were additionally substituted with tryptophan. Amino acid 1 corresponds to His, bases 115–118 according to Redl (10).
To characterize the ligand binding sites, 44 single Trp mutants that cover all β-strands of the cavity as well as loops at the open end of the cavity were tested (Figure 1). W17Y mutant that has been characterized previously was used as a template. Single Trp mutants of TL include the following: W17Y/M22W (for simplicity denoted as W22); W17Y/V24W (W24); W17Y/D25W (W25); W17Y/R26W (W26); W17Y/E27W (W27); W17Y/ F28W (W28); W17Y/E30W (W30); W17Y/L33W (W33); W17Y/E34W (W34); W17Y/S35W (W35); W17Y/V36W (W36); W17Y/T37W (W37); W17Y/M39W (W39); W17Y/ L41W (W41); W17Y/L49W (W49); W17Y/V53W (W53); W17Y/M55W (W55); W17Y/I57W (W57); W17Y/G59W (W59); W17Y/R60W (W60); W17Y/Q62W (W62); W17Y/ V64W (W64); W17Y/A66W (W66); W17Y/L68W (W68); W17Y/Y77W (W77); W17Y/A79W (W79); W17Y/D80W (W80); W17Y/G81W (W81); W17Y/G82W (W82); W17Y/ H84W (W84); W17Y/A86W (W86); W17Y/I88W (W88); W17Y/Y97W (W97); W17Y/F99W (W99); W17Y/C101W (W101); W17Y/G103W (W103); W17Y/L105W (W105); W17Y/H106W (W106); W17Y/G107W (W107); W17Y/ K108W (W108); W17Y/V110W (W110); W17Y/G112W (W112); W17Y/K114W (W114); W17Y/V116W (W116). All mutants, which have previously been characterized by far-UV circular dichroism, have the native fold. In addition, ligand binding properties of all mutant proteins (see below) indicate that they possess the function of native TL.
FIGURE 1.
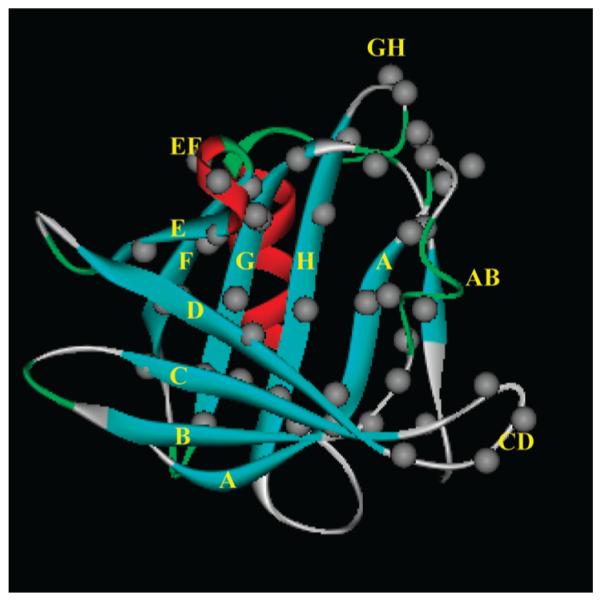
Positions of the residues that were sequentially substituted with Trp to explore the binding energy landscape of TL. Gray circles show locations of the Cαatoms of the amino acid residues. The side chains of all β-strand residues are oriented inside the cavity. Single and double letters denote the identities of the β-strands and loops, respectively. The ribbon diagram (blue, β-strands; red, R-helix; green, turns; gray, loops) of TL was generated from PDB 1XKI.
Expression and Purification of Mutant Proteins
The mutant plasmids were transformed in Escherichia coli, BL21-(DE3); cells were cultured, and proteins were expressed according to the manufacturer’s protocol (Novagen). Following cell lysis (23), the supernatant was treated with methanol (40% final concentration) at 4 °C for 2.5 h. Alternatively, mutant proteins expressed in inclusion bodies were dissolved in 8 M urea at room temperature for 2 h. The resulting suspension was centrifuged at 3000g for 30 min. The supernatant was dialyzed against 50 mM Tris-HCl, pH 8.4. The dialysate was treated with ammonium sulfate 45–75% saturation. The resulting precipitate was dissolved in 50 mM Tris-HCl, pH 8.4, and applied to a Sephadex G-100 column (2.5×100 cm) equilibrated with 50 mM Tris-HCl and 100 mM NaCl, pH 8.4. The fraction containing the mutant protein was dialyzed against 50 mM Tris-HCl, pH 8.4, and applied to a DEAE-Sephadex A-25 column. Bound protein was eluted with a 0–0.8 M NaCl gradient. Eluted fractions containing mutant proteins were centrifugally concentrated (Amicon, Centricon-10). Purity of mutant proteins was verified by SDS-tricine gel electrophoresis (2). The protein concentrations of stock solutions were determined by the biuret method (24), and dilute protein concentrations were determined by the Lowry method. In addition, to avoid any discrepancy, absorbance of each protein solution at 280 nm was measured.
Steady-State Fluorescence
Steady-state fluorescence measurements were made on a Jobin Yvon-SPEX (Edison, NJ) Fluorolog tau-3 spectrofluorometer; bandwidths for excitation and emission monochromators were 2 and 3 nm, respectively. The excitation λ of 295 nm was used to ensure that fluorescence originates entirely from tryptophanyl groups. All measurements were conducted in 10 mM sodium phosphate buffer (pH 7.3) at room temperature. The fluorescence spectra were corrected for light scattering from buffer.
Fluorescence Binding Assays
The samples of the mutant proteins (about 14 μM) in 10 mM sodium phosphate at pH 7.3 were titrated by addition of PA or C12SL, and the fluorescence was measured at emission λmax. Following each addition of ligand, the solution was mixed and allowed to equilibrate for 2 min. At the end of each titration experiment, the ethanol concentration did not exceed 2%. The ligand binding data were analyzed with the following formula for one binding site using OriginPro 8 software (OriginLab Corp., Northampton, MA):
where F is a fluorescence scaling factor, Kd is the apparent dissociation constant, P is total protein concentration, Lt is the total ligand concentration, and n is the stoichiometry.
Competitive Displacement Assays To Identify Which Fatty Acid Is Homologous to C12SL in Ligand Binding to TL
To compare binding affinities of the ligands to TL, competitive displacement assays were performed with LA, PA, and C12SL. DAUDA exhibits low fluorescence intensity in buffer but shows enhancement of fluorescence and a blue shift when bound to tear lipocalin (22). The displacement of bound DAUDA by the ligands accompanies decreased fluorescence intensity. Samples of TL (4 μM) were incubated with DAUDA (2 μM) in 10 mM sodium phosphate, pH 7.3, and competitors were added in increasing concentration. The IC50 parameter for each ligand was extrapolated from the graph of fluorescence intensity of bound DAUDA versus competitor concentration using a model for one binding site with OriginPro 8 software.
Fluorescence Lifetime Measurements
The fluorescence intensity decays were measured using a LaserStrobe fluorescence lifetime instrument (Photon Technology International, Inc., Birmingham, NJ), which consists of a nitrogen laser (GL-3300) linked to a dye laser (GL 302), a frequency doubler (GL 303), and a stroboscopic detector. Rhodamine 6G (Exciton, Inc., Dayton, OH) dye solution was used to obtain a wavelength of 295 nm. The 295 nm pulses (fwhm ~ 1.5 ns) were used for the excitation of the single Trp mutants. The decay curves were analyzed at the wavelengths of the respective emission maxima. The emission monochromator slit was 3–5 nm. All measurements were conducted at room temperature. The IRF (instrument response function) was determined by measuring scattered light from a solution of glycogen. A DPU-15 optical depolarizer (Optics for Research, Caldwell, NJ) was placed before the emission monochromator to eliminate polarization dependence of the detection train. Each data point on a lifetime decay curve represents the average of at least nine laser flashes, and each decay represents 300 of these data points evenly spaced over the collection time interval.
