

Abstract
As part of an ongoing project aimed at identifying protective capsular polysaccharide epitopes for the development of vaccine candidates against the fungal pathogen Cryptococcus neoformans, the synthesis and glycosylation properties of a naphthalenylmethyl (NAP) orthogonally protected trisaccharide thioglycoside, a common building block for construction of serotype B and C capsular polysaccharide structures, were investigated. Ethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyl‐ uronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside was prepared and used both as a donor and an acceptor in glycosylation reactions to obtain spacer equipped hexa‐ and heptasaccharide structures suitable either for continued elongation or for deprotection and printing onto a glycan array or conjugation to a carrier protein. The glycosylation reactions proceeded with high yields and α‐selectivity, proving the viability of the building block approach also for construction of 4‐O‐xylosyl‐containing C. neoformans CPS structures.
Keywords: block synthesis, capsular polysaccharide, carbohydrates, Cryptococcus neoformans, thioglycosides
Introduction
Cryptococcus neoformans is a fungal pathogen which causes severe infections in immunocompromised individuals, for example, AIDS patients and patients going through organ transplantation.1 C. neoformans is surrounded by capsular polysaccharides (CPSs), primarily the glucurono‐xylo‐mannan (GXM)‐polysaccharide comprising 90–95 % of the total capsule mass. The structure of the GXM is believed to be built up of triads, that is, substituted α‐(1→3) linked trisaccharide mannans, as depicted in Figure 1. β‐glucuronic acid residues are linked to position 2 of the mannose backbone, together with a heterogeneous pattern of 2‐ and/or 4‐β‐xylose substituents. The amount of xylose substitution is the major determinant for the serotyping, with serotype B and C being the more substituted. GXM is also heterogeneously acetylated with the acetate positioned at the 6‐OH of the mannose backbone but not present in the residues carrying 4‐O‐xylose.2 The acetylation is believed to be important for virulence for Serotypes A and D.3
Figure 1.
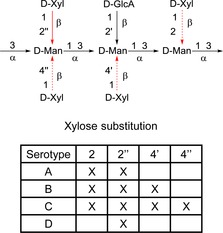
Suggested structures of C. neoformans GXM serotype triads.
Our main focus is on Serotypes A and D, which are the most common ones in human infections, but we are also interested in 4‐O‐xylosyl containing motifs usually attributed to Serotypes B and C, since these structures, owing to the large heterogeneity of the CPS, are present in minor quantities also in CPSs serotyped as A or D. Furthermore, Serotype C structures have the lowest degree of acetylation, which should simplify structure–activity interpretation of immunological results. A building block synthetic strategy for Serotype A and D structures using 2‐O‐substituted disaccharides (I–III, Figure 2)4, 5, 6, 7, 8, 9 has been developed, but the use of 2,4‐di‐O‐substituted trisaccharide building blocks has not been investigated in detail. The syntheses of both the 2,4‐di‐O‐Xyl (IV, Figure 2)10 and the 2‐O‐GlcA‐[4‐O‐Xyl] (V, Figure 2)6, 11 substituted trisaccharide thioglycoside building blocks have been published, but only the former has been used in glycosylations and then only as a donor. The potential use of block V as donor or as acceptor to form heavily branched 2,3,4‐tri‐substituted mannose motifs has not been explored.
Figure 2.
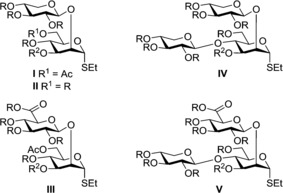
Desired thioglycoside building blocks. R=persistent protecting group, orthoghonal to the acetyl group; R2=temporary protecting group, orthoghonal to R and acetyl groups.
A slightly different approach to the one we are pursuing has been investigated by Zhao and Kong12, 13 on the syntheses of non‐acetylated methyl glycoside structures of Serotype B. These capsular polysaccharide fragments were prepared following a mixed convergent‐linear strategy. In particular, the xylose‐substituted trisaccharide mannan backbone was constructed before, and the glucuronic acid (GlcA) was introduced in the final step.
