

Abstract
Sparse energy functions that ignore long range interactions between residue pairs are frequently used by protein design algorithms to reduce computational cost. Current dynamic programming algorithms that fully exploit the optimal substructure produced by these energy functions only compute the GMEC. This disproportionately favors the sequence of a single, static conformation and overlooks better binding sequences with multiple low-energy conformations. Provable, ensemble-based algorithms such as A* avoid this problem, but A* cannot guarantee better performance than exhaustive enumeration. We propose a novel, provable, dynamic programming algorithm called Branch-Width Minimization* (BWM*) to enumerate a gap-free ensemble of conformations in order of increasing energy. Given a branch-decomposition of branch-width w for an n-residue protein design with at most q discrete side-chain conformations per residue, BWM* returns the sparse GMEC in O(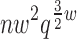 ) time and enumerates each additional conformation in merely O(
) time and enumerates each additional conformation in merely O(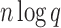 ) time. We define a new measure, Total Effective Search Space (TESS), which can be computed efficiently a priori before BWM* or A* is run. We ran BWM* on 67 protein design problems and found that TESS discriminated between BWM*-efficient and A*-efficient cases with 100% accuracy. As predicted by TESS and validated experimentally, BWM* outperforms A* in 73% of the cases and computes the full ensemble or a close approximation faster than A*, enumerating each additional conformation in milliseconds. Unlike A*, the performance of BWM* can be predicted in polynomial time before running the algorithm, which gives protein designers the power to choose the most efficient algorithm for their particular design problem.
) time. We define a new measure, Total Effective Search Space (TESS), which can be computed efficiently a priori before BWM* or A* is run. We ran BWM* on 67 protein design problems and found that TESS discriminated between BWM*-efficient and A*-efficient cases with 100% accuracy. As predicted by TESS and validated experimentally, BWM* outperforms A* in 73% of the cases and computes the full ensemble or a close approximation faster than A*, enumerating each additional conformation in milliseconds. Unlike A*, the performance of BWM* can be predicted in polynomial time before running the algorithm, which gives protein designers the power to choose the most efficient algorithm for their particular design problem.
Key words: : protein design, sparse residue interaction graphs, provable algorithms, branch-decomposition, dynamic programming, ensemble-based algorithms, OSPREY
1. Introduction
Computational structure-based protein design is a transformative field that can advance both basic science and translational medical research. Several protein design algorithms have successfully predicted protein sequences that fold and bind the desired target in vitro (Frey et al., 2010; Roberts et al., 2012; Rudicell et al., 2014; Stevens et al., 2006; Georgiev et al., 2012; Georgiev and Donald, 2007; Georgiev et al., 2014; Donald, 2011), and even in vivo (Reeve et al., 2015; Roberts et al., 2012; Rudicell et al., 2014; Georgiev et al., 2012; Georgiev et al., 2014; Donald, 2011). However, protein design is NP-hard (Kingsford et al., 2005), making algorithms that guarantee optimality expensive for larger designs where many residues are allowed to mutate simultaneously. Therefore, researchers have developed tractable approximations of the protein design problem to obtain provably good approximate solutions (Leach and Lemon, 1998; Roberts et al., 2012; Chen et al., 2009; Lilien et al., 2005; Georgiev and Donald, 2007; Donald, 2011, Smadbeck et al., 2014), or employed heuristic approaches to rapidly generate candidate solutions (Lee and Subbiah, 1991; Kuhlman and Baker, 2000; Jones, 1994; Desjarlais and Handel, 1995; Koehl and Delarue, 1994; Jiang et al., 2000; Donald, 2011). Heuristic sampling of sequences quickly generates locally optimal candidate sequences, whereas provable algorithms are guaranteed to return the global minimum energy conformation (GMEC). However, algorithms that compute only the GMEC have been shown to overlook sequences with better binding affinity, because proteins exist as a thermodynamic ensemble and not just as a single low-energy conformation (Roberts et al., 2012; Lilien et al., 2005; Chen et al., 2009). Provable, ensemble-based algorithms ameliorate this issue (Roberts et al., 2012; Lilien et al., 2005; Silver et al., 2013).
One provable, ensemble-based algorithm is osprey's K* (Roberts et al., 2012; Lilien et al., 2005), which has been used to provably approximate the binding constant Ka. By explicitly modeling proteins as a thermodynamic ensemble of molecular conformations, osprey/K* has successfully designed sequences that have performed well both in vitro (Roberts et al., 2012; Chen et al., 2009; Frey et al., 2010; Rudicell et al., 2014; Georgiev et al., 2012; Gorczynski et al., 2007; Stevens et al., 2006) and in vivo (Roberts et al., 2012; Gorczynski et al., 2007; Rudicell et al., 2014; Frey et al., 2010), as well as in non-human primates (Rudicell et al., 2014). K* accomplishes this by using dead end elimination followed by A* (DEE/A*) (Leach and Lemon, 1998; Goldstein, 1994) to provably compute a gap-free list of conformations within an energy window Ew of the GMEC, and provably approximate partition functions over molecular ensembles. A* search uses a lower-bound scoring function, which allows it to outperform exhaustive search in practice, but cannot guarantee any improvement over exhaustive search. In the worst case, A* must explore a significant part of the exponentially large space of possible sequences and conformations to guarantee that the first conformation returned is the GMEC. In addition, enumeration of each successive conformation is also worst-case exponential time. Hence, enumerating a gap-free list with A* can be prohibitively expensive.
