

Abstract
Identification of antigenic variants is the key to a successful influenza vaccination program. The empirical serological methods to determine influenza antigenic properties require viral propagation. Here a novel quantitative PCR-based antigenic characterization method using polyclonal antibody and proximity ligation assays, or so-called polyPLA, was developed and validated. This method can detect a viral titer that is less than 1000 TCID50/mL. Not only can this method differentiate between different HA subtypes of influenza viruses but also effectively identify antigenic drift events within the same HA subtype of influenza viruses. Applications in H3N2 seasonal influenza data showed that the results from this novel method are consistent with those from the conventional serological assays. This method is not limited to the detection of antigenic variants in influenza but also other pathogens. It has the potential to be applied through a large-scale platform in disease surveillance requiring minimal biosafety and directly using clinical samples.
Keywords: Influenza virus, Antigenic drift, Antigenic variant, HI, Neutralization
Introduction
The influenza virus is a perpetual threat to public health. Seasonal influenza infections are associated with thousands of deaths every year in the United States (Thompson et al., 2010). A worldwide pandemic could increase the death toll to millions in a short period of time. The hallmark of the influenza virus is antigenic variation, which comes in two forms: antigenic drift and antigenic shift, leading to the recurrence of influenza virus infections (Katz et al., 1987). Mutations in the hemagglutinin and neuraminidase glycoproteins cause antigenic drift. Meanwhile, antigenic shift is caused by the replacement of a new subtype of hemagglutinin and sometimes neuraminidase through genetic reassortment.
The influenza vaccine is the most viable option in counteracting and reducing the impact of influenza outbreaks (Harper et al., 2004). Since influenza viruses are continuously changing their antigenicity in order to escape the host immunity (Nobusawa and Nakajima, 1988; Webster et al., 1982), the vaccine strains need to be updated almost annually to obtain antigenic matches between the vaccine strain and the strain potentially causing future outbreaks (Ampofo et al., 2012; Gerdil, 2003). Identification of influenza antigenic variants is the key to a successful influenza vaccination program for both pandemic preparedness as well as seasonal influenza prevention and control (Katz et al., 1987).
Routinely, immunological tests, such as hemagglutination inhibition (HI) assays and microneutralization (MN) assays, have been relied upon to identify antigenic variants among the circulating strains (Medeiros et al., 2001). The HI assay is an experiment to measure how a test influenza antigen and a reference antigen (e.g. a current vaccine strain) match through the immunological reaction between the test antigen and the reference antiserum. This reference antiserum is usually generated in ferrets using the reference antigen. HI assays are limited due to their use of red blood cells (RBCs), e.g. turkey red blood cells, as indicators for the binding affinity of antigen and antiserum (Kendal et al., 1982). A higher interaction between antigen and antisera will lead to less hemagglutination of RBCs (Hirst, 1941). Compared to HI assays, MN assays seem to be more sensitive and specific but are much more time-consuming. Moreover, for influenza viruses requiring biosafety-level 3 (BSL-3) or higher, MN assays are difficult to perform (Grund et al., 2011). For this reason, HI assays have been one of the routine procedures used to identify influenza antigenic variants for vaccine strain selection while MN assays are generally used to validate the results from HI assays.
However, the data from HI assays are notoriously noisy, and HI experiments are affected by many factors. For example, RBCs used from different species and even variation in RBC sialic acid receptors can produce varied results (Medeiros et al., 2001). The data are subjective interpretations and the HI assays have difficulty in automating and standardizing operations. Minor antigenic variants within a heterogeneous population cannot be assessed by the serological method of HI (Patterson and Oxford, 1986). More importantly, mutations of the receptor binding site in HA (Nobusawa and Nakajima, 1988) (antigenic drift) are causing human seasonal H1N1 (Azzi et al., 1993; Morishita et al., 1993) and H3N2 influenza A viruses (Nobusawa et al., 2000) to lose the ability to bind to RBCs. For example, the mutations at residues 193, 196, 197, and 225 in the human epidemic H1N1 influenza A viruses in 1988 or later caused the loss of their abilities to agglutinate chicken RBCs due to four amino acid changes (Morishita et al., 1996). For H3N2 viruses, the Gly190Asp substitution has been correlated to the loss of the ability to agglutinate chicken erythrocytes (Cox and Bender, 1995; Fitch et al., 1997; Lindstrom et al., 1998, 1996; Medeiros et al., 2001; Mori et al., 1999; Nobusawa et al., 2000). Since 2000, human seasonal H1N1 and H3N2 influenza A viruses have been losing their binding abilities to turkey red blood cells (Medeiros et al., 2001; Oh et al., 2008). This may be attributed to a reduced affinity for sialic acid-linked receptors (particularly α2-6-linked receptors), which are at lower levels on chicken and turkey RBC compared to levels on guinea pig RBC (Medeiros et al., 2001; Oh et al., 2008). Consequently, a critical demand exists for the development of a red blood cell independent assay for influenza antigenic variation.
Proximity ligation assay utilizes quantitative real time PCR (qRT-PCR) for the detection of antigen–antibody interaction (Schlingemann et al., 2010). For this assay: (1) oligonucleotide-linked monoclonal antibodies are incubated with the analyte in question; (2) if in close proximity, the oligonucleotides can be ligated together; and (3) presence of analyte will be shown by amplification of ligated products with qRT-PCR. The assay reporter signal is dependent on a proximal and dual recognition of each target analyte providing high specificity (Fredriksson et al., 2007).
In this study, we developed a novel antigenic characterization method using polyclonal antibody-based proximity ligation assays (polyPLA). This method was found to be useful in detecting influenza antigenic variants in clinical samples.
Results
PolyPLA for influenza antigenic variant detection
PolyPLA quantifies the antibody antigen binding avidity using the amplification signals in quantitative PCR (qPCR) from the pairs of primers attached to a reference polyclonal antiserum. The first step of this experiment is to biotinylate a reference polyclonal antiserum (Fig. 1A), which will be then labeled with sodium azide-linked 5′ and 3′ oligonucleotides (Fig. 1B). The ligation efficiency will be assessed with qPCR. A labeled polyclonal antiserum with ΔCt ≥ 8.5 in the ligation efficiency test is then incubated with a reference antigen (virus) or a testing antigen (Fig. 1C), followed by the proximity ligation of the two oligos (Fig. 1D). The antibody antigen binding avidity is quantified using the amplification signals ΔCt in qPCR (Fig. 1E). The ΔCt values among the polyclonal antisera and antigens can be compared to assess antigenic differences among these tested antigens. These ΔCt values can be viewed as similar to the serological titers, such as HI and neutralization titers, from conventional serological assays (Fig. 1F).