The fluorescence intensity decay data were analyzed by the multiexponential decay law, using the software supplied with the PTI instrument:
where I is fluorescence intensity and αi and τi are the normalized preexponential factors and decay time, respectively. The intensity averaged lifetime was used for Stern–Volmer plots of τ0/τ. fi, the fractional contribution of each lifetime component to steady-state fluorescence intensity, is defined as .
Quenching Experiments by Steady-State and Time-Resolved Fluorescence
Steady-state quenching data reflect both dynamic and static quenching. To segregate the dynamics and static components, quenching of the fluorescence by PA or C12SL in single Trp mutants of TL was performed with both steady-state and time-resolved fluorescence methods. However, static quenching does not impact the fluorescence lifetime. Both steady-state and time-resolved fluorescence quenching data were simultaneously fitted to and , respectively, in Global analyses (OriginPro 8). and τav are intensity-averaged fluorescence lifetimes in the absence and presence of quencher, respectively. F0 and F are steady-state fluorescence intensity in the absence and presence of quencher [Q], respecrespectively. KD and KS are dynamic and static quenching constants, respectively. The bimolecular quenching constant was calculated as . Apparent κq values exceed the rate constant for diffusion-limited reactions about 3 orders of magnitude. To estimate realistic κq values, ligand distribution was limited to the volume of the protein (14 μM). The volume of TL (PDB code 1XKI) was taken as 104 Å3 (DS Visualizer; Accelrys Software Inc.).
To eliminate the possible effect of mutation-derived variation in binding both the dynamic and static quenching constants were normalized to the same Kd value, 0.5 μM. The rationalization for normalization is that unlike a freely diffusing quencher where only the static quenching constants are used to calculate association constants, our quencher is an intracavitary ligand. The dynamic quenching originates from that subset of the bound ligands which undergo intracavitary nanosecond motion. This subset is identified from the nanosecond time window of the lifetime of Trp fluorescence. The remaining subset of the ligands, the nitroxide moiety that complexes with the Trp side chain at the time of excitation, gives rise to static quenching. The approach ensures that, for the purpose of comparing intracavitary site-specific quenching, the number of bound ligands, which produce both dynamic and static quenching, is considered the same for each mutation.
In quenching experiments, fluorescence measurements were performed at wavelengths of emission maxima.
Construction of the Binding Energy Landscape for TL
The fraction of the ligands with static quenching is regarded as bound. This approach is justified because only static quenching reflects association, i.e., complex formation, of the ligand with a particular site. Since static quenching constants manifest the ligand binding constants for particular sites, apparent ΔGi were calculated by the formula apparent ΔGi = RT ln , where represents static quenching constants for the particular site. Normalized fractions of the ligand population within the cavity of TL were calculated according to the Boltzmann distribution:
RESULTS AND DISCUSSION
Strategy To Construct the Binding Energy Landscape of TL
To access the ligand binding sites, 44 single Trp mutants that cover all β-strands of the cavity as well as loops at the open end of the cavity were tested (Figure 1). Nitroxide effectively quenches Trp fluorescence by direct contact (25). Therefore, the fluorescence quenching of single Trp mutants by C12SL will identify the amino acid residue positions, which have direct contact with the ligand’s nitroxide group.
In order to select the proper nonquencher analogue for the C12SL, competitive binding experiments were performed with LA and PA, both native ligands of TL. Fluorescence properties of DAUDA are very sensitive to its immediate environment. The intensity of the fluorescence spectrum of DAUDA increases significantly and exhibits a blue shift of the fluorescence λmax when bound to TL (22). The DAUDA and fatty acids bind to the same binding site of TL. Consequently, the addition of fatty acids displaces bound DAUDA from the protein and results in decreased fluorescence intensity and a red shift of fluorescence λmax. Binding affinities (IC50) from competitive binding experiments show that PA, which has about equal number of methyl plus methylene groups, more closely resembles C12SL than does LA (Figure 2). Therefore, PA was chosen in conjunction with C12SL to determine which sites of TL have direct collisions and/or form ground-state complexes with the nitroxide group. The carboxylate group does not significantly quench indole fluorescence (26, 27). Therefore, tryptophan quenching observed with PA is derived mainly from indirect interactions with amino side chains and peptide bonds that are quenchers. Substitutions of native amino acids with Trp do not alter binding affinities of ligands to TL for most sites. Dissociation constants for the majority of the mutants are within the 0.3–1.0 μM range (Table 1).
FIGURE 2.

Displacement of the DAUDA from the DAUDA–TL complex by various ligands. The DAUDA–TL complex was prepared as the mixture of TL (4 μM) and DAUDA (2 μM). IC50 values (concentration of a ligand that displaces 50% of bound DAUDA) for LA, C12SL, and PA are 12.6, 1.9, and 2.3 μM, respectively.
Table 1.
Dissociation Constants for Ligand Binding to Single Trp Mutants of Human Tear Lipocalin
| β-strand or loop | mutant | Kd (μM), C12SL | Kd (μM), C16 |
|---|---|---|---|
| β-strand A | W22 | 0.4 ± 0.1 | 0.9 ± 0.2 |
| β-strand A | W24 | 0.4 ± 0.1 | naa |
| loop AB | W25 | 0.5 ± 0.1 | na |
| loop AB | W26 | 1.6 ± 0.2 | na |
| loop AB | W27 | 2.5 ± 0.1 | na |
| loop AB | W28 | 0.4 ± 0.2 | 0.7 ± 0.4 |
| loop AB | W30 | 0.5 ± 0.1 | na |
| loop AB | W33 | 5.9 ± 0.4 | na |
| loop AB | W34 | 0.4 ± 0.1 | na |
| loop AB | W35 | 3.5 ± 0.3 | na |
| loop AB | W36 | 0.4 ± 0.1 | na |
| loop AB | W37 | 4.7 ± 0.5 | 5.8 ± 0.6 |
| β-strand B | W39 | 3.1 ± 0.3 | na |
| β-strand B | W41 | na | na |
| β-strand C | W49 | 2.4 ± 0.2 | 2.3 ± 0.2 |
| β-strand C | W53 | 0.3 ± 0.1 | na |
| β-strand C | W55 | 0.3 ± 0.1 | na |
| β-strand C | W57 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| β-strand D | W60 | 1.9 ± 0.5 | na |
| β-strand D | W62 | 0.3 ± 0.1 | na |
| β-strand D | W64 | 0.3 ± 0.1 | na |
| β-strand D | W66 | 0.3 ± 0.1 | na |
| β-strand D | W68 | 0.7 ± 0.1 | 0.3 ± 0.1 |
| β-strand E | W77 | 1.0 ± 0.5 | na |
| β-strand E | W79 | 0.5 ± 0.1 | na |
| β-strand E | W80 | 0.9 ± 0.4 | na |
| β-strand F | W84 | 0.5 ± 0.1 | na |
| β-strand F | W86 | 1.0 ± 0.3 | na |
| β-strand F | W88 | 0.5 ± 0.2 | 0.5 ± 0.2 |
| β-strand G | W97 | 0.4 ± 0.1 | 0.4 ± 0.1 |
| β-strand G | W99 | 0.3 ± 0.1 | na |
| β-strand G | W101 | 0.4 ± 0.2 | 2.8 ± 0.5 |
| β-strand G | W103 | 0.3 ± 0.1 | 0.3 ± 0.1 |
| β-strand G | W105 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| β-strand H | W110 | 0.7 ± 0.1 | na |
| β-strand H | W112 | 0.4 ± 0.1 | na |
| β-strand H | W114 | 0.4 ± 0.1 | 1.1 ± 0.6 |
| β-strand H | W116 | 0.5 ± 0.2 | na |
Not applicable due to the absence of quenching.