1.1. Design with sparse energy functions
Because protein design is computationally expensive, many protein design algorithms use sparse energy functions that omit interaction energy between sufficiently distant atoms (Desmet et al., 2002; Fleishman et al., 2011; Zhang and Lange, 2013; Robertson and Varani, 2007; Privett et al., 2012; Leaver-Fay et al., 2011; Lazaridis and Karplus, 1999; Jiang et al., 2000; Jones, 1994; Kaufmann et al., 2010; Kilambi and Gray, 2012; King et al., 2014; Kingsford et al., 2005; Koehl and Delarue, 1994; Kortemme et al., 2003; Krivov et al.; 2009). These sparse energy functions not only reduce the time to compute conformational energy, but also define a different energy landscape: the omitted terms eliminate energy differences between conformations, introducing optimal substructure to the energy landscape. Dynamic programming algorithms use concepts such as tree decomposition and tree width to exploit this optimal substructure to compute the corresponding GMEC more efficiently (Leaver-Fay et al., 2005; Xu and Berger, 2006; Krivov et al., 2009). However, these algorithms compute only the GMEC and do not enumerate a gap-free list of conformations. Naïve extensions to do this are worst-case exponential time in enumerating additional conformations.
We propose a novel, dynamic programming algorithm called Branch-Width Minimization* (BWM*) to efficiently and provably enumerate a gap-free ensemble of conformations, in order of increasing sparse energy. To exploit the optimal substructure, our algorithm uses the concepts of branch-decomposition with recursive heaps. Like tree decompositions, branch-decompositions have also been used in dynamic programming approaches for discrete optimization problems (Fomin, 2003; Hicks et al., 2005; Hlinẽný et al., 2008). Our algorithm treats the protein backbone as rigid and models side-chain flexibility using frequently observed, low-energy discrete conformations called rotamers (Lovell et al., 2000; Donald, 2011). Given a branch-decomposition of branch-width w for an n-residue design with at most q rotamers per residue, our algorithm computes the corresponding GMEC, called the sparse GMEC, in O(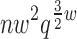 ) time and O(
) time and O(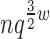 ) space, and enumerates each additional conformation in merely O(n log q) time and O(n) space. Because BWM* enumerates conformations in order of increasing sparse energy, the sparse ensemble of all conformations within an energy window Ew of the sparse GMEC may contain different conformations from those in the full ensemble of all conformations within Ew of the GMEC. For a given sparse energy function, we can compute a bound ɛB on the difference in energy between the sparse GMEC and GMEC. Using this bound, we prove that a sparse ensemble of all conformations within an expanded energy window Ew + ɛB of the sparse GMEC contains all conformations within Ew of the GMEC. Thus, BWM* is guaranteed to compute a sparse ensemble which contains the full ensemble.
) space, and enumerates each additional conformation in merely O(n log q) time and O(n) space. Because BWM* enumerates conformations in order of increasing sparse energy, the sparse ensemble of all conformations within an energy window Ew of the sparse GMEC may contain different conformations from those in the full ensemble of all conformations within Ew of the GMEC. For a given sparse energy function, we can compute a bound ɛB on the difference in energy between the sparse GMEC and GMEC. Using this bound, we prove that a sparse ensemble of all conformations within an expanded energy window Ew + ɛB of the sparse GMEC contains all conformations within Ew of the GMEC. Thus, BWM* is guaranteed to compute a sparse ensemble which contains the full ensemble.
Since BWM* is a dynamic programming algorithm, we should be able to distinguish between problems for which BWM* is more efficient, and those for which A* is more efficient. To test this hypothesis, we defined a new measure, Total Effective Search Space (TESS), which can be computed in polynomial time a priori before BWM* is run. Next, we ran BWM* and A* on 67 different protein design problems and found that TESS perfectly discriminates between BWM*-efficient cases and A*-efficient cases. For the 73% of cases in which BWM* is predicted by TESS to outperform A*, it computes the full ensemble or a close approximation faster than A*, enumerating each additional conformation in milliseconds, and computes the full ensemble in seconds, up to 315 times faster than A*. Of the other 27%, BWM* could enumerate the sparse ensemble for some problems by using a smaller energy window and sparser energy functions, while other problems were, as predicted, more suitable for a GMEC-based method or A*. In practice, we found that branch-width w (used to calculate TESS) can be small irrespective of n, making TESS much smaller than the worst-case bounds of O(qn) for A*.
This article makes the following contributions:
1. A new dynamic programming algorithm called BWM*, which exploits branch-decompositions for protein designs with sparse energy functions, and proof of its asymptotic time and space complexity bounds.
2. Proof that BWM* is guaranteed to enumerate a gap-free list containing all conformations within a user-specified energy window Ew of the GMEC.
3. Definition of a new measure, Total Effective Search Space (TESS), which can be computed a priori in polynomial time before running BWM*, and reliably predicts BWM* performance, allowing selection of the most efficient algorithm for a particular design problem.
4. Comparison between A* and BWM* on 67 protein design problems showing that in 73% of the cases, BWM* (as predicted) is superior to A* in both worst-case bounds and empirical enumeration time.
