

Supplemental Digital Content is available in the text.
Keywords: arrhythmias, cardiac; calcium; calcium signaling; sodium-calcium exchanger
Abstract
Background—
In catecholaminergic polymorphic ventricular tachycardia (CPVT), cardiac Purkinje cells (PCs) appear more susceptible to Ca2+ dysfunction than ventricular myocytes (VMs). The underlying mechanisms remain unknown. Using a CPVT mouse (RyR2R4496C+/Cx40eGFP), we tested whether PC intracellular Ca2+ ([Ca2+]i) dysregulation results from a constitutive [Na+]i surplus relative to VMs.
Methods and Results—
Simultaneous optical mapping of voltage and [Ca2+]i in CPVT hearts showed that spontaneous Ca2+ release preceded pacing-induced triggered activity at subendocardial PCs. On simultaneous current-clamp and Ca2+ imaging, early and delayed afterdepolarizations trailed spontaneous Ca2+ release and were more frequent in CPVT PCs than CPVT VMs. As a result of increased activity of mutant ryanodine receptor type 2 channels, sarcoplasmic reticulum Ca2+ load, measured by caffeine-induced Ca2+ transients, was lower in CPVT VMs and PCs than respective controls, and sarcoplasmic reticulum fractional release was greater in both CPVT PCs and VMs than respective controls. [Na+]i was higher in both control and CPVT PCs than VMs, whereas the density of the Na+/Ca2+ exchanger current was not different between PCs and VMs. Computer simulations using a PC model predicted that the elevated [Na+]i of PCs promoted delayed afterdepolarizations, which were always preceded by spontaneous Ca2+ release events from hyperactive ryanodine receptor type 2 channels. Increasing [Na+]i monotonically increased delayed afterdepolarization frequency. Confocal imaging experiments showed that postpacing Ca2+ spark frequency was highest in intact CPVT PCs, but such differences were reversed on saponin-induced membrane permeabilization, indicating that differences in [Na+]i played a central role.
Conclusions—
In CPVT mice, the constitutive [Na+]i excess of PCs promotes triggered activity and arrhythmogenesis at lower levels of stress than VMs.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inheritable, proarrhythmogenic syndrome that affects ≈1 in 10 000 children.1 CPVT is characterized by adrenergically mediated bidirectional VT or polymorphic VT that may lead to sudden cardiac death in the absence of structural heart disease.2 CPVT may be caused by mutations in genes coding calcium handling proteins, including the cardiac sarcoplasmic reticulum (SR) Ca2+ release (ryanodine receptor type 2 [RyR2]) channel (RYR2),3 calsequestrin (CASQ2),4 triadin (TRDN),5 and calmodulin (CaM).6 The focus of this article is RYR2. Arrhythmias produced by gain-of-function mutations in RyR2 are postulated to result from destabilization of the channel with increased diastolic SR Ca2+ leak in ventricular myocytes (VMs), leading to delayed afterdepolarizations (DADs) and triggered activity via the Na+/Ca2+ exchanger (NCX) current.7,8
Clinical Perspective on p 2359
Using a knock-in mouse model with a point mutation in RyR2 (RyR2R4496C), we and others have postulated that Purkinje cells (PCs), not VMs, are the major source of triggered arrhythmogenic activity, including biventricular tachycardia.9–11 There is also evidence supporting a prominent role for the PC conducting system in triggered activity and arrhythmogenesis in the CASQ2−/− model of CPVT.12 These findings have spawned increasing interest in the mechanisms of intracellular Ca2+ [Ca2+]i homeostasis of the cardiac PCs in both diseased and healthy hearts. PCs have an increased incidence of early afterdepolarizations (EADs) and DADs compared with VMs.13 PCs have greater sensitivity to arrhythmogenesis in conditions of Ca2+ overload, produced by agents such as digitalis compounds that block the membrane Na/K ATPase, perhaps in part as a result of reduced transcript abundance of that protein in PCs versus VMs.14 PCs from CPVT mice have a shorter latency and a lower threshold to develop spontaneous Ca2+ release (SCR) events than VMs.11 Whether this applies also to the whole heart and whether SCR triggers premature ventricular complexes that propagate from the Purkinje system to the ventricles remain to be determined. Most important, the question of what makes PCs more susceptible to arrhythmogenesis in CPVT than VMs has never been addressed. Answering the above questions may lead to novel and more effective antiarrhythmic therapeutic strategies in CPVT and other cardiac diseases associated with Ca2+-linked ventricular arrhythmias.
Our main objective in this study was 2-fold: to test whether Ca2+ dysregulation from within the Purkinje network underlies focal triggered activity in CPVT hearts (RyR2R4496C+/−) and to establish whether a higher [Na+]i in mouse PCs than VMs exacerbates [Ca2+]i dysregulation in CPVT and explains their increased susceptibility to EADs, DADs, and triggered activity compared with VMs.
Methods
Expanded versions of the Methods are presented in the online-only Data Supplement.
Animals
The study was approved by the University Committee on the Use and Care of Animals at the University of Michigan. RyR2+/Cx40eGFP+/− (hereafter control) and RyR2R4496C+/Cx40eGFP (hereafter CPVT) mice were used to enable visualization of the Purkinje network and isolated PCs via green fluorescent protein expression (Figure I in the online-only Data Supplement).11
Optical Mapping
Optical mapping of the subendocardium was conducted in Langendorff-perfused hearts with di-4-ANBDQPQ and rhod-2 am dyes, a single charge-coupled device camera, and a custom-made LED system.15 Lead I ECGs were acquired and digitized at 1 kHz.16 The protocol included 4 groups: baseline sinus rhythm, high-frequency (12 Hz) pacing, adrenergic stimulation (3.6 mmol/L Ca2+, 160 nmol/L isoproterenol), and high-frequency pacing plus adrenergic stimulation.
Isolation of PCs and VMs
PCs and VMs were dissociated as previously described.11,17,18 For permeabilized membrane experiments, cells were kept in Ca2+ free stopping buffer. Cells were used for electrophysiological recording within 4 to 6 hours after isolation.
Immunohistochemistry
Immunofluorescence was examined with a Nikon Eclipse Ti inverted confocal microscope with a ×63/1.2-NA oil objective (Nikon Inc, Tokyo, Japan) at ambient temperature. Rabbit anti-CaV1.2 (Alomone, Jerusalem, Israel) and mouse anti-RyR2 (Iowa Hybridoma Core, University of Iowa) antibodies were used.
Simultaneous Action Potential and Ca2+ Transient Measurements
Cells were perfused with Tyrode’s solution (37°C) and patched with 2- to 3-MΩ glass pipettes filled with intracellular solution at pH 7.2. Ca2+ signals were recorded with a whole-cell fluorescence photometry acquisition system (IonOptix). Fluorescent signals were synchronized to action potential (AP) recordings. Cells were stimulated at increasing frequencies (1, 3, 5 Hz) by current injection via patch pipette.
Ca2+ Transients and Ca2+ Sparks
Cells were incubated for 30 minutes in Tyrode’s solution containing 10 µmol/L rhod-2 am, washed with a fluorophore-free solution, and de-esterified for 15 minutes. Cells were then perfused with Tyrode’s solution at 37° C. Measurements were acquired with a Nikon A1R confocal microscope in line scan mode. For Ca2+ transients, cells were field stimulated at 1, 3, and 5 Hz. For Ca2+ spark analysis, a 2-second line scan image window was selected 3 seconds after a steady-state Ca2+ transient had decreased to baseline level.
Permeabilized Cellular Ca2+ Spark Recordings
VMs and PCs were perfused with 50 nmol/L free Ca2+ internal solution plus saponin 0.005% for 1 minute for membrane permeabilization. Then, cells were perfused for 5 minutes with internal solution containing 2.5 µmol/L fluo-4 penta-K+ (LifeTechnologies). Confocal line scan recordings (4-second duration) were obtained and analyzed within 10 minutes after dye loading as described above.
SR Ca2+ Load
Cells were loaded with rhod-2 (Life Technologies) as described above. Cells were perfused with 37°C Tyrode’s solution containing 1 mmol/L Ca2+ and paced at 3 Hz for 10 seconds. One hundred milliseconds after pacing, cells were quickly perfused with Tyrode’s solution containing caffeine (10 mmol/L). Ca2+ transient amplitude was a fraction of the caffeine-induced Ca2+ transient amplitude. Time to 50% decay was estimated from time at maximal caffeine-induced Ca2+ transient amplitude.
