

Abstract
Senescence is defined as a stable cell growth arrest. Oncogene-induced senescence (OIS) occurs in normal primary human cells after activation of an oncogene in the absence of other cooperating oncogenic stimuli. OIS is therefore considered a bona fide tumor suppression mechanism in vivo. Indeed, overcoming OIS-associated stable cell growth arrest can lead to tumorigenesis. Although cells that have undergone OIS do not replicate their DNA, they remain metabolically active. A number of recent studies report significant changes in cellular metabolism during OIS, including alterations in nucleotide, glucose, and mitochondrial metabolism and autophagy. These alterations may be necessary for stable senescence-associated cell growth arrest, and overcoming these shifts in metabolism may lead to tumorigenesis. This review highlights what is currently known about alterations in cellular metabolism during OIS and the implication of OIS-associated metabolic changes in cellular transformation and the development of cancer therapeutic strategies.
Keywords: autophagy, lipogenesis, metabolism, oncogene-induced senescence, oxidative phosphorylation, RRM2, glycolysis, TCA cycle, The Warburg Effect
Abbreviations
- 3-AP
3-aminopyridine-2-carboxaldehyde thiosemicarbazone
- ACC
acetyl-CoA carboxylase
- ACLY
ATP-dependent citrate lyase
- CPT1
carnitine-palmitoyl transferase I
- dNDP
deoxyribonucleoside diphosphate
- dNTP
deoxyribonucleotide triphosphate
- FASN
fatty acid synthase
- GPC
L-a-glycerophosphocholine
- MDH
malate dehydrogenase
- ME
malic enzyme
- NAD
nicotinamide adenine dinucleotide
- NDP
ribonucleoside diphosphate
- NTP
ribonucleoside triphosphate
- OIS
oncogene-induced senescence
- PC
phosphocholine
- PDH
pyruvate dehydrogenase kinase
- PDK
pyruvate dehydrogenase kinase
- PDP2
pyruvate dehydrogenase phosphatase 2
- PKM2
pyruvate kinase M2
- PML
promyelocytic leukemia
- PPAR
peroxisome proliferator-activated receptor
- PPP
pentose phosphate pathway
- SA-B-Gal
senescence-associated β-galactosidase
- RISP
Rieske iron sulfur protein
- RNR
ribonucleotide reductase
- ROS
reactive oxygen species
- RRM1
ribonucleotide reductase subunit M1
- RRM2
ribonucleotide reductase subunit M2
- RRM2B/p53R2
ribonucleotide reductase subunit M2B
- SASP
senescence-associated secretory phenotype
- SDHA
succinate dehydrogenase
- TCA
tricarboxylic acid
Introduction
Senescence
Senescence was first described in 1961 by Leonard Hayflick and Paul Moorhead while working at The Wistar Institute in Philadelphia.1 Specifically, these scientists found that normal cells in culture could only double a limited number of times, after which the cells would exit the cell cycle (termed “senescence”). In the decades since this discovery, a number of other stimuli have been shown to induce senescence, including oncogene activation, DNA damage, oxidative damage, and certain chemotherapeutic agents.2 Although the cells do not continue to replicate their DNA, it is well appreciated that senescent cells remain metabolically active, and that these metabolic changes play a role in both senescence-associated cell growth arrest and human disease.
Oncogene-induced senescence
Oncogene-induced senescence (OIS) occurs when an oncogene (such as RAS, BRAF, MYC, etc.) becomes activated in a normal diploid cell.3 This leads to a paradoxical stable cell cycle exit, which arrests cell growth.4 Therefore, OIS is considered a bona fide tumor suppressor mechanism in vivo.4,5 For instance, benign nevi, which are often characterized by mutations in BRAF and NRAS, have been shown to be senescent lesions.4
OIS is characterized by a number of phenotypic and molecular alterations. Phenotypically, cells that have undergone OIS exhibit a large, flat morphology and are positive for increased β-galactosidase activity (termed senescence-associated β-galactosidase [SA-β-Gal]).6 Additionally, OIS is often accompanied by accumulation of DNA damage, in particular DNA double strand breaks, and a robust DNA damage response.7-10 This damage is largely due to the aberrant DNA replication observed during OIS.9,11 Further, OIS is characterized by upregulation of the p53/p21 and pRb/p16 pathways,12 which both play a role in inhibition of cell cycle progression. Many recent reports demonstrate that cells undergoing OIS secrete a number of inflammatory mediators, including cytokines and chemokines, termed the senescence-associated secretory phenotype (SASP).13 The SASP is thought to play a role in both the establishment and maintenance of the senescence-associated cell growth arrest.14 Taken together, cells undergoing OIS activate a number of cellular signaling pathways to ensure a stable cell growth arrest.
Although senescent cells do not replicate their DNA, they remain metabolically active.2,15 It is now known that a number of changes in cellular metabolism occur during OIS.16 This review will focus on the changes in cellular metabolism that accompany OIS, including changes in nucleotide, glucose, mitochondrial, and lipid metabolism and autophagy. We will also discuss how these pathways may be circumvented during transformation and tumorigenesis.