2. Background
2.1. Sparse residue interaction graphs
Let G = (V, 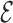 ) be a residue interaction graph (Fig. 1B) corresponding to a protein design problem (Fig. 1A), with a vertex for every mutable residue, and an edge e for every pairwise residue interaction. The energy of a conformation c can be computed as
) be a residue interaction graph (Fig. 1B) corresponding to a protein design problem (Fig. 1A), with a vertex for every mutable residue, and an edge e for every pairwise residue interaction. The energy of a conformation c can be computed as 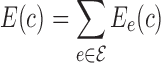 where Ee(c) denotes the interaction energy of the residue pair in c represented by e. By omitting interaction energies between certain residue pairs, the edge set
where Ee(c) denotes the interaction energy of the residue pair in c represented by e. By omitting interaction energies between certain residue pairs, the edge set 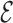 ′ can be deleted from G, producing a sparse graph G′ = (V,
′ can be deleted from G, producing a sparse graph G′ = (V, 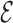 −
− 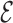 ′), shown in Fig. 1C. The energy function E′(c) corresponding to G′ can then be defined to be
′), shown in Fig. 1C. The energy function E′(c) corresponding to G′ can then be defined to be 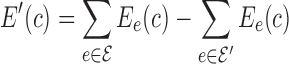 . In this way, the sparse energy of c is represented as the difference between the full energy E(c) and the energy terms missing from E′(c).
. In this way, the sparse energy of c is represented as the difference between the full energy E(c) and the energy terms missing from E′(c).
FIG. 1.

(A) A sample protein design problem represented as a residue interaction graph (B), with residues as vertices and pairwise interactions as edges. (C) The sparse residue interaction graph generated by deleting (a, d), (a, e), (b, d), and (b, e), shown as red crosses in (A) and (B).
2.2. Branch-decomposition in protein design
Let the branch-decomposition of G′ be an unrooted binary tree T = (D, P) with tree nodes D and tree edges P, such as the one in Fig. 2A. T satisfies the following property: For every edge 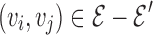 , there exists a leaf node in T that corresponds to (vi, vj). This can be seen explicitly in the rooted branch-decomposition, shown in Fig. 2B. For every edge p ∈P, we arbitrarily define the two subtrees separated by removing p as SL and SR, and the respective sparse graph vertices contained in each subtree as SL(p) and SR(p). This separation divides the vertex set V into three sets L(p), M(p), and R(p): M(p) = SR(p) ∩ SL(p), L(p) = SL(p) − M(p), and R(p) = SR(p) − M(p). By definition, this means that there are no edges between L(p) and R(p) in G′. Finally, the branch-width w of a branch-decomposition is defined to be
, there exists a leaf node in T that corresponds to (vi, vj). This can be seen explicitly in the rooted branch-decomposition, shown in Fig. 2B. For every edge p ∈P, we arbitrarily define the two subtrees separated by removing p as SL and SR, and the respective sparse graph vertices contained in each subtree as SL(p) and SR(p). This separation divides the vertex set V into three sets L(p), M(p), and R(p): M(p) = SR(p) ∩ SL(p), L(p) = SL(p) − M(p), and R(p) = SR(p) − M(p). By definition, this means that there are no edges between L(p) and R(p) in G′. Finally, the branch-width w of a branch-decomposition is defined to be 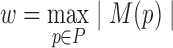 . Branch-decompositions and similar concepts have previously been used in dynamic programming algorithms for other discrete optimization problems (Fomin, 2003; Hicks et al., 2005; Hlinẽný et al., 2008; Nilsson, 1998; Dechter and Mateescu, 2007). The following section describes how BWM* uses the branch-decomposition T for protein design with sparse energy functions.
. Branch-decompositions and similar concepts have previously been used in dynamic programming algorithms for other discrete optimization problems (Fomin, 2003; Hicks et al., 2005; Hlinẽný et al., 2008; Nilsson, 1998; Dechter and Mateescu, 2007). The following section describes how BWM* uses the branch-decomposition T for protein design with sparse energy functions.
FIG. 2.

An example branch-decomposition. (A) The edges of the sparse graph correspond to a node in the branch-decomposition tree. Along the highlighted edge of the branch-decomposition, the mutable residues are separated into three sets: the L-set, which exists only in leaves to the left of the edge; the R-set, which exists only in leaves to the right of that edge; and the M-set, which can be found on both sides of the edge. (B) This tree is arbitrarily rooted for use by BWM*.
3. Methods
We now arbitrarily root T, producing a tree such as the one shown in Fig. 2B. For every internal edge p ∈P, let its two child edges be c1 and c2. We define the λ-set of p λ(p) = L(p) ∩ M(c1) ∩ M(c2). Note that since w is the size of the largest M(p) for any p ∈P, |λ(p)| ≤ w.
3.1. Total Effective Search Space
We can now define a new measure Total Effective Search Space (TESS), which predicts BWM* performance. Let the set of mutable residues be R, and the number of unpruned rotamers for a residue r ∈R be qr. For each nonempty λ-set λ and its corresponding M-set Mλ, Mλ∪λ ⊆ R and the number of all unpruned conformations for a particular subproblem can be computed as 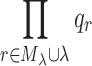 . This value corresponds to the total number of conformations enumerated by BWM* for each Mλ ∪λ, and summing over all nonempty λ-sets is a deterministic measure of the time and space complexity of BWM*. We define the sum of products
. This value corresponds to the total number of conformations enumerated by BWM* for each Mλ ∪λ, and summing over all nonempty λ-sets is a deterministic measure of the time and space complexity of BWM*. We define the sum of products 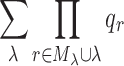 as the Total Effective Search Space. Because computing the smallest-width branch-decomposition is NP-complete, polynomial-time approximation algorithms that typically do not return the optimal branch-decomposition are used in practice. We now show that when using an algorithm that provably computes a branch-decomposition in polynomial time βt = nO(1), TESS can be computed in polynomial time. The proof of Theorem 1 is provided in section A.1 of the supplementary information (SI) in Jou et al. (2015).
as the Total Effective Search Space. Because computing the smallest-width branch-decomposition is NP-complete, polynomial-time approximation algorithms that typically do not return the optimal branch-decomposition are used in practice. We now show that when using an algorithm that provably computes a branch-decomposition in polynomial time βt = nO(1), TESS can be computed in polynomial time. The proof of Theorem 1 is provided in section A.1 of the supplementary information (SI) in Jou et al. (2015).