When the mannose residue involved in the glycosylation was at the nonreducing end in a hexasaccharide acceptor (Figure 3 A), no reaction was observed with methyl 2,3,4‐tri‐O‐acetyl bromo‐ or trichloroacetimidate GlcA donors.12
Figure 3.
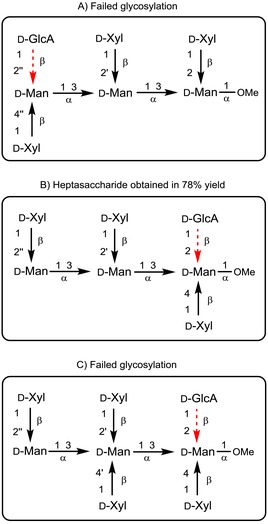
Schematic representation of GlcA glycosylation of xylose‐substituted trisaccharide mannan backbone, reported previously.12, 13, 14
However, when the mannose residue was at the reducing end of the hexasaccharide acceptor (Figure 3 B), the glycosylation proceeded smoothly (78 % yield), affording the target non‐acetylated heptasaccharide structural motif.13 Noteworthy, this latter strategy failed when the same glycosylation was performed on Serotype C heptasaccharide acceptor (Figure 3, C).14 The conflicting outcomes of GlcA glycosylation decrease the attractiveness of this strategy in the preparation of larger GXM fragments.
Herein, our recent efforts in the preparation of the orthogonally protected building block type V are reported together with its use in the construction of larger spacer‐containing acetylated part structures following a convergent approach.
Results and Discussion
In our previous synthesis,6 building block V was prepared with an allyl group as the temporary protecting group in position 3. However, in light of results obtained in attempts to remove the allyl group on disaccharide thioglycosides, showing its incompatibility with the thioethyl group,9 the allyl group was changed into a naphthalenylmethyl protecting group (NAP) in the new synthesis (Scheme 1).
Scheme 1.
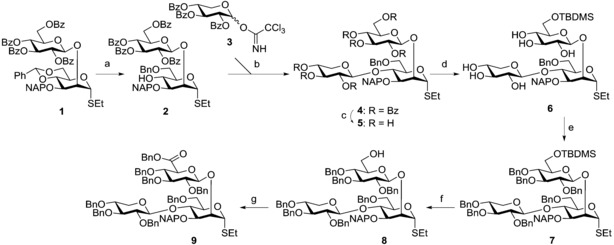
Reagents and conditions: a) NaCNBH3, HCl (1 m in Et2O), THF, pH 1–2, 20 °C, 30 min, 93 %; b) TMSOTf, CH2Cl2, AW‐300 MS, −78 °C→0 °C, o/n, 70 %; c) 1. NaOMe, MeOH, 20 °C, o/n; 2. Dowex H+ ion‐exchange resin, 80 %; d) TBDMSCl, pyridine, DMAP, 20 °C, 4 h, 76 %; e) NaH, BnBr, DMF, 0 °C→20 °C, o/n, 77 %; f) TBAF trihydrate, THF, 20 °C, 2 h, 96 %; g) 1) TEMPO, BAIB, CH2Cl2/H2O (2:1), 20 °C; 2) Cs2CO3, BnBr, DMF, 0 °C→20 °C, 2 h, 60 %.
Starting from compound 1,7 the benzylidene ring was opened regioselectively with NaCNBH3/HCl15 obtaining compound 4‐OH acceptor 2 in 93 % yield. The coupling of 2 and 3 16 was carried out using trimethylsilyl trifluoromethanesulfonate (TMSOTf) in the presence of (commercial) acid‐washed molecular sieves to prevent orthoester formation and afford compound 4 in 70 % yield. At this stage, benzoyl groups, which ensured the stereoselective course of the glycosylation, were removed and the primary OH was selectively protected by reaction with tert‐butyldimethylsilyl chloride (TBDMSCl) giving 6 in 61 % yield over two steps. The remaining hydroxy groups were per‐benzylated (→7, 77 %) before compound 8 was obtained in an almost quantitative yield (96 %) by reaction with tetra‐n‐butylammonium fluoride (TBAF).