Na+/Ca2+ Exchange Current
Cells were perfused with external solution containing nifedipine (10 µmol/L), ouabain (1 mmol/L), and niflumic acid (10 µmol/L). Free Ca2+ in the pipette solution was 205 nmol/L, Current was elicited with a descending-ascending voltage ramp from a holding potential of −75 mV. Na+/Ca2+ exchange current (INCX) density was determined as the NiCl2 (1 mmol/L)-sensitive current.
[Na]i Measurements
[Na]i was measured with SBFI-AM as previously described.19
Statistical Analyses
Data are presented as mean±SEM, and values of P<0.05 were considered significant. The unpaired Student t test, Fisher exact test, Mann-Whitney U test, or Kruskal-Wallis test with Dunn posttest were performed when appropriate to compare experimental groups. Statistical analysis was carried out with GraphPad Prism software (version 6.0).
Biophysical Model of Murine PC
The model described in Vaidyanathan et al18 was modified to include cytosolic Ca2+ waves and late sodium current (INaL). Two-dimensional cytosolic Ca2+ diffusion was implemented to reproduce the 2-component Ca2+ transients in canine PCs.20 Details are given in the online-only Data Supplement (Tables I–V and Figures II–IV in the online-only Data Supplement).
Computer Simulations
[Na+]i was increased by reducing the magnitude of Na+-K+ pump current (INaK) as summarized in Table III in the online-only Data Supplement on the basis of earlier work (but also see Limitations).19,21 The functional effects of RyR2 mutations were implemented by lowering the threshold of SR Ca2+ release.22 Isoproterenol effects were implemented by altering the maximum conductance of Ca2+ currents (ICaL and ICaT),23,24 inward rectifier current (Ik1), and INaK.25 Table IV in the online-only Data Supplement provides details.
Results
Subendocardial Origin of Postpacing Triggered Beats
Previously, we demonstrated that polymorphic VT in the CPVT mouse model is attributable to focal discharges at multiple locations within the specialized conduction system.9 Here, we provide direct evidence of both the origin and the triggering mechanisms of such ectopic activity. Abnormal electric activity was elicited in 6 of 7 (85.7%) CPVT hearts and in 3 of 7 (42.8%) control hearts treated with Tyrode’s solution containing high Ca2+ and isoproterenol after burst pacing. Figure 1A shows a representative volume-conducted ECG tracing from a CPVT heart before and after burst pacing. Rapid electric pacing in the presence of high Ca2+ and isoproterenol generated a series of ectopic beats immediately after pacing cessation. Each ectopic beat is identified by a number (1–9) in the red box in Figure 1A.
Figure 1.
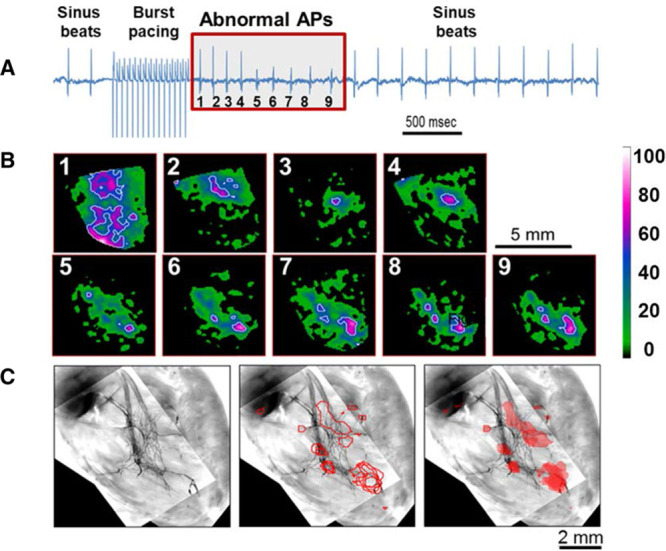
Subendocardial origins of spontaneous beats. A, Volume-conducted ECG recording of a catecholaminergic polymorphic ventricular tachycardia heart with 3.6 mmol/L Ca2+ and 160 nmol/L isoproterenol before, during, and after 12-Hz burst pacing. Nine abnormal action potentials (APs) were induced after pacing (red box, numbered). B, Earliest right ventricular endocardial AP wave breakthroughs of abnormal beats (1–9, each in a separate box) shown by white and magenta spots (color index bar represents percent from maximal signal in each frame). C, Image of green fluorescent protein–mapped area of the Purkinje network (left) superimposed with white contour lines from B (middle and right). Note colocalization of triggered beats with branches of the Purkinje conduction system.
The precise origin for each of the 9 abnormal beats was identified in endocardial optical maps (Figure 1B), with earliest breakthrough points represented by white and magenta regions. These 9 beats were overlaid onto a high-resolution fluorescent image of the heart displaying green fluorescent protein–expressing Purkinje fibers. Initial breakthroughs colocalized with Purkinje fiber sites within the subendocardial conducting network (Figure 1C), suggesting that such fibers were the source of arrhythmogenic focal activity.
Spontaneous Ca2+ Release Precedes Triggered Activity in Subendocardial PCs of the CPVT Mouse
We then examined the temporal relationship between [Ca2+]i release and membrane voltage (Vm) with respect to triggered activity in the subendocardium of CPVT mouse hearts. Hearts were loaded with the Ca2+ indicator dye rhod-2 am and the voltage sensitive dye di-4-ANBDQPQ. In Figure 2A, simultaneous single pixel Ca2+ (red) and voltage (black) signals at a Purkinje fiber location (asterisk in Figure 2C) are displayed with paired ECG recordings (blue). Continuous rapid pacing (dotted line) in the presence of high Ca2+ and isoproterenol elicited a run of VT (box in Figure 2A). During VT, the sequence of Ca2+ and Vm activation signals was opposite sinus rhythm (before pacing). In Figure 2B, examples of 1 sinus discharge (left) and 1 VT discharge (right) emphasize that difference. During the abnormal beat, SCR preceded membrane depolarization, giving rise to a triggered discharge. In Figure 2C, the optical signals were analyzed beat by beat at high spatiotemporal resolution in endocardial Vm-Ca2+ difference maps. In sharp contrast with sinus and recovery beats, the Ca2+ transient preceded depolarization during each triggered discharge and the mean map.
Figure 2.
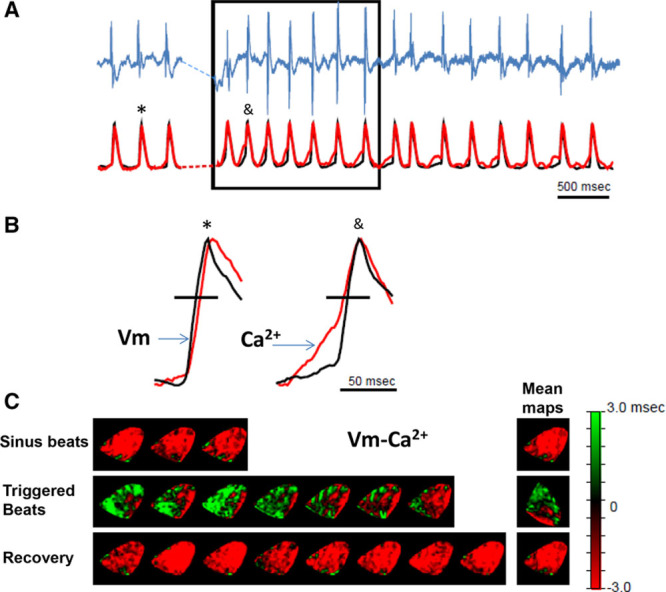
Spontaneous Ca2+ release precedes voltage depolarization during triggered activity in the subendocardium of the catecholaminergic polymorphic ventricular tachycardia heart. The heart was perfused with 3.6 mmol/L Ca2+ and 160 nmol/L isoproterenol. A, Volume-conducted ECG (top, blue) and optical signals obtained from a single pixel (black, voltage; red, Ca2+) at the earliest breakthrough on a subendocardial Purkinje fiber location. B, Enlarged representative traces for sinus (left) and triggered (right) beats. Symbols on top identify the specific beats selected from A. C, Beat-to-beat (left) and mean (right) Vm-Ca2+ timing difference maps for sinus (top), triggered (middle), and recovery (bottom) beats measured at 60% maximum depolarization (horizontal black lines in B).