Theorem 1. TESS can be computed in O(n2 + βt) time, where βt is the time taken to compute the branch-decomposition of a sparse graph.
3.2. Algorithm: preprocessing and enumeration
The preprocessing phase of BWM* computes the energy of the sparse GMEC and constructs a data structure for efficient enumeration of conformations in order of increasing sparse energy. This data structure is called a recursive heap. Let a recursive heap H be a canonical min heap satisfying the heap property, with the following two additional properties:
1. For every heap node in H, there are zero, one, or two child heaps. These child heaps are also recurisve heaps, and their heap nodes have the same properties.
2. The sort key of nodes in H are the sum of the their own self key and the smallest keys of their two child heaps, that is, the root keys of its two child heaps.
During preprocessing, BWM* performs a post-order traversal of T. For each edge with a nonempty λ-set, the following operations are performed:
1. Enumerate all possible rotamer assignments to the residues in M ∪ λ, and for each assignment look up optimal assignments for the residues in L − λ, which were previously computed in its child edges.
2. For each assignment to the residues in M, store all assignments to the residues in λ in a canonical min heap, called a λ-heap. The key of each node is its energy, and the data is the assignment to the residues of the λ-set.
3. Construct a recursive heap for each rotamer assignment to the M-set using the λ-heaps from step 2 and the previously constructed child heaps from its two child edges. These were constructed earlier as a consequence of post order traversal.
For every assignment in the M-set, we enumerate all assignments of the λ-set and look up all remaining assignments to the (L − λ)-set from the child edges. Therefore, at the end of this procedure we have the energy of the optimal assignment to the L-set for each assignment to the M-set in each recursive heap. Since the residues of the L-set interact only with each other and the residues of the M-set, the optimal assignment to the L-set is determined at the end of this procedure. As the L-set of the root edge contains all n mutable residues, once the traversal has returned to the root edge the optimal solution for every residue has been calculated, and the energy of the sparse GMEC is contained in the root node of the recursive heap constructed at the root edge. The sparse GMEC can then be calculated by finding the lowest-energy partial conformation in the λ-set at the root, and recursively looking up the optimal assignments in its children to reassemble the corresponding full conformation. After the sparse GMEC has been returned, the heap must be updated to return the next best conformation. This procedure has two steps:
1. Call this procedure recursively on its two child heaps, and update the energy of the root node with the new energies at the roots of its two child heaps.
2. After the energy of the root node is updated, it may no longer contain the minimum energy of the heap and must be bubbled down to restore the heap property.
After these steps are completed, the resulting root of the heap now contains the energy of the next best conformation. This procedure is called repeatedly to enumerate additional conformations.
We now give an upper bound on the size of |M ∪λ|, and analysis of the time and space complexity of the preprocessing and enumeration steps of BWM*. The proofs of the theorems, along with details of the construction of recursive heaps and enumeration phase are provided in section A.2 and section A.3 of the SI in Jou et al. (2015).
Theorem 2. The maximum subproblem M ∪ λ is bounded by the relation
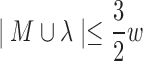 .
.
Theorem 3. BWM* takes O(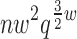 ) time and O(
) time and O(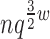 ) space to preprocess and return the sparse GMEC.
) space to preprocess and return the sparse GMEC.
Theorem 4. Enumerating the next best conformation takes O(n log q) time and O(n) space to remove the minimum-energy conformation from H.
3.3. Sparse error bounds
To show the bounds on error introduced by sparse graphs, we first define two terms to bound the largest possible difference between sparse and full energy for any conformation c from the set of all allowed conformations C. Let 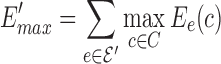 and
and 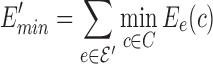 bound the positive and negative energy change for c, respectively. While these bounds are loose, they have the benefit of being computable in polynomial time. With these two terms, we can bound the total energy difference between the GMEC c* and the sparse GMEC c′, and show the relationship between the sparse ensemble and full ensemble. The following lemma bounds the sparse energy difference between the sparse GMEC and GMEC. The proof for lemma 1 is provided in section A.4 of the SI in Jou et al. (2015).
bound the positive and negative energy change for c, respectively. While these bounds are loose, they have the benefit of being computable in polynomial time. With these two terms, we can bound the total energy difference between the GMEC c* and the sparse GMEC c′, and show the relationship between the sparse ensemble and full ensemble. The following lemma bounds the sparse energy difference between the sparse GMEC and GMEC. The proof for lemma 1 is provided in section A.4 of the SI in Jou et al. (2015).
Lemma 1. The difference in sparse energy between the sparse GMEC c′ and the GMEC c* is bounded by the relationship |E′(c*) − E′(c′)| < E′max − E′min.
With these results we can now show that a sparse ensemble S′ of conformations within Ew + E′max − E′min of the sparse GMEC is guaranteed to contain the full ensemble S* of all conformations within Ew of the GMEC. The proof for Theorem 5 is provided in section A.4 of the SI in Jou et al. (2015).