Trp fluorescence, which is very sensitive to its local environment, has been used extensively to gain local structural information regarding solvent accessibility, dynamics, relaxation, and distance between the donor–acceptor pair, etc. (28). The rotamer populations of a Trp side chain depend on backbone conformation and exhibit distinct lifetimes (29). The rates of excited state electron transfer from tryptophan to the peptide bonds determine the intramolecular quenching of fluorescence and the lifetime for each rotamer of Trp (30). Therefore, fluorescence intensity and lifetime of Trp are sensitive to its local conformation. In addition, side chains of several amino acids are electron acceptors or proton donors and quench Trp fluorescence (26). Consequently, in some particular instances Trp fluorescence can be sensitive to the conformations that have tertiary interactions with another side chain.
It is widely accepted that proteins in an unbound state exist in large number of conformations. Recent evidence suggests that only a small subset of protein conformations, which have higher energies and, therefore, are sparsely populated, are active for ligand binding. Although never absolute, a mechanism of population shift to the active conformations (excited protein states), rather than induced fit by the ligand, is more appropriate for the description of the protein binding to small ligands (1). The preferred ligand binding positions in the cavity reflect the excited conformational states selected. Changes in conformation are reported as site-specific environmental alterations that can be observed with fluorescent quenching parameters. To detect conformational changes that originate from increased populations of the protein in excited states, steady-state fluorescence intensities and fluorescence lifetimes of single Trp mutants were measured at various ligand concentrations.
Figure 3 illustrates the fluorescence intensity decay for W37 at various C12SL concentrations that manifest dynamic quenching by the ligand. Steady-state fluorescence quenching, however, reports both dynamic and static quenching. As examples of the fluorescence quenching by single Trp mutants by C12SL, W37 and W62, mutants, are shown in Figure 4. Fatty acids are unable to diffuse freely into the protein cavity. Therefore, only the bound ligand can quench the fluorescence. The Stern-Volmer plots, F0/F (static and dynamic quenching) and τ0/τ (dynamic quenching only) for ligand quenching, can be used to segregate the static and dynamic quenching (see Materials and Methods). Fluorescence quenching parameters for both PA and C12SL binding to single Trp mutants of TL are shown in Table 2.
FIGURE 3.
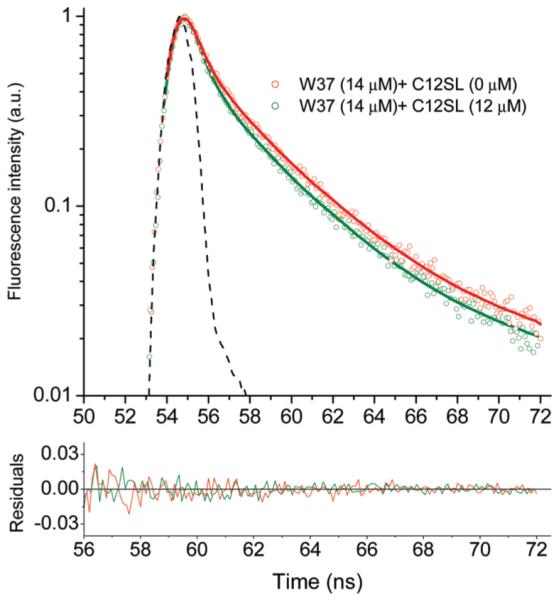
Fluorescence intensity decay curves for W37 with various concentrations of C12SL. Dots represent experimental data points. Dashed line is IRF. Solid curves are generated by fitting the data to multiexponential decay (see Materials and Methods).
FIGURE 4.
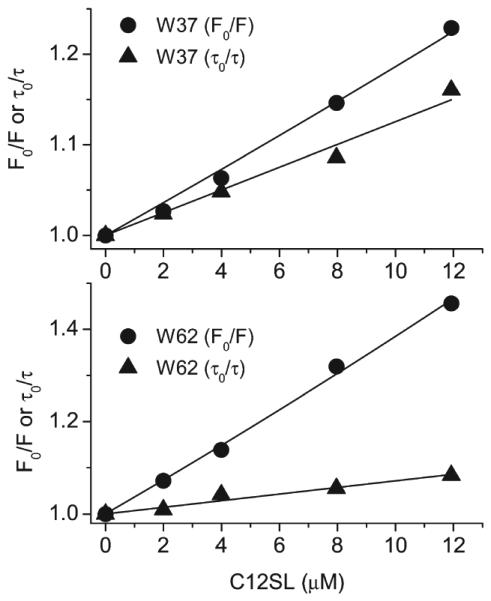
Stern–Volmer plots of F0/F (steady-state intensity) and τ0/τ (intensity averaged fluorescence lifetime) for W37 and W62 with C12SL. Steady-state data reflect both static and dynamic quenching. However, fluorescence lifetime data result only from dynamic quenching.
Table 2.
Fluorescence Quenching Parameters for Palmitic Acid and C12SL Binding to Single Trp Mutants of Human Tear Lipocalina
| C12SL |
PA |
|||||||
|---|---|---|---|---|---|---|---|---|
| Trp mutant |
KSVapp × 10−3
(M−1) |
KD × 10−3
(M−1) |
kq × 10−12
(M−1 s−1) |
KS × 10−3
(M−1) |
KSVapp × 10−3
(M−1) |
KD × 10−3
(M−1) |
kq × 10−12
(M−1 s−1) |
KS × 10−3
(M−1) |
| W22 | 36.2 (1.0) | 15.4 (1.2) | 3.6 | 17.5 (1.6) | 12.9 (0.8) | 10.2 (0.7) | 2.4 | 2.1 (1.0) |
| W24 | 35.8 (0.3) | 13.1 (0.5) | 4.0 | 20.1 (0.8) | 3.8 (0.9) | 3.8 (0.9) | 1.1 | 0 |
| W25 | 17.1 (0.9) | 10.3 (0.9) | 2.3 | 6.0 (1.2) | 2.7 (0.7) | 2.7 (0.7) | 0.6 | 0 |
| W26 | 24.2 (0.8) | 7.1 (1.0) | 1.8 | 15.8 (1.4) | 3.5 (0.7) | 3.5 (0.7) | 0.8 | 0 |
| W27 | 9.5 (0.9) | 9.5 (0.9) | 1.7 | 0 | 0 | 0 | 0 | 0 |
| W28 | 51.9 (1.5) | 22.3 (1.9) | 5.4 | 24.2 (2.5) | 23.1 (2.2) | 12.3 (1.9) | 3.0 | 9.8 (2.5) |
| W30 | 23.4 (0.6) | 8.5 (1.1) | 3.3 | 13.8 (1.6) | 0 | 0 | 0 | 0 |