NMR analysis of compound 8 at 25 °C in CDCl3 gave unexpected results, which included broad peaks in the 1H NMR spectrum and even missing carbon peaks in the 13C NMR spectrum (Figure 4).
Figure 4.
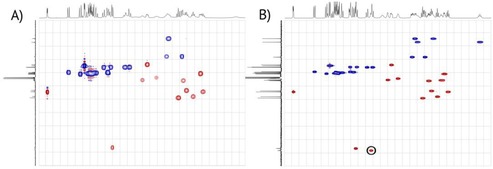
A) 1H−13C HSQC spectrum of compound 8 at 25 °C; B) 1H−13C HSQC spectrum of compound 8 at 50 °C; the now visible anomeric cross peak is highlighted.
By carrying out the NMR experiments at 50 °C (Figure 4 B), sharper peaks (1H NMR) and expected peaks (13C NMR) were observed. In particular, the cross peak for C−1′′ as well as the two anomeric carbons for C−1′ and C−1′′ were visible. Still, no signal was observed for position 4 of the mannose residue. This behaviour is in agreement with hindered rotation for compound 8.17 Interestingly, the NMRs of the related compounds 7 and 9 do not show this behaviour.
Final oxidation with the (2,2,6,6‐tetramethylpiperidin‐1‐yl)oxyl‐[bis(acetoxy)iodo]benzene (TEMPO‐BAIB)18 system followed by benzylation of the crude (Cs2CO3, BnBr) afforded the desired building block 9 in satisfactory 60 % yield.
Prepared trisaccharide 9 was tested in a dimethyl(methylthio)sulfonium trifluoromethansulfonate (DMTST)‐promoted glycosylation with the spacer‐containing derivative 10 10 (Scheme 2) and afforded α‐linked tetrasaccharide 11 as the sole product in 95 % yield, demonstrating excellent donor properties of trisaccharide 9 to monosaccharide acceptors.
Scheme 2.
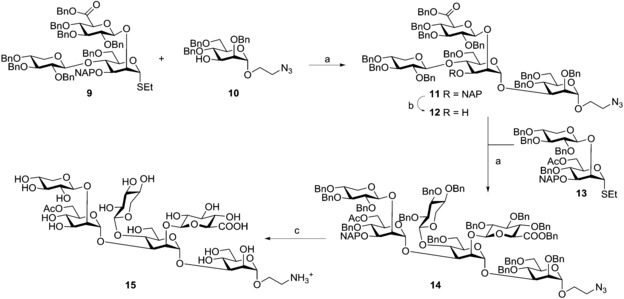
Reagents and conditions: a) DMTST, Et2O, 0 °C→20 °C, 1.5 h, 95 % for 11, 79 % for 14; b) DDQ, CH2Cl2/H2O (10:1), 20 °C, 60 min, 74 %; c) H2/Pd−C (30 bar), EtOAc, H2O, AcOH, 67 %.
The removal of the naphthalenylmethyl protecting group by reaction with 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ) in a mixture of dichloromethane/water proceeded in 74 % yield to obtain acceptor 12 ready for testing the possibility of obtaining 2,3,4‐tri‐O‐glycosylated structures. Thus, acceptor 12 was reacted with disaccharide thioglycoside 13 9 again using DMTST as promoter. Satisfactorily, hexasaccharide 14 was obtained with complete α‐selectivity and in high yield (79 %) and was then completely deprotected by means of hydrogenolysis to afford 15 in 67 % yield. This rather unusual GXM structural motif was reported recently by Nimrichter et al.19 who characterised a substituted triad from encapsulated cells that had only been described in polysaccharide fractions from a hypocapsular mutant. NMR data for 15 are in good agreement with the one reported for the polysaccharide: anomeric reported values: 4.45 ppm (GlcA), 4.35 ppm (2‐O‐Xyl), 4.25 ppm (4‐O‐Xyl); found for 15 4.47 ppm (GlcA), 4.40 ppm (2‐O‐Xyl), 4.31 ppm (4‐O‐Xyl).
