

Abstract
The pathological basis of neonatal hypoxia–ischemia (HI) brain damage is characterized by neuronal cell loss. Oxidative stress is thought to be one of the main causes of HI-induced neuronal cell death. The p38 mitogen-activated protein kinase (MAPK) is activated under conditions of cell stress. However, its pathogenic role in regulating the oxidative stress associated with HI injury in the brain is not well understood. Thus, this study was conducted to examine the role of p38 MAPK signaling in neonatal HI brain injury using neonatal rat hippocampal slice cultures exposed to oxygen / glucose deprivation (OGD). Our results indicate that OGD led to a transient increase in p38 MAPK activation that preceded increases in superoxide generation and neuronal death. This increase in neuronal cell death correlated with an increase in the activation of caspase-3 and the appearance of apoptotic neuronal cells. Pre-treatment of slice cultures with the p38 MAPK inhibitor, SB203580, or the expression of an antisense p38 MAPK construct only in neuronal cells, through a Synapsin I-1-driven adeno-associated virus vector, inhibited p38 MAPK activity and exerted a neuroprotective effect as demonstrated by decreases in OGD-mediated oxidative stress, caspase activation and neuronal cell death. Thus, we conclude that the activation of p38 MAPK in neuronal cells plays a key role in the oxidative stress and neuronal cell death associated with OGD.
Keywords: apoptosis, hypoxia–ischemia, neonate brain, neuronal cell death, rat, superoxide
Introduction
Peri-natal hypoxia–ischemia (HI) remains an important cause of acute neonatal mortality and chronic morbidity in infants and children. The neurologic consequences of injury include mental retardation, epilepsy, cerebral palsy and blindness (Smith et al., 2008). The lack of effective treatment severely hampers the clinical options in children with HI. The mechanisms underlying the damage associated with HI are only partly understood (Lipton, 1999) but probably involve the activation of multiple signal transduction cascades.
The p38 mitogen-activated protein kinase (MAPK), a member of the MAPK pathway, was first isolated as a 38 kDa protein that is rapidly tyrosine phosphorylated in response to lipopolysaccharide stimulation (Han et al., 1993, 1994). p38 MAPK is activated in response to various physical and chemical stresses, such as oxidative stress, UV irritation, hypoxia, ischemia and various cytokines (Chen et al., 2001; Kyriakis & Avruch, 2001; Junttila et al., 2008). Previous studies, mainly from cell culture-based assays, indicate that p38 MAPK plays a role in regulating neuronal death on various insults (Harper & LoGrasso, 2001). In neuronal cells or cell lines, a number of stimulations have been reported to activate p38 MAPK (Xia et al., 1995; Heidenreich & Kummer, 1996; Horstmann et al., 1998; Park et al., 2002) and studies have reported activated p38 MAPK in rat and mouse models of neonatal HI brain injury (Hee Han et al., 2002; Bu et al., 2007). However, there are no data regarding the role of p38 MAPK activation in the increased generation of reactive oxygen species associated with the neuronal death in the neonatal brain exposed to HI. Thus, the aim of the present study was to investigate p38 MAPK activation and its relationship with the oxidative stress associated with neonatal brain injury after HI and to evaluate its role in both the necrotic and apoptotic pathways of neuronal cell death associated with oxygen / glucose deprivation (OGD).
Materials and methods
Hippocampal slice culture and oxygen / glucose deprivation exposure
Neonatal rats (Sprague–Dawley) at post-natal day (P)7 were decapitated and the hippocampi dissected under sterile conditions. Each hippocampus was sliced into 400 μm slices using a Mcllwain tissue chopper (Science Products GmbH, Switzerland). Slices were then cultured on permeable membrane Millicell inserts (Millipore, Billerica, MA, USA) (0.4 μm pore size) in six-well plates for 6 days at 37 °C in 5% CO2. For the first 2 days, the slices were maintained in a primary plating medium – 50% Opti-Mem (Gibco, Grand Island, NY, USA), 25% Hank's Buffered Salt Solution, 25% heat-inactivated horse serum, 5 mg / mL d-glucose (Sigma, St Louis, MO, USA) and 1.5% PenStrep / Fungizone (Gibco). The primary plating medium was changed at 24 h. After 48 h, the slices were switched to neurobasal-A medium (Gibco), with 1 mm glutamax, 1% penstrep / fungizone (Gibco) and 2% B27 (Gibco) supplemented with antioxidants for a further 4 days. At 24 h before exposure to OGD the culture medium was changed to neurobasal-A and B27 supplement without antioxidants. Just prior to OGD, a sucrose balanced salt solution (120 mm NaCl, 5 mm KCl, 1.25 mm NaH2PO4, 2 mm MgSO4, 2 mm CaCl2, 25 mm NaHCO3, 20 mm HEPES, 25 mm sucrose, pH 7.3) was infused for 1 h with 5% CO2 and 10 L / h nitrogen gas. The inserts were then transferred into deoxygenated sucrose balanced salt solution, placed in a ProOxC system chamber with an oxygen controller (BioSpherix) and exposed to 0.1% O2, 5%CO2 and 94.4% nitrogen for 90 min at 37 °C. The slices were then returned to oxygenated serum-free neurobasal medium with B27 supplement. The p38 MAPK inhibitor, SB203580 (Calbiochem, Gibbstown, NJ, USA), was dissolved in dimethyl sulphoxide (50 μm) and added to the medium at 2 h before OGD. Control experiments contained equivalent amounts of dimethyl sulphoxide, which did not exceed 0.2% (v / v). All protocols and procedures were approved by the Committees on Animal Research of Georgia Health Sciences University and the University of Montana.