| W33 | 42.0 (0.9) | 11.0 (1.1) | 3.6 | 29.0 (1.9) | 0 | 0 | 0 | 0 |
| W34 | 21.1 (1.2) | 4.9 (1.2) | 1.5 | 15.5 (1.2) | 7.0 (0.8) | 7.0 (0.8) | 2.1 | 0 |
| W35 | 45.6 (1.1) | 12.2 (1.1) | 3.0 | 30.7 (1.6) | 0 | 0 | 0 | 0 |
| W36 | 24.6 (0.6) | 13.6 (1.2) | 3.3 | 9.6 (1.1) | 0 | 0 | 0 | 0 |
| W37 | 28.5 (0.6) | 19.1 (0.5) | 6.3 | 8.3 (0.7) | 20.1 (0.5) | 7.6 (0.5) | 2.4 | 11.8 (0.8) |
| W39 | 64.9 (1.0) | 14.9 (1.2) | 3.7 | 45.1 (1.9) | 0 | 0 | 0 | 0 |
| W41 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| W49 | 14.3 (0.3) | 7.6 (1.6) | 3.1 | 4.9 (1.8) | 5.6 (0.5) | 0 | 0 | 5.6 (0.5) |
| W53 | 8.5 (0.3) | 1.1 (0.9) | 0.5 | 7.4 (1.4) | 0 | 0 | 0 | 0 |
| W55 | 28.3 (1.3) | 9.6 (2.4) | 2.6 | 17.1 (2.8) | 0 | 0 | 0 | 0 |
| W57 | 27.2 (0.6) | 14.7 (1.7) | 4.6 | 10.9 (2.2) | 19.5 (0.5) | 10.9 (0.5) | 3.7 | 7.8 (0.7) |
| W59 | 5.2 (0.5) | 5.2 (0.5) | 1.6 | 0 | 2.8 (0.3) | 2.8 (0.3) | 0.9 | 0 |
| W60 | 11.1 (1.2) | 11.1 (1.2) | 3.5 | 0 | 1.3 (0.5) | 1.3 (0.5) | 0.4 | 0 |
| W62 | 37.7 (0.7) | 7.1 (0.6) | 2.2 | 28.6 (0.9) | 5.5 (0.6) | 5.5 (0.6) | 1.7 | 0 |
| W64 | 23.4 (0.5) | 0 | 0 | 23.4 (0.5) | 0 | 0 | 0 | 0 |
| W66 | 17.8 (0.2) | 8.8 (1.4) | 2.8 | 8.3 (1.8) | 0 | 0 | 0 | 0 |
| W68 | 17.5 (2.2) | 17.5 (2.2) | 5.7 | 0 | 11.0 (1.8) | 11.0 (1.8) | 3.5 | 0 |
| W77 | 3.2 (0.5) | 0 | 0 | 3.2 (0.5) | 0 | 0 | 0 | 0 |
| W79 | 21.5 (0.8) | 5.2 (1.1) | 2.0 | 15.3 (1.4) | 1.1 (0.6) | 1.1 (0.6) | 0.4 | 0 |
| W80 | 7.7 (0.5) | 7.7 (0.5) | 1.1 | 0 | 0 | 0 | 0 | 0 |
| W81 | 6.9 (0.5) | 6.9 (0.5) | 1.8 | 0 | 0 | 0 | 0 | 0 |
| W82 | 18.1 (0.4) | 2.5 (0.6) | 0.8 | 15.3 (0.9) | 0 | 0 | 0 | 0 |
| W84 | 21.5 (0.8) | 3.7 (1.0) | 1.5 | 17.1 (1.5) | 2.6 (1.0) | 2.6 (1.0) | 1.1 | 0 |
| W86 | 35.8 (1.0) | 9.9 (1.3) | 2.5 | 23.1 (2.4) | 1.6 (0.4) | 1.6 (0.4) | 0.4 | 0 |
| W88 | 2.2 (0.3) | 6.8 (0.3) | 2.1 | −4.3 (0.5) | −23.0 (1.4) | 6.3 (1.3) | 1.9 | 27.0 (1.5) |
| W97 | 17.5 (0.9) | 6.2 (0.9) | 2.4 | 10.5 (1.3) | 31.4 (1.0) | 11.6 (1.0) | 4.5 | 17.4 (1.5) |
| W99 | 9.5 (0.6) | 3.2 (0.6) | 1.0 | 5.9 (0.8) | 0 | 0 | 0 | 0 |
| W101 | 82.4 (3.2) | 13.3 (2.4) | 3.2 | 64.7 (3.8) | 24.8 (1.5) | 33.2 (1.5) | 8.0 | 0 |
| W103 | 36.2 (1.9) | 4.4 (1.2) | 1.9 | 32.1 (2.8) | 4.7 (0.3) | 4.6 (0.3) | 1.7 | 0 |
| W105 | 69.9 (1.3) | 2.7 (1.2) | 1.0 | 63.8 (2.4) | 25.2 (0.3) | 4.6 (1.2) | 1.9 | 19.1 1.2) |
| W106 | 36.1 (0.8) | 12.9 (0.9) | 4.0 | 7.3 (0.6) | 6.5 (0.6) | 7.3 (0.6) | 2.4 | 0 |
| W107 | 27.0 (1.4) | 5.0 (1.4) | 1.0 | 21.0 (2.1) | 10.8 (0.7) | 0 | 0 | 10.8 (0.7) |
| W108 | −7.3 (0.3) | 5.2 (0.5) | 1.2 | −12.0 (0.6) | −12.1 (0.9) | 4.1 (0.8) | 0.9 | 19.6 (1.0) |
| W110 | 51.5 (1.7) | 10.3 (1.2) | 3.3 | 39.2 (1.9) | 0 | 0 | 0 | 0 |
| W112 | 31.6 (1.9) | 6.6 (1.5) | 2.3 | 23.0 (2.1) | 7.3 (0.9) | 2.9 (0.5) | 1.0 | 4.3 (0.9) |
| W114 | 59.8 (1.9) | 15.8 (1.4) | 3.8 | 37.9 (2.1) | 7.6 (0.6) | 7.6 (0.6) | 1.8 | 0 |
| W116 | 6.5 (0.3) | 3.3 (0.6) | 1.1 | 2.9 (0.7) | 0 | 0 | 0 | 0 |
Errors for quenching parameters are reported as the standard deviation (numbers in parentheses) from the mean value determined from multiple experimental data. Negative numbers reflect increased fluorescence upon ligand binding. KSVapp: apparent Stern-Volmer quenching constant. KD: dynamic
Because of the instrument limitations, it is possible that dynamic quenching of a short lifetime below 100 ps may be mistaken as static quenching. However, the fluorescence lifetime data at various concentrations of C12SL validate the assignment of static quenching (Table 3). Lifetime data for W101 and W105 with various concentrations of C12SL show very low dynamic quenching despite very high static quenching (Table 2). In addition, the average lifetimes of Trp fluorescence for most mutants are decreased. If dynamic quenching of short lifetime components of Trp fluorescence occurred, the average lifetime would be expected to increase (very short lifetimes will be invisible). Finally, contributions of short lifetime components to the steady-state fluorescence intensity (αiτi) are much lower compared to that of long lifetime components. Therefore, a significant decrease observed in steady-state fluorescence (for example, 2.2 and 1.5 times for W101 and W105, respectively, at a concentration of 12 μM C12SL) cannot be explained by dynamic quenching of short lifetime components.
Table 3.