Subcellular Distribution of the L-type Ca2+ Channel and RyR2 in Mouse PCs
The previously demonstrated presence of T tubules in mouse PCs26 suggests that dyadic structures exist within these cells. Therefore, we assessed whether mouse PCs contain 2 key Ca2+ channels, L-type Ca2+ channel (α subunit, CaV1.2) and RyR2, in the dyad. Intracellular areas of RyR2 and CaV1.2 colocalizations were observed in both VMs and PCs isolated from control hearts; however, they were more heterogeneous in PCs than in VMs (Figure V in the online-only Data Supplement). Both proteins exhibited a striated staining pattern with significant colocalization at the sarcolemma (Figure Vb–Vd in the online-only Data Supplement) and at the expected T-tubule locations (Figure Ve and Vf in the online-only Data Supplement).
CPVT PCs Have a Lower Threshold for Triggered Activity Than VMs and Correlate With Abnormal Ca2+ Release
Figure 3 shows representative APs and Ca2+ transients recorded simultaneously from cells paced at 1 and 5 Hz in the absence and presence of isoproterenol. The addition of isoproterenol increased the Ca2+ transient amplitude in all cell types. Only CPVT VM and CPVT PC are shown because neither control VM nor control PC presented with abnormal beats during steady-state pacing (n=4 in each group). At steady-state baseline conditions (1-Hz pacing), no cells presented with abnormal Ca2+ handling or triggered activity, and each pacing-induced VM or PC AP was followed by a single Ca2+ transient (Figure 3A and 3B, left, paced beats indicated by black arrows). On addition of 30 nmol/L isoproterenol, only CPVT PCs exhibited triggered activity, mainly in the form of EADs (Figure 3A and 3B, right, abnormal beats indicated by blue arrows). At a faster pacing frequency (5 Hz), which leads to elevated SR Ca2+ load, CPVT PCs developed triggered activity even in the absence of isoproterenol (Figure 3D, left). In PCs paced at 5 Hz, spontaneous triggered activity and SCR increased significantly with 30 nmol/L isoproterenol (Figure 3D, right), which led to the appearance of both EADs and DADs coupled with early and late after Ca2+ transients. Altogether, CPVT PCs had shorter latencies and a higher incidence and number of events than CPVT VMs, as previously described.9,11
Figure 3.
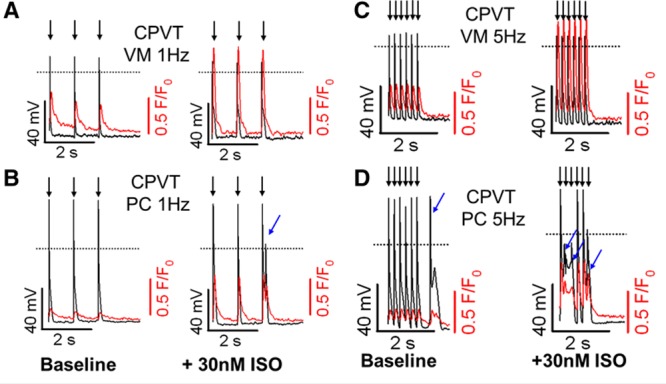
Catecholaminergic polymorphic ventricular tachycardia (CPVT) Purkinje cells (PCs) have lower threshold for Ca2+-linked triggered activity. Representative simultaneous action potential (black) and Ca2+ fluorescence (red) recordings are presented on the same time scale. A and B, Steady-state 1-Hz pacing traces at baseline (left) and after 30 nmol/L isoproterenol (ISO) perfusion (right) for CPVT ventricular myocytes (VMs) and CPVT PCs, respectively. C and D, Steady-state 5-Hz pacing traces at baseline (left) and after 30 nmol/L isoproterenol perfusion (right) for CPVT VMs and CPVT PCs, respectively. Addition of isoproterenol significantly increases Ca2+ transient amplitude in both cell types. Neither control VMs nor control PCs developed abnormal beats under the same conditions (not shown). Black arrows indicate paced beats; blue arrows, spontaneous triggered activity.
CPVT PCs Have a Higher Diastolic Ca2+ Leak
Three distinct patterns of SCR were observed in all cells: Ca2+ waves, early after- Ca2+ transients, and delayed after-Ca2+ transients (Figure VI in the online-only Data Supplement). As expected from previous reports showing a higher incidence of DADs, CPVT PCs had the highest incidence of SCR among the 4 cell types (Figure VII in the online-only Data Supplement).9–11
Representative baseline and post-isoproterenol Ca2+ spark 3-dimensional surface plots for CPVT VMs and PCs are shown in Figure 4A, demonstrating a larger number of Ca2+ sparks in CPVT PCs. In Figure 4B, quantification of Ca2+ spark recordings revealed that CPVT PCs had a significantly higher baseline Ca2+ spark frequency (CaSpF) after steady-state pacing compared with control VMs, control PCs, or CPVT VMs (10.4±2.4 versus 2.3±0.6, 4.4±0.9, and 4.4±1.3 sparks per 100 µm/s at 3 Hz, respectively; P=0.0002, P=0.0174, and P=0.0162). In Figure 4C, addition of isoproterenol (10 nmol/L) significantly and progressively increased CaSpF in all groups, with CPVT PCs having the highest CaSpF at all the time points recorded.
Figure 4.
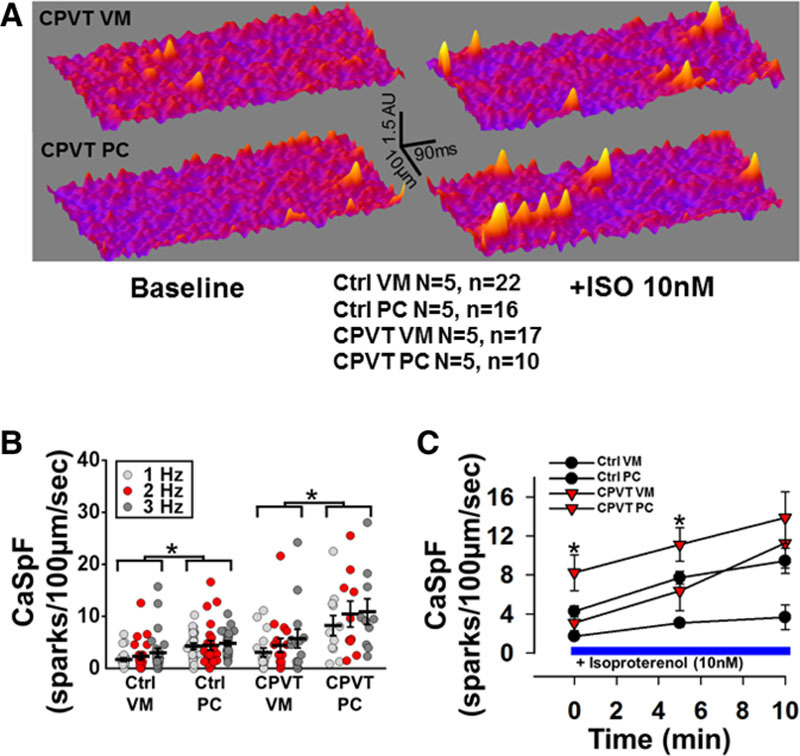
Ca2+ spark frequency (CaSpF) is higher in control and catecholaminergic polymorphic ventricular tachycardia (CPVT) Purkinje cells (PCs) than in ventricular myocytes (VMs). A, Three-dimensional surface plots of representative line scan images for a CPVT VM and CPVT PC for baseline (left) and 10 nM/L isoproterenol (right). B, Mean CaSpF at 1-, 3-, and 5-Hz stimulation. *P<0.05 vs control VMs at the same pacing frequency. #P<0.05 vs all other cell groups at the same pacing frequency. C, Time course of responses of CPVT VMs and CPVT PCs to isoproterenol (ISO; 10 nmol/L). *P<0.05 in Purkinje CPVT myocytes vs all other cell groups at same time point after isoproterenol.