A more complex acceptor than the monosaccharide 10 was then prepared to further investigate the donor properties of trisaccharide 9 (Scheme 3 and 4). Spacer‐equipped acceptor 17, obtained from 16 10 in 88 % yield, was coupled with trichloroacetimidate donor 3 16 using tert‐butyldimethylsilyl trifluoromethanesulfonate (TBDMSOTf) in the presence of acid‐washed molecular sieves yielding disaccharide 18 in 86 % yield. Benzoyl groups, again utilised for their anchimeric assistance in the glycosylation reaction, were exchanged for benzyl groups affording 20 in 77 % yield over two steps. The opening of the benzylidene ring was this time performed using Bu2BOTf/BH3 20 to afford the opposite regioselectivity to compound 1, giving the 6‐OH compound 21 (76 %), which was either acetylated (→22, 93 %) or benzylated (→23, 95 %). The 6‐O‐acetyl disaccharide 22 was then chosen for the preparation of a tetrasaccharide acceptor for the consecutive construction of the spacer‐containing C. neoformans Serotype B heptasaccharide triad motif which followed our standard sequence of reactions: NAP removal followed by DMTST‐promoted glycosylation (Scheme 4).
Scheme 3.
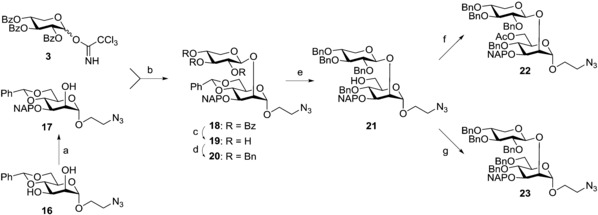
Reagents and conditions: a) Bu2SnO, Bu4NBr, NapBr, toluene, reflux, 3 h, 88 %; b) TBDMSOTf, CH2Cl2, AW‐300 MS, −78 °C→20 °C, o/n, 86 %; c) 1. NaOMe, MeOH, 20 °C; 2. Dowex H+ ion‐exchange resin, 86 %; d) NaH, BnBr, DMF, 0 °C→20 °C, 3 h, 90 %; e) Bu2BOTf, BH3 (1 m in THF), CH2Cl2, 0 °C, 90 min, 76 %; f) Ac2O, pyridine, 20 °C, 3 h, 93 %; g) NaH, BnBr, DMF, 0 °C→20 °C, 2 h, 95 %.
Scheme 4.
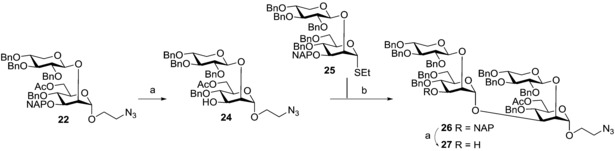
Reagents and conditions: a) DDQ, CH2Cl2/H2O (10:1), 20 °C, 60 min, 73 % for 24, 80 % for 27; b) DMTST, Et2O, 0 °C→20 °C, 3.5 h, 85 %.
The first reaction gave disaccharide acceptor 24 in 73 % yield, while the glycosylation with 25 9 afforded tetrasaccharide 26 in 85 % yield, which was in turn converted into the new acceptor 27 (80 % yield). As mentioned in the introduction, acetates are heterogenously present on the 6‐OH of mannose residues if no 4‐O‐xylose substituents are present. By choosing the correct combination from the set of thioglycoside donors (13 and 25) and spacer‐containing acceptors (24 and the one that can be prepared from 23 after NAP removal), and by following the same sequence of reactions reported in Scheme 4, all possible acetylation patterns on the tetrasaccharide motif can be obtained, permitting following investigation into the effect of the acetylation pattern on the immune response.
Finally, building block 9 was used in the glycosylation reaction with 27 to prepare the desired structural motif (Scheme 5).
Scheme 5.
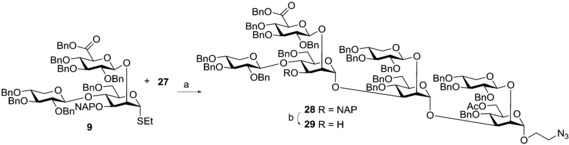
Reagents and conditions: a) DMTST, Et2O, DMTST, Et2O, 0 °C→20 °C, 3.5 h, 83 %; b) DDQ, CH2Cl2/tBuOH (10:1), 20 °C, 1.5 h, 68 %.