Fluorescence Lifetime Parameters for the Mutants W101 and W105 at Varying Concentrations of C12SL
| Trp mutant | C12SL (μM) | α 1 | α 2 | τ1 (ns) | τ2 (ns) | τav (ns) | χ 2 |
|---|---|---|---|---|---|---|---|
| W101 | 0 | 0.31 | 0.69 | 0.66 | 4.35 | 4.12 | 1.01 |
| 2 | 0.35 | 0.65 | 0.97 | 4.36 | 4.00 | 1.04 | |
| 4 | 0.39 | 0.61 | 1.16 | 4.37 | 3.90 | 0.97 | |
| 8 | 0.39 | 0.61 | 1.12 | 4.28 | 3.83 | 0.94 | |
| 11.9 | 0.46 | 0.54 | 0.98 | 4.06 | 3.54 | 0.91 | |
| 15.6 | 0.56 | 0.44 | 0.93 | 3.93 | 3.24 | 0.99 | |
| W105 | 0 | 0.65 | 0.35 | 0.78 | 3.43 | 2.65 | 1.08 |
| 2 | 0.63 | 0.37 | 0.74 | 3.33 | 2.62 | 1.12 | |
| 4 | 0.65 | 0.35 | 0.74 | 3.34 | 2.59 | 1.10 | |
| 8 | 0.65 | 0.35 | 0.81 | 3.35 | 2.55 | 1.17 | |
| 11.9 | 0.68 | 0.32 | 0.80 | 3.44 | 2.56 | 0.88 | |
| 15.6 | 0.67 | 0.33 | 0.72 | 3.33 | 2.52 | 1.12 |
Side Chains and Ligand Dynamics from Dynamic Fluorescence Quenching
Quenching of Trp fluorescence can result from population reshuffling among the rotamers (conformational change), changes in local environment, etc. Additional strong fluorescence quenching observed with C12SL above that of PA can be related to direct collisions between the nitroxide moiety of the ligand and the side chain of Trp (Figure 5). Previously, it has been shown that the rotational correlation time of the nitroxide moiety of the bound C12SL is lower than the overall rotational correlation time of the protein. Therefore, the nitroxide moiety of C12SL has additional motion relative to that of the protein (31); the dynamic quenching of Trp fluorescence by ligand can be expected at multiple sites (32). It should be emphasized that the dynamic quenching originates from intracavitary dynamics of the bound ligand. Static quenching of fluorescence originates from the association of the nitroxide moiety of C12SL with the Trp in the ground state, a nonfluorescent complex. The chromophore–quencher complexes occur as a prefluorescence state and are invisible in fluorescence lifetime measurements. However, these complexes, which absorb excitation light, decrease steady-state fluorescence intensity. The bound ligands (nitroxide moiety), which form a complex with a particular Trp side chain at the moment of excitation, comprise the subset of the bound ligand with static quenching. The time scale of the motion of the ligand or the Trp side chain is irrelevant for static quenching. The quenching parameters, particularly static quenching, are significantly higher for C12SL compared to that of PA (Figures 5 and 6 and Table 2). The fluorescence lifetime parameters, which were used to calculate kq values, are shown in Table 4.
FIGURE 5.

Dynamic quenching of fluorescence of single Trp mutants by PA or C12SL and distribution of the quenching sites in the structure of TL. (A) Dynamic quenching constants and (B) rate constants for bimolecular collisional quenching for single Trp mutants by PA (red) or C12SL (black) in TL. (A, B) ▲ and ■ symbols represent β-stand and loop regions, respectively. Single and double letters denote the identities of the β-strands and loop, respectively. (C) Ribbon representation of TL. Residues are colored according to the rate constants of dynamic quenching observed with C12SL. Inset: color coding.
FIGURE 6.

Static quenching of fluorescence of single Trp mutants by PA or C12SL and distribution of the quenching sites in the structure of TL. (A) Static quenching (or enhancement) constants for single Trp mutants by PA (red) or C12SL (black) in TL. ▲ and ■ symbols are the same as in Figure 5. (B) Solvent-accessible surface representation of TL. Residues are colored according to the static quenching constants observed with C12SL. Inset: color coding.
Table 4.
Fluorescence Lifetime Parameters for Single Trp Mutants of TL
| Trp mutant | α 1 | α 2 | τ1 (ns) | τ2 (ns) | αav (ns) | χ 2 |
|---|---|---|---|---|---|---|
| W22 | 0.18 | 0.82 | 1.41 | 4.47 | 4.28 | 0.97 |
| W24 | 0.37 | 0.63 | 1.02 | 3.68 | 3.31 | 1.01 |
| W25 | 0.49 | 0.51 | 2.10 | 5.16 | 4.30 | 1.10 |
| W26 | 0.62 | 0.38 | 2.12 | 5.39 | 4.12 | 0.96 |
| W27 | 0.37 | 0.63 | 1.57 | 6.34 | 5.73 | 1.07 |
| W28 | 0.40 | 0.60 | 0.93 | 4.50 | 4.07 | 1.05 |
| W30 | 0.65 | 0.35 | 0.80 | 3.38 | 2.59 | 1.03 |
| W33 | 0.50 | 0.50 | 1.19 | 3.70 | 3.08 | 0.97 |
| W34 | 0.43 | 0.57 | 0.92 | 3.83 | 3.37 | 1.19 |
| W35 | 0.27 | 0.73 | 1.1 | 4.28 | 4.01 | 0.94 |
| W36 | 0.45 | 0.55 | 1.40 | 4.73 | 4.08 | 1.09 |
| W37 | 0.65 | 0.35 | 0.77 | 3.89 | 3.04 | 1.10 |
| W39 | 0.38 | 0.62 | 0.28 | 4.07 | 3.92 | 0.99 |
| W49 | 0.58 | 0.42 | 0.93 | 3.12 | 2.48 | 1.09 |
| W53 | 0.50 | 0.50 | 0.81 | 2.81 | 2.37 | 1.08 |
| W55 | 0.39 | 0.61 | 0.85 | 4.02 | 3.65 | 1.10 |
| W57 | 0.48 | 0.52 | 0.99 | 3.70 | 3.17 | 1.05 |
| W59 | 0.43 | 0.57 | 1.05 | 3.71 | 3.25 | 1.18 |
| W60 | 0.40 | 0.60 | 0.85 | 3.48 | 3.11 | 1.08 |
| W62 | 0.29 | 0.71 | 0.88 | 3.48 | 3.23 | 1.17 |
| W64 | 0.52 | 0.48 | 0.82 | 3.69 | 3.13 | 1.12 |
| W66 | 0.51 | 0.49 | 0.92 | 3.62 | 3.11 | 0.94 |
| W68 | 0.68 | 0.32 | 1.02 | 4.14 | 3.07 | 1.17 |
| W77 | 0.77 | 0.23 | 0.73 | 2.88 | 1.89 | 0.89 |
| W79 | 0.67 | 0.33 | 0.96 | 3.47 | 2.58 | 1.01 |
| W80 | 0.16 | 0.84 | 0.36 | 6.92 | 6.86 | 1.16 |
| W81 | 0.67 | 0.33 | 0.14 | 4.01 | 3.75 | 1.06 |
| W82 | 0.49 | 0.51 | 0.43 | 3.43 | 3.10 | 0.99 |
| W84 | 0.68 | 0.32 | 0.96 | 3.33 | 2.44 | 0.94 |
| W86 | 0.25 | 0.75 | 1.12 | 4.2 | 3.94 | 0.83 |
| W88 | 0.43 | 0.57 | 0.77 | 3.65 | 3.26 | 0.85 |
| W97 | 0.63 | 0.37 | 0.51 | 3.22 | 2.64 | 1.04 |
| W99 | 0.37 | 0.63 | 0.82 | 3.53 | 3.20 | 1.13 |
| W101 | 0.31 | 0.69 | 0.66 | 4.35 | 4.12 | 1.01 |
| W103 | 0.64 | 0.36 | 0.72 | 3.00 | 2.32 | 1.08 |
| W105 | 0.65 | 0.35 | 0.78 | 3.43 | 2.65 | 1.08 |
| W106 | 0.38 | 0.62 | 1.16 | 3.56 | 3.16 | 0.90 |
| W107 | 0.44 | 0.56 | 1.08 | 5.07 | 4.50 | 0.93 |
| W108 | 0.41 | 0.59 | 0.80 | 4.55 | 4.14 | 0.96 |
| W110 | 0.42 | 0.58 | 0.85 | 3.51 | 3.11 | 0.96 |
| W112 | 0.69 | 0.31 | 0.80 | 3.78 | 2.82 | 1.10 |
| W114 | 0.23 | 0.79 | 0.35 | 4.27 | 4.18 | 0.82 |
| W116 | 0.55 | 0.45 | 0.72 | 3.47 | 2.91 | 0.91 |
The variations of static and dynamic quenching constants in the single Trp mutants are not the result of differences in Kd values. Normalization resulted in only minor changes for the quenching parameters of most mutants (Figures 5 and 6 and Table 2) and did not alter the results significantly or the conclusions.