Quantification of slice culture cell death
Propidium iodide (PI) (1 μg / mL; Sigma) was added to the culture medium at 24 h prior to OGD. Slice cultures were then examined prior to OGD with an inverted fluorescence microscope (Olympus IX51; Japan) using an excitation wavelength of 510 nm and an emission wavelength of 590 nm. Slices showing distinct PI intake were excluded from further study. Slice culture images were obtained at 0, 4, 8 and 24 h after OGD using a 10-bit monochrome fluorescence camera (Digital Camera C4742-95; Hamamatsu, Japan). Images were processed using Image-Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD, USA). The exposure time was set at 200 ms, using 4 × magnification to capture the entire slice. The evaluation of cell death was performed using a modification of the method of Cronberg et al. (2004). The fluorescence intensity of the whole slice area, as well as the CA1, CA3 and dentate gyrus (DG) sub-regions was quantified with Image-Pro (Media Cybernetics).
Lactate dehydrogenase cytotoxicity assay
Cytotoxicity was evaluated by measuring lactate dehydrogenase (LDH) released into the slice culture medium using a Cytotoxicity Detection kit (Roche Applied Science, Mannheim, Germany). Samples were analyzed at 0, 4, 8 and 24 h after OGD. All LDH measurements were divided by their protein levels (Bradford protein assay; Bio-Rad Laboratories, CA, USA) to normalize for possible variability in tissue levels between inserts. Data were presented as LDH absorbance divided by microgram protein.
Histologic evaluations
Slice cultures were washed in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde (room temperature, 1 h), then in 30% sucrose (room temperature, 1 h), embedded in O.C.T. embedding medium (Tissue-Tek; Sakura Finetechnical, Tokyo, Japan) and stored at −80 °C overnight. Embedded slices were sectioned to 15 μm thickness and each cryosection was mounted on slides and stored at −80 °C until use. Sections were analyzed for the presence of apoptotic nuclei using the DeadEnd Fluorometric Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) System (Promega, Madison, WI, USA). TUNEL-positive nuclei (green) and PI-stained total nuclei (red) were quantitated by ImagePro Plus v.5.0 imaging software (Media Cybernetics), and the percentage of TUNEL-positive nuclei was presented. Fluorescent immunohistochemical staining combined with TUNEL labeling was also performed. After blocking with 1% bovine serum albumin in PBS for 30 min at room temperature, cryosections were incubated overnight at 4 °C with a mouse anti-neuronal nuclei (NeuN) (to label neurons) (1 : 500; Chemicon, Gibbstown, NJ, USA) or rabbit anti-glial fibrillary acidic protein (GFAP) (to label astrocytes and glia) (1 : 100; Dako, Denmark). After washing in PBS, cryosections were exposed to goat anti-mouse (Thermo Scientific, Rockford, IL, USA) or anti-rabbit (Thermo Scientific) secondary antibodies conjugated with fluorescent dye Cy3 (red). Both were used at 1 : 1000 for 1 h at room temperature. The sections were again washed with PBS and TUNEL staining was performed as described above but without the PI co-staining. In order to ensure the specificity of the signals from the primary antibodies, normal rat lung tissue sections were stained with anti-NeuN or anti-GFAP followed by secondary antibody conjugates as described above. In this case no fluorescence was observed for either NeuN or GFAP. To demonstrate that fluorescent signals were not due to secondary antibody artifacts, sections were also incubated with the secondary antibodies alone. In all cases, fluorescent-stained cells were observed using an inverted fluorescence microscope (Olympus IX51; Japan).
Immunoblot analysis
After experimental treatment, slice cultures were washed with ice-cold PBS, homogenized in lysis buffer containing 1% Triton X-100, 20 mm Tris, pH 7.4, 100 mm NaCl, with 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail (Sigma). Lysates were centrifuged at 13 000 g for 10 min at 4 °C to precipitate the debris, and the protein content in the supernatant was determined by the Bio-Rad protein assay (Bio-Rad Laboratories). Lysate protein (20 μg / lane) was separated using 4–20% gradient gels (Thermo Scientific) and transferred to polyvinylidene fluoride membranes. The blots were then probed with the appropriate antibody overnight at 4 °C. The primary antibodies used were anti-phospho-p38 MAPK and p38 MAPK (Santa Cruz, CA, USA), anti-caspase-3 and anti-cleaved caspase-3 (Cell Signaling, Danvers, MA, USA). Blots were washed in 1× Tris Buffered saline-Tween (3 × 15 min) and the appropriate secondary antibodies conjugated to horse radish peroxidase were then added for 1 h at room temperature (Thermo Scientific). After further washing in Tris Buffered saline-Tween (3 × 15 min), bands were visualized by chemiluminescence (West-Femto; Pierce, Rockford, IL, USA) and quantified using a Molecular Imaging System (Kodak, Rochester, NY, USA).
Measurement of superoxide levels
Superoxide production was measured using electron paramagnetic resonance (EPR) spectroscopy as we have previously described (Shiino et al., 1998b; Wiseman et al., 2007). Briefly, slices were homogenized in EPR buffer (7.5 μm desferrioxamine and 25 μm diethyldithiocarbamate in 1× PBS) on ice. The homogenate protein level was then determined and adjusted to the same concentration using EPR buffer. Each homogenate was then mixed 1 / 5 (v / v) with spin trap stock solution (25 mg / mL 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl) (Alexis Biochemicals, San Diego, CA, USA) and incubated on ice for 1 h. Aliquots (35 μL) of each incubated homogenate were then added to a 50 μL capillary tube and analyzed using a MiniScope MS200 EPR (Magnettech, Berlin, Germany) at a microwave power of 40 mW, modulation amplitude of 3000 mG and modulation frequency of 100 kHz. EPR spectra were analyzed by measurement of amplitude using analysis software (version 2.02). To convert the waveform amplitude into a concentration of superoxide per milligram of protein, we also performed a standard reaction using a known amount of the superoxide-generating enzyme, xanthine oxidase in the presence of xanthine and 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine. HCl, as we have described previously (Oishi et al., 2008).