Metabolic Changes During OIS
Nucleotide metabolism is decreased during OIS
Deoxyribonucleotide triphosphates (dNTPs) are necessary for both nuclear and mitochondrial DNA replication and repair.17,18 Alterations in dNTP levels are known to play a pathogenic role in a number of human diseases, including cancer.9,11 dNTPs can be synthesized through the de novo pathway or the salvage pathway.19 The rate-limiting step in the synthesis of dNTPs during the de novo pathway is reduction of ribonucleoside di- or tri- phosphates (NDPs/NTPs) to deoxyribonucleotide di- or tri- phosphates (dNDPs/dNTPS) by ribonucleotide reductase (RNR).17,18 RNR is a tetrameric complex comprised of 2 large catalytic subunits (R1: ribonucleotide reductase M1 [RRM1]) and 2 small regulatory subunits (R2: ribonucleotide reductase M2 [RRM2] or RRM2B/p53R2). Although RRM1 is expressed during all phases of the cell cycle, RRM2 and p53R2 are cell-cycle regulated.20 RRM2 is expressed during S phase, when dNTPs are needed for DNA replication,20 whereas p53R2 is expressed during G0/G1 and is important for DNA repair and mitochondrial DNA synthesis.21 Changes in the expression or activity of RNR can therefore lead to altered DNA synthesis and repair.
Our group and others have shown that the levels of all 4 dNTPs are significantly decreased during OIS.9,11,22 We found that this is specifically due to a decrease in expression of RRM2 (Fig. 1), but not RRM1 or p53R2.9 Interestingly, the decrease in dNTP levels via suppression of RRM2 is a driver, not a consequence, of the cell cycle exit.9 Increasing dNTP levels by ectopic expression of RRM2 or addition of exogenous nucleosides in oncogene-expressing normal diploid cells is able to overcome the aberrant DNA replication, DNA damage accumulation, and senescence-associated stable cell growth arrest induced by oncogenic RAS or BRAF.9,22 Additionally, decreasing nucleotide metabolism using short hairpin RNA-mediated specific knockdown of RRM2 expression can induce the senescence-associated stable cell growth arrest via aberrant DNA replication and DNA damage accumulation.9 These results demonstrate that nucleotide metabolism is decreased during OIS and is a key driver of the OIS phenotype.
Figure 1.
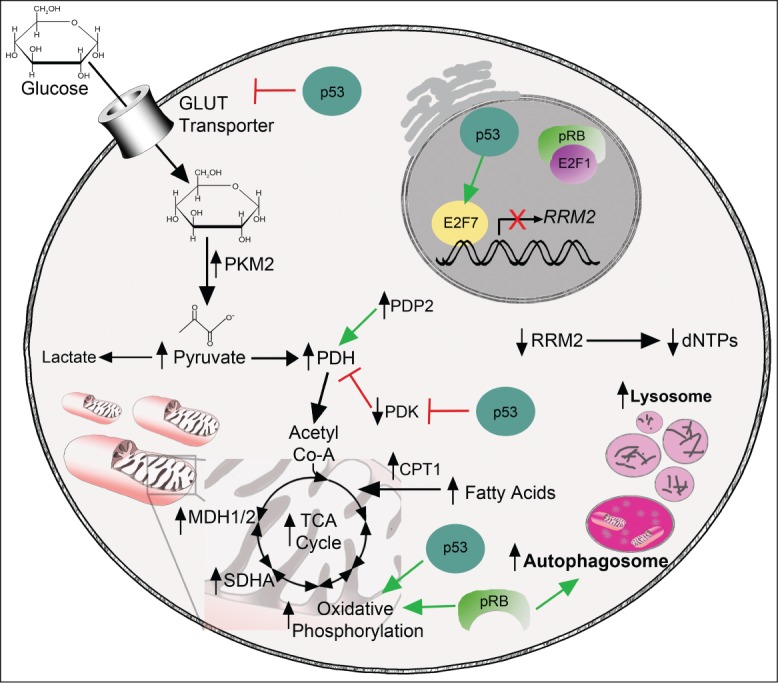
Alterations in metabolic pathways during oncogene-induced senescence. Glucose enters the cell through glucose transporters. p53 inhibits glucose uptake via negative regulation of glucose transporters.45 During glycolysis, glucose is metabolized into pyruvate. One glycolytic enzyme, PKM2, is significantly upregulated during OIS.40 Additionally, pyruvate levels are higher in cells that have undergone OIS.40 The pyruvate produced by metabolism of glucose can be shunted into multiple pathways including fermentation, which leads to lactate production, or into the TCA cycle after further processing by PDH. PDH and its positive regulator PDP2 are upregulated during OIS, whereas the negative regulator of PDH (PDK1) is downregulated,42,43 possibly in part through inhibition mediated by p53.53 This suggests that pyruvate is preferentially shunted into the TCA cycle and away from fermentation. Levels of TCA cycle metabolites are increased during OIS, as are a number of TCA cycle enzymes, including SDHA and MDH1/2.43 Oxidative phosphorylation is also increased during OIS through increased expression of all complexes in the electron transport chain.43 p53 and pRb also positively regulate oxidative phosphorylation.30,53,54 Nucleotide metabolism is decreased in cells that have undergone OIS as a result of suppression of E2F1-mediated transcription of RRM2 and increased repression by the p53 target E2F7.9 Free fatty acid levels are increased in cells undergoing OIS, most likely due to an increase in fatty acid oxidation.52 Finally, both autophagy and lysosomal activity are increased during OIS,68,75,76 which may be in part occur via positive regulation through pRb.79
We recently reported that RRM2, and therefore nucleotide metabolism, is decreased in human benign nevi with mutations in BRAF or NRAS.9 Decreased RRM2 expression was significantly correlated with increased p16 expression, suggesting that these cells have undergone OIS. Additionally, melanoma cell lines and primary melanoma samples with mutations in BRAF or NRAS display a marked increase in RRM2 expression. This suggests that overcoming the suppression of nucleotide metabolism that occurs during OIS may allow cells to re-enter the cell cycle, gain additional mutagenic hits, and become transformed. Indeed addition of exogenous nucleosides or ectopic RRM2 could overcome full senescence induced by RAS.9 Additionally, supraphysiological levels of RRM2 or dNTPs can lead to DNA damage and genomic instability,9,23 which is a hallmark of cancer cells.24,25 RRM2 is transcriptionally regulated by E2F1 (Fig. 1).9,26,27 E2F1 is negatively regulated by pRb (Fig. 1),28,29 which is an important effector of the senescence phenotype.12 In some contexts, loss of pRb can suppress the OIS phenotype.12 When pRb is lost, E2F1 is no longer repressed and can induce transcription of a number of genes that regulate metabolism, including RRM2.30 Additionally, loss of p53 can suppress OIS and can overcome OIS in cells with low p16 expression.10,31,32 Interestingly, a repressive E2F, E2F7, is a p53 target gene.33 E2F7 cooperates with pRb to promote senescence, thus coupling the p53 and pRb pathways. We have previously shown that E2F7 replaces E2F1 on the RRM2 promoter during OIS to suppress RRM2 transcription (Fig. 1).9 This suggests that p53 and pRb act in concert to suppress nucleotide metabolism during OIS, and loss of these tumor suppressors may overcome OIS due to an increase in nucleotide metabolism, thereby leading to cancer formation.