In single Trp mutants of TL, the rate constants for bimolecular collisional quenching of Trp fluorescence by the nitroxide moiety of the ligand are of the order of ~1012 M−1 s−1. That is 3 orders of magnitude higher than expected for the diffusion-limited rate constant. PA induces dynamic quenching, the rate constants of which are of the same order of magnitude as C12SL. This can be misleading. For estimation of Kd and, therefore, κq values (Figures 5A,B and Table 2), the quencher (PA and C12SL) concentrations were calculated using sample volume, which is not the reaction volume in this case. Because the reaction volume is limited to the cavity volumes, effective concentrations are much higher. Therefore, the data reflect the intracavitary dynamics of the ligands. Assuming that the total volume of the protein is the effective volume for the ligand, one can estimate actual κq. The estimated κq values are of the order of 12×109 M−1 s−1 (see Materials and Methods). The implication is that ligand and/or side chain motions evident from dynamic quenching occur in the nanosecond range.
The sites of the protein that show the most dynamic quenching by PA as well as C12SL are located at loop AB and at the tips of β-strands, except position 101 (Figure 5). Since the dynamic quenching of fluorescence by PA is indirect, these sites exhibit dynamic conformational changes associated with intracavitary ligand dynamics. Additional quenching of the fluorescence observed with C12SL, compared to that of PA, reveals side chains that have direct collision with the nitroxide moiety of the ligand (Figure 5A,B). Assuming that the interaction sites of the protein for PA and C12SL are the same, it is evident that conformational changes are not realized by every collision. For example, W39 exhibits dynamic quenching with C12SL; the corresponding rate constant is 3.7 x 1012 M−1 s−1. However, PA does not induce any observable quenching at the same site.
Conformational Selections with Ligand Binding from Static Quenching
Unlike dynamic quenching, static fluorescence quenching by the ligands reveals the protein sites at which the side chains of Trp form nonfluorescent ground-state complexes with the “quencher” groups. In contrast to the dynamic fluorescence quenching data, only a few sites, which are located at loops AB and GH as well as at the tips of β-strands (F and G), exhibit static alterations (both quenching and enhancement) associated with PA binding (Figure 6A). As indicated for dynamic quenching, static quenching of Trp fluorescence induced by PA is not direct. Static quenching of fluorescence takes place when the side chain of Trp forms the ground-state complex with the “quencher” side chain, which acts as an electron acceptor or a proton donor (28). Therefore, static quenching is reported in Trp sites when ligand binding selects the conformation with such complexes. However, static fluorescence enhancement occurs if the ligand binding is associated with release of the ground-state complexes between Trp and the “quencher” side chain. Because the fluorescence lifetime is not affected by static quenching, the contribution of the dynamic and static quenching can easily be recognized by comparing the Stern–Volmer plots for the fluorescence lifetime and intensity data (Figure 4). Large differences between such plots are a sign of static alteration. The much deviated plots observed for W62 compared to that of W37 manifest higher static quenching (Figure 4 and Table 2).
The interactions that generate static quenching are very insightful. The Trp side chain at position 88, which shows the biggest enhancement with PA binding, is in close proximity to the Tyr97 side chain (Figures 6 and 7A), which quenches Trp fluorescence by an excited-state proton transfer mechanism (26). On the other hand, the Trp side chain at position 97 is in close proximity to the side chains of Arg90 and Arg118, which also could quench Trp fluorescence by the same mechanism (26). Previously, similar quenching of Trp fluorescence at position 97 was observed with LA (32). The data suggest that PA binding results in the altered conformation in the tip of hairpin FG, in which the Trp88–quencher complex is broken. It should be noted that the tip of hairpin FG is located at the closed end of the TL barrel. Because hairpin FG does not provide access to the cavity, the observed altered conformation can be considered as induced rather than selected by ligand binding. In addition, as previously suggested loop FG may be involved in the receptor binding for holo-TL (32). In loop GH, three (Glu104, His106, and Lys108) of the five residues have side chains that can quench Trp fluorescence (Figures 6 and 7B). Only the protonated Glu (pKa =4.5 for solvent-exposed residue) residue can quench Trp fluorescence (26). The protonation state of Glu depends on its environment. Therefore, the static quenching (positions 105 and 107) or enhancement (position 108) of fluorescence observed in the loop GH residues (Figures 6A and 7B) strongly suggests that in excited protein states the conformation of loop GH is changed. This, in turn, will rearrange side chains of the loop residues, perhaps, necessary for ligand binding.
FIGURE 7.
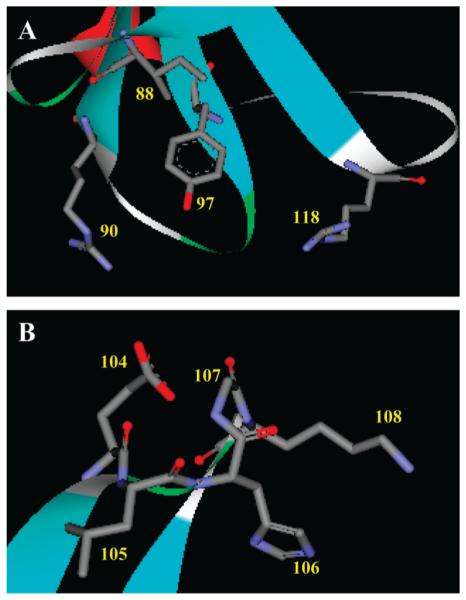
Residues at the tip of hairpin FG (A) or loop GH (B). Color coding: gray, carbon; red, oxygen; blue, nitrogen.
Loop AB also shows the conformational changes in the ligand-bound state compared to that of the ligand-free state. Obviously, Trp fluorescence intensity is most sensitive to tertiary interaction when its counterpart is a fluorescence quencher. In loop AB, only Trp in positions 28 and 37 shows static quenching with PA. Trp28 is flanked with Arg26, Glu27, and Glu30 that can potentially quench fluorescence. Similarly, Trp37 is in close proximity to the quencher, side chain of Tyr18. Loop AB is the longest of the loops in all of the proteins of the lipocalin family and also serves as a receptor binding site for some lipocalins. Loop AB is a putative gate region for ligand binding (16). Indeed, the quenching data (Figure 6) are consistent with that proposal; loop AB together with neighboring loop GH may be the gate region in TL for the ligand binding. It should be noted that intracavitary ligand dynamics are more complex than we described here. Additional experimental data are necessary to address full complexity of ligand dynamics.
Binding Energy Landscape for TL
The static quenching data obtained with C12SL are immensely different from the data observed with PA (Figure 6A and Table 2). The number of the protein sites, which show static fluorescence quenching with C12SL, is increased about 3-fold compared to that obtained with PA. The distribution of the quenching sites of the protein (Figures 1 and 6) is well beyond the interaction distance (about 4–6 Å) between the indole ring of a tryptophan and the nitroxide (25). Because the nitroxide moiety of C12SL quenches Trp fluorescence by collision, the strong static quenching observed with C12SL compared to that of PA reflects nonfluorescent complex formation between a nitroxide moiety of the ligand and the side chains at the respective positions in the protein. The bound fraction of C12SL, which collides with the Trp side chain during its fluorescence lifetime, will produce dynamic quenching. In contrast, static quenching reveals the fraction of bound ligand, which forms a nonfluorescence complex with a particular Trp side chain at the moment of excitation. This fraction can be envisioned as statically bound. The ligand association sites are distributed widely within the cavity of TL (Figure 6). The majority are localized at the adjacent β-strands G and H and loop AB. The static quenching constant is directly related to the association constant between the chromophore and the quencher. With this definition, apparent ΔG for each site can be calculated. The distribution of the ligand within the cavity can then be assigned (Figure 8). This method unravels the ensemble average of the protein–ligand complexes. About 70% of the bound ligands are distributed around β-strands G and H and loop AB. The roughness of the binding energy landscape is about 2.5kBT (6 kJ/mol), which is close to the value (1.7kBT) determined for a diffusion-limited loop enclosure in small peptides (33). Taken together, all of the results indicate that the excited protein states result primarily from conformational selections of loops AB and GH. Thus, the conformations of loops AB and GH not only provide access for the ligand but also dictate its positions in the cavity.