Generation of an antisense p38 mitogen-activated protein kinase adeno-associated virus
Virus production
Recombinant adeno-associated virus (AAV)1 was packaged in cultures of human embryonic kidney (HEK) 293T cells. Approximately 1.5 × 107 HEK293T cells were seeded into 150 cm dishes in complete Dulbecco's Modified Eagle Medium (Cellgro) supplemented with 10% fetal bovine serum, 1 mm Modified Eagle Medium sodium pyruvate, 0.1 mm Modified Eagle Medium nonessential amino acids solution and 0.05% penicillin–streptomycin (5000 units / mL). At 24 h after seeding, the medium was changed to culture medium containing 5% fetal bovine serum and cells were transfected with three separate plasmids – adeno helper plasmid (pFΔ6), AAV helper encoding serotype 1 (H21) and AAV transgene expression cassette containing a polylinker (null) or the p38 MAPK gene sequence in the antisense orientation all flanked by the AAV2 inverted terminal repeats. Plasmids were transfected into HEK293T cells using polyfect according to the manufacturers' conditions (Qiagen, Valencia, CA, USA). Cultures were incubated at 37 °C in 5% CO2 for 72 h, harvested and pelleted by centrifugation. The pellets were resuspended in 10 mm Tris, pH 8.0, and chilled on ice. Cells were lysed by three repeated freeze–thaw cycles in a dry ice–ethanol bath followed by sonication and treatment with 50 U benzonase (Novagen) and 0.5% sodium deoxycholate for 30 min at 37 °C. Virus was purified by density gradient centrifugation in iodixinol according to the method of Zolotukhin et al. (1999). Purified virus preparations were concentrated and desalted in artificial cerebrospinal fluid by centrifugation in Ultrafree 15 filter devices (Millipore). The titer of each virus (genomic particles / mL) was determined by real-time-polymerase chain reaction using primers and a probe specific for the Woodchuck hepatitis virus posttranscriptional regulatory element sequence.
Virus delivery
Freshly harvested hippocampal slice cultures (9–12 slices in 1 mL of medium) were transduced with either AAV-Synaptin I (SYN)-1-p38 MAPK-anti-sense (AS) or AAV-SYN-1null vector (as a control) using 1 × 1011 genomic particles for 1 h at room temperature. During virus adsorption, a steady stream of sterilized O2 was bubbled into the medium. After 1 h, slices were placed on Millipore membranes and cultured at least for 1 week to allow significant downregulation of p38 MAPK expression prior to exposure to OGD.
Statistical analysis
The variance of many of the measures (superoxide level, PI uptake, and total and cleaved caspase-3) increased as the amount of OGD exposure increased, violating the homogeneity of variance assumption of anova. A rank transformation (Conover, 1981) was used prior to analysis for these measures. The effect of OGD on activated p38 MAPK was tested using a two Treatment (vehicle vs. SB203580) × 3 OGD exposure (0, 2 and 4 h) anova. The effect of OGD on superoxide levels, PI uptake in whole slice or CA1, CA3 or DG sub-regions, LDH release, and total and cleaved caspase-3 was tested using a two Treatment (vehicle vs. SB203580) × 4 OGD exposure (0, 4, 8 and 24 h) anova. The analysis of the effect of OGD on apoptotic nuclei required a two Treatment (vehicle vs. SB203580) × 3 OGD exposure (4, 8 and 24 h) anova as TUNEL values were zero with no OGD exposure. An interaction between treatment and OGD exposure in these analyses would indicate a differential effect of OGD that is dependent on the p38 MAPK inhibitor. The comparisons of interest were the within-treatment comparison of all levels of OGD exposure with no OGD exposure, the within-treatment comparison of each time-point with the previous time-point, and the within-time-point comparison of treatments. The analysis for the effects of targeted decreases in p38 MAPK expression in neuronal cells was performed using a three Virus (none, AAV-SYN-1null and AAV-SYN-1-p38 MAPK-AS) one-way anova. The effect of OGD on targeted decreases in p38 MAPK expression in neuronal cells for superoxide levels, LDH release, PI uptake and cleaved caspase-3 was tested using a three Virus (none, AAV-SYN-1null and AAV-SYN-1-p38 MAPKAS) × 2 OGD (no vs. yes) anova. The analysis of the effect of OGD on targeted decreases in p38 MAPK expression in neuronal cells for apoptotic nuclei required a three Virus (none, AAV-SYN-1null and AAV-SYN-1-p38 MAPK-AS) one-way anova as TUNEL values were zero with no OGD exposure. An interaction between virus transduction and OGD exposure in these analyses would indicate a differential effect of the virus that is dependent on OGD. Comparisons of interest for these analyses are the within-virus comparison of OGD exposure and the comparison of all OGD-exposed virus groups with each other. A Tukey's test was used to adjust for multiple comparisons when determining differences for comparisons of interest for significant anova effects. Descriptive statistics are presented as mean ± SD unless otherwise noted. Effects were considered statistically significant at P < 0.05. SAS® (SAS Institute, Inc., Cary, NC, USA) version 9.2 was used for all analyses.
Results
Oxygen / glucose deprivation increases p38 mitogen-activated protein kinase activation in rat hippocampal slice cultures
Initially, slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm). The effect of OGD on the activation of p38 MAPK was analyzed using western blot analysis to determine the ratio of phosphorylated (active) to total p38 MAPK. Our data indicate that phospho-p38 MAPK levels are increased at 2 h after OGD and the activation significantly declines by 4 h post-OGD (Fig. 1). SB203580 significantly inhibits the activation of p38 MAPK by OGD and has no effect without OGD exposure (F2,18 = 8.41, P = 0.0026; Fig. 1).