Glucose metabolism is altered during OIS
Glucose is one of the main cellular nutrients and is used in a variety of cellular metabolic processes such as the pentose phosphate pathway (PPP) and the tricarboxylic acid (TCA) cycle.34,35 Glucose metabolites are necessary for nucleotide, amino acid, and lipid biogenesis. Under normal conditions, cells take up glucose through glucose transporters. Intracellular glucose is then metabolized during a process called glycolysis, which has an end product of pyruvate and produces 2 ATP molecules. The pyruvate can then be further metabolized. In normal cells under anaerobic conditions, pyruvate is generally metabolized into lactate (fermentation), during which reduced nicotinamide adenine dinucleotide (NAD+) is produced. This NAD+ is important for cells to continue glycolysis. Under aerobic conditions, pyruvate dehydrogenase (PDH) metabolizes pyruvate into Acetyl Co-A to be further used in the TCA cycle and oxidative phosphorylation to produce 36 ATP molecules.
Cancer cells preferentially use glycolysis and subsequent fermentation to produce ATP even under aerobic conditions.24,34,36 First observed by Otto Warburg in 1924,37 this is now known as the Warburg effect. Although glycolysis is a much less efficient way of producing ATP, cancer cells have a high demand for biomass to continue proliferation.36 To keep up with both the bioenergetic and biomass needs of a highly proliferative state, tumor cells increase their glucose uptake and metabolism through a variety of mechanisms, including increased expression of glucose transporters and glycolytic enzymes.34,35 This allows for the high rate of growth and proliferation that is a hallmark of cancer cells.24
A number of recent studies have indicated that glucose metabolism is altered during OIS but with conflicting findings. Some studies have indicated an increase in glucose uptake during OIS.38-40 However, a number of other studies have observed either no change or a significant decrease in glucose uptake.41-43 The basis for the discrepancy among these studies remains to be determined, although it may be due to differences in cell type, oncogenes, or timing. Regardless, all of these studies have shown changes in glucose metabolism when cells undergo OIS.
Two reports have demonstrated that cells undergoing OIS have increased pyruvate levels,40,42 demonstrating an increase in glycolysis. This is most likely due to increased expression of a number of enzymes involved in pyruvate synthesis, such as pyruvate kinase (PKM2),40 and pyruvate metabolism, including the gatekeeper enzyme pyruvate dehydrogenase (PDH) (Fig. 1).42,43 Because PDH expression is increased, the pyruvate that is produced during glycolysis is shunted into the TCA cycle, thereby decreasing shunting into the fermentation arm that leads to lactate production. Taken together, these studies reveal an overall shift in glucose metabolism in cells undergoing OIS toward the mitochondrial TCA cycle and away from fermentation and the Warburg effect. This is consistent with the fact that OIS is a tumor suppression mechanism.
It is thought that the Warburg effect is a consequence of increased cellular proliferation during tumorigenesis, and not necessarily a cause of transformation itself.44 However, a number of important senescence effectors are known to play an important role in metabolism. For instance, loss of p53 can suppress OIS in some contexts.10,31,32 p53 is known to transcriptionally repress glucose transporters (GLUT-1, GLUT-3, GLUT-4) and a number of glycolytic enzymes.45 Additionally, p53 positively regulates oxidative phosphorylation (Fig. 1),46 thus loss of p53 can lead to a switch from oxidative phosphorylation to increased aerobic glycolysis.46 Therefore, suppressing OIS could lead to a change in glucose metabolism that primes cells to become cancerous.
Mitochondrial metabolism is altered during OIS
Mitochondria are the cell's powerhouses. Under normal oxygen conditions, glucose is broken down into pyruvate, which is then shunted through the TCA cycle. The TCA cycle starts when pyruvate dehydrogenase metabolizes pyruvate into acetyl-CoA.47 Acetyl-CoA can also be produced from fatty acid oxidation (discussed below).48 A series of enzymatic reactions then occurs to produce 3 molecules of NADH, which can then fuel oxidative phosphorylation (oxphos). Oxphos occurs in the inner mitochondrial membrane through 5 enzyme complexes49 and is the most efficient way for a cell to produce ATP as it generates 36 ATP molecules for each glucose molecule.36
A number of recent studies have indicated a significant shift in mitochondrial metabolism in OIS cells. Firstly, a number of studies have demonstrated that cells undergoing OIS display mitochondrial dysfunction. Studies of RAS- or HER2-induced senescence reveal that cells undergoing OIS have decreased mitochondrial membrane potential,39,50 indicating mitochondrial dysfunction. Interestingly, these studies report conflicting results in terms of mitochondrial mass and mitochondrial DNA content, likely due to differences in cell type and the oncogene used. Cadenas et al. reported that mitochondrial mass is decreased and that mitochondria are found in vacuole-rich areas, likely demonstrating that these dysfunctional mitochondria are targeted for degradation.50 In contrast, Moiseeva et al. found that mitochondrial mass and DNA content is increased in cells undergoing OIS.39 These authors also observed a marked increase in mitochondrial superoxides, indicating a significant change in mitochondrial metabolism. Indeed, senescence can be directly induced by interference with normal mitochondrial metabolism using specific knockdown of the important mitochondrial protein Rieske iron sulfur protein (RISP) or the pharmacologic inhibitors rotenone and oligomycin.39 Although studies have shown some conflicting results, it is clear that mitochondrial metabolism is markedly changed during OIS and may regulate senescence.