FIGURE 8.
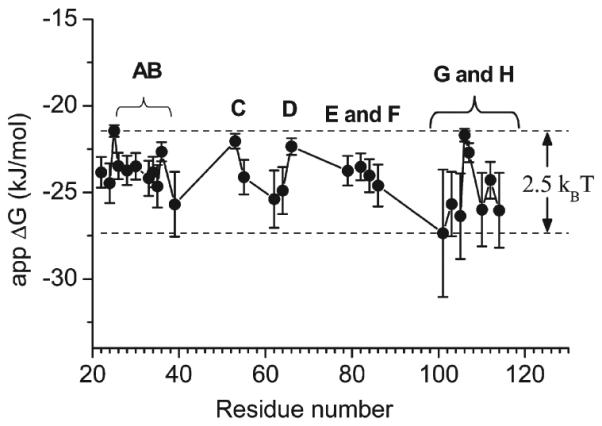
Binding energy landscape of TL deduced from static quenching constants of single Trp mutants of TL by C12SL. Vertical bars are normalized fractions of the ligands populated within the cavity of TL according to the Boltzmann distribution. Single and double letters denote the identities of the β-strands and loop, respectively.
Conclusion
The strategy developed in this study to investigate the ligand binding mechanism can be compared to the NMR chemical shift perturbation method. Unlike NMR chemical shift perturbation studies (34) where chemical shifts are monitored for backbone amide atoms, in this study ligand interactions are monitored at the side chains of individual amino acid residues. Comparison of the results obtained in this study is relevant with those for other lipocalins. The lipocalin-type prostaglandin D synthase (L-PGDS), besides its unique enzymatic activity in the lipocalin family, binds a variety of ligands with high affinities (34–39). The ligand-induced chemical shift changes in backbone resonances (1H and 15N) of L-PGDS in complexes with all-trans retinoic acid and U-46619 have been observed for about 90 and 120 backbone sites, respectively (34). The perturbation sites are widely distributed around the β-barrel. In L-PGDS-ligand complexes, the distribution of the residues that exhibit the chemical shift perturbation can be compared with the sites of TL, which show dynamic and static quenching upon ligand binding. These residues are scattered widely in the cavities of both respective lipocalins. Docking studies reveal the unique positions for the all-trans retinoid acid and U-46619 in the cavity of L-PGDS; the contact sites show large changes in chemical shifts. However, the docking models do not elucidate the origin of the moderate or small changes in chemical shifts. Commonly, a unique solution for docking studies is presumed. When the docking studies yield multiple solutions for a ligand with similar binding energy, the result that better fits experimental data is chosen to represent the protein–ligand complex (for example, in TL (40), in β-lactoglobulin (41)). Interestingly, although interaction energies have not been reported, retinoic acid could be docked to TL in two orientations (17). While the β-ionone ring of retinoic acid resides approximately at the same position, the terminal carboxylate group can take two different positions (17). In addition, fatty acid binding with two opposite orientations, albeit with significant differences in Kd values, has previously been suggested for TL (40).
Site-directed tryptophan fluorescence in combination with ligand quenchers provides an exciting tool for identifying ligand binding dynamics, conformational shifts, and ligand distribution for many proteins. This methodology should prove applicable to engineer mutant proteins with improved ligand binding characteristics and to develop algorithms for docking ligands.
Footnotes
Supported by U.S. Public Health Service Grants NIH EY11224 and EY00331 as well as the Edith and Lew Wasserman Endowed Professorship in Ophthalmology.
- C12SL
- spin-labeled analogue of palmitic acid
- DAUDA
- 11-((5-dimethylaminonaphthalene-1-sulfonyl)amino)undecanoic acid
- IRF
- instrument response function
- LA
- lauric acid
- LCN1
- lipocalin 1
- PA
- palmitic acid
- SDTF
- site-directed tryptophan fluorescence
- TL
- human tear lipocalin
REFERENCES
- 1.Okazaki K, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: induced-fit versus population-shift mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glasgow BJ, Abduragimov AR, Farahbakhsh ZT, Faull KF, Hubbell WL. Tear lipocalins bind a broad array of lipid ligands. Curr. Eye Res. 1995;14:363–372. doi: 10.3109/02713689508999934. [DOI] [PubMed] [Google Scholar]
- 3.Scalfari F, Castagna M, Fattori B, Andreini I, Maremmani C, Pelosi P. Expression of a lipocalin in human nasal mucosa. Comp. Biochem. Physiol. 1997;118B:819–824. [Google Scholar]
- 4.Lacazette E, Gachon AM, Pitiot G. A novel human odorant-binding protein gene family resulting from genomic duplicons at 9q34: differential expression in the oral and genital spheres. Hum. Mol. Genet. 2000;9:289–301. doi: 10.1093/hmg/9.2.289. [DOI] [PubMed] [Google Scholar]
- 5.Redl B. Human tear lipocalin. Biochim. Biophys. Acta. 2000;1482:241–248. doi: 10.1016/s0167-4838(00)00142-4. [DOI] [PubMed] [Google Scholar]
- 6.Wojnar P, Dirnhofer S, Ladurner P, Berger P, Redl B. Human lipocalin-1, a physiological scavenger of lipophilic compounds, is produced by corticotrophs of the pituitary gland. J. Histochem. Cytochem. 2002;50:433–435. doi: 10.1177/002215540205000314. [DOI] [PubMed] [Google Scholar]
- 7.Selsted ME, Martinez RJ. Isolation and purification of bactericides from human tears. Exp. Eye Res. 1982;34:305–318. doi: 10.1016/0014-4835(82)90079-3. [DOI] [PubMed] [Google Scholar]
- 8.van’t Hof W, Blankenvoorde MF, Veerman EC, Amerongen AV. The salivary lipocalin von Ebner’s gland protein is a cysteine proteinase inhibitor. J. Biol. Chem. 1997;272:1837–1841. doi: 10.1074/jbc.272.3.1837. [DOI] [PubMed] [Google Scholar]
- 9.Blaker M, Kock K, Ahlers C, Buck F, Schmale H. Molecular cloning of human von Ebner’s gland protein, a member of the lipocalin superfamily highly expressed in lingual salivary glands. Biochim. Biophys. Acta. 1993;1172:131–137. doi: 10.1016/0167-4781(93)90279-m. [DOI] [PubMed] [Google Scholar]
- 10.Redl B, Holzfeind P, Lottspeich F. cDNA cloning and sequencing reveals human tear prealbumin to be a member of the lipophilic-ligand carrier protein superfamily. J. Biol. Chem. 1992;267:20282–20287. [PubMed] [Google Scholar]
- 11.Lechner M, Wojnar P, Redl B. Human tear lipocalin acts as an oxidative-stress-induced scavenger of potentially harmful lipid peroxidation products in a cell culture system. Biochem. J. 2001;356:129–135. doi: 10.1042/0264-6021:3560129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glasgow BJ, Abduragimov AR, Gassymov OK, Yusifov TN, Ruth EC, Faull KF. Vitamin E associated with the lipocalin fraction of human tears. Adv. Exp. Med. Biol. 2002;506:567–572. doi: 10.1007/978-1-4615-0717-8_79. [DOI] [PubMed] [Google Scholar]
- 13.Yusifov TN, Abduragimov AR, Gasymov OK, Glasgow BJ. Endonuclease activity in lipocalins. Biochem. J. 2000;347:815–819. Part 3. [PMC free article] [PubMed] [Google Scholar]