Fig. 1.
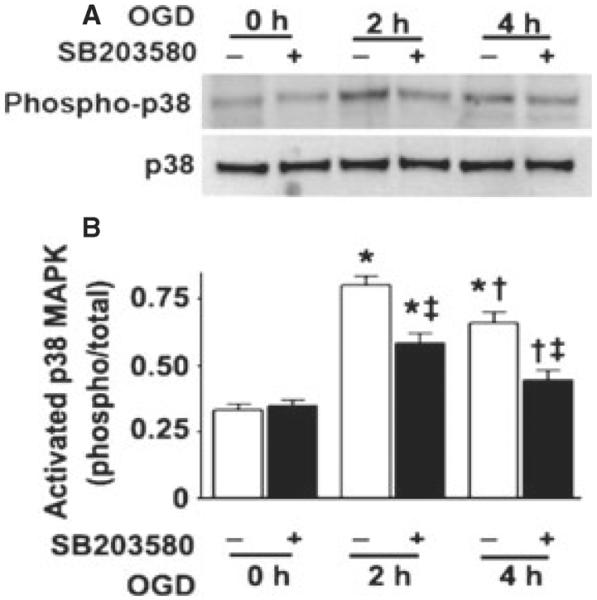
OGD rapidly activates p38 MAPK in rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). Slices were harvested at 0, 2 and 4 h after OGD and subjected to western blot analysis to determine the effects on total MAPK (p38) and phospho-p38 MAPK (phospho-p38). A representative blot is shown (A). Relative phospho-p38 MAPK levels were determined as the ratio of phospho-p38 to total p38 MAPK (B). Data are presented as mean + SE from four independent experiments using 12 pooled slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. no SB203580 at the same time-point.
p38 mitogen-activated protein kinase inhibition attenuates the increase in superoxide generation associated with oxygen / glucose deprivation in rat hippocampal slice cultures
To determine the effect of p38 MAPK inhibition on the oxidative stress associated with OGD, we utilized EPR spectroscopy and spin trapping to detect superoxide generation in hippocampal slices. OGD induced a time-dependent increase in superoxide generation (Fig. 2) and this increase was significantly attenuated by SB203580 at longer exposures (F3,56 = 3.90, P = 0.013; Fig. 2).
Fig. 2.

OGD increases p38 MAPK-dependent increases in superoxide generation in rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). Slices were harvested at 0, 4, 8 and 24 h after OGD and subjected to EPR using the spin-trap compound 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl to determine superoxide levels. Representative EPR waveforms are shown (A). Absolute levels of superoxide generation were then determined as nmol superoxide generated / min per mg protein (B). Values are presented as mean + SE from four independent experiments using 12 pooled slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. without SB20358 at the same time-point.
p38 mitogen-activated protein kinase inhibition attenuates the neuronal cell death associated with oxygen / glucose deprivation in rat hippocampal slice cultures
To examine the effect of p38 MAPK inhibition on the cell death associated with OGD we quantified PI uptake in the whole hippocampal slice and in the hippocampal sub-regions – CA1, CA3 and the DG. Our data indicate that OGD caused a time-dependent increase in PI uptake, indicative of increased cell death (all P < 0.0001; Fig. 3A). The severity of cell death in the CA1 region (Fig. 3B) was higher than in the CA3 and DG regions at 4 h (Fig. 3C and D), indicating that CA1 is more vulnerable to hypoxia than CA3 and the DG at early time-points post-OGD. In addition, we found that p38 MAPK inhibition significantly decreased cell death in the whole slice (F1,88 = 24.50, P < 0.0001; Fig. 3A). Protection was noted in the CA1 (F1,88 = 22.76, P < 0.0001) and DG (F1,88 = 11.30, P = 0.0011) sub-regions (Fig. 3B and D) but not in the CA3 (F1,88 = 0.0, P = 0.98; Fig. 3C). After OGD exposure, we also identified a time-dependent increase in LDH release (Fig. 4) that was significantly attenuated by p38 MAPK inhibition at longer exposure (F3,56 = 6.50, P = 0.0008; Fig. 4).
Fig. 3.

OGD induces regional cell injury in rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). The effect on cell injury was then quantified by measuring changes in PI uptake fluorescence either in the whole slice (A) or the CA1 (B), CA3 (C) or DG (D) sub-regions over a 24 h period after OGD. Representative images are shown for the PI uptake in the entire slice (A). PI uptake in the whole slice exhibited a time-dependent increase and SB203580 pre-treatment significantly decreased PI uptake (A). Similar effects were found in the CA1 (B) and DG sub-region (D). However, in the CA3 area, SB203580 pre-treatment did not show a significant decrease in PI uptake (C). Data are presented as mean + SE from four independent experiments using 12 slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. no SB203580 at the same time-point.
Fig. 4.