In addition to changes in mitochondria themselves, flux through the mitochondrial TCA cycle is increased in cells undergoing OIS. One study has demonstrated a significant increase in TCA cycle metabolites in OIS cells (Fig. 1).42 This is likely due to both an increase in the mitochondrial pyruvate gatekeeper enzyme PDH42,43 and an increase in enzymes in the TCA cycle such as malate dehydrogenase (MDH1/2) and succinate dehydrogenase (SDHA).43 Increased PDH activity results from decreased expression of the PDH suppressive pyruvate dehydrogenase kinase 1 (PDK1) and increased expression of the PDH activating pyruvate dehydrogenase phosphatase 2 (PDP2) (Fig. 1).42,51
Oxidative phosphorylation is one of the most important biological processes of the mitochondrion.47 A number of studies have shown that oxphos is increased during OIS, leading to an increase in oxygen consumption (Fig. 1).38,42,52 This is likely due to increased TCA cycle metabolites and enzyme expression42,43 in addition to increased expression of all complexes of the electron transport chain.43 Increased oxygen consumption and oxphos complex enzyme expression should lead to an increase in ATP levels. However, a number of studies have shown a decrease in ATP levels in cells undergoing OIS.39-41 This may be due to an increased usage of ATP by senescent cells for other highly energetic metabolic processes.
As discussed above, cancer cells show a marked decrease in TCA cycle and oxidative phosphorylation compared to normal cells. However, this is not due to mitochondrial defects in tumor cells,35 but instead is most likely due to an increase in glucose uptake and glycolytic enzymes in addition to an increase in the PDH inhibitory PDK enzymes.34,44 This leads to hyperactivation of aerobic glycolysis and less pyruvate entering the TCA cycle. As discussed above, loss of p53 can suppress OIS,10,32 and p53 is known to be important for mitochondrial respiration and as a negative transcriptional regulator of aerobic glycolysis (Fig. 1). In part, this occurs through negative regulation of PDK2 (Fig. 1).53 Loss of p53 thereby leads to an increase in PDK2, which in turn reduces the amount of pyruvate entering the mitochondria for the TCA cycle.44 Additionally, loss of pRB, which can suppress OIS,12 leads to decreased mitochondrial oxidative phosphorylation.30,54 This indicates that suppression of OIS through loss of p53 and/or pRb can switch metabolism from the mitochondria to a more Warbug effect type of phenotype.
Lipid metabolism is altered during OIS
Lipids are important for a number of cellular processes, including energy storage and signaling, and as components of cellular membranes.55 Lipids can be grouped into 8 different categories including 2 lipids that will be discussed below: fatty acids and phospholipids. Importantly, fatty acids can be used as a major source of energy during a process called fatty acid oxidation (or β-oxidation) in which these lipids are catabolized into acetyl-CoA to be used in the TCA cycle. During β-oxidation, fatty acids are transported across the outer mitochondrial membrane by carnitine-palmitoyl transferase I (CPT1), which is rate-limiting for this process.56 Although phospholipids are generally thought to be most important as a major component of cellular membranes, they can also be signaling messengers. Phospholipids are generated from fatty acids and can have different head groups; in particular, choline is the head group of phosphatidylcholine, which is the main constituent of cell membranes.57 These phospholipids can be catabolized into free fatty acids to generate ATP under periods of cell stress.55
Recent studies have shown a change in lipid metabolism in cells undergoing OIS. Quijano et al. recently demonstrated that senescent cells display an elevation in free fatty acid levels (Fig. 1).52 However, this is not due to an increase in lipid metabolism because these authors showed a decrease in acetyl-CoA carboxylase (ACC), the rate-limiting enzyme in lipid metabolism, in cells undergoing OIS. Instead, an increase in fatty acid oxidation was found to be the dominant factor in the increase in oxygen consumption in these cells. This may be another reason why senescent cells display an increase in oxygen consumption but no appreciable increase in ATP levels. The mechanism by which increased fatty acid levels are increased in OIS cells is unclear but may be due to promyelocytic leukemia (PML) activation of the fatty acid oxidation pathway through peroxisome proliferator-activated receptor (PPAR) signaling.58 PML and PML nuclear bodies are known to play a major role during OIS.59,60 Increased levels of fatty acids and their subsequent degradation, leading to increased NADPH levels, may occur because of an increased need for antioxidants to combat the high levels of reactive oxygen species (ROS) observed during OIS.39,61 Interestingly, changes in fatty acid oxidation were shown to play a role in the senescence-associated secretory phenotype (SASP). Knockdown of CPT1, the rate-limiting enzyme in fatty acid oxidation, decreased the SASP during OIS.52 This suggests that fatty acid oxidation is necessary for SASP expression during OIS. Another recent study also indicates an alteration in lipid metabolism during OIS. Gey et al. found a specific decrease in phosphocholine (PC) and an increase in L-a-glycerophosphocholine (GPC), suggesting catabolism of phospholipids via phospholipase A1 and/or A2 and lysophospholipase.62 Indeed, another study showed alterations specifically in mitochondrial phospholipids,50 which may play a role in the mitochondrial dysfunction observed during OIS. These studies suggest that the changes in lipid metabolism observed during OIS may play a specific role in the senescence phenotype, especially the SASP.