Fig.1.
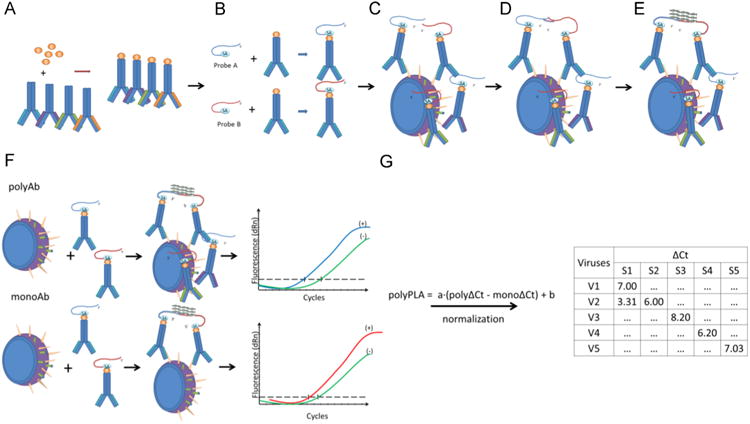
The simplified diagram of polyPLA. PolyPLA quantifies the antibody antigen binding avidity using the amplification signals in quantitative PCR (qPCR) from the pairs of primers attached to a reference polyclonal antiserum. First, polyPLA biotinylates a reference polyclonal antiserum (A), which will be then labeled with sodium azide-linked 5′ and 3′ oligonucleotides (B). A labeled polyclonal antiserum with ΔCt≥8.5 in the ligation efficiency test is then incubated with a reference antigen (virus) or a testing antigen (C), followed by the approximate ligation of the two oligos (D). The antibody antigen binding avidity is quantified using the amplification signals ΔCt in qPCR (E). The ΔCt values among the polyclonal antisera and antigens can be compared to assess antigenic differences among these tested antigens, and these ΔCt values can be viewed as similar as the serological titers, such as HI and neutralization titers, from conventional serological assays (F). The polyPLA units were normalized by its ΔCt values for polyclonal antiserum (polyΔCt) with its ΔCt values for monoclonal antibody against NP (monoΔCt) (G).
To make the ΔCt values comparable across reference sera, we have to ensure that the testing antigens have the same quantities across quantification assays. In HI assays, we usually standardize the antigens to be 4 units of hemaglutinnation titer before HI; in neutralization assays, we usually standardize antigen quantities using TCID50 (2013). In this assay, we use the quantities of nucleoproteins (NPs) to normalize the amount of viruses in the analyses. For data consistency, we used a monoclonal antibody targeting conserved regions of NPs in the proximity ligation assay (Schlingemann et al., 2010). Thus, for a testing antigen, the polyPLA units were normalized by its ΔCt values for polyclonal antiserum (polyΔCt) with its ΔCt values for monoclonal antibody against NP (monoΔCt) (Fig. 1G).
Viral particles must be completely lysed to release NPs and allow for an accurate measure of these protein quantities. We compared two commonly used methods for viral lysis: freeze/thaw and treatment with lysis buffer. The results showed that lysis buffer treated virus had a significantly higher ΔCt value of 7.46 (± 0.45) for A/Sydney/05/1997 (SY/05), p < 0.05 (Fig. 2) compared to the freeze/thaw method of viral lysis. However, for A/Sichuan/2/1987 (SI/2), lysis buffer treated virus did not have a significantly different ΔCt value compared to that of the freeze/thaw method of viral lysis. In the following assays, all the samples used in normalization were treated with lysis buffer.
Fig. 2.
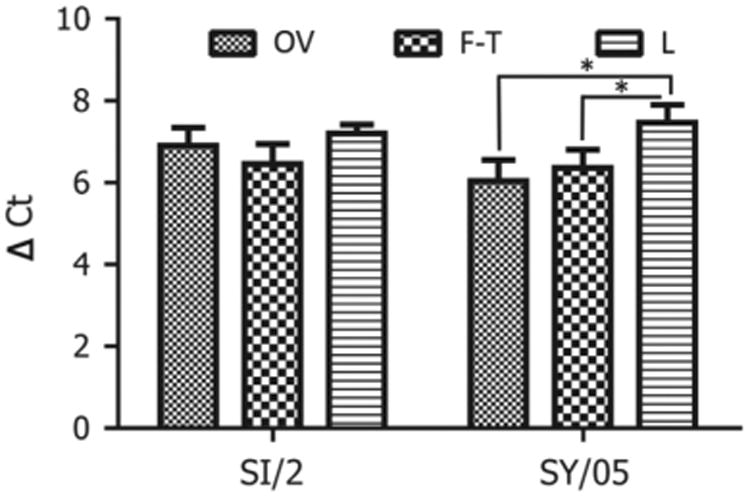
Optimization of the methods in detecting NP proteins using proximity ligation assays. OV denotes the control the control viruses, which were harvested directly after viral propagation in MDCK cells; F–T denotes the viruses, which were frozen and thawed 5 times; L denotes the viruses, which were treated with lysis buffer.
HA specific IgG predominates polyclonal antisera
PolyPLA quantifies the interactions between influenza viral proteins and all IgG present in the polyclonal antisera. To assess the impacts of NA and other internal proteins on polyPLA, we constructed three reassortants between SY/05 and PR8, including SY/05xPR8 (H3N2), SY/05xPR8(H3N1), and SY/05xPR8(H1N2). The signals from NPC were used as the control to calculate ΔCt value from proximity ligation assays. Our results showed that SY/05, SY/05xPR8(H3N2), and SY/05xPR8(H3N1) had ΔCt values of 5.40(± 0.74), 5.67(± 0.17), and 5.26(± 0.34), respectively (Fig. 3). The ΔCt values from PR8 and the reassortant SY/05xPR8(H1N2) were negligible.
Fig. 3.