Theorem 5. For any conformation c, if E(c) ≤ E(c*) + Ew then E′(c) ≤ E′(c′) + Ew + E′max − E′min
We can now show the total complexity to enumerate a gap free list of k conformations, starting from the sparse GMEC. The proof for Theorem 6 is provided in section A of the SI in Jou et al. (2015).
Theorem 6. BWM* provably computes the sparse GMEC and an ensemble of the top k conformations in O(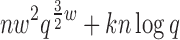 ) time and O(
) time and O(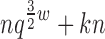 ) space, which is guaranteed to contain all conformations within Ew of the GMEC when all conformations within Ew + E′max − E′min
have been enumerated.
) space, which is guaranteed to contain all conformations within Ew of the GMEC when all conformations within Ew + E′max − E′min
have been enumerated.
4. Computational Experiments
4.1. Experimental methods
We implemented BWM* in our laboratory's open source osprey (Gainza et al., 2013) protein design package. To test the hypothesis that TESS could predict whether BWM* or A* would be faster, and also determine empirical enumeration times, we ran BWM* and A* on 67 different design problems taken from Gainza et al., (2012). Each protein design problem consists of a rigid backbone design with 4–16 mutable residues and 5–10 allowed amino acid mutations per residue. Side-chain flexibility was modeled using a rigid rotamer library with 153 modal rotamers (Lovell et al., 2000). For each protein design problem, DEE was run with energy windows Ew of both 0.5 kcal/mol and 1 kcal/mol, followed by either A* or BWM* with different distance and energy cutoffs: distance cutoffs (deleting edges whose minimum distance between all unpruned rotamer pairs exceeds the cutoff distance) of 7 Å, 8 Å, and energy cutoffs (deleting edges whose maximum absolute interaction energy between all unpruned rotamer pairs is less than cutoff energy) of 0.1 kcal/mol and 0.2 kcal/mol. Then the performance was compared with the TESS predictions. Information on the 67 protein design problems, along with details of the computational experiments performed, are provided in section C of the SI in Jou et al. (2015).
4.2. Total Effective Search Space predictions
To determine if TESS could be used to predict BWM* performance, we plotted TESS for all 67 protein design problems against the number of unpruned conformations after DEE, as shown in Fig. 3. Shown in green dots is the TESS for 39 out of 67 design problems for which BWM* enumerated the sparse ensemble for an energy cutoff of 0.1 kcal/mol and an energy window of 1 kcal/mol. Ten more design problems (shown as light green dots) were solved efficiently by increasing the energy cutoff to 0.2 kcal/mol to make the residue interaction graph sparser, and decreasing the energy window to 0.5 kcal/mol to prune additional conformations from the design search space. The TESS for these 49 problems in which BWM* outperforms A* is less than 8.97 × 108 conformations (shown with a line in Fig. 3). TESS remains larger for the remaining 18 problems for which BWM* exceeded memory limits during preprocessing. Using a larger energy cutoff (0.3–0.5 kcal/mol) reduced the TESS for 6 of these 18 problems (yellow triangles) below 8.97 × 108 (blue triangles), and BWM* was able to compute the sparse ensemble, as seen in Fig. 3B. For 11 additional problems (orange diamonds), reducing the energy window to 0 kcal/mol reduced their TESS below 1.57 × 1011 conformations (blue diamonds), which was sufficient to compute the sparse GMEC but not the sparse ensemble. In one case neither the sparse ensemble nor the sparse GMEC could be computed, which is shown as a red dot and red cross, respectively. In this case the periplasmic copper/silver-binding protein (PDB id: 2qcp) contains a set of residues with large interaction energy greater than 0.5 kcal/mol and many unpruned rotamers, producing a dense subgraph with a large TESS.
FIG. 3.

Total Effective Search Space predicts BWM* performance. x-axis shows TESS for each design problem using their respective cutoff and energy window. y-axis shows unpruned conformations for each design problem after DEE pruning with their respective energy window. (A) Dark green points: full ensemble with energy cutoff 0.1 kcal/mol, energy window 1 kcal/mol. Light green points: full ensemble with energy cutoff 0.2 kcal/mol, energy window 0.5 kcal/mol. The yellow triangles, orange diamonds, and red dot have x values of TESS using unpruned conformations with energy cutoff 0.1 kcal/mol, energy window 1 kcal/mol for cases where larger energy cutoffs or smaller energy windows were necessary to finish preprocessing. (B) Blue triangles: x values are TESS for calculating only the sparse ensemble (energy cutoff 0.3 kcal/mol or higher, energy window 0.5 kcal/mol). (C) Blue diamonds: x values are TESS for calculating only the sparse GMEC (energy cutoff between 0.1 and 0.5 kcal/mol, energy window 0 kcal/mol). Red cross: x value is TESS for computing the sparse GMEC of 2qcp (see text).
Based on our analysis, the plot contains three distinct regions: all protein design problems with TESS below 8.97 × 108 conformations are tractable for BWM*, and in this region it will always outperform A*. For protein design problems with TESS values above 1.57 × 1011 conformations, BWM* attempts to enumerate all conformations of a prohibitively large subproblem even when computing only the sparse GMEC, and A* will have the advantage. For the problems that lie between these two thresholds, increasing the energy cutoff and reducing the energy window allows BWM* to return the sparse GMEC and possibly the sparse ensemble as well. This shows that the performance of BWM* can be reliably predicted with TESS. The next section compares the performance of BWM* and A* for the 49 problems on which BWM* is predicted to perform better.