It has become apparent in recent years that lipid biogenesis is altered in cancer cells.48 In contrast to cells that have undergone OIS, which display increased fatty acid catabolism, many cancer cells show a significant increase in de novo fatty acid synthesis.48 This is likely due to increased expression of key enzymes in this pathway, including ATP-dependent citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN).34,48 A number of the fatty acid pathway enzymes are regulated by AKT.63 Interestingly, activation of AKT or suppression of PTEN in cells that have undergone OIS can overcome the cell growth arrest.64,65 Although AKT/PTEN is involved in a number of different pathways, these data suggest that overcoming OIS through upregulation of these pathways may be in part due to a change in lipid biogenesis.
Autophagy and lysosomal activity is increased during OIS
Autophagy is literally the process by which a cell eats itself.66 Macroautophagy, which is the process discussed in this review, delivers cytoplasmic materials to an autolysosome via an autophagosome. Therefore, autophagy and the lysosomal compartment are intimately linked. Under nutrient-rich conditions, the mTORC1 complex generally inhibits autophagy.67 Under nutrient-poor conditions, mTORC1 is inhibited, leading to upregulation of a number of autophagy-related genes (Atg genes) and initiation of the phagophore. Autophagy is thought to be a cell survival mechanism and has been implicated in several human diseases including cancer.66
A number of studies have indicated that autophagy is upregulated during OIS. LC-III, a marker of autophagy, and the number of autophagic vesicles are both significantly increased in cells undergoing OIS (Fig. 1).68 Indeed, long-lived proteins are degraded at an increased rate in OIS cells compared to proliferating cells. Autophagy is likely increased during OIS as a result of repression of the mTORC1 and mTORC2 complexes through a negative feedback loop.69 Furthermore, induction of autophagy was found to be necessary for OIS as knockdown of key autophagic proteins such as ATG5 or ATG7 bypassed senescence induced by RAS, BRAF, or the KSHV v-cyclin A protein.68,70-73 It is known that senescent cells display an increase in protein content.43,74 Therefore, during senescence, autophagy is probably activated to increase the amino acid content in order to cope with the higher rate of protein translation.75
Similar to the increase in autophagy, a number of studies have observed an increase in lysosomal activity in cells undergoing OIS. This is likely because autophagy and lysosomes go hand-in-hand to quickly degrade proteins and other biomolecules within the cell. Young et al. observed a significant increase in lysosomal genes in cells undergoing OIS.68 These authors also observed an increased lysosomal compartment using LysoTracker and electron microscopy (Fig. 1). The increase in lysosomal gene expression directly correlates with the timing of the increase in autophagy genes, indicating coordinated upregulation of both cellular processes during OIS. A more recent study further identified the mechanism of upregulation of lysosomal genes. Urbanelli et al. demonstrated a significant increase in the transcription factor TFEB that leads to upregulation of HEXA and HEXB, which encode lysosomal glycohydrolase β-hexosaminidase.76 Taken together, these studies indicate an increase in both autophagy and lysosomal activity in cells undergoing OIS.
Subversion of the autophagic process can overcome OIS68 and potentially lead to transformation of cells. Indeed, autophagy is thought to be a tumor suppressive mechanism.66 Autophagy is negatively regulated by mTOR,77 which is induced during AKT signaling.78 As discussed above, increased AKT signaling through activated AKT or inactivated PTEN can overcome the OIS-associated cell growth arrest.64,65 In addition to the role of this pathway in other metabolic processes, this may also be due to inhibition of autophagy. Autophagy is also positively regulated by pRb (Fig. 1),79 and loss of pRb can suppress OIS,68 which may in part be due to a decrease in autophagy. Together, these studies suggest that overcoming OIS through suppression of autophagy could lead to transformation and tumorigenesis.
Targeting the metabolome as a pro-senescence cancer therapeutic strategy
Senescence is now thought to be a viable outcome for cancer therapy.80 Additionally, targeting the altered metabolome in cancer cells has attracted great interest in recent years.47,81,82 Numerous compounds that have been developed to target different metabolic pathways are reviewed elsewhere.47,81,82 Here, we will review recent studies that have shown that shifting metabolism toward that seen during OIS could be a viable strategy to induce cancer cell senescence and/or regression of tumors.
Targeting nucleotide metabolism as a pro-senescence cancer therapy
Cells that have undergone OIS show a marked decrease in dNTP levels and the rate-limiting enzyme RRM2.9,22 Overcoming the decrease in nucleotide metabolism can lead to re-entry of fully senescent cells into the cell cycle.9 To sustain a high level of proliferation, cancer cells require elevated levels of dNTPs.24 It is possible to exploit this need by targeting this pathway to induce cancer cell senescence. A number of inhibitors of the nucleotide metabolic pathway are currently being used in the clinic.11,81 Studies from our laboratory have shown that inhibition of nucleotide metabolism through specific knockdown or inhibition of RRM2 using 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP)83 can induce senescence in multiple cancer cell models with oncogenic signaling activation.9,84 Notably, senescence induced by RRM2 inhibition is independent of p53 and p16 and correlates with the DNA damage response.9,84 This supports the premise that inhibition of this pathway in cancer cells is a viable pro-senescence therapeutic strategy. The dNTP metabolic pathway has been exploited for cancer treatment for decades. New insights suggest that it remains an important target for the development of novel cancer therapeutic strategies using senescence as a primary tumor suppression mechanism.