The reaction proceeded smoothly (83 %) and permitted, for the first time, the synthesis of monoacetyl heptasaccharide 28. This result, together with the preparation of tetrasaccharide 11, confirmed the versatility of the proposed convergent strategy which allows for installation of the GlcA containing trisaccharide in every position of the mannan triad thus overcoming the problems encountered previously with different strategies. Derivative 28 could be completely deprotected by hydrogenolysis, as shown for 14, and used to prepare a candidate vaccine after conjugation with an immunogenic protein, or could be employed as an acceptor after removal of the NAP group (→29, 68 %), to further elongate the GXM fragment.
Conclusions
In conclusion, an efficient synthesis of a GlcA‐containing trisaccharide thioglycoside building block corresponding to Cryptococcus neoformans Serotype B and C glucurono‐xylo‐mannan (GXM) oligosaccharide structure has been developed. The naphthalenylmethyl (NAP)‐protected building block was shown to be a most efficient glycosyl donor to both monosaccharide and more complex acceptors in dimethyl(methylthio)sulfonium‐trifluoromethansulfonate (DMSTS)‐promoted glycosylation reactions which proceeded with high yields and complete α‐selectivity. Subsequent removal of the 2‐NAP temporary protecting group converted obtained saccharides into new acceptors, which were shown to work well in following glycosylation reactions allowing effective construction of heavily branched 2,3,4‐subtituted motifs. Thus, the presented strategy permitted the preparation of both a hexasaccharide (14) and a Serotype B heptasaccharide structural motif, and the results demonstrate the possible synthesis of any C. neoformans GXM structure from the various available mono‐, di‐, and trisaccharide building blocks.
Experimental Section
General: Thin‐layer chromatography (TLC) was carried out on precoated 60 F254 silica gel alumina plates (Merck) using UV light and/or 8 % H2SO4 and/or AMC‐solution (ammonium molybdate, cerium (IV) sulphate, 10 % H2SO4 [5:0.1:100, w/w/v] for visualisation. Flash column chromatography was performed on silica gel (Merck, pore size 60 Å, particle size 40–63 μm). NMR spectra were recorded in CDCl3 (internal Me4Si d=0.00 ppm) at 25 °C on a Varian instrument (500 MHz for 1H and 125 MHz for 13C or 600 MHz for 1H and 150 MHz for 13C, VNMRS 500 MHz or 600 MHz, Palo Alto, USA). Coupling constants are given in Hertz (Hz). High‐resolution mass spectrometry (HRMS) spectra were recorded on a Micromass LCT instrument (Waters, Milford, USA) using electrospray ionisation (ESI) in either the positive or negative modes. Optical rotations were measured with a PerkinElmer 343 polarimeter (Waltham, USA) at the sodium D‐line (589 nm) at 20 °C using a 1 dm cell. All reactions containing air‐ and moisture‐sensitive reagents were carried out under an Ar atmosphere. Organic phases were dried over MgSO4 before evaporation, which was performed under reduced pressure at temperatures not exceeding 40 °C.