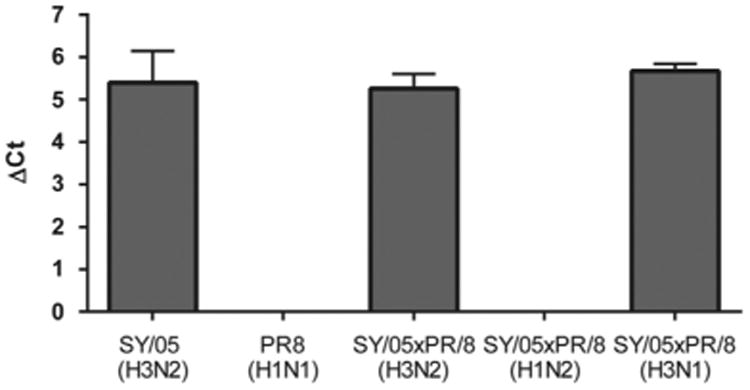
PolyPLA detects predominant IgG against HA gene. The ΔCt of proximity ligation assays for SY/05(H3N2), PR8(H1N1), and three reassortants SY05xPR8 (H3N2), SY05xPR8(H1N2), and SY05xPR8(H3N1) were measured using reference polyclonal sera against SY/05(H3N2).
Viral quantities are linearly correlated with ΔCt values
To assess the sensitivity of polyPLA, we performed PLA on influenza A viruses with serial dilutions. Regression analyses demonstrated that the polyΔCt values are linearly correlated with the influenza viral quantities, with Pearson's coefficient R = 0.98 for the testing strain SY/05 (p < 0.001) (Fig. 4). The cutoff ΔCt value 3.00 was equivalent to 4.90 × 104 TCID50/mL against its homologous polyclonal antibodies. Similarly, the mono-ΔCt values were also linearly correlated with HA titers, and the R was 0.92 for SY/05 (p < 0.001). The cutoff ΔCt value was equivalent to 9.80 × 104 TCID50/mL against the NP-specific monoclonal antibody. Similar linear correlations were also observed in A/Johannesburg/33/1994(H3N2) (JO/33) and A/Nanchang/933/1995 (H3N2) (NA/933) (data not shown). Linear correlation between viral quantities and ΔCt allows us to normalize the viral titers by using a simple equation such as a × (polyΔCt–monoΔCt)+b, where a and b are constant parameters. This normalization method enables us to compare the antigenic properties between the testing antigens (viruses) without justifying the viral quantities before measuring polyΔCt, having been used in HI and neutralization assays to ensure the equivalency of the viral quantities before assays.
Fig. 4.
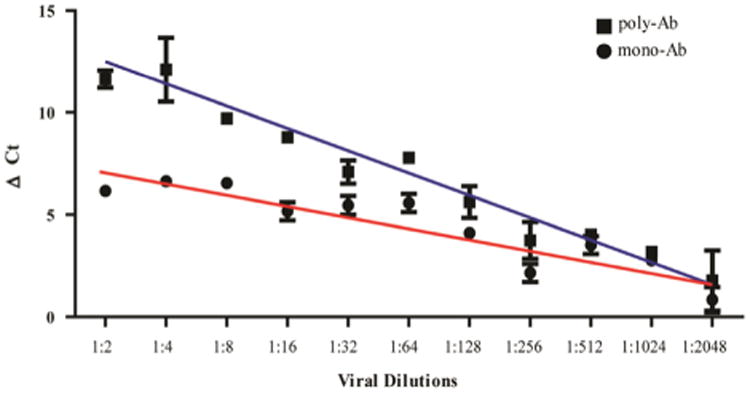
The linear correlation of polyΔCt (R = 0.98) and monoΔCt (R = 0.92) with viral quantities. The cutoff ΔCt value 3.00 was equivalent to 4.90 × 104 TCID50/mL against its homologous polyclonal antibodies and 9.80 × 104 TCID50/mL against NP specific monoclonal antibody.
Sensitivities of polyPLA
To test the sensitivity of polyPLA, we evaluated the viral loads from nasal swabs collected from ferrets infected with A/swine/K6/2011 (H6N6). After only the first day of infection, a polyΔCt titer of 3.20 (± 0.06, standard deviation) was obtained, corresponding to a TCID50 titer of 1.00 × 103 for the infected ferret (Fig. 5). After two days of infection, a polyΔCt value of 5.19 (± 0.06) was obtained, corresponding to a TCID50 titer of 1.00 × 104. A higher polyΔCt titer corresponded to a higher TCID50 titer among the nasal wash samples post-infection. All the samples collected from the control ferrets had polyΔCt titers of less than 3.00, and no viruses were recovered from these control ferrets (Fig. 5). Thus, this method is sensitive sufficiently to detect not only the viruses propagated from the laboratory, but also those in animal specimens. The detection limit is approximately 103 TCID50/mL, which is much less than the viral loads from most patients at the peak time of virus shedding. For example, man can shed 2.6, 5.0, 5.1, 4.9, 3.8, and 1.9 TCID50/mL from one through six days post-inoculation of H1N1 seasonal influenza virus, respectively (Baccam et al., 2006).
Fig. 5.
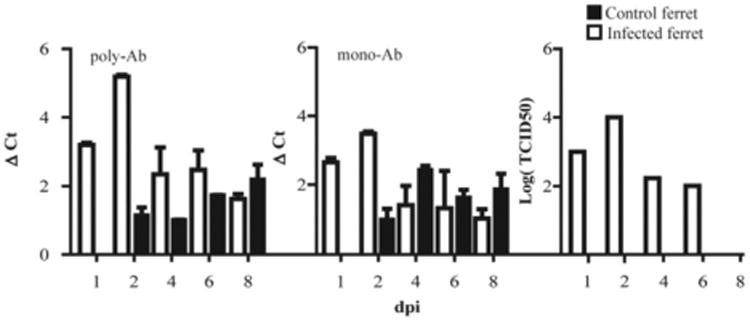
Sensitivity of polyPLA. Comparison of polyΔCt and monoΔCt titers with viral culture titers using the nasal swabs collected from the ferrets infected with A/swine/Guangdong/K6/2010(H6N6). After only the first day of infection, a polyΔCt value of 3.20 (± 0.06, standard deviation) was obtained, corresponding to a TCID50 titer of 1.00 × 103 for the infected ferret. After two days of infection, a polyΔCt value of 5.19 (± 0.06) was obtained, corresponding to a TCID50 titer of 1.00 × 104.