Targeting glucose metabolism as a pro-senescence cancer therapy
During OIS, cells display a significant shift in glucose metabolism toward the mitochondrial TCA and oxidative phosphorylation pathways and away from the Warburg effect and fermentation (discussed above). Increased aerobic glycolysis and fermentation are hallmarks of cancer cells.24 Therefore, suppression of OIS may alter glucose metabolism to a more Warburg effect-like phenotype. Because aerobic glycolysis is not observed in normal cells, targeting this pathway has been of major interest in recent years; indeed, a number of agents are being developed to target this pathway for cancer therapy.81,82 For example, a small molecule of the GLUT1 transporter (WZB117) induces senescence and inhibits tumor growth in vivo.85 Although a number of other inhibitors of glycolysis have been developed, most have not moved past Phase I/II clinical trials and their development has been discontinued.82 This is most often due to unacceptable toxicities.81 Future inhibitors will need to be more specific to limit off-target effects. Nevertheless, these data support the hypothesis that shifting metabolism away from the Warburg effect is a viable pro-senescence cancer therapeutic strategy.
Targeting mitochondrial metabolism
OIS cells display a significant increase in TCA cycle intermediates and oxidative phosphorylation (discussed above). Although cancer cells have functional mitochondria,35 most of their ATP is produced through glycolysis.36 Therefore, drugs that shift metabolism toward the mitochondria may induce senescence of cancer cells. For instance, upregulation of PDH by knockdown of PDK1, which would shunt pyruvate into the TCA cycle,44 can cause regression of melanoma tumors with oncogenic BRAF mutations.42 In addition, knockdown of malic enzymes (ME1/2) can induce senescence of tumor cells.86 ME1/2 metabolize malate into pyruvate, thereby producing NADPH, which is necessary for a number of metabolic processes including nucleic acid synthesis.44 Thus, targeting the mitochondrial pathways can exert an effect on other metabolic pathways known to play a role in senescence.
Concluding Remarks
In recent years, it has becoming increasingly clear that OIS is accompanied by a significant alteration in cellular metabolism. Given that OIS is a tumor suppressor mechanism,4,5 the changes in cellular metabolism are mostly opposite to those observed during tumorigenesis. For example, during OIS glucose metabolites are mainly shunted toward the TCA cycle and oxidative phosphorylation. This is in contrast to tumor metabolism, which shunts glucose either to fermentation to produce lactate or the PPP, thereby increasing the level of nucleotides and other necessary biomolecules for proliferation. This indicates that these pathways play a role in the tumor suppressive phenotype of OIS. Understanding these alterations in the metabolome may give researchers further insight into how cells are able to circumvent the OIS phenotype to become cancerous. Indeed, a number of key senescence effectors such as p53 and pRb also play major roles in metabolism.30,34,44,46,53,54,79 This underscores the importance of further understanding these pathways to potentially prevent the bypass of OIS. Additionally, this knowledge may allow us to exploit cancer cells' metabolic weaknesses as a therapeutic strategy. Interestingly, a recent study found that exploiting metabolic changes during therapy-induced senescence of cancer cells could induce a synthetic lethality.38 This suggests that these changes can be targeted in cancer patients to improve cancer therapy. Further studies are therefore warranted to fully understand the metabolic alterations accompanying OIS and how these are changed when cells overcome the OIS-associated cell cycle exit.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a NIH/NCI grant (R01CA160331 to R.Z.), a DoD Ovarian Cancer Academy Award (OC093420 to R.Z.) and an NIH/NCI training grant (T32CA9171–35 to K.M.A.). Support of Core Facilities used in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
References
- 1. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:585-621; PMID:13905658; http://dx.doi.org/ 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- 2. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/ 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 3. Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell 2007; 128:233-4; PMID:17254959; http://dx.doi.org/ 10.1016/j.cell.2007.01.005 [DOI] [PubMed] [Google Scholar]
- 4. Mooi WJ, Peeper DS. Oncogene-induced cell senescence–halting on the road to cancer. N Engl J Med 2006; 355:1037-46; PMID:16957149; http://dx.doi.org/ 10.1056/NEJMra062285 [DOI] [PubMed] [Google Scholar]
- 5. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436:660-5; PMID:16079837; http://dx.doi.org/ 10.1038/nature03841 [DOI] [PubMed] [Google Scholar]
- 6. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. . A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-7; PMID:7568133; http://dx.doi.org/ 10.1073/pnas.92.20.9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26:7773-9; PMID:18066090; http://dx.doi.org/ 10.1038/sj.onc.1210881 [DOI] [PubMed] [Google Scholar]
- 8. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. . Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633-7; PMID:17136093; http://dx.doi.org/ 10.1038/nature05268 [DOI] [PubMed] [Google Scholar]
- 9. Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, et al. . Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep 2013; 3:1252-65; PMID:23562156; http://dx.doi.org/ 10.1016/j.celrep.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. . Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638-42; PMID:17136094; http://dx.doi.org/ 10.1038/nature05327 [DOI] [PubMed] [Google Scholar]
- 11. Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett 2014; PMID:24486217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/ 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- 13. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99-118; PMID:20078217; http://dx.doi.org/ 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133:1019-31; PMID:18555778; http://dx.doi.org/ 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 15. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10:51-7; PMID:20029423; http://dx.doi.org/ 10.1038/nrc2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev 2014; 28:99-114; PMID:24449267; http://dx.doi.org/ 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem 1988; 57:349-74; PMID:3052277; http://dx.doi.org/ 10.1146/annurev.bi.57.070188.002025 [DOI] [PubMed] [Google Scholar]