4.3. Ensemble enumeration time
BWM* was able to provably compute the full ensemble in 45 out of 49 protein design problems for at least one combination of energy window, distance, and energy cutoffs. For the 36 design problems where BWM* enumerates the full ensemble with Ew = 1 kcal/mol, energy cutoff 0.1 kcal/mol, Fig. 4A shows the maximum observed problem size |M ∪ λ| and branch-width w compared to the number of mutable residues n. The maximum observed problem size never exceeds 6, and its upper bound never goes beyond 7.5 (w ≤ 5). The fact that the maximum observed subproblem is small irrespective of n makes TESS much smaller than the search space of A* (O(qn)), as shown by the solid line in Fig. 4C. This reduced effective search space allows BWM* to search more efficiently than A*, and as plotted in Fig. 4B, BWM* enumerates not only the sparse ensemble but also the full ensemble faster than A*. In cases where BWM* cannot fully enumerate the full ensemble (because the error bounds calculated according to Lemma 1 are large), the sparse ensemble contains more than 93% of all conformations in the full ensemble. One such case is the protein YciI from H. influenzae (PDB id: 1mwq) with energy window of 1 kcal/mol and an energy cutoff of 0.1 kcal/mol, where the number of conformations enumerated by BWM* exceeded 10,000 before it provably enumerated the full ensemble. Fig. 5 compares the sequence-specific ensembles for this case, and shows that the sparse ensemble contains almost all low-energy conformations in the full ensemble. The missing conformations introduce less than a 3% difference in the Boltzmann-weighted partition function (Roberts et al., 2012; Georgiev et al., 2008b). A detailed description of the high similarity between sparse and full ensemble, as well as additional runtime comparisons for the remaining nine cases in which BWM* computes the full ensemble can be found in section B of the SI in Jou et al. (2015).
FIG. 4.

Both actual and worst-case bounds for BWM* are better than A*.
(A) Comparison of the number of mutable residues considered. The maximum observed subproblem size is |M ∪ λ| and worst-case problem size is 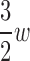 , which bounds |M ∪ λ| from above. (B) Times to enumerate full ensemble for all design problems with energy window of 1 kcal/mol and energy cutoff of 0.1 kcal/mol. (C) Actual search space size vs. worst-case bounds. x-Axis is the 36 design problems in (A). Problems are ranked by A* worst-case problem size (qn); q and n measure the size of the design problem and are shown below the line; n increases from 4 to 16, shown in orange, and q is between 3 and 41, shown in red. Solid green line: Actual number (
, which bounds |M ∪ λ| from above. (B) Times to enumerate full ensemble for all design problems with energy window of 1 kcal/mol and energy cutoff of 0.1 kcal/mol. (C) Actual search space size vs. worst-case bounds. x-Axis is the 36 design problems in (A). Problems are ranked by A* worst-case problem size (qn); q and n measure the size of the design problem and are shown below the line; n increases from 4 to 16, shown in orange, and q is between 3 and 41, shown in red. Solid green line: Actual number (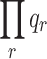 ) of unpruned conformations after DEE, where qr is the number of unpruned rotamers for mutable residue r and
) of unpruned conformations after DEE, where qr is the number of unpruned rotamers for mutable residue r and 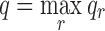 . Solid blue: TESS. Dotted blue: BWM worst-case problem size bounds q|M∪λ|. Dotted green: A* worst-case problem size bounds qn.
. Solid blue: TESS. Dotted blue: BWM worst-case problem size bounds q|M∪λ|. Dotted green: A* worst-case problem size bounds qn.
FIG. 5.

The sparse ensemble closely approximates the full ensemble. Figures shown for YciI protein from H. influenzae (PDB id: 1mwq) with energy cutoff of 0.1 kcal/mol and energy window of 1 kcal/mol. For each sequence, the x-axis plots the full energy of each conformation of the ensemble, and the y-axis plots the percent contribution of each conformation to the partition function Z (called q in Georgiev et al., 2008b). Conformations in both the full and sparse ensembles are colored green. Conformations in the full ensemble that are not in the sparse ensemble are in red. Conformations in the sparse ensemble that are not in the full ensemble are in blue. GMEC is circled in green.
Fig. 6 shows the individual and cumulative time taken by BWM* and A* to return each additional conformation in the gap-free list for two representative design problems. For the aortic preferentially expressed protein-1 (PDB id:1u2h), BWM* computes the full ensemble before A* even returns the first conformation. For cases like the third KH domain of heterogeneous nuclear ribonucleoprotein K (PDB id: 1zzk), the preprocessing time of BWM* can be significant, but once BWM* begins enumerating conformations it rapidly enumerates more conformations than A* in less time. This is because BWM* requires only O(n log q) time (milliseconds, empirically) for each additional conformation generated, as shown in Fig. 6B and D. Simply modifying A* to use the sparse energy function does not change the worst-case complexity of A*, and BWM* is still faster. A comparison of runtimes showing superior performance of BWM* compared to A* with a sparse energy function are provided as Fig. 3 in section B of the SI in Jou et al. (2015).
FIG. 6.