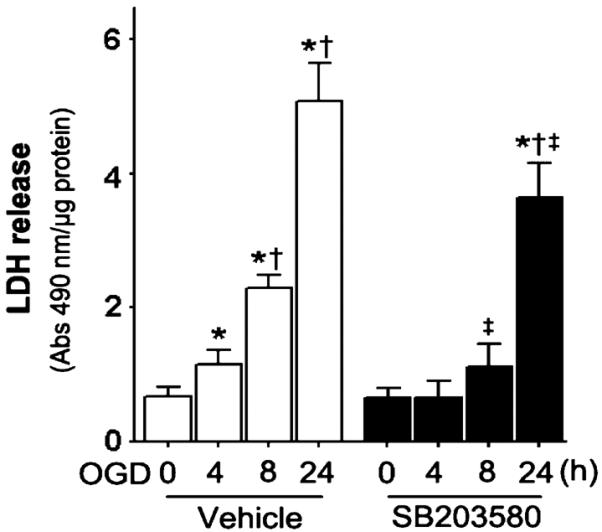
OGD increases LDH release in rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). The time-dependent effect on LDH release was then determined. The LDH absorbance at 490 nm was divided by the protein content to determine relative LDH release. Values are presented as mean + SE from four independent experiments using 12 slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. no SB203580 at the same time-point.
p38 mitogen-activated protein kinase inhibition attenuates neuronal cell apoptosis in rat hippocampal slice cultures exposed to oxygen / glucose deprivation
Utilizing western blot analysis we evaluated two apoptosis signal pathway proteins – caspase-3 and cleaved caspase-3. We found that total caspase-3 levels were decreased after OGD (F3,16 = 24.37, P < 0.0001; Fig. 5A and B) and this corresponded to a significant increase in the levels of cleaved caspase-3 (F3,24 = 24.00, P < 0.0001; Fig. 5C and D). The decrease in caspase-3 (Fig. 5A and B) and the cleavage of caspase-3 were significantly attenuated by p38 MAPK inhibition (F1,16 = 14.32, P = 0.0016 and F1,24 = 9.74, P = 0.0046, respectively) (Fig. 5C and D). The alterations in cleaved caspase also correlated with TUNEL-positive nuclei (Fig. 6A and B). There was a time-dependent increase in TUNEL-positive nuclei (F2,72 = 168.46, P < 0.0001) in the CA1 sub-region (4 h = 14.8 ± 2.1%, 8 h = 31.7 ± 6.4% and 24 h = 43.0 ± 7.0%, respectively). The p38 MAPK inhibitor, SB203580, significantly reduced (F1,72 = 86.547, P < 0.0001) the percentage of apoptotic cells at all time-points examined (4 h = 6.2 ± 0.8%, 8 h = 21.6 ± 3.1% and 24 h = 27.6 ± 5.6%, respectively). By staining slices with both TUNEL and anti-NeuN (to identify neurons) or anti-GFAP (to identify astrocytes and glia) we identified the apoptotic cells as neuronal (Fig. 6C and D).
Fig. 5.

OGD increases caspase activation in rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). Slices were harvested at 0, 4, 8 and 24 h after OGD and subjected to western blot analysis to determine effects on total caspase-3 (A, B) and cleaved caspase-3 (C, D). Representative blots are shown (A, C). Total caspase-3 levels are decreased by OGD, whereas cleaved caspase-3 levels are increased (A–D). SB203580 pre-treatment attenuates the changes in caspase-3 activation. Data are presented as mean + SE from four independent experiments using 12 pooled slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. no SB203580 at the same time-point.
Fig. 6.

OGD increases apoptotic nuclei in the neurons of rat hippocampal slice cultures. Rat hippocampal slice cultures were exposed to OGD in the presence or absence of the p38 MAPK inhibitor, SB203580 (50 μm, 2 h prior to OGD). Slices were harvested at 0, 4, 8 and 24 h after OGD and subjected to TUNEL analysis. Representative images are shown demonstrating that TUNEL staining of apoptotic cells (green) co-localized with PI staining of all of the nuclei (red) resulting in more yellow (merged) nuclei than in SB203580-pre-treated slices (A). The magnifications used were 10 × and 40 ×. Quantification of the percentage of apoptotic nuclei to total nuclei was carried out (B). In addition, at 8 h, co-staining of TUNEL (green) with NeuN (red) shows neuronal cell apoptosis (yellow merge), whereas co-staining with GFAP (red) shows no astrocyte or glial apoptosis (without yellow merge). Controls included sections treated without secondary antibody (Ab) conjugated with fluorescent dye Cy3 alone (control 1) and rat lung sections stained with anti-NeuN or anti-GFAP (control 2). In both cases, no fluorescence was observed (C, D). Data are presented as mean + SE from six independent experiments using eight slices per experiment. *P < 0.05 vs. 0 h, †P < 0.05 vs. previous time-point, ‡P < 0.05 vs. no SB203580 at the same time-point. For interpretation of color references in figure legend, please refer to the Web version of this article.
Neuronal-specific downregulation of p38 mitogen-activated protein kinase signaling
Pharmacologic inhibition always has the potential for unappreciated off-target effects. In addition, our analyses could not determine if the increases in p38 MAPK activation were specific to neurons in the mixed cell population present in our slice cultures. Thus, we followed up our inhibitor studies with a molecular analysis targeted at the p38 MAPK specifically expressed in neurons. To accomplish this we utilized an AAV delivery system using the neuronal-specific promoter, SYN-1, to specifically express an antisense construct of p38 MAPK in neurons. Our initial studies indicated that the AAV-SYN-1-p38 MAPK-AS construct significantly reduced (F2,9 = 103.76, P < 0.0001) the levels of p38 MAPK protein (Fig. 7A and B). This decrease in p38 MAPK also significantly attenuated the increase in superoxide generation induced by OGD (F2,18 = 4.30, P = 0.030; Fig. 7C and D). Further, the expression of the AAV-SYN-1-p38 MAPK-AS construct significantly attenuated LDH release (F2,18 = 15.25, P = 0.0001; Fig. 8A), PI uptake (F2,66 = 6.82, P = 0.0020; Fig. 8B) and the increases in cleaved caspase-3 (F2,24 = 12.87, P = 0.0002; Fig. 8C), and reduced the number of TUNEL-positive nuclei (F2,12 = 13.67, P = 0.0008; Fig. 8D).