Decreased SR Ca2+ Load and Increased SR Fractional Release in CPVT
We next characterized the state of the SR Ca2+ load in CPVT PCs. Representative caffeine-induced Ca2+ transients after 3 Hz of steady-state pacing are illustrated in Figure 5A. In Figure 5B, the maximum amplitude of the caffeine-induced Ca2+ transient, an index of SR Ca2+ load, was significantly lower in each CPVT cell type compared with its control counterpart (CPVT VM versus control VM: ΔF/F0, 4.5±0.8 versus 8.6±1.7, P=0.0359; and CPVT PC versus control PCs: ΔF/F0, 2.7±0.4 versus 5.1±1.7, P=0.0416). Steady-state Ca2+ transient amplitude was presented as a fraction of the caffeine-induced Ca2+ transient amplitude, indicating fractional release. There was a significant increase in SR fractional release in both CPVT VM versus control VM (50.2±4.2% versus 31.2±3.0%; P=0.0011; Figure 5C) and CPVT PC versus control PC (59.2±11.3% versus 33.1±5.7%; P=0.0449), suggesting that Ca2+ gain was similarly altered by RyR2 gain of function in CPVT. Thus, the CPVT RyR2 mutation increased CaSpF, especially in PCs (Figure 4), despite a reduction in SR Ca2+ load.
Figure 5.
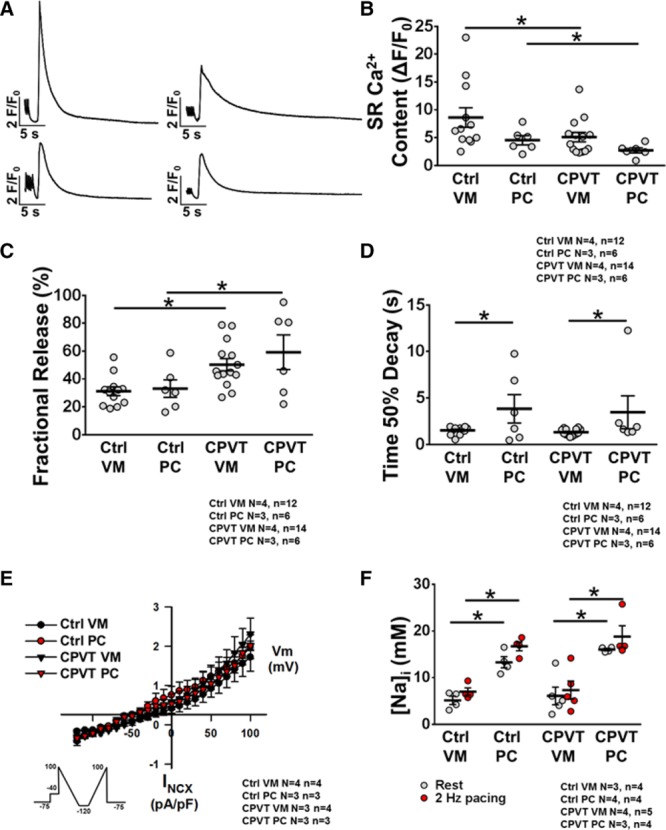
Sarcoplasmic reticulum (SR) Ca2+ handling and high [Na+]i in Purkinje cells (PCs). A, Representative traces of caffeine-induced Ca2+ transient from control ventricular myocytes (VMs; top left) and PCs (bottom left) and catecholaminergic polymorphic ventricular tachycardia VMs (top right) and PCs (bottom right) recorded at 3-Hz stimulation with 10 mmol/L caffeine. B, Mean peak SR Ca2+ content in each cell type. C, SR fractional release and (D) time to 50% decay from caffeine-induced Ca2+ transient VMs. E, Average NiCl2-sensitive Na+/Ca2+ exchanger current/voltage relationships (voltage ramp protocol shown in the inset). F, Mean [Na+] concentration. *P<0.05.
Slower Cellular Ca2+ Extrusion in PCs Compared With VMs Is Attributable to Sarcolemmal Ionic Gradients
Time to 50% decay of the caffeine-induced Ca2+ transient, an indirect measure of NCX activity, was significantly prolonged in control PCs compared with control VMs (3.83±1.39 versus 1.51±0.12 seconds; P=0.0218; Figure 5D) and in CPVT PCs compared with CPVT VMs (3.46±1.61 versus 1.31±0.09 seconds; P=0.0359). There was no significant difference in this parameter between ventricular groups and between Purkinje groups. Whole-cell patch-clamp experiments revealed that INCX was similar in all 4 cell groups (INCX at 100 mV in control VMs, 1.7±0.4 pA/pF; CPVT VMs, 2.3±0.5 pA/pF; control PCs, 1.9±0.3 pA/pF; CPVT PCs, 2.0±0.2 pA/pF; P=0.9158; Figure 5E). Thus, although a slower caffeine-induced Ca2+ transient decay in control and CPTV PCs indicates that PCs have a reduced level of NCX functional expression than VMs, the voltage-clamp results failed to provide support for this idea. Both approaches measure function of NCX as opposed to Western blots or transcriptional levels of NCX. However, the intracellular milieus at which the 2 functional assays measure NCX activity are vastly different from each other. One (caffeine-induced Ca2+ transient assay) is more physiological than the other.
Higher [Na+]i in PCs at Rest and During Stimulation
[Na+]i was measured in all 4 cellular groups at rest and after 2-Hz pacing. Figure 5F shows the average calibrated measurements for each group, and representative examples can be found in Figure VIII in the online-only Data Supplement. Control PCs compared with control VMs had significantly higher [Na+]i at baseline (13.3±1.0 versus 5.2±0.8 mmol/L; P=0.0286) and under 2-Hz pacing (16.7±0.8 versus 7.0±0.7 mmol/L; P=0.0286). Likewise, CPVT PCs had significantly higher [Na+]i both at baseline (16.0±0.2 versus 6.1±1.6 mmol/L; P=0.0159) and at 2-Hz pacing (18.8±2.0 versus 7.4±1.7 mmol/L; P=0.0159) compared with CPVT VMs. There were no differences in [Na+]i between control VMs and CPVT VMs or between control PCs and CPVT PCs. Our calibrated [Na]i measurements with SBFI were conducted at physiological temperatures (37°C) and most likely underlie the reduced values of [Na+]I in VMs (5–7 mmol/L) compared with the values shown by others in experiments conducted at room temperature, in the range of 11 to 15 mmol/L.27 This is in line with the studies of Chapman28 showing the relationship between intracellular Na activity and its sensitivity to temperature.
Higher [Na+]i in PCs Increases SR Ca2+ Leak and Yields More Frequent DADs
To test the hypothesis that a constitutive excess in [Na+]i in PCs promotes RyR2 leak and arrhythmias at lower levels of stress than VMs, we conducted computer simulations in a well-tested model of the murine PC. The model accurately reproduces AP morphology, AP duration (APD) at 90% of repolarization (APD90), and maximum dV/dt values obtained in experiments (Table II and Figure IV in the online-only Data Supplement).18 Moreover, the model was able to reproduce the experimentally observed biphasic calcium transients resulting from both radial and longitudinal diffusion components (Figure IVC and IVD in the online-only Data Supplement).
In Figure 6A, the PC model with SCRs enabled was burst paced at 5 Hz for 10 seconds to overload the SR with calcium. At the end of pacing, a triggered AP was followed by a DAD, both of which were mediated via the cytosolic calcium waves. Similar to what happened in the experiments (Figure 3), simulating the effects of isoproterenol in the PC model significantly increased the frequency of SCR, thereby producing sustained DAD-induced triggered AP, as shown in Figure 6B. These results are in full agreement with our previously reported results in CPVT mice9,11 and the experimental data presented above.
Figure 6.