BWM* enumeration is orders of magnitude faster than A*. Figures shown for aortic preferentially expressed protein-1 (PDB id:1u2h) with energy cutoff 0.1 kcal/mol, and third KH domain of heterogeneous nuclear ribonucleoprotein K (PDB id: 1zzk) with distance cutoff of 7 Å. Energy window was 1 kcal/mol for both. Blue: BWM* time. Green: A* time. (A) and (C) show cumulative time against total conformations enumerated. (B) and (D) show enumeration time per conformation.
Conclusion
We have presented a novel dynamic programming algorithm called BWM* for use with sparse energy functions that provably computes the sparse GMEC, and also (unlike other sparse-GMEC-based algorithms) enumerates a gap-free list of conformations in order of increasing sparse energy. BWM* exploits the optimal substructure of the sparse energy landscape to efficiently enumerate conformations in merely O(n log q) time. In contrast, A* cannot guarantee better performance than exhaustive enumeration. We defined a new measure, TESS, which can be computed a priori in polynomial time to predict BWM* peformance, and showed that A* can be substituted when BWM* is not favorable. In the cases where BWM* is predicted to perform better, BWM* enumerates the full ensemble faster than A*. Our results indicate that the branch-width w (and a fortiori, the worst-case bounds of O(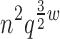 ) of our algorithm) can be small irrespective of the total number of mutable residues n, thereby making TESS much smaller than the worst-case bounds for A*. This, combined with the predictable performance of BWM*, gives protein designers the power to choose beforehand the most efficient algorithm for their particular protein design problem. Similarly to our previous algorithms, BWM* can also be extended to incorporate continuous backbone and side-chain flexiblity (Gainza et al., 2012; Hallen et al., 2013; Georgiev and Donald, 2007, Georgiev et al., 2008a).
) of our algorithm) can be small irrespective of the total number of mutable residues n, thereby making TESS much smaller than the worst-case bounds for A*. This, combined with the predictable performance of BWM*, gives protein designers the power to choose beforehand the most efficient algorithm for their particular protein design problem. Similarly to our previous algorithms, BWM* can also be extended to incorporate continuous backbone and side-chain flexiblity (Gainza et al., 2012; Hallen et al., 2013; Georgiev and Donald, 2007, Georgiev et al., 2008a).
Availability
The design software is available as open-source software online (Jou et al. 2015), or by contacting the authors.
Acknowledgments
We thank all members of the Donald lab, and Prof. Jane Richardson and Prof. David Richardson for helpful discussion and comments. This work is supported by the following grants from the National Institutes of Health: R01-GM-78031 to B.R.D, R01-GM073919 and R01-GM073930 to D.C.R.
Author Disclosure Statement
No competing financial interests exist.
References
- Chen C.-Y., Georgiev I., Anderson A.C., and Donald B.R. 2009. Computational structure-based redesign of enzyme activity. Proc. Natl. Acad. Sci. U.S.A. 106, 3764–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechter R., and Mateescu R. 2007. AND/OR search spaces for graphical models. Artif. Intell. 171, 73–106 [Google Scholar]
- Desjarlais J.R., and Handel T.M. 1995. De novo design of the hydrophobic cores of proteins. Prot. Sci. 4, 2006–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet J., Spriet J., and Lasters I. 2002. Fast and accurate side-chain topology and energy refinement (FASTER) as a new method for protein structure optimization. Proteins 48, 31–43 [DOI] [PubMed] [Google Scholar]
- Donald B.R. 2011. Algorithms in Structural Molecular Biology. MIT Press, New York [Google Scholar]
- Fomin F.V., Thilikos D.M. 2003. Dominating sets in planar graphs: Branch-width and exponential speedup, 168–177. In SODA ’03 Proceedings of the Fourteenth Annual ACM-SIAM Symposium on Discrete Algorithms Society for Industrial and Applied Mathematics Philadelphia, Philadelphia, PA [Google Scholar]
- Fleishman S.J., Leaver-Fay A., Corn J.E., et al. 2011. RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey K.M., Georgiev I., Donald B.R., and Anderson A.C. 2010. Predicting resistance mutations using protein design algorithms. Proc. Natl. Acad. Sci. U.S.A. 107, 13707–13712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainza P., Roberts K.E., and Donald B.R. 2012. Protein design using continuous rotamers. PLoS Comput. Biol. 8, e1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainza P., Roberts K.E., Georgiev I., et al. 2013. OSPREY: Protein design with ensembles, flexibility, and provable algorithms. Methods Enzymol. 523, 87–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev I., and Donald B.R. 2007. Dead-end elimination with backbone flexibility. Bioinformatics 23, i185–i194 [DOI] [PubMed] [Google Scholar]
- Georgiev I., Acharya P., Schmidt S.D., et al. 2012. Design of epitope-specific probes for sera analysis and antibody isolation. Retrovirology 9, P50 [Google Scholar]
- Georgiev I., Keedy D., Richardson J., et al. 2008a. Algorithm for backrub motions in protein design. Bioinformatics 24, i196–i204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev I., Lilien R.H., and Donald B.R. 2008b. The minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. J. Comput. Chem. 29, 1527–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev I.S., Rudicell R.S., Saunders K.O., et al. 2014. Antibodies VRC01 and 10e8 neutralize HIV-1 with high breadth and potency even with Ig-framework regions substantially reverted to germline. J. Immunol. 192, 1100–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein R.F. 1994. Efficient rotamer elimination applied to protein side-chains and related spin glasses. Biophys. J. 66, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczynski M.J., Grembecka J., Zhou Y., et al. 2007. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFβ. Chem. Biol. 14, 1186–1197 [DOI] [PubMed] [Google Scholar]