Fig. 7.

Targeted decreases in p38 MAPK expression in neuronal cells attenuate superoxide generation in rat hippocampal slice cultures. Rat hippocampal slice cultures were transduced with either the AAV-SYN-1-p38 MAPK-AS or the AAV-SYN-1null constructs or were untransduced. After 7 days, slices were harvested and subjected to western blot analysis to determine the effects on p38 MAPK protein levels. A representative image is shown (A). The AAV-SYN-1-p38 MAPK-AS construct significantly decreases p38 MAPK levels (B). Slices were also exposed to OGD, harvested at 8 h and then subjected to EPR using the spin-trap compound 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl to determine superoxide levels. Representative EPR waveforms are shown (C). Absolute levels of superoxide generation were then determined as nanomols superoxide generated / min per mg protein (D). Values are presented as mean + SE from four independent experiments using 12 pooled slices per experiment. *P < 0.05 vs. uninfected, †P < 0.05 vs. AAV-SYN-1null-exposed slices, ‡P < 0.05 vs. OGD-exposed untransduced slices.
Fig. 8.

Targeted decreases in p38 MAPK expression in neuronal cells attenuate neuronal cell death in rat hippocampal slice cultures. Rat hippocampal slice cultures were transduced with either the AAV-SYN-1-p38 MAPK-AS or the AAV-SYN-1null constructs or were untransduced. After 7 days, slices were exposed to OGD and the effect on LDH release (A), PI uptake (B), cleaved caspase-3 (C) and TUNEL-positive nuclei (D) was determined. Representative images are shown. Values are presented as mean + SE from four independent experiments using 12 pooled slices per experiment. *P < 0.05 vs. uninfected, †P < 0.05 vs. AAVSYN-1null-exposed slices, ‡P < 0.05 vs. OGD-exposed untransduced slices.
Discussion
Our data demonstrate that the activation of p38 MAPK by OGD is an early and transient event. This transient activation has been previously shown in other model systems exposed to ischemia–reperfusion. The exposure of cardiomyocytes to hypoxia has been shown to increase the activity of p38 MAPK within 1 h and this activation persisted for only 3 h (Kulisz et al., 2002). Further phospho-p38 MAPK levels in the hippocampus are increased immediately following HI and return to basal levels after 6 h in a neonatal rat model of HI (Hee Han et al., 2002). Studies have also shown that p38 MAPK can be activated in response to oxidative stress (Clerk et al., 1998; Kulisz et al., 2002). However, the role of p38 MAPK in stimulating oxidative stress is less clear. Using EPR, we have found that p38 MAPK activation preceded the increase in superoxide, whereas the p38 MAPK inhibition led to a significant decrease in OGD-induced superoxide generation. Thus, our data suggest that p38 MAPK signaling lies upstream of superoxide generation. Our data do not identify the downstream target of the p38 MAPK. It is possible that p38 MAPK specifically activates a superoxide generating system such as Nicotinamide adenine dinucleotide phosphate oxidase (Chan et al., 2005; Sakamoto et al., 2006; Wang et al., 2007) or xanthine oxidase (Abdulnour et al., 2006). It is also possible that p38 MAPK signaling is involved in the mitochondrial dysfunction associated with HI (Kang & Lee, 2008). Studies have also identified inflammatory processes as risk factors for ischemic stroke (Bova et al., 1996; Lindsberg & Grau, 2003). Cytokines such as tumor necrosis factor-α, Interleukin 1-1β, Interleukin 1–6 and inducible nitric oxide synthase are all induced in a variety of cell types in the brain including endothelial cells, microglia and neurons (Huang et al., 2006). The activation of p38 MAPK has been linked to inflammatory cytokine production (Kaushal & Schlichter, 2008; Coulthard et al., 2009) and cell death (Thornton & Rincon, 2009). Thus, it is possible that the neuronal protection that we observe by attenuating p38 MAPK signaling may be mediated by a reduction in inflammatory cytokine expression (Barone et al., 2001; Legos et al., 2001; Piao et al., 2003) as well as through reductions in oxidative stress. However, further studies will be required to identify the downstream target(s) of p38 MAPK.
Our data also demonstrate that cell death, as measured by PI uptake, progressively increases after OGD. However, there is variable vulnerability within the hippocampal sub-regions. The CA1 region is more vulnerable to HI than either the CA3 or DG. The hippocampal sub-region analysis also revealed that p38 MAPK inhibition was more effective in protecting neurons in the CA1 and DG than in CA3. These data are in agreement with previous studies that found the CA1 region to be highly vulnerable to damage compared with the CA3 and DG (Schmidt-Kastner & Freund, 1991; Kreisman et al., 2000; Bonde et al., 2005). This regional vulnerability in response to HI may be due to differences in cellular homogeneity (Shiino et al., 1998b; Newrzella et al., 2007) or that, during ischemia, the CA1 experiences more pronounced hypoxia than less vulnerable regions (Shiino et al., 1998a). It has also been reported that the CA1 has a low capillary density compared with the neighboring CA3 and the CA1 vessels exhibit greater blood–brain barrier leakage than CA3 after HI (Cavaglia et al., 2001). These differences, either singly or in tandem, could explain the enhanced vulnerability of CA1 to HI and the region-specific neuroprotective effects of p38 MAPK inhibition. This variable regional protection has not been previously identified and may be important for clinical prognosis and for developing new therapies.