- 14.Gouveia SM, Tiffany JM. Human tear viscosity: an interactive role for proteins and lipids. Biochim. Biophys. Acta. 2005;1753:155–163. doi: 10.1016/j.bbapap.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 15.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Site-directed tryptophan fluorescence reveals the solution structure of tear lipocalin: evidence for features that confer promiscuity in ligand binding. Biochemistry. 2001;40:14754–14762. doi: 10.1021/bi0110342. [DOI] [PubMed] [Google Scholar]
- 16.Flower DR. The lipocalin protein family: structure and function. Biochem. J. 1996;318:1–14. doi: 10.1042/bj3180001. Part 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breustedt DA, Korndorfer IP, Redl B, Skerra A. The 1.8-Å crystal structure of human tear lipocalin reveals an extended branched cavity with capacity for multiple ligands. J. Biol. Chem. 2005;280:484–493. doi: 10.1074/jbc.M410466200. [DOI] [PubMed] [Google Scholar]
- 18.Marvin JS, Hellinga HW. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 2001;8:795–798. doi: 10.1038/nsb0901-795. [DOI] [PubMed] [Google Scholar]
- 19.Glasgow BJ, Heinzmann C, Kojis T, Sparkes RS, Mohandas T, Bateman JB. Assignment of tear lipocalin gene to human chromosome 9q34-9qter. Curr. Eye Res. 1993;12:1019–1023. doi: 10.3109/02713689309029229. [DOI] [PubMed] [Google Scholar]
- 20.Glasgow BJ. Tissue expression of lipocalins in human lacrimal and von Ebner’s glands: colocalization with lysozyme. Graefes Arch. Clin. Exp. Ophthalmol. 1995;233:513–522. doi: 10.1007/BF00183433. [DOI] [PubMed] [Google Scholar]
- 21.Cormack B. in Current Protocol in Molecular Biology. Suppl. Vol. 15. Greene Publishing Associates and Wiley-Interscience; New York, NY: 1987. [Google Scholar]
- 22.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Binding studies of tear lipocalin: the role of the conserved tryptophan in maintaining structure, stability and ligand affinity. Biochim. Biophys. Acta. 1999;1433:307–320. doi: 10.1016/s0167-4838(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 23.Marston FAO. In: The purification of eukaryotic polypeptides expresses in Eschericia coli, in DNA Cloning: a practical approach. Glover DM, editor. IRL Press; Oxford, England: 1987. pp. 59–88. [Google Scholar]
- 24.Bozimowski D, Artiss JD, Zak B. The variable reagent blank: protein determination as a model. J. Clin. Chem. Clin. Biochem. 1985;23:683–689. doi: 10.1515/cclm.1985.23.10.683. [DOI] [PubMed] [Google Scholar]
- 25.Green JA, II, Singer LA, Parks JH. Fluorescence quenching by the stable free radical di-t-butylnitroxide. J. Chem. Phys. 1973;58:2690–2695. [Google Scholar]
- 26.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 27.Yu HT, Colucci WJ, McLaughlin ML, Barkley MD. Fluorescence quenching in indoles by excited-state proton transfer. J. Am. Chem. Soc. 1992;114:8449–8454. [Google Scholar]
- 28.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd Springer; New York: 2006. [Google Scholar]
- 29.Pan CP, Barkley MD. Conformational effects on tryptophan fluorescence in cyclic hexapeptides. Biophys. J. 2004;86:3828–3835. doi: 10.1529/biophysj.103.038901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams PD, Chen Y, Ma K, Zagorski MG, Sonnichsen FD, McLaughlin ML, Barkley MD. Intramolecular quenching of tryptophan fluorescence by the peptide bond in cyclic hexapeptides. J. Am. Chem. Soc. 2002;124:9278–9286. doi: 10.1021/ja0167710. [DOI] [PubMed] [Google Scholar]
- 31.Glasgow BJ, Gasymov OK, Abduragimov AR, Yusifov TN, Altenbach C, Hubbell WL. Side chain mobility and ligand interactions of the G strand of tear lipocalins by site-directed spin labeling. Biochemistry. 1999;38:13707–13716. doi: 10.1021/bi9913449. [DOI] [PubMed] [Google Scholar]
- 32.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Resolution of ligand positions by site-directed tryptophan fluorescence in tear lipocalin. Protein Sci. 2000;9:325–331. doi: 10.1110/ps.9.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapidus LJ, Eaton WA, Hofrichter J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimamoto S, Yoshida T, Inui T, Gohda K, Kobayashi Y, Fujimori K, Tsurumura T, Aritake K, Urade Y, Ohkubo T. NMR solution structure of lipocalin-type prostaglandin D synthase: evidence for partial overlapping of catalytic pocket and retinoic acid-binding pocket within the central cavity. J. Biol. Chem. 2007;282:31373–31379. doi: 10.1074/jbc.M700123200. [DOI] [PubMed] [Google Scholar]
- 35.Beuckmann CT, Aoyagi M, Okazaki I, Hiroike T, Toh H, Hayaishi O, Urade Y. Binding of biliverdin, bilirubin, and thyroid hormones to lipocalin-type prostaglandin D synthase. Biochemistry. 1999;38:8006–8013. doi: 10.1021/bi990261p. [DOI] [PubMed] [Google Scholar]
- 36.Inui T, Ohkubo T, Emi M, Irikura D, Hayaishi O, Urade Y. Characterization of the unfolding process of lipocalin-type prostaglandin D synthase. J. Biol. Chem. 2003;278:2845–2852. doi: 10.1074/jbc.M209934200. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka T, Urade Y, Kimura H, Eguchi N, Nishikawa A, Hayaishi O. Lipocalin-type prostaglandin D synthase (beta-trace) is a newly recognized type of retinoid transporter. J. Biol. Chem. 1997;272:15789–15795. doi: 10.1074/jbc.272.25.15789. [DOI] [PubMed] [Google Scholar]
- 38.Kanekiyo T, Ban T, Aritake K, Huang ZL, Qu WM, Okazaki I, Mohri I, Murayama S, Ozono K, Taniike M, Goto Y, Urade Y. Lipocalin-type prostaglandin D synthase/ beta-trace is a major amyloid beta-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 2007;104:6412–6417. doi: 10.1073/pnas.0701585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, Fukumoto N, Eguchi N, Kushi A, Sasai H, Kanaoka Y, Ozono K, Narumiya S, Suzuki K, Urade Y. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 2006;26:4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasymov OK, Abduragimov AR, Glasgow BJ. Ligand binding site of tear lipocalin: contribution of a trigonal cluster of charged residues probed by 8-anilino-1-naphthalenesulfonic acid. Biochemistry. 2008;47:1414–1424. doi: 10.1021/bi701955e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collini M, D’Alfonso L, Molinari H, Ragona L, Catalano M, Baldini G. Competitive binding of fatty acids and the fluorescent probe 1-8-anilinonaphthalene sulfonate to bovine beta-lactoglobulin. Protein Sci. 2003;12:1596–1603. doi: 10.1110/ps.0304403. [DOI] [PMC free article] [PubMed] [Google Scholar]