- 18. Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem 2006; 75:681-706; PMID:16756507; http://dx.doi.org/ 10.1146/annurev.biochem.75.103004.142443 [DOI] [PubMed] [Google Scholar]
- 19. Blakley RL, Vitols E. The control of nucleotide biosynthesis. Annu Rev Biochem 1968; 37:201-24; PMID:4875716; http://dx.doi.org/ 10.1146/annurev.bi.37.070168.001221 [DOI] [PubMed] [Google Scholar]
- 20. Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem 1985; 260:9114-6; PMID:3894352 [PubMed] [Google Scholar]
- 21. Hakansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem 2006; 281:7834-41; PMID:16436374; http://dx.doi.org/ 10.1074/jbc.M512894200 [DOI] [PubMed] [Google Scholar]
- 22. Mannava S, Moparthy KC, Wheeler LJ, Natarajan V, Zucker SN, Fink EE, Im M, Flanagan S, Burhans WC, Zeitouni NC, et al. . Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing oncogene-induced senescence. Am J Pathol 2013; 182:142-51; PMID:23245831; http://dx.doi.org/ 10.1016/j.ajpath.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012; 149:1023-34; PMID:22632967; http://dx.doi.org/ 10.1016/j.cell.2012.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 25. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220-8; PMID:20177397; http://dx.doi.org/ 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- 26. Chabes AL, Bjorklund S, Thelander L. S Phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region. J Biol Chem 2004; 279:10796-807; PMID:14688249; http://dx.doi.org/ 10.1074/jbc.M312482200 [DOI] [PubMed] [Google Scholar]
- 27. DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol 1995; 15:4215-24; PMID:7623816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flemington EK, Speck SH, Kaelin WG, Jr. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci U S A 1993; 90:6914-8; PMID:8346196; http://dx.doi.org/ 10.1073/pnas.90.15.6914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol 1993; 13:6501-8; PMID:8413249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nicolay BN, Dyson NJ. The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 2013; 25:735-40; PMID:23916769; http://dx.doi.org/ 10.1016/j.ceb.2013.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:2853-68; PMID:19053174; http://dx.doi.org/ 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, Dal Zuffo R, Matti V, d'Ario G, Montani E, et al. . Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 2011; 13:292-302; PMID:21336312; http://dx.doi.org/ 10.1038/ncb2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, Lowe SW. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 2012; 26:1546-57; PMID:22802529; http://dx.doi.org/ 10.1101/gad.196238.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romero-Garcia S, Lopez-Gonzalez JS, Baez-Viveros JL, Aguilar-Cazares D, Prado-Garcia H. Tumor cell metabolism: an integral view. Cancer Biol Ther 2011; 12:939-48; PMID:22057267; http://dx.doi.org/ 10.4161/cbt.12.11.18140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/ 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/ 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Warburg O. On the origin of cancer cells. Science 1956; 123:309-14; PMID:13298683; http://dx.doi.org/ 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 38. Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. . Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013; 501:421-5; PMID:23945590; http://dx.doi.org/ 10.1038/nature12437 [DOI] [PubMed] [Google Scholar]
- 39. Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol 2009; 29:4495-507; PMID:19528227; http://dx.doi.org/ 10.1128/MCB.01868-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mazurek S, Zwerschke W, Jansen-Durr P, Eigenbrodt E. Metabolic cooperation between different oncogenes during cell transformation: interaction between activated ras and HPV-16 E7. Oncogene 2001; 20:6891-8; PMID:11687968; http://dx.doi.org/ 10.1038/sj.onc.1204792 [DOI] [PubMed] [Google Scholar]
- 41. Gitenay D, Wiel C, Lallet-Daher H, Vindrieux D, Aubert S, Payen L, Simonnet H, Bernard D. Glucose metabolism and hexosamine pathway regulate oncogene-induced senescence. Cell Death Disease 2014; 5:e1089; PMID:24577087; http://dx.doi.org/ 10.1038/cddis.2014.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. . A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013; 498:109-12; PMID:23685455; http://dx.doi.org/ 10.1038/nature12154 [DOI] [PubMed] [Google Scholar]
- 43. Li M, Durbin KR, Sweet SM, Tipton JD, Zheng Y, Kelleher NL. Oncogene-induced cellular senescence elicits an anti-Warburg effect. Proteomics 2013; 13:2585-96; PMID:23798001; http://dx.doi.org/ 10.1002/pmic.201200298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85-95; PMID:21258394; http://dx.doi.org/ 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- 45. Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta 2012; 1826:370-84; PMID:22750268 [DOI] [PubMed] [Google Scholar]
- 46. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science 2006; 312:1650-3; PMID:16728594; http://dx.doi.org/ 10.1126/science.1126863 [DOI] [PubMed] [Google Scholar]
- 47. Jones NP, Schulze A. Targeting cancer metabolism–aiming at a tumour's sweet-spot. Drug Discov Today 2012; 17:232-41; PMID:22207221; http://dx.doi.org/ 10.1016/j.drudis.2011.12.017 [DOI] [PubMed] [Google Scholar]
- 48. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J 2012; 279:2610-23; PMID:22621751; http://dx.doi.org/ 10.1111/j.1742-4658.2012.08644.x [DOI] [PubMed] [Google Scholar]
- 49. Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, Petruzzella V. The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol 2012; 942:3-37; PMID:22399416; http://dx.doi.org/ 10.1007/978-94-007-2869-1_1 [DOI] [PubMed] [Google Scholar]
- 50. Cadenas C, Vosbeck S, Hein EM, Hellwig B, Langer A, Hayen H, Franckenstein D, Buttner B, Hammad S, Marchan R, et al. . Glycerophospholipid profile in oncogene-induced senescence. Biochim Biophys Acta 2012; 1821:1256-68; PMID:22178194; http://dx.doi.org/ 10.1016/j.bbalip.2011.11.008 [DOI] [PubMed] [Google Scholar]