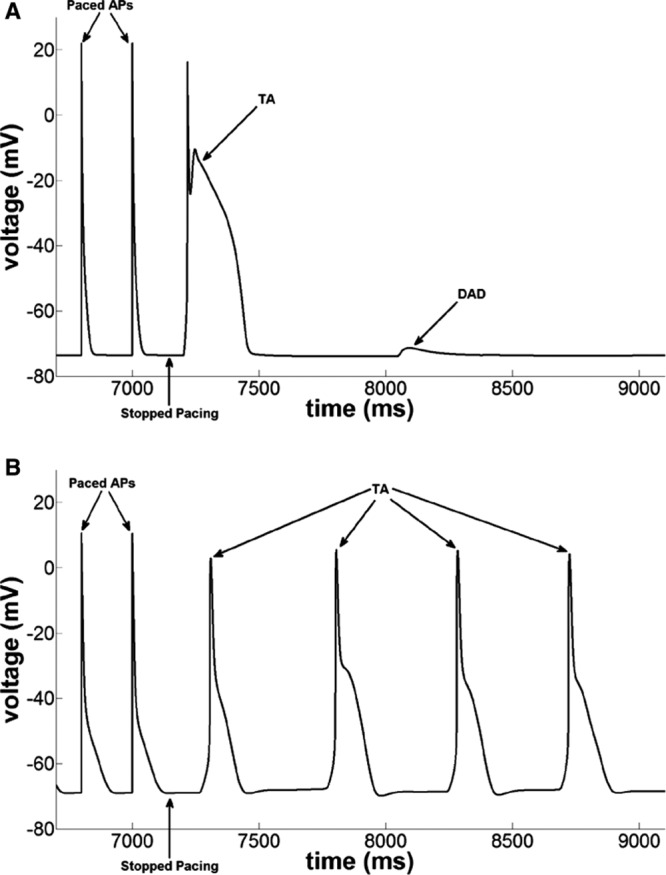
Pacing induced delayed afterdepolarizations (DADs) and triggered activity (TA) in the numeric mouse Purkinje cell model with spontaneous Ca2+ release enabled (catecholaminergic polymorphic ventricular tachycardia model; A) control case and (B) in the presence of isoproterenol effects followed by burst pacing at 5 Hz.
The [Na+]i levels in the PC model were elevated by scaling INaK to 85%, 75%, and 65% of the control values (Table III in the online-only Data Supplement ). Figure IXA and IXB in the online-only Data Supplement clearly shows that the APs were prolonged as a result of increased [Na+]i. Furthermore, the increased [Na+]i was associated with elevated levels of averaged [Ca2+] in the model with progressively slower Ca2+ extrusion (Figure IXC in the online-only Data Supplement), which significantly increased the inward component of the NCX (Figure IXD in the online-only Data Supplement), leading to APD prolongation at higher [Na+]i. We did not observe noticeable changes in the resting membrane potential (Vrest) at elevated levels of [Na]i (Table VI in the online-only Data Supplement). Thus, the fast and slow inactivation gates, namely h and j, in the sodium current formulation did not change significantly (Figure X and Table VI in the online-only Data Supplement), indicating that the inactivation kinetics of sodium channels was not altered at the elevated [Na]i levels. We also investigated the effects of blocking INaL in our model. As listed in Table VII and shown in Figure XI in the online-only Data Supplement, blocking INaL abbreviated the APD (both APD50 and APD90) at all levels of [Na]i; however, dV/dtmax and Vrest were not altered significantly. When SCRs were implemented in these models of elevated [Na+]i, we observed frequent DADs (Figure 7A). The DADs were always preceded by SCR events from the leaky RyR channels (Figure 7B). These RyR2 leaks initiated cytosolic Ca2+ waves, thus elevating the local Ca2+ concentration in the subsarcolemmal region. The elevated Ca2+ levels in the subsarcolemmal region activated the inward component of NCX, thereby producing DADs.
Figure 7.
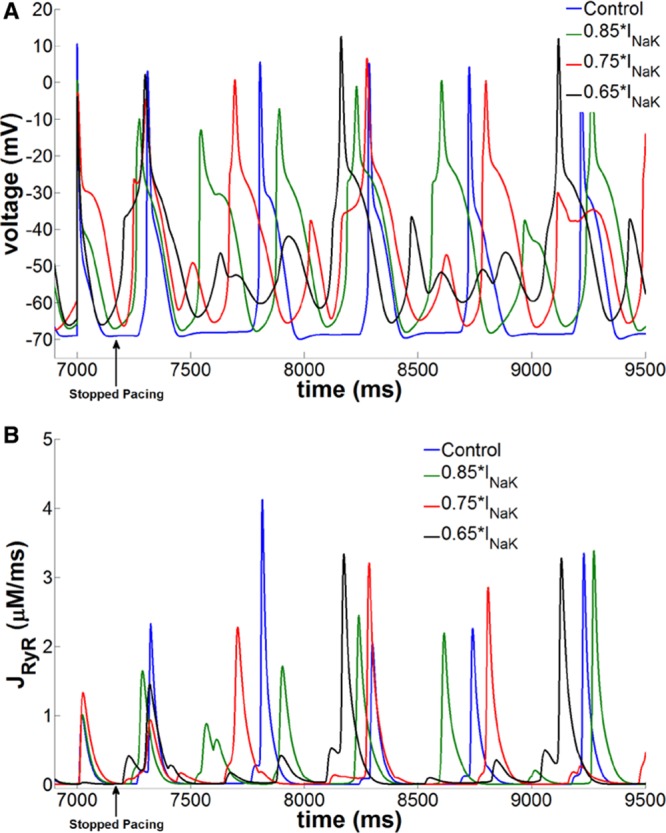
Ca2+-linked arrhythmogenesis in the [Na]i-overloaded numeric Purkinje cell model with isoproterenol effects. A, Generation of frequent delayed afterdepolarizations and triggered activity in the catecholaminergic polymorphic ventricular tachycardia model at various elevated levels of [Na+]i obtained by reducing INaK density. B, Corresponding Ca2+ release flux from the ryanodine channels shows large-magnitude spontaneous Ca2+ release events preceding the membrane depolarizations.
Membrane Permeabilization Reverses the Differential CaSpF in PCs Versus VMs
To provide further direct evidence that a higher [Na]i makes PCs more susceptible to RyR2 dysfunction than VMs, we measured Ca2+ sparks in saponin-permeabilized PCs and VMs. Figure 8A through 8D shows representative confocal line scan recordings of Ca2+ sparks in saponin-permeabilized control VMs, control PCs, CPVT VMs, and CPVT PCs with 50 nmol/L free Ca2+. The CPVT mutation increased CaSpF significantly in CPVT VMs compared with control VMs (11.5±0.5 versus 5.8±0.5 sparks per 100 µm/s, respectively; P<0.0001; Figure 8E) and CPVT PCs to control PCs (8.0±0.7 versus 4.8±0.6 sparks per 100 µm/s, respectively; P=0.0317; Figure 8E). However, unlike intact cells (Figure 4), the CaSpF in control PCs was similar to that in control VMs (4.8±0.6 versus 5.8±0.5 sparks per 100 µm/s, respectively; P=0.3087; Figure 8E). Most important, CPVT PCs had lower CaSpF compared with CPVT VMs (8.0±0.7 versus 11.5±0.5 sparks per 100 µm/s, respectively; P=0.0087; Figure 8E).
Figure 8.