- Hallen M.A., Keedy D.A., and Donald B.R. 2013. Dead-end elimination with perturbations (DEEPer): A provable protein design algorithm with continuous sidechain and backbone flexibility. Proteins 81, 18–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks I.V., Koster A., and Kolotoglu E. 2005. Branch and Tree Decomposition Techniques for Discrete Optimization. TutORials in Operation Research: INFORMS, New Orleans, 2005, pp. 1–9 [Google Scholar]
- Hlinẽný P., Oum S., Seese D., and Gottlob G. 2008. Width parameters beyond tree-width and their applications. Comput. J. 51, 326–362 [Google Scholar]
- Jiang X., Farid H., Pistor E., and Farid R.S. 2000. A new approach to the design of uniquely folded thermally stable proteins. Prot. Sci. 9, 403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T. 1994. De novo protein design using pairwise potentials and a genetic algorithm. Prot. Sci. 3, 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou J.D., Jain S., Georgiev I.S., and Donald B.R. 2015. Supplementary information: BWM*: A novel, provable, ensemble-based dynamic programming algorithm for sparse approximations of computational protein design. Available at: www.cs.duke.edu/donaldlab/Supplementary/jcb15/bwmstar [DOI] [PMC free article] [PubMed]
- Kaufmann K.W., Lemmon G.H., DeLuca S.L., et al. 2010. Practically useful: What the Rosetta protein modeling suite can do for you. Biochemistry 49, 2987–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilambi K.P., and Gray J.J. 2012. Rapid calculation of protein pKa values using Rosetta. Biophys. J. 103, 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C., Garza E.N., Mazor R., et al. 2014. Removing Tcell epitopes with computational protein design. Proc. Natl. Acad. Sci. U.S.A. 111, 8577–8582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsford C.L., Chazelle B., and Singh M. 2005. Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics 21, 1028–1039 [DOI] [PubMed] [Google Scholar]
- Koehl P., and Delarue M. 1994. Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J. Mol. Biol. 239, 249–275 [DOI] [PubMed] [Google Scholar]
- Kortemme T., Morozov A.V., and Baker D. 2003. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 326, 1239–1259 [DOI] [PubMed] [Google Scholar]
- Krivov G.G., Shapovalov M.V., and Dunbrack R.L. 2009. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77, 778–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., and Baker D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T., and Karplus M. 1999. Effective energy function for proteins in solution. Proteins 35, 133–152 [DOI] [PubMed] [Google Scholar]
- Leach A.R., and Lemon A.P. 1998. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins 33, 227–239 [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A., Kuhlman B., and Snoeyink J. 2005. An adaptive dynamic programming algorithm for the side chain placement problem. Pac. Symp. Biocomput. 2005, 16–27 [PubMed] [Google Scholar]
- Leaver-Fay A., Tyka M., Lewis S.M., et al. 2011. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C., and Subbiah S. 1991. Prediction of protein side-chain conformation by packing optimization. J. Mol. Biol. 217, 373–388 [DOI] [PubMed] [Google Scholar]
- Lilien R.H., Stevens B.W., Anderson A.C., and Donald B.R. 2005. A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Comput. Biol. 12, 740–761 [DOI] [PubMed] [Google Scholar]
- Lovell S.C., Word J.M., Richardson J.S., and Richardson D.C. 2000. The penultimate rotamer library. Prot. Struct. Funct. Bioinform. 40, 389–408 [PubMed] [Google Scholar]
- Nilsson D. 1998. An efficient algorithm for finding the M most probable configurations in probabilistic expert systems. Stat. Comput. 8, 159–173 [Google Scholar]
- Privett H.K., Kiss G., Lee T.M., et al. 2012. Iterative approach to computational enzyme design. Proc. Natl. Acad. Sci. U.S.A. 109, 3790–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve S.M., Gainza P., Frey K.M., et al. 2015. Protein design algorithms predict viable resistance to an experimental antifolate. Proc. Natl. Acad. Sci. U.S.A. 112, 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K.E., Cushing P.R., Boisguerin P., et al. 2012. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput. Biol. 8, e1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson T.A., and Varani G. 2007. An all-atom, distance-dependent scoring function for the prediction of protein-DNA interactions from structure. Prot. Struct. Funct. Bioinform. 66, 359–374 [DOI] [PubMed] [Google Scholar]
- Rudicell R.S., Kwon Y.D., Ko S.-Y., et al. 2014. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J. Virol. 88, 12669–12682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver N.W., King B.M., Nalam M.N.L., et al. 2013. Efficient computation of small-molecule configurational binding entropy and free energy changes by ensemble enumeration. J. Chem. Theory Comput. 9, 5098–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadbeck J., Chan K.H., Khoury G.A., et al. 2014. De novo design and experimental characterization of ultrashort self-associating peptides. PLoS Comput. Biol. 10, e1003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B.W., Lilien R.H., Georgiev I., et al. 2006. Redesigning the PheA domain of gramicidin synthetase leads to a new understanding of the enzyme's mechanism and selectivity. Biochemistry 45, 15495–15504 [DOI] [PubMed] [Google Scholar]
- Xu J., and Berger B. 2006. Fast and accurate algorithms for protein side-chain packing. J. ACM 53, 533–557 [Google Scholar]
- Zhang Z., and Lange O.F. 2013. Replica exchange improves sampling in low-resolution docking stage of RosettaDock. PLoS ONE 8, e72096. [DOI] [PMC free article] [PubMed] [Google Scholar]