Detecting antigenic variants of H3N2 historical seasonal influenza viruses
The H3N2 viruses have been causing seasonal epidemic outbreaks since its first introduction into the human population, resulting in the pandemic of 1968. During the past four decades, at least 12 antigenic drift events have been detected; six of which occurred from 1997 to 2010 (Skowronski et al., 2007; Sun et al., 2013). In this study, six historical H3N2 isolates, JO/33, NA/933, SY/05, A/Brisbane/10/2007 (BR/10), A/Perth/16/2009 (PE/16), and A/Victoria/361/2011(VI/361) representing antigenic cluster BE92, WU95, SY97, BR07, PE09, and VI11 (Sun et al., 2013), respectively, and their corresponding homologous ferret antisera were used to validate polyPLA. Our method was expected to identify significant differences in homologous and heterologous polyPLA titers (Table 1).
Table 1.
The H3N2 influenza A viruses used in this study.
| Virus | Abbreviation | Antigenic clustera |
|---|---|---|
| A/Sichuan/2/87(H3N2) | SI/2 | ND |
| A/Johannesburg/33/94(H3N2) | JO/33 | BE92 |
| A/Nanchang/933/95(H3N2) | NA/933 | WU95 |
| A/Sydney/05/97(H3N2) | SY/05 | SY97 |
| A/Brisbane/10/07(H3N2) | BR/10 | BR07 |
| A/Perth/16/09(H3N2) | PE/16 | PE09 |
| A/Victoria/361/11(H3N2) | VI/361 | VI11 |
The antigenic cluster was described in Sun et al. (2013), and ND=not determined.
Our results showed that the homologous titers were approximately 10.00 polyPLA units. The polyPLA titers for NA/933 and SY/05 against JO/33 antisera were 9.37 (±0.22) and 6.725 (±0.32) polyPLA units, respectively, which were significantly less than the homologous JO/33 titer 11.43 (±0.01) units, p < 0.0001 (Fig. 6A, Table 2). The homologous titer for NA/933 was 11.71 (±0.28) whereas the titers for JO/33 and SY/05 against NA/933 antisera were 8.44 (±0.32) and 8.16 (±0.43) units, respectively; the titers for JO/33 against NA/933 antisera were significantly less than the homologous titers for JO/33, p < 0.001. The homologous titer for SY/05 was 13.19 (±0.06) units whereas the titers for JO/33 and NA/933 against SY/05 antisera were 10.59 (±0.15) and 8.21 (±0.07) units, respectively; the titers for both JO/33 and NA/933 against SY/05 antisera were significantly less than the homologous titers for SY/05, p < 0.0001.
Fig. 6.
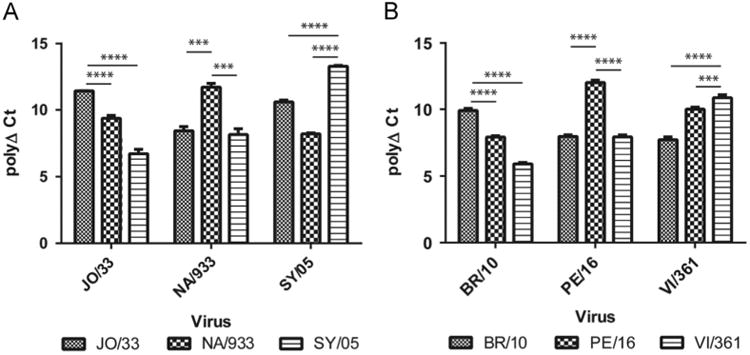
Detecting antigenic variants of H3N2 historical seasonal influenza viruses. JO/33, NA/933, SY/05, BR/10, PE/16, VI/361 representing antigenic cluster BE92, WU95, SY97, BR07, PE09, and VI11, respectively. The homologous polyPLA titers were significantly higher than the heterologous titers for both the antigenic drift event BE92>WU95>SY97 (A) and BR07>PE09>VI11(B). These results were consistent with those measured from conventional HI and neutralization assays (Tables 2 and 3).
Table 2.
The correlation among the titers from polyPLA and those from HI and MN assays for JO/33, NA/33, and SY/05.
| Virus | Ferret antiseraa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| JO/33 | NA/933 | SY/05 | |||||||
|
|
|
|
|||||||
| HI | MN | PolyΔCt (standard deviation) | HI | MN | PolyΔCt (standard deviation) | HI | MN | PolyΔCt (standard deviation) | |
| JO/33 | 640 | ND | 11.43 (0.01) | 40 | ND | 8.44 (0.32) | <10 | 40 | 10.59 (0.15) |
| NA/933 | 160 | ND | 9.37 (0.22) | 1280 | ND | 11.71 (0.28) | 160 | 30 | 8.21 (0.07) |
| SY/05 | <10 | ND | 6.725 (0.32) | <10 | ND | 8.16 (0.43) | 1280 | 1280 | 13.29 (0.06) |
The number in bold is the homologous titer.
Similarly, the homologous titers for BR/10 were 9.91 (±0.16) whereas the titers for PE/16 and VI/361 against BR/10 sera were 7.97 (±0.12) and 7.72 (±0.21), respectively (Fig. 6B); the titers for PE/16 and VI/361 against BR/10 sera were significantly less than the homologous titers for BR/10, p < 0.001. PE/16 had the highest polyΔCt value of 12.00 (±0.20) against PE/16 sera whereas the titers for BR/10 and VI/361 against PE/16 sera were 7.92 (±0.10) and 10.01 (±0.15), respectively; the titers for BR/10 and VI/361 against PE/16 sera were significantly less than the homologous titers for PE/16, p < 0.001. The homologous titer for VI/361 was 10.87 (±0.22) whereas the titers for BR/10 and PE/09 against VI/361 sera were 5.90 (±0.10) and 7.93 (±0.15), respectively; the titers for BR/10 and PE/09 against VI/361 sera were significantly less than the homologous titers for VI/361, p < 0.001.
Detecting antigenic variants in human clinical specimens
To test the applicability of polyPLA in clinical samples, we applied the same method to characterize antigenic profiles of H3N2 influenza A viruses, which came directly from clinical samples collected in the 2012–2013 influenza season. A total of 100 nasal swabs were collected from September 2012 to April 2013, and confirmed as H3N2 positive using quantitative RT-PCR.
About 50% of these samples had at least a 3-polyPLA-unit decrease compared to BR/10 homologous titers and that about 18% and 1% of these samples had at least 3-polyPLA-unit decrease when compared to PE/16 and VI/361 homologous titers, respectively (Fig. 7A). The polyPLA titers of the majority clinical samples were highest when using VI/361 sera, followed by PE/16 and BR/10. We compared the polyΔCt titers of the clinical samples with the HI titers for these 21 isolates, and the HI titers were positively correlated with polyΔCt titers (Fig. 7B).