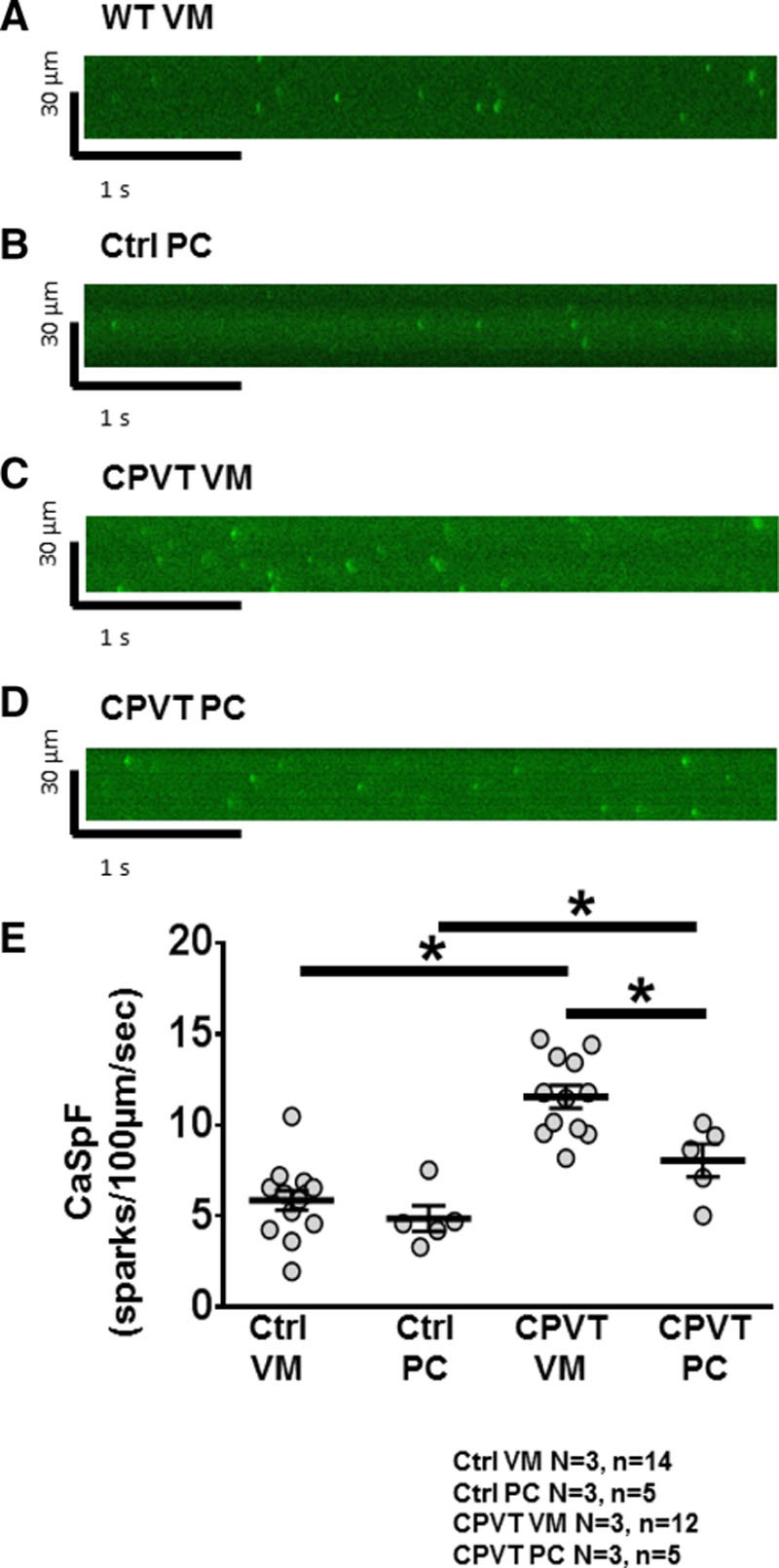
Membrane permeabilization terminates differential Ca2+ spark frequency (CaSpF) in Purkinje cells (PCs) compared with ventricular myocytes (VMs). A through D, Representative confocal line scan recordings showing Ca2+ sparks under 50 nmol/L free Ca2+ in permeabilized control VMs, control PCs, catecholaminergic polymorphic ventricular tachycardia (CPVT) VMs, and CPVT PCs. E, Average CaSpF among all groups. Note that there is no significant difference in CaSpF between control VMs and control PCs and that there is a significant decrease in CaSpF between CPVT VMs and CPVT PCs. *P<0.05.
Discussion
The major findings of this work may be summarized as follows. First, SCR preceded triggered activity in RyR2R4496C+/− CPVT hearts, whereas initial AP breakthrough points colocalized with the Purkinje conduction network, suggesting that PCs are the source of triggered activity in CPVT hearts. Second, in control and CPVT cardiomyocytes, CaSpF was greater in PCs than VMs at baseline and under β-adrenergic stress, reflecting increased activity of RyR2 channels. Third, CPVT mutation decreased SR Ca2+ load and increased fractional shortening in both PCs and VMs compared with control, suggesting that RyR2 Ca2+ sensitivity was equally increased by the CPVT mutation in PCs and VMs. Fourth, caffeine-induced Ca2+ transient decay was slower in control and CPVT PCs compared with VMs. Fifth, compared with VMs, both control and CPVT PCs had higher [Na+]i at rest and during steady-state pacing. Sixth, in computer simulations, increasing [Na+]i in a PC model cell greatly promoted RyR2 leak and arrhythmias. Finally, membrane permeabilization reversed the different PC versus VM postpacing CaSpF. Together, these results demonstrate for the first time the essential role of the constitutively higher PC [Na+]i in promoting greater arrhythmogenicity than VMs in CPVT mice.
Subcellular Expression and Distribution of the L-type Ca2+ Channel and RyR2 Channels in Mouse PCs
The observation that (at least some) mouse PCs contain T tubules suggests the presence of normal dyadic structures that form at the sites of Ca2+ release at the junctional SR/T tubule membrane interfaces. Using fluorescent immunocytochemistry, we found that both control and CPVT mouse PCs express CaV1.2 and that this protein colocalizes with RyR2 to the same discrete subcellular locale, although with higher intercellular and intracellular heterogeneity than in VMs (Figure V in the online-only Data Supplement). These findings suggest that the main protein components of the classic Ca2+-induced Ca2+ release process in VMs are present but are more heterogeneous in PCs.
Ca2+ Handling in Cardiac PCs
Although Ca2+ handling in VMs has been extensively studied and is relatively well understood, such is not the case for cardiac PCs. Previous studies have demonstrated that PCs are different from VMs, both morphologically and electrophysiologically.9,18,29 We investigated whether inherent differences between the 2 cell types affect Ca2+ regulation and underlie the enhanced vulnerability of cardiac PCs over VMs to RyR2 dysfunction in CPVT. PCs are more arrhythmogenic than VMs in CPVT mice, and previous studies support the hypothesis that arrhythmias in the RyR2R4496C+/− mouse are caused by enhanced/abnormal diastolic Ca2+ release through defective RyR2 channels.9,11 In this context, we sought to determine in the whole heart the role that cellular Ca2+ leak plays in RyR2R4496C+/− PCs. First, by measuring Vm and [Ca2+]i simultaneously, we confirmed that PCs have a lower threshold for triggered activity, both during high-frequency pacing and under adrenergic stress (Figure 3). Most important, we determined that all DADs and EADs observed within mutant PCs correlated temporally with a synchronous rise in Ca2+-mediated fluorescence. Then, to explore the mechanism for such findings, we dived further into the molecular basis of the Ca2+ handling machinery in mouse PCs.
Ca2+ sparks are stochastic activations of a cluster of RyR2s that form a Ca2+ release unit. In VMs, the synchronized opening of Ca2+ release units during Ca2+-induced Ca2+ release is thought to generate a global Ca2+ transient in the cytosol. Although the Ca2+ release unit is incompletely understood in PCs, the general consensus is that in both VMs and PCs, Ca2+ sparks reflect cellular in situ activity of the RyR2. Therefore, it is well known that RyR2 Ca2+ sensitivity within Ca2+ release units correlates with a higher CaSpF. As shown in Figure 4, diastolic Ca2+ leak was higher in RyR2R4496C+/− mutant PCs compared with all other cell types, both at baseline and under β-adrenergic stress. To the best of our knowledge, this is the first time that Ca2+ sparks have been recorded in isolated murine PCs. An increased CaSpF resulting from increased RyR2 sensitivity is also in accordance with a decrease in SR Ca2+ load and an increase in SR fractional release, both of which were observed in CPVT PCs (Figure 5). Taken together, our results support the hypothesis that enhanced diastolic Ca2+ leak in PCs is responsible for triggered activity arising from the conduction system. Furthermore, we found that permeabilization of the membrane terminates this cell type–dependent difference in CaSpF (Figure 8). Together, these data suggest that a difference in the intracellular milieu that results from different transporters at the sarcolemmal membrane is an essential factor in making the PC more vulnerable to Ca2+ dysfunction than the VM.
What Is the Mechanism of the Increased Ca2+ Leak in the PCs?
We found that the caffeine-induced Ca2+ transient time to 50% decay was significantly prolonged in PCs. The caffeine transient decay time provides an indirect measure of NCX-mediated cytosolic Ca2+ removal. Therefore, this would suggest that, at least after pacing, PCs in general have decreased activity of NCX compared with VMs. Through patch-clamp experiments, we determined that this is not attributable to decreased functional levels of NCX because INCX was not different among all 4 cell groups studied (Figure 5E). We postulate that the slower caffeine-induced Ca2+ transient decay time observed in control and CPVT PCs likely reflects changes in ionic gradients, particularly Na+ gradients, which would slow NCX extrusion. Given that in the mouse heart PCs have a reduced density of T tubules26 and larger INa density and are hyperpolarized compared with VMs,18 it is reasonable to suggest that they are prone to Na+ overload, which could also decrease postpacing NCX Ca2+ extrusion. We measured [Na+]i and found that it was increased in PCs compared with VMs (Figure 5F), which is consistent with what has been shown in canine PCs.30 Higher Na+ load in PCs was also found in cells from CPVT hearts. These findings likely explain the increased caffeine-induced Ca2+ transient decay time observed in PCs compared with VMs because higher [Na+]i would impair Ca2+ extrusion and lead to increased [Ca2+]i, as predicted by previous mathematical modeling31 and by our own simulations (Figures 6 and 7). To the best of our knowledge, these results have not been reported previously in mouse PCs or in the context of inherited arrhythmogenic disease.