- 51. Olenchock BA, Vander Heiden MG. Pyruvate as a pivot point for oncogene-induced senescence. Cell 2013; 153:1429-30; PMID:23791173; http://dx.doi.org/ 10.1016/j.cell.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, Rovira II, Mohney RP, Karoly ED, Finkel T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012; 11:1383-92; PMID:22421146; http://dx.doi.org/ 10.4161/cc.19800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res 2012; 72:560-7; PMID:22123926; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1215 [DOI] [PubMed] [Google Scholar]
- 54. Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev 2008; 22:463-75; PMID:18258751; http://dx.doi.org/ 10.1101/gad.1627208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism 2014; 63:895-902; PMID:24814684; http://dx.doi.org/ 10.1016/j.metabol.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 56. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem 2006; 281:37372-80; PMID:17030509; http://dx.doi.org/ 10.1074/jbc.M608372200 [DOI] [PubMed] [Google Scholar]
- 57. Gibellini F, Smith TK. The Kennedy pathway–De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010; 62:414-28; PMID:20503434; http://dx.doi.org/ 10.1002/iub.354 [DOI] [PubMed] [Google Scholar]
- 58. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K, et al. . A metabolic prosurvival role for PML in breast cancer. J Clin Invest 2012; 122:3088-100; PMID:22886304; http://dx.doi.org/ 10.1172/JCI62129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, et al. . PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000; 406:207-10; PMID:10910364; http://dx.doi.org/ 10.1038/35021000 [DOI] [PubMed] [Google Scholar]
- 60. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:2015-27; PMID:10950866 [PMC free article] [PubMed] [Google Scholar]
- 61. Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 1999; 274:7936-40; PMID:10075689; http://dx.doi.org/ 10.1074/jbc.274.12.7936 [DOI] [PubMed] [Google Scholar]
- 62. Gey C, Seeger K. Metabolic changes during cellular senescence investigated by proton NMR-spectroscopy. Mech Ageing Dev 2013; 134:130-8; PMID:23416267; http://dx.doi.org/ 10.1016/j.mad.2013.02.002 [DOI] [PubMed] [Google Scholar]
- 63. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005; 24:6465-81; PMID:16007182 [DOI] [PubMed] [Google Scholar]
- 64. Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, Ajouaou A, Kortman PC, Dankort D, McMahon M, et al. . Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012; 26:1055-69; PMID:22549727; http://dx.doi.org/ 10.1101/gad.187252.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, et al. . Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell 2011; 42:36-49; PMID:21474066; http://dx.doi.org/ 10.1016/j.molcel.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/ 10.1038/nrc1692 [DOI] [PubMed] [Google Scholar]
- 67. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 68. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al. . Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798-803; PMID:19279323; http://dx.doi.org/ 10.1101/gad.519709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006; 10:459-72; PMID:17157787; http://dx.doi.org/ 10.1016/j.ccr.2006.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012; 11:167-80; PMID:22341465; http://dx.doi.org/ 10.1016/j.chom.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 71. Leidal AM, Lee PW, McCormick C. Viral subversion of autophagy impairs oncogene-induced senescence. Autophagy 2012; 8:1138-40; PMID:22735194; http://dx.doi.org/ 10.4161/auto.20340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu H, He Z, Simon HU. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014; 10:372-3; PMID:24300435; http://dx.doi.org/ 10.4161/auto.27163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu H, He Z, von Rutte T, Yousefi S, Hunger RE, Simon HU. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 2013; 5:202ra123; PMID:24027027; http://dx.doi.org/ 10.1126/scitranslmed.3005864 [DOI] [PubMed] [Google Scholar]
- 74. De Cecco M, Jeyapalan J, Zhao X, Tamamori-Adachi M, Sedivy JM. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY) 2011; 3:955-67; PMID:22006542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy 2009; 5:1046-7; PMID:19652542; http://dx.doi.org/ 10.4161/auto.5.7.9444 [DOI] [PubMed] [Google Scholar]
- 76. Urbanelli L, Magini A, Ercolani L, Sagini K, Polchi A, Tancini B, Brozzi A, Armeni T, Principato G, Emiliani C. Oncogenic H-Ras up-regulates acid beta-hexosaminidase by a mechanism dependent on the autophagy regulator TFEB. PLoS One 2014; 9:e89485; PMID:24586816; http://dx.doi.org/ 10.1371/journal.pone.0089485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. . Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942-6; PMID:20526321; http://dx.doi.org/ 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 2000; 103:253-62; PMID:11057898; http://dx.doi.org/ 10.1016/S0092-8674(00)00117-3 [DOI] [PubMed] [Google Scholar]
- 79. Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E, Xu J, McDonnell TJ, Shinojima N, Fueyo J. The RB-E2F1 pathway regulates autophagy. Cancer Res 2010; 70:7882-93; PMID:20807803; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011; 11:503-11; PMID:21701512; http://dx.doi.org/ 10.1038/nrc3057 [DOI] [PubMed] [Google Scholar]
- 81. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 2011; 10:671-84; PMID:21878982; http://dx.doi.org/ 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 82. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov 2013; 12:829-46; PMID:24113830; http://dx.doi.org/ 10.1038/nrd4145 [DOI] [PubMed] [Google Scholar]
- 83. Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul 1999; 39:3-12; PMID:10470363; http://dx.doi.org/ 10.1016/S0065-2571(98)00017-X [DOI] [PubMed] [Google Scholar]
- 84. Aird KM, Li H, Xin F, Konstantinopoulos PA, Zhang R. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle 2014; 13:199-207; PMID:24200970; http://dx.doi.org/ 10.4161/cc.26953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, et al. . A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 2012; 11:1672-82; PMID:22689530; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0131 [DOI] [PubMed] [Google Scholar]
- 86. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013; 493:689-93; PMID:23334421; http://dx.doi.org/ 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]