Fig. 7.
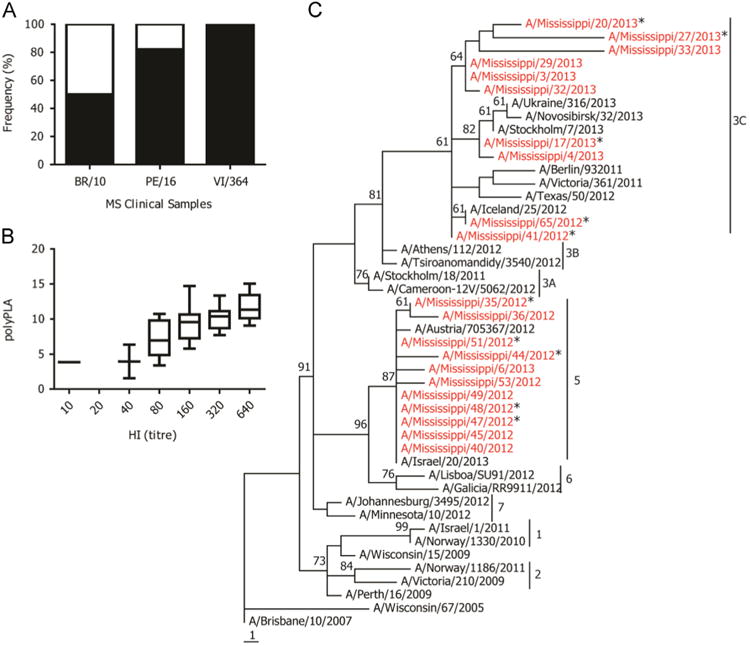
Detecting antigenic variants in human clinical specimens. (A) The percentile of antigenic variants (white portion) compared to their corresponding homologous titers, and a 3-polyPLA-unit threshold was used; (B) the correlation between polyPLA units and HI titers, which was measured using PE/16 polyclonal antisera; (C) phylogenetic tree of HA protein sequences of H3N2 seasonal influenza viruses. The 21 isolates recovered from the 2012–2013 influenza season from Mississippi were marked in red, and the antigenic variants proposed by polyPLA assays using clinical samples against PE/16 were marked with stars. The phylogenetic tree was constructed by maximum parsimony based on HA protein sequences, and genetic clusters were defined based on the reports from Community Network of Reference Laboratories (CNRL) for Human Influenza in Europe (http://www.ecdc.europa.eu/en/publications/Publications/Influenza-visus-characterisation-June-2012.pdf).
To better understand the antigenic and genetic background of the H3N2 viruses in these samples, 21 samples were randomly selected and subjected to viral isolation using MDCK cells. A total of 21 viruses were recovered, and the HA sequences of these viruses were sequenced. The sequence analyses showed that no consistent mutations at the reported antibody binding sites (Wilson and Cox, 1990) were observed in the isolates we recovered from this study (Table 4).
Table 4.
Molecular characterization of amino acid changes in the HA1 proteins of H3N2 isolates recovered from 2012–2013 season.
| Isolate | Amino acid positiona | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||
| 2 | 8 | 33 | 45 (C) | 48 (C) | 53 (C) | 94 (E) | 128 (B) | 142 (A) | 144 (A) | 145 (A) | 158 (B) | 198 (B) | 199 | 223 | 278 (C) | 280 (C) | 312 (C) | |
| WI/67 | K | N | Q | S | T | D | Y | T | R | N | N | K | A | S | I | N | E | N |
| BR/10 | K | N | Q | S | T | D | Y | T | R | N | N | K | A | S | V | N | E | N |
| PE/16 | K | N | Q | S | T | D | Y | T | R | K | N | N | A | S | V | N | E | N |
| VI/361 | K | N | Q | N | I | D | Y | T | R | N | N | N | S | S | I | N | E | S |
| TX/50 | K | N | R | N | I | D | Y | N | R | N | N | N | P | S | I | K | E | S |
| MS/3 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/4 | K | N | R | N | I | D | Y | A | G | N | S | N | S | S | I | K | E | S |
| MS/17 | K | N | R | N | I | D | Y | A | G | N | S | N | S | S | I | K | E | S |
| MS/20 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/27 | N | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/29 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/32 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/33 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/35 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/36 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/40 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/41 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/44 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/45 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/47 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/48 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/49 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/51 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/53 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
| MS/65 | K | N | R | N | I | D | Y | T | R | N | S | N | S | S | I | K | E | S |
| MS/6 | E | D | Q | S | T | N | H | T | R | N | N | N | A | S | V | N | A | N |
The antibody binding sites were annotated based on previous report (Wilson and Cox, 1990).
Since 2007, the H3N2 seasonal influenza viruses have six genetic clusters (2013). The genetic clusters 1 and 2 were PE/16 like-viruses. The viruses from 2010 were scattered into genetic clusters 3, 4, 5, and 6. The phylogenetic analyses showed that these 21 viruses belonged to genetic cluster 3C and 5 (Fig. 7C).
Discussion
Identification of antigenic variants in disease surveillance is essential, and an ideal antigenic characterization platform should meet the following four criteria: (1) robust, the results should be repeatable; (2) simple and economic, the methods can be carried out in a common diagnosis laboratory; (3) high throughput, the method should be able to perform on a large-scale; and (4) sensitive, the method will have optimal performance in identifying antigenic variants directly from clinical samples, avoiding viral isolation. For many diseases, the pathogen isolation process is not only time-consuming but also can change virus antigenic properties, resulting in data that does not accurately represent those antigenic properties in circulating viruses. Furthermore, some pathogens cannot even be recovered from the specimen. The polyPLA developed in this study was designed to meet these four criteria, as confirmed in influenza antigenic variant identification in this study.
The polyPLA quantifies antibody antigen interactions for influenza viral proteins, including both HA and NA, against their corresponding antibodies in the antisera. The principle of this method is more similar to neutralization assays rather than HI assays. More importantly, it can avoid the red blood cell binding problems usually seen in HI assay. For example, egg-adaptation substitutions affect the architecture of the HA receptor-binding site and alter the interactions of the HA with the terminal sialic acid moiety (Gambaryan et al., 1999).