Intracellular Ca2+ Handling in Mouse PCs
PCs are different from VMs both morphologically and electrophysiologically.10,11,18,32 In addition, [Na+]i and Ca2+ homeostasis mechanisms are different in PCs and VMs.33 In larger mammalian species, it has been shown that PCs have 2 types of SR, junctional and corbular,34 and that this may underlie a Ca2+ activation process that is peculiar to the cell type and perhaps to the species.35 For example, studies in canine PCs have shown that on electric stimulation there is an initial uniform Ca2+ rise under the cell membrane that propagates to the central core region via Ca2+-induced Ca2+ release.36,37 On the other hand, in CPVT, leaky RyR2 channels can initiate a Ca2+ wave that propagates from the core of a PC back to the membrane and causes depolarization. This reverse propagation of Ca2+ as a wave may then travel through the single PC, between cells, and along a fiber.20
On the basis of all the above, we propose that the constitutive excess of [Na+]i in PCs with respect to VMs leads to PC [Ca2+]i overload–induced SCR and triggered activity through an Na+ overload–dependent decreased cytosolic Ca2+ extrusion mechanism. This potential mechanism becomes even more feasible when we consider that bidirectional VT has thus far been strongly associated only with CPVT and digitalis toxicity.1,2,38,39 Indeed, the mechanism of action of digitalis is thought to be that of Na+-dependent [Ca2+]i overload through impaired NCX-mediated Ca2+ extrusion occurring as a consequence of Na+/K+ ATPase inhibition.40 The functional density of Na+/K+ ATPase in the T tubules of VMs is 3- to 3.5-fold higher than in the lateral sarcolemma, and VMs have a ≈4- to 20-fold higher density of T tubules than PCs, depending on species, which may explain the higher conduction velocities in PC networks than VM.21,26 Although the biophysical characterization of Na+/K+ ATPase in PCs is beyond the scope of this study, our measurements of [Na+]I, together with our computer simulations showing a strong dependence of SR Ca2+ leak on INaK, strongly support our conjecture that a PC [Na+]i surplus drives calcium-linked ventricular arrhythmogenesis in mice. Nevertheless, further studies are warranted to elucidate the relationship between this important protein and triggered activity in PCs.
Limitations
We have not conducted direct measurements of [Na+]i using Na+-selective microelectrodes,41 but our SFBI measurements were calibrated at the end of each experiment according to previous studies.27 Some research groups have proposed a “triple layer” of Ca2+ activation in canine PCs involving IP3 receptors.20 However, this mechanism may not hold true across different mammalian species. Although permeabilization of the cell membrane likely abolished the differences in [Na+]i between PCs and VMs, at this point, we cannot exclude the possibility that the loss of an intracellular soluble factor contributed to the reversal of the different PC versus VM postpacing CaSpF. Obviously, although data on [Na+]i differences in ventricular cells and Purkinje fibers obtained in mice and computer simulations can provide great insight into molecular and ionic mechanisms of inheritable cardiac diseases such as CPVT, extrapolation to other species and the human condition needs to be conducted with extreme caution. In our computer simulations, we have modeled elevated [Na+]i on the basis of a reduced density of the INaK in PCs versus ventricular cells as in earlier studies.31 However, whether differences in this current or those of other important regulators of [Na+]i homeostasis such as the sodium-hydrogen exchanger and sodium-bicarbonate symporter42 are important is unknown and will have to be investigated systematically in future studies. We have not accounted for alterations in both the hydrogen ion homeostasis and cell-coupling effects of the elevated Na values in PCs seen in our present study, which will need to be examined in the future. This is important because earlier studies have shown an important and complex relationship between changes in pH, [Na+]i, and rate of stimulation,43 which may have implications for the generation of DADs.44 Finally, unlike in other experimental models such as dogs,45 mouse Purkinje fibers exhibit a similar density of the inward rectifier K+ current (IK1) in PCs and VMs, as shown in our earlier study.19 Thus, IK1 does not play a role in the preferential formation of DADs in PCs compared with VMs, at least in mice, but this cannot be ruled out for other species, including humans.
Conclusions and Clinical Implications
CPVT is a congenital, proarrhythmogenic syndrome characterized by dangerous ventricular arrhythmias that appear under stress. CPVT has been linked to mutations in cardiac Ca2+ handling proteins, yet new evidence has shown that the cardiac Purkinje network appears to be involved in the initiation of bidirectional VT and PVT in this disease. This work has shed new light on the mechanisms of Ca2+-linked ventricular arrhythmias arising from the cardiac Purkinje network in CPVT mice. Although translation from mouse model to humans is not trivial, our results present PC Na+ and Ca2+ regulation as a new potential treatment avenue in CPVT and other cardiac diseases associated with Ca2+-linked arrhythmias.
Acknowledgments
We thank Nulang Wang for her technical assistance.
Sources of Funding
This work was supported by National Heart, Lung, and Blood Institute grants P01-HL039707, P01-HL087226, and R01-HL122352; the Leducq Foundation: Transatlantic Network of Excellence Program on “Structural Alterations in the Myocardium and the Substrate for Cardiac Fibrillation” (Dr Jalife); grants HL055438 and HL120108 (to Dr Valdivia); and American Heart Association grant 12SDG11480010 (Dr Deo).
Disclosures
None.
Supplementary Material
Footnotes
Drs Willis and Pandit contributed equally to this work.
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.116.021936/-/DC1.
CLINICAL PERSPECTIVE
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly malignant, inheritable, proarrhythmogenic syndrome characterized by life-threatening ventricular arrhythmias that appear under stress. The work presented here sheds new light on the mechanisms of Ca2+-linked ventricular arrhythmias in CPVT by demonstrating for the first time the essential role of the constitutively higher intracellular sodium of the specialized Purkinje cell in promoting greater arrhythmogenesis than ventricular myocytes in CPVT mice. CPVT has been linked to human mutations in cardiac Ca2+ handling proteins, yet new evidence has shown that the Purkinje network appears to be involved in the initiation of bidirectional and polymorphic ventricular arrhythmias in this disease. Although translation from the mouse model to the human should be done with caution, our results introduce the Purkinje cell Na+ and Ca2+ regulation as a new potential target in CPVT and other cardiac diseases associated with Ca2+-linked arrhythmias.