The inhibition of p38 MAPK reduced LDH release in OGD slice cultures, suggesting its involvement in necrotic cell death. This is in agreement with earlier work from Holtzman's group who found that p38 MAPK inhibition reduced necrotic cell death in a neonatal rat model of HI (Hee Han et al., 2002). In addition to necrotic injury, increased apoptosis has been found in vivo in rodent models of HI brain injury. These apoptotic events include activation of caspase-3 and increased TUNEL-positive cells (Pulera et al., 1998; Nakajima et al., 2000; Manabat et al., 2003; de Pina-Benabou et al., 2005). Further, increased caspase-3 activation has been identified in brain sections from children who died after experiencing HI (Rossiter et al., 2002). Apoptosis may account for a significant portion of neuronal cell loss associated with ischemic events (Nakatomi et al., 2002). In agreement with this, the CA1 region TUNEL-positive cells account for 18, 35 and 43% of total cell numbers at 4, 8 and 24 h after OGD, respectively. Further, our data indicate that the apoptotic process involves the activation of caspase-3 and the identified apoptotic cells are limited to neurons. This suggests that the neurons are more vulnerable to HI injury especially at an early stage of the insult. The attenuation of p38 MAPK signaling significantly reduced the number of apoptotic-positive cells after OGD, correlating with a significant decrease in the activation of caspase-3. The role of p38 MAPK in apoptotic signaling during HI is far from clear. The p38 MAPK inhibitor, SB203580, has been shown to reduce the activation of caspase-3 in cultured neonatal rat cardiomyocytes exposed to ischemia (Mackay & Mochly-Rosen, 1999; Wu et al., 2007), whereas in neuronal cell culture p38 MAPK inhibition reduces the cell death associated with N-methyl-d-aspartate exposure (Chen et al., 2005). However, although p38 MAPK inhibition has been shown to significantly reduce the brain injury associated with HI, this did not correlate with a reduction in apoptotic signaling (Hee Han et al., 2002). These differences in response to p38 MAPK inhibition are not easily explained. The differences may be explained by the model studied (in vivo vs. in vitro) or the limited time-point of observation in the in vivo study. Further, in the in vivo studies, p38 MAPK inhibitors have been delivered using different approaches – oral vs. intracerebroventricular. Thus, targeting of the drug to the hippocampal neurons may be sub-optimal. Our data show that specifically reducing the neuronal levels of p38 MAPK decreases neuronal cell death in hippocampal slice cultures exposed to OGD. Our results suggest that targeting p38 MAPK signaling has therapeutic potential. It should be noted that our data do not enable us to determine how the activation of p38 MAPK leads to the activation of caspase-3. However, it may be due to the increased oxidative stress induced by p38 MAPK activation.
In conclusion, we show that p38 MAPK is rapidly and transiently activated in rat hippocampal slice cultures exposed to OGD. Both global and neuronal-specific inhibition of p38 MAPK signaling exert a significant neuroprotective effect. This protective effect was associated with a decrease in superoxide generation, suggesting that the mediators of oxidative stress lie downstream of p38 MAPK activation. Further, we found that p38 MAPK signaling is involved in the signaling pathways that lead to both necrotic and apoptotic cell death. We speculate that targeting p38 MAPK signaling in neurons could have a therapeutic benefit in infants and children exposed to HI.
Abbreviations
- AAV
adeno-associated virus
- DG
dentate gyrus
- EPR
electron paramagnetic resonance
- GFAP
glial fibrillary acidic protein
- HI
hypoxia–ischemia
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- NeuN
neuronal nuclei
- OGD
oxygen / glucose deprivation
- PBS
phosphate-buffered saline
- PI
propidium iodide
- SYN
Synapsin I
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
References
- Abdulnour RE, Peng X, Finigan JH, Han EJ, Hasan EJ, Birukov KG, Reddy SP, Watkins JE, 3rd, Kayyali US, Garcia JG, Tuder RM, Hassoun PM. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L345–L353. doi: 10.1152/ajplung.00453.2005. [DOI] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001;21:129–145. doi: 10.1002/1098-1128(200103)21:2<129::aid-med1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Bonde C, Noraberg J, Noer H, Zimmer J. Lonotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen-glucose deprivation of hippocampal slice cultures. Neuroscience. 2005;136:779–794. doi: 10.1016/j.neuroscience.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Bova IY, Bornstein NM, Korczyn AD. Acute infection as a risk factor for ischemic stroke. Stroke. 1996;27:2204–2206. doi: 10.1161/01.str.27.12.2204. [DOI] [PubMed] [Google Scholar]
- Bu X, Huang P, Qi Z, Zhang N, Han S, Fang L, Li J. Cell type-specific activation of p38 MAPK in the brain regions of hypoxic preconditioned mice. Neurochem. Int. 2007;51:459–466. doi: 10.1016/j.neuint.2007.04.028. [DOI] [PubMed] [Google Scholar]
- Cavaglia M, Dombrowski SM, Drazba J, Vasanji A, Bokesch PM, Janigro D. Regional variation in brain capillary density and vascular response to ischemia. Brain Res. 2001;910:81–93. doi: 10.1016/s0006-8993(01)02637-3. [DOI] [PubMed] [Google Scholar]
- Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ. Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem. Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Chen J, Errico SL, Freed WJ. Reactive oxygen species and p38 phosphorylation regulate the protective effect of Delta9-tetrahydrocannabinol in the apoptotic response to NMDA. Neurosci. Lett. 2005;389:99–103. doi: 10.1016/j.neulet.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases / c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J. Biol. Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- Conover WJ, Imran RL. Rank transformations as a bridge between parametric and nonparametric statistics. Am. Stat. 1981;35:124–129. [Google Scholar]
- Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009;15:369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronberg T, Rytter A, Asztely F, Soder A, Wieloch T. Glucose but not lactate in combination with acidosis aggravates ischemic neuronal death in vitro. Stroke. 2004;35:753–757. doi: 10.1161/01.STR.0000117576.09512.32. [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Tobias PS, Ulevitch RJ. Endotoxin induces rapid protein tyrosine phosphorylation in 70Z / 3 cells expressing CD14. J. Biol. Chem. 1993;268:25009–25014. [PubMed] [Google Scholar]
- Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- Harper SJ, LoGrasso P. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell. Signal. 2001;13:299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- Hee Han B, Choi J, Holtzman DM. Evidence that p38 mitogen-activated protein kinase contributes to neonatal hypoxic-ischemic brain injury. Dev. Neurosci. 2002;24:405–410. doi: 10.1159/000069046. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Kummer JL. Inhibition of p38 mitogen-activated protein kinase by insulin in cultured fetal neurons. J. Biol. Chem. 1996;271:9891–9894. doi: 10.1074/jbc.271.17.9891. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kahle PJ, Borasio GD. Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J. Neurosci. Res. 1998;52:483–490. doi: 10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg. Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Kang YH, Lee SJ. The role of p38 MAPK and JNK in Arsenic trioxide-induced mitochondrial cell death in human cervical cancer cells. J. Cell. Physiol. 2008;217:23–33. doi: 10.1002/jcp.21470. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisman NR, Soliman S, Gozal D. Regional differences in hypoxic depolarization and swelling in hippocampal slices. J. Neurophysiol. 2000;83:1031–1038. doi: 10.1152/jn.2000.83.2.1031. [DOI] [PubMed] [Google Scholar]
- Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L1324–L1329. doi: 10.1152/ajplung.00326.2001. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 2001;892:70–77. doi: 10.1016/s0006-8993(00)03228-5. [DOI] [PubMed] [Google Scholar]
- Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke. 2003;34:2518–2532. doi: 10.1161/01.STR.0000089015.51603.CC. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Mackay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J. Biol. Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J. Neurosci. 2000;20:7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Newrzella D, Pahlavan PS, Kruger C, Boehm C, Sorgenfrei O, Schrock H, Eisenhardt G, Bischoff N, Vogt G, Wafzig O, Rossner M, Maurer MH, Hiemisch H, Bach A, Kuschinsky W, Schneider A. The functional genome of CA1 and CA3 neurons under native conditions and in response to ischemia. BMC Genomics. 2007;8:370. doi: 10.1186/1471-2164-8-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi PE, Wiseman DA, Sharma S, Kumar S, Hou Y, Datar SA, Azakie A, Johengen MJ, Harmon C, Fratz S, Fineman JR, Black SM. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: a role for oxidative stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;295:L756–L766. doi: 10.1152/ajplung.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Lee H, Hur J, Kim SY, Kim H, Park JH, Cha S, Kang SS, Cho GJ, Choi WS, Suk K. Hypoxia induces nitric oxide production in mouse microglia via p38 mitogen-activated protein kinase pathway. Brain Res. Mol. Brain Res. 2002;107:9–16. doi: 10.1016/s0169-328x(02)00421-7. [DOI] [PubMed] [Google Scholar]
- Piao CS, Kim JB, Han PL, Lee JK. Administration of the p38 MAPK inhibitor SB203580 affords brain protection with a wide therapeutic window against focal ischemic insult. J. Neurosci. Res. 2003;73:537–544. doi: 10.1002/jnr.10671. [DOI] [PubMed] [Google Scholar]
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke. 2005;36:2232–2237. doi: 10.1161/01.STR.0000182239.75969.d8. [DOI] [PubMed] [Google Scholar]
- Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG, del Zoppo GJ. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia? Editorial comment. Stroke. 1998;29:2622–2630. doi: 10.1161/01.str.29.12.2622. [DOI] [PubMed] [Google Scholar]
- Rossiter JP, Anderson LL, Yang F, Cole GM. Caspase-3 activation and caspase-like proteolytic activity in human perinatal hypoxic-ischemic brain injury. Acta Neuropathol. 2002;103:66–73. doi: 10.1007/s004010100432. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Kuribayashi F, Nakamura M, Takeshige K. Involvement of p38 MAP kinase in not only activation of the phagocyte NADPH oxidase induced by formyl-methionyl-leucyl-phenylalanine but also determination of the extent of the activity. J. Biochem. 2006;140:739–745. doi: 10.1093/jb/mvj204. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- Shiino A, Matsuda M, Handa J, Chance B. Poor recovery of mitochondrial redox state in CA1 after transient forebrain ischemia in gerbils. Stroke. 1998a;29:2421–2424. doi: 10.1161/01.str.29.11.2421. discussion 2425. [DOI] [PubMed] [Google Scholar]
- Shiino A, Matsuda M, Handa J, Chance B, Kontos HA. Poor recovery of mitochondrial redox state in CA1 after transient forebrain ischemia in gerbils? Editorial comment. Stroke. 1998b;29:2421–2425. doi: 10.1161/01.str.29.11.2421. [DOI] [PubMed] [Google Scholar]
- Smith KE, Keeney S, Zhang L, Perez-Polo JR, Rassin DK. The association of early blood oxygenation with child development in preterm infants with acute respiratory disorders. Int. J. Dev. Neurosci. 2008;26:125–131. doi: 10.1016/j.ijdevneu.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, Tridandapani S, Sen CK, Marsh CB. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte / macrophage survival. Am. J. Respir. Cell Mol. Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman DA, Wells SM, Hubbard M, Welker JE, Black SM. Alterations in zinc homeostasis underlie endothelial cell death induced by oxidative stress from acute exposure to hydrogen peroxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L165–L177. doi: 10.1152/ajplung.00459.2005. [DOI] [PubMed] [Google Scholar]
- Wu X, Liu X, Zhu X, Tang C. Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation. Shock. 2007;27:572–577. doi: 10.1097/01.shk.0000246901.58068.a8. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]