The high concentration of influenza A viruses could lead to the saturation of viruses over antibody, which is 200 nM used in this study. For example, when the undiluted, MDCK cell derived NA/933 virus was used, there was no significant difference between the ΔCt values against NA/933 antibody and those against JO/33 antibody (data not shown). After the NA/933 viruses were diluted to 1:40, there was significant difference between the ΔCt values against NA/933 and those against JO/33 antibodies (Tables 2 and 3 and Fig. 6). However, we do not believe this potential limitation will affect the application of this method in clinical samples, from which the virus loads are much smaller than a single isolate.1
Table 3.
The correlation among the titers from polyPLA and those from HI and MN assays for BR/10, PE/16, and VI/361.
| Virus | Ferret antiseraa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| BR/10 | PE/16 | VI/361 | |||||||
|
|
|
|
|||||||
| HI | MN | PolyΔCt (standard deviation) | HI | MN | PolyΔCt (standard deviation) | HI | MN | PolyΔCt (standard deviation) | |
| WI/67 | ND | ND | 40 | 160 | 320 | 320 | |||
| BR/10 | 1280 | 2560 | 9.91(0.16) | 40 | 160 | 7.92(0.10) | 40 | 40 | 5.90(0.10) |
| PE/16 | 80 | 80 | 7.97(0.12) | 640 | 640 | 12.00(0.20) | 160 | 160 | 7.93(0.15) |
| VI/361 | 20 | 20 | 7.72(0.21) | 320 | 640 | 10.01(0.15) | 640 | 640 | 10.87(0.22) |
The number in bold is the homologous titer.
Compared to seasonal influenza virus surveillance, the anti-genic characterization of emerging pathogens for pandemic pre-paredness, especially those pathogens in areas without sufficient biosafety facilities, has been challenging. Propagation of these emerging viruses usually requires a high biosafety containment such as BSL-3 and even BSL-4. In most cases, the specimens need to be shipped to a laboratory with appropriate biosafety containment, and sometimes even the paperwork for collaborative agreements, especially among countries, can cause delays when an outbreak occurs. Because the polyPLA can use the clinical samples directly and also use a common qRT-PCR platform, it can perform large-scale analyses with minimal biosafety requirements in laboratory conditions. For example, biosafety level 2 will meet for the polyPLA in influenza surveillance (Engelhardt et al. 2013). Thus, this method will be very useful in detecting antigenic variants for the H5N1 highly pathogenic avian influenza viruses and emerging H7N9 low pathogenic avian influenza viruses. In resource limited areas, although the RBCs in conventional serological assays such as HI can be more accessible, it would be much easier and economic to set up a qRT-PCR platform than a high containment environment for viral propagation.
In summary, polyPLA is a simple method quantifying the binding avidity of antibody antigen interactions. Beyond influenza applications, this method can also be used in other pathogens including those cannot be propagated in laboratory.
Materials and methods
Viruses and antibodies
The H3N2 viruses used in this study were obtained from the Centers of Disease Control and Prevention, Department of Health & Human Services and BEI Research Resources Repository (http://www.beiresources.org/) (Table 1), and the monoclonal antibodies against nucleoprotein (NP) from Millipore, United States. The viruses were propagated at Madin–Darby Canine Kidney (MDCK) cells and stored at −80 °C before usage. The polyclonal antisera were generated using 4- to 6-month-old ferrets (Triple F Farm, PA). The ferrets were anesthetized with isoflurane and inoculated intranasally with 106 50% egg infectious doses (EID50) of a challenging virus. The ferret sera were collected three weeks post-infection. The viral isolation was performed using MDCK cells.
Labeling of antibodies
IgG were purified from ferret polyclonal antisera and mice monoclonal antibodies using Pierce Chromatography Cartridges Protein A/G according to the manufacturer's instruction (Pierce, Rockford, IL) and then biotinylated with Biotin-XX Microscale Protein Labeling Kit according to manufacturer directions (Life Technologies, Carlsbad, CA). To remove the free biotin, Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific Pierce, Rockford, IL) were used; dialysis was performed at 4 °C in cold 1X PBS (pH 7.4), and the buffer was changed at least 5 times each 2 h.
Forced proximity probe test
An aliquot of biotinylated antibody stock solution was diluted to 200 nM (30 μg/mL); 2 μL of diluted biotinylated antibody was added to 2 μL of equal mixture of 200 nM 3′ and 5′ TaqMan Prox-Oligo, designated probe A and probe B (Life Technologies, Carlsbad, CA), and incubated at room temperature for 60 min. A negative control was made replacing diluted biotinylated antibody with 2 μL antibody dilution buffer. After incubation, 396 μL of assay probe dilution buffer was added and incubated for another 30 min at room temperature. Then 96 μL of ligation solution was added to 4 μL of the probe and virus mixture, incubated again at 37 °C for 10 min, and cooled at 4 °C for 10 min. After incubation, 9 μL was transferred to a new 0.2 mL microcentrifuge tube with 11 μL qPCR mix (10 μL Fast Master Mix, 2X plus 1 μL Univeral PCR Assay, 20X) and was briefly centrifuged. The qPCR cycling was as follows: 95 °C for 2 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min.
The change in threshold cycle (ΔCt) values was calculated for each biotinylated antibody: average Ct (negative control)–average Ct (forced proximity probe). If the ΔCt ≥ 8.5, the test biotinylated antibody was considered suitable for use in the PLA.
Probe preparation
Two assay probes for each antibody were prepared by combining the biotinylated antibodies with either 3′ or 5′ TaqMan Prox-Oligo (probe A and probe B). For example, for each probe, 2.5 μL 200 nM biotinylated antibody was combined with 2.5 μL of either 200 nM 3′ prox-Oligo or 200 nM 5′ prox-Oligo, and the mixture was incubated at room temperature for 60 min. Then, 45 μL Assay Probe Storage Buffer was added, briefly centrifuged, and incubated at room temperature for 20 min.
PLA and quantification of polyΔCt and monoΔCt
The Taqman PLA was performed by first diluting equal parts of probes A and B mixture 1:10 with phosphate-buffered saline (PBS, pH 7.4). For the non-protein control (NPC), 2 μL was combined with 2 μL diluted virus (virus lysed for NP detection) or 2 μL 1X PBS, pH 7.4. The mixture was centrifuged briefly and incubated at 37 °C for 1 h. Then, 96 μL of ligation solution was added to 4 μL of the probe and virus mixture, incubated again at 37 °C for 10 min, and cooled at 4 °C up to 10 min. After incubation, 2 μL of 1X protease was added to each ligation reaction and incubated at 37 °C for 10 min, 95 °C for 5 min, and 4 °C for holding. Lastly, 9 μL of the product was transferred to a new 0.2 mL microcentrifuge tube with 11 μL qPCR mix (10 μL Fast Master Mix, 2X plus 1 μL Univeral PCR Assay, 20X) and was briefly centrifuged. The qPCR cycling was as follows: 95 °C for 2 min, 45 cycles of 95 °C for 15 s, and 60 °C for 1 min.