References
- 1.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children: a 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 2.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93. doi: 10.1038/nrcardio.2012.93. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 4.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le Marec H, Lucet V, Mabo P, Probst V, Monnier N, Ray PF, Santoni E, Trémeaux P, Lacampagne A, Fauré J, Lunardi J, Marty I. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Søndergaard MT, Sorensen AB, Skov LL, Kjaer-Sorensen K, Bauer MC, Nyegaard M, Linse S, Oxvig C, Overgaard MT. Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 2015;282:803–816. doi: 10.1111/febs.13184. doi: 10.1111/febs.13184. [DOI] [PubMed] [Google Scholar]
- 7.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 8.Faggioni M, Knollmann BC. Calsequestrin 2 and arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302:H1250–H1260. doi: 10.1152/ajpheart.00779.2011. doi: 10.1152/ajpheart.00779.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG, Fishman GI. Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res. 2010;107:512–519. doi: 10.1161/CIRCRESAHA.110.221481. doi: 10.1161/CIRCRESAHA.110.221481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herron TJ, Milstein ML, Anumonwo J, Priori SG, Jalife J. Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2010;7:1122–1128. doi: 10.1016/j.hrthm.2010.06.010. doi: 10.1016/j.hrthm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mezu UL, Singh P, Shusterman V, Hwang HS, Knollmann BC, Němec J. Accelerated junctional rhythm and nonalternans repolarization lability precede ventricular tachycardia in Casq2-/- mice. J Cardiovasc Electrophysiol. 2012;23:1355–1363. doi: 10.1111/j.1540-8167.2012.02406.x. doi: 10.1111/j.1540-8167.2012.02406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyden PA, Hirose M, Dun W. Cardiac Purkinje cells. Heart Rhythm. 2010;7:127–135. doi: 10.1016/j.hrthm.2009.09.017. doi: 10.1016/j.hrthm.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 14.Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582(pt 2):675–693. doi: 10.1113/jphysiol.2006.126714. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee P, Klos M, Bollensdorff C, Hou L, Ewart P, Kamp TJ, Zhang J, Bizy A, Guerrero-Serna G, Kohl P, Jalife J, Herron TJ. Simultaneous voltage and calcium mapping of genetically purified human induced pluripotent stem cell-derived cardiac myocyte monolayers. Circ Res. 2012;110:1556–1563. doi: 10.1161/CIRCRESAHA.111.262535. doi: 10.1161/CIRCRESAHA.111.262535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578(Pt 1):315–326. doi: 10.1113/jphysiol.2006.121475. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anumonwo JM, Tallini YN, Vetter FJ, Jalife J. Action potential characteristics and arrhythmogenic properties of the cardiac conduction system of the murine heart. Circ Res. 2001;89:329–335. doi: 10.1161/hh1601.095894. [DOI] [PubMed] [Google Scholar]
- 18.Vaidyanathan R, O’Connell RP, Deo M, Milstein ML, Furspan P, Herron TJ, Pandit SV, Musa H, Berenfeld O, Jalife J, Anumonwo JM. The ionic bases of the action potential in isolated mouse cardiac Purkinje cell. Heart Rhythm. 2013;10:80–87. doi: 10.1016/j.hrthm.2012.10.002. doi: 10.1016/j.hrthm.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 20.Stuyvers BD, Dun W, Matkovich S, Sorrentino V, Boyden PA, ter Keurs HE. Ca2+ sparks and waves in canine Purkinje cells: a triple layered system of Ca2+ activation. Circ Res. 2005;97:35–43. doi: 10.1161/01.RES.0000173375.26489.fe. doi: 10.1161/01.RES.0000173375.26489.fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Despa S, Brette F, Orchard CH, Bers DM. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J. 2003;85:3388–3396. doi: 10.1016/S0006-3495(03)74758-4. doi: 10.1016/S0006-3495(03)74758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 23.Gaur N, Rudy Y, Hool L. Contributions of ion channel currents to ventricular action potential changes and induction of early afterdepolarizations during acute hypoxia. Circ Res. 2009;105:1196–1203. doi: 10.1161/CIRCRESAHA.109.202267. doi: 10.1161/CIRCRESAHA.109.202267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang LM, Wang Z, Nattel S. Effects of sustained beta-adrenergic stimulation on ionic currents of cultured adult guinea pig cardiomyocytes. Am J Physiol Heart Circ Physiol. 2002;282:H880–H889. doi: 10.1152/ajpheart.01138.2000. doi: 10.1152/ajpheart.01138.2000. [DOI] [PubMed] [Google Scholar]
- 25.Faber GM, Rudy Y. Calsequestrin mutation and catecholaminergic polymorphic ventricular tachycardia: a simulation study of cellular mechanism. Cardiovasc Res. 2007;75:79–88. doi: 10.1016/j.cardiores.2007.04.010. doi: 10.1016/j.cardiores.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Maio A, Ter Keurs HE, Franzini-Armstrong C. T-tubule profiles in Purkinje fibres of mammalian myocardium. J Muscle Res Cell Motil. 2007;28:115–121. doi: 10.1007/s10974-007-9109-6. doi: 10.1007/s10974-007-9109-6. [DOI] [PubMed] [Google Scholar]
- 27.Despa S, Tucker AL, Bers DM. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation. 2008;117:1849–1855. doi: 10.1161/CIRCULATIONAHA.107.754051. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman RA. Sodium/calcium exchange and intracellular calcium buffering in ferret myocardium: an ion-sensitive micro-electrode study. J Physiol. 1986;373:163–179. doi: 10.1113/jphysiol.1986.sp016040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 30.Lee CO, Dagostino M. Effect of strophanthidin on intracellular Na ion activity and twitch tension of constantly driven canine cardiac Purkinje fibers. Biophys J. 1982;40:185–198. doi: 10.1016/S0006-3495(82)84474-3. doi: 10.1016/S0006-3495(82)84474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li P, Rudy Y. A model of canine Purkinje cell electrophysiology and Ca(2+) cycling: rate dependence, triggered activity, and comparison to ventricular myocytes. Circ Res. 2011;109:71–79. doi: 10.1161/CIRCRESAHA.111.246512. doi: 10.1161/CIRCRESAHA.111.246512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI. Contactin-2 expression in the cardiac Purkinje fiber network. Circ Arrhythm Electrophysiol. 2010;3:186–194. doi: 10.1161/CIRCEP.109.928820. doi: 10.1161/CIRCEP.109.928820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vassalle M, Lin CI. Calcium overload and cardiac function. J Biomed Sci. 2004;11:542–565. doi: 10.1007/BF02256119. doi: 10.1159/000079666. [DOI] [PubMed] [Google Scholar]
- 34.Sommer JR, Johnson EA. Cardiac muscle: a comparative study of Purkinje fibers and ventricular fibers. J Cell Biol. 1968;36:497–526. doi: 10.1083/jcb.36.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rysevaite K, Saburkina I, Pauziene N, Vaitkevicius R, Noujaim SF, Jalife J, Pauza DH. Immunohistochemical characterization of the intrinsic cardiac neural plexus in whole-mount mouse heart preparations. Heart Rhythm. 2011;8:731–738. doi: 10.1016/j.hrthm.2011.01.013. doi: 10.1016/j.hrthm.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 37.Boyden PA, Pu J, Pinto J, Keurs HE. Ca(2+) transients and Ca(2+) waves in Purkinje cells: role in action potential initiation. Circ Res. 2000;86:448–455. doi: 10.1161/01.res.86.4.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kummer JL, Nair R, Krishnan SC. Images in cardiovascular medicine. Bidirectional ventricular tachycardia caused by digitalis toxicity. Circulation. 2006;113:e156–e157. doi: 10.1161/CIRCULATIONAHA.105.557561. doi: 10.1161/CIRCULATIONAHA.105.557561. [DOI] [PubMed] [Google Scholar]
- 39.Valent S, Kelly P. Images in clinical medicine: digoxin-induced bidirectional ventricular tachycardia. N Engl J Med. 1997;336:550. doi: 10.1056/NEJM199702203360805. doi: 10.1056/NEJM199702203360805. [DOI] [PubMed] [Google Scholar]
- 40.Hauptman PJ, Kelly RA. Digitalis. Circulation. 1999;99:1265–1270. doi: 10.1161/01.cir.99.9.1265. [DOI] [PubMed] [Google Scholar]
- 41.Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol. 1989;256(pt 1):C813–C822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- 42.Clancy CE, Chen-Izu Y, Bers DM, Belardinelli L, Boyden PA, Csernoch L, Despa S, Fermini B, Hool LC, Izu L, Kass RS, Lederer WJ, Louch WE, Maack C, Matiazzi A, Qu Z, Rajamani S, Rippinger CM, Sejersted OM, O’Rourke B, Weiss JN, Varró A, Zaza A. Deranged sodium to sudden death. J Physiol. 2015;593:1331–1345. doi: 10.1113/jphysiol.2014.281204. doi: 10.1113/jphysiol.2014.281204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bountra C, Kaila K, Vaughan-Jones RD. Effect of repetitive activity upon intracellular pH, sodium and contraction in sheep cardiac Purkinje fibres. J Physiol. 1988;398:341–360. doi: 10.1113/jphysiol.1988.sp017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yano T, Hotokebuchi N, Morioka T, Nishi K. Acidosis depresses delayed afterdepolarization in guinea pig myocardium. Am J Physiol. 1989;257(pt 2):H996–1004. doi: 10.1152/ajpheart.1989.257.3.H996. [DOI] [PubMed] [Google Scholar]
- 45.Cordeiro JM, Zeina T, Goodrow R, Kaplan AD, Thomas LM, Nesterenko VV, Treat JA, Hawel L, 3rd, Byus C, Bett GC, Rasmusson RL, Panama BK. Regional variation of the inwardly rectifying potassium current in the canine heart and the contributions to differences in action potential repolarization. J Mol Cell Cardiol. 2015;84:52–60. doi: 10.1016/j.yjmcc.2015.04.010. doi: 10.1016/j.yjmcc.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]