The NPC was used as a reference background and the threshold cycle (Ct) value given dictated the non-target ligation signal noise of the assay. A total of three replicates for each sample and control were performed. To calculate the ΔCt values: average Ct (NPC)– average Ct (sample), which represented the true target-mediated signal above background. The cutoff of ΔCt ≥ 3.00 was used for qualitative analysis of viral and antibody binding, as according to the Taqman Protein Assays Sample Prep and Protocol (2013).
HI and virus neutralization assays
In the HI assay, the receptor destroying enzyme (RDE, Denka Seiken Co., Japan) was used to treat the ferret sera in the ratio of 1:3 (RDE: sera, volume: volume) for 18 h at 37 °C, then heat inactivated at 56 °C for 30 min. The treated ferret sera were diluted to 1:10 with phosphate-buffered saline (PBS) then 2-fold serial diluted and reacted with 4-hemmagglutination-units viruses. The HI titers were expressed as the reciprocal of the highest dilution at which virus binding to the 0.5% turkey red blood cells (RBC) was blocked.
For the virus neutralization assay, serially diluted ferret sera were first incubated with 100 TCID50 viruses at 37 °C for 1 h. The virus-sera mixtures were then adsorbed to MDCK cells for 1 h. The infected cells were washed twice with PBS buffer and replenished with Opti-Mem Reduced Serum Media (Life Technologies, US). The supernatants from the infected cells were harvested 4 days postinfection and were analyzed using hemagglutination assay.
Data analysis
To compare the antigenic properties across the testing antigens (viruses), we computed the polyPLA unit between antigen (virus) and antibody using the following equation:
To improve our computation, we simply used a = 1.00 and b = 10.00. The b = 10.00 enabled us to avoid negative numbers. If monoΔCt < 3.00, polyPLA will be assigned as “< 0”, meaning that the viral loads were too low for analyses. As mentioned above, in general, polyPLA is sensitive in detecting viral loads of approximately 103 TCID50/mL
Genomic sequencing and Genbank accession number
The HA genes of 21H3N2 isolates recovered from human clinical specimens were sequenced using Sanger Sequencing, and they were deposited in Genbank with the accession numbers KM244531–KM244551.
Molecular characterization and phylogenetic analyses
The multiple sequence alignments were conducted using the MUSCLE software package (Edgar, 2004). The phylogenetic analyses were performed using maximum likelihood by GARLI version (Zwickl, 2006), and bootstrap resampling analyses were conducted with 1000 runs using PAUP* 4.0 Beta (Swofford, 1998) with a neighborhood joining method, as previously described (Wan et al., 2008) (Table 4).
Supplementary Material
Acknowledgments
We are thankful for Nan Zhao, Chun-Kai Yang, Jeff Eells, and Matthew Ross providing helpful discussion and comments and Xiyan Xu supplying the H3N2 influenza A viruses and ferret sera used in this study. We also thank Dena Pruett and Julie Lopez for editorial assistance.
Footnotes
Author contributions: XFW initiated this project; BM, KJ, and CH performed the experiments; BM, KJ, HS, and XFW performed data analysis; BM, KJ, and XFW wrote this paper.
Appendix A. Supporting information: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2014.11.029.
References
- Thompson MG, Shay DK, Zhou H, Bridges CB, Cheng PY, Burns E, Cox JJ. Estimates of deaths associated with seasonal influenza-United States, 1976-2007. MMWR Morb Mortal Wkly Rep. 2010;59:1057–1062. [PubMed] [Google Scholar]
- Engelhardt OG, Graham ML, Grohmann G, Itamura S, Katz JM, Li X, Summermatter K, Webby R, Weir J, Zimmer G. Update of WHO biosafety risk assessment and guidelines for the production and quality control of human influenza vaccines against avian influenza A(H7N9) virus. 2013 http://www.who.int/biologicals/areas/vaccines/influenza/biosafety_risk_assessment_10may2013.pdf.
- Ampofo WK, Baylor N, Cobey S, Cox NJ, Daves S, Edwards S, Ferguson N, Grohmann G, Hay A, Katz J, Kullabutr K, Lambert L, Levandowski R, Mishra AC, Monto A, Siqueira M, Tashiro M, Waddell AL, Wairagkar N, Wood J, Zambon M, Zhang W. Improving influenza vaccine virus selection: report of a WHO informal consultation held at WHO headquarters, Geneva, Switzerland, 14-16 June 2010. Influenza Other Respir Viruses. 2012;6:142–152. [Google Scholar]
- Azzi A, Bartolomei-Corsi O, Zakrzewska K, Corcoran T, Newman R, Robertson JS, Yates P, Oxford JS. The haemagglutinins of influenza A (H1N1) viruses in the ‘O’ or ‘D’ phases exhibit biological and antigenic differences. Epidemiol Infect. 1993;111:135–142. doi: 10.1017/s0950268800056752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccam P, Beauchemin C, Macken CA, Hayden FG, Perelson AS. Kinetics of influenza A virus infection in humans. J Virol. 2006;80:7590–7599. doi: 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox NJ, Bender CA. The molecular epidemiology of influenza viruses. Semin Virol. 1995;6:359–370. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM, Bush RM, Bender CA, Cox NJ. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA. 1997;94:7712–7718. doi: 10.1073/pnas.94.15.7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Dixon W, Ji H, Koong AC, Mindrinos M, Davis RW. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nat Methods. 2007;4:327–329. doi: 10.1038/nmeth1020. [DOI] [PubMed] [Google Scholar]
- Gambaryan AS, Robertson JS, Matrosovich MN. Effects of egg-adaptation on the receptor-binding properties of human influenza A and B viruses. Virology. 1999;258:232–239. doi: 10.1006/viro.1999.9732. [DOI] [PubMed] [Google Scholar]
- Gerdil C. The annual production cycle for influenza vaccine. Vaccine. 2003;21:1776–1779. doi: 10.1016/s0264-410x(03)00071-9. [DOI] [PubMed] [Google Scholar]
- Grund S, Adams O, Wahlisch S, Schweiger B. Comparison of hemagglutination inhibition assay, an ELISA-based micro-neutralization assay and colorimetric microneutralization assay to detect antibody responses to vaccination against influenza A H1N1 2009 virus. J Virol Methods. 2011;171:369–373. doi: 10.1016/j.jviromet.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Harper SA, Fukuda K, Uyeki TM, Cox NJ, Bridges CB. Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2004;53:1–40. [PubMed] [Google Scholar]
- Hirst GK. The agglutination of red cells by allantoic fluid of chick embryos infected with influenza virus. Science. 1941;94:22–23. doi: 10.1126/science.94.2427.22. [DOI] [PubMed] [Google Scholar]
- Katz JM, Naeve CW, Webster RG. Host cell-mediated variation in H3N2 influenza viruses. Virology. 1987;156:386–395. doi: 10.1016/0042-6822(87)90418-1. [DOI] [PubMed] [Google Scholar]
- Lindstrom S, Endo A, Sugita S, Pecoraro M, Hiromoto Y, Kamada M, Takahashi T, Nerome K. Phylogenetic analyses of the matrix and non-structural genes of equine influenza viruses. Arch Virol. 1998;143:1585–1598. doi: 10.1007/s007050050400. [DOI] [PubMed] [Google Scholar]
- Lindstrom S, Sugita S, Endo A, Ishida M, Huang P, Xi SH, Nerome K. Evolutionary characterization of recent human H3N2 influenza A isolates from Japan and China: novel changes in the receptor binding domain. Arch Virol. 1996;141:1349–1355. doi: 10.1007/BF01718836. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Escriou N, Naffakh N, Manuguerra JC, van der Werf S. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology. 2001;289:74–85. doi: 10.1006/viro.2001.1121. [DOI] [PubMed] [Google Scholar]
- Mori SI, Nagashima M, Sasaki Y, Mori K, Tabei Y, Yoshida Y, Yamazaki K, Hirata I, Sekine H, Ito T, Suzuki S. A novel amino acid substitution at the receptor-binding site on the hemagglutinin of H3N2 influenza A viruses isolated from 6 cases with acute encephalopathy during the 1997–1998 season in Tokyo. Arch Virol. 1999;144:147–155. doi: 10.1007/s007050050491. [DOI] [PubMed] [Google Scholar]
- Morishita T, Kobayashi S, Miyake T, Ishihara Y, Nakajima S, Nakajima K. Host-specific hemagglutination of influenza A (H1N1) virus. Microbiol Immunol. 1993;37:661–665. doi: 10.1111/j.1348-0421.1993.tb01689.x. [DOI] [PubMed] [Google Scholar]
- Morishita T, Nobusawa E, Nakajima K, Nakajima S. Studies on the molecular basis for loss of the ability of recent influenza A (H1N1) virus strains to agglutinate chicken erythrocytes. J Gen Virol. 1996;77(Pt 10):2499–2506. doi: 10.1099/0022-1317-77-10-2499. [DOI] [PubMed] [Google Scholar]
- Nobusawa E, Ishihara H, Morishita T, Sato K, Nakajima K. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acid change in hemagglutinin altered its recognition of sialyloligosaccharides. Virology. 2000;278:587–596. doi: 10.1006/viro.2000.0679. [DOI] [PubMed] [Google Scholar]
- Nobusawa E, Nakajima K. Amino acid substitution at position 226 of the hemagglutinin molecule of influenza (H1N1) virus affects receptor binding activity but not fusion activity. Virology. 1988;167:8–14. doi: 10.1016/0042-6822(88)90048-7. [DOI] [PubMed] [Google Scholar]
- Oh DY, Barr IG, Mosse JA, Laurie KL. MDCK-SIAT1 cells show improved isolation rates for recent human influenza viruses compared to conventional MDCK cells. J Clin Microbiol. 2008;46:2189–2194. doi: 10.1128/JCM.00398-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S, Oxford JS. Analysis of antigenic determinants on internal and external proteins of influenza virus and identification of antigenic subpopulations of virions in recent field isolates using monoclonal antibodies and immunogold labelling. Arch Virol. 1986;88:189–202. doi: 10.1007/BF01310874. [DOI] [PubMed] [Google Scholar]
- Schlingemann J, Leijon M, Yacoub A, Schlingemann H, Zohari S, Matyi-Toth A, Kiss I, Holmquist G, Nordengrahn A, Landegren U, Ekstrom B, Belak S. Novel means of viral antigen identification: improved detection of avian influenza viruses by proximity ligation. J virol Methods. 2010;163:116–122. doi: 10.1016/j.jviromet.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Kendal AP, Pereira MS, Skehel JJ. Hemagglutination inhibition. In: Kendal AP, Pereira MS, Skehel JJ, editors. Concepts and procedures for laboratory-based influenza surveillance. Centers for Disease Control; Atlanta, GA: 1982. pp. B17–B35. [Google Scholar]
- Skowronski DM, Masaro C, Kwindt TL, Mak A, Petric M, Li Y, Sebastian R, Chong M, Tam T, De Serres G. Estimating vaccine effectiveness against laboratory-confirmed influenza using a sentinel physician network: results from the 2005–2006 season of dual A and B vaccine mismatch in Canada. Vaccine. 2007;25:2842–2851. doi: 10.1016/j.vaccine.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Sun H, Yang J, Zhang T, Long LP, Jia K, Yang G, Webby R, Wan XF. Inferring influenza virus antigenicity using sequence data. mBio. 2013;4:4. doi: 10.1128/mBio.00230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*: Phylogenic Analysis Using Parsimony. Sinauer; Sunderland, Massachusetts: 1998. [Google Scholar]
- Wan XF, Nguyen T, Davis CT, Smith CB, Zhao ZM, Carrel M, Inui K, Do HT, Mai DT, Jadhao S, Balish A, Shu B, Luo F, Emch M, Matsuoka Y, Lindstrom SE, Cox NJ, Nguyen CV, Klimov A, Donis RO. Evolution of highly pathogenic H5N1 avian influenza viruses in Vietnam between 2001 and 2007. PLoS One. 2008;3:e3462. doi: 10.1371/journal.pone.0003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Laver WG, Air GM, Schild GC. Molecular mechanisms of variation in influenza viruses. Nature. 1982;296:115–121. doi: 10.1038/296115a0. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. The University of Texas; Austin: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.