Ethyl 2,3,4,6‐tetra‐O‐benzoyl‐β‐d‐glucopyranosyl‐(1→2)‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (2). Sodium cyanoborohydride (223 mg, 3.56 mmol) was added to a solution of acetal 1 (612 mg, 0.59 mmol) in dry tetrahydrofuran (THF, 25 mL) containing crushed molecular sieves (3 Å, 150 mg). A 1 m solution of HCl in Et2O (9.0 mL, 9.0 mmol) was added dropwise at 20 °C (until pH 1–2). The reaction mixture was stirred until no starting material was detected by TLC (toluene/EtOAc, 6:1). After 30 min, Et3N (2.5 mL, 17.80 mmol) was added, followed by dropwise addition of MeOH (10 mL). CH2Cl2 (20 mL) was added, the solids were removed by filtration through a short pad of Celite, concentrated in vacuo, and then redissolved and co‐evaporated with MeOH (3×50 mL). Purification by flash column chromatography (SiO2, 100 mL, 4.5 cm, toluene→toluene/EtOAc, 96:4→93:7→90:10→87:13→85:15→80:20→75:25) gave 2 (571 mg, 93 %) as a colourless, amorphous solid: R f=0.32 (toluene/EtOAc, 6:1); [α]D 20+6.7 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.96–7.88 (m, 6 H), 7.87–7.81 (m, 2 H), 7.79–7.66 (m, 4 H), 7.54–7.38 (m, 7 H), 7.36–7.32 (m, 2 H), 7.31–7.14 (m, 11 H), 5.93 (t, J 9.7, 1 H), 5.70–5.61 (m, 1 H), 5.27 (d, J 1.0, 1 H), 5.01 (d, J 7.9, 1 H), 4.91 (d, J 11.5, 1 H), 4.65 (dd, J 3.1, J 12.1, 1 H), 4.57 (d, J 11.5, 1 H), 4.48 (dd, J 5.7, J 12.1, 1 H), 4.32 (dd, J 1.7, J 2.9, 1 H), 4.26 (s, 2 H), 4.21–4.15 (m, 1 H), 3.96 (ddd, J 3.3, J 6.6, J 9.7, 1 H), 3.81 (t, J 9.4, 1 H), 3.68 (dd, J 3.1, J 9.2, 1 H), 3.58 (dd, J 3.3, J 10.7, 1 H), 3.27 (dd, J 6.6, J 10.7, 1 H), 2.55–2.38 (m, 3 H), 1.15 ppm (t, J 7.4, 3 H); 13C NMR (125 MHz, CDCl3): δ=166.2, 166.0, 165.3, 164.9, 138.5, 135.2, 133.6, 133.4, 133.3, 133.2, 133.2, 133.1, 130.0, 129.9, 129.9, 129.8, 129.7, 129.6, 129.0, 128.9, 128.6, 128.5, 128.4, 128.4, 128.3, 128.1, 127.8, 127.5, 127.5, 127.1, 126.2, 126.1, 126.0, 99.8, 81.2, 77.9, 75.5, 73.4, 73.0, 72.7, 71.9, 71.86, 70.9, 70.9, 69.9 68.1, 63.4, 25.4, 14.8; HRMS (ESI): [M−H]− m/z calcd for C60H55O14S: 1031.3313, found: 1031.3279; Anal. calcd for C60H55O14S: C 69.75, H 5.46, S 3.10, found: C 69.43, H 5.43, S 3.28.
Ethyl 2,3,4,6‐tetra‐O‐benzoyl‐β‐d‐glucopyranosyl‐(1→2)‐[2,3,4‐tri‐O‐benzoyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (4). A catalytic amount of TMSOTf (20 μL, 11 μmol) was added to a solution of donor 3 16 (88 mg, 0.145 mmol) and acceptor 2 (116 mg, 0.113 mmol) in dry CH2Cl2 (5 mL) containing crushed molecular sieves (AW‐300, 40 mg) kept at −78 °C in an atmosphere of nitrogen. The temperature was then allowed to rise to 20 °C overnight (TLC, toluene‐EtOAc, 6:1). The reaction mixture was neutralised with Et3N (16 μL, 0.113 mmol), the solids were removed by filtration, and the filtrate was concentrated in vacuo to a yellowish foam. Purification by flash column chromatography (SiO2, 100 mL, 4.5 cm, toluene→toluene/tOAc, 98:2→96:4→94:6 →92:8→90:10→88:12→86:14) gave 4 (118 mg, 70 %) as a colourless foam: R f=0.46 (toluene/EtOAc, 9:1); [α]D 20+5.6 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.93–7.83 (m, 14 H), 7.77–7.69 (m, 4 H), 7.56–7.42 (m, 7 H), 7.39–7.19 (m, 20 H), 7.10–7.03 (m, 2 H), 5.90 (t, J 9.7 Hz, 1 H), 5.71–5.62 (m, 2 H), 5.53 (t, J 8.2 Hz, 1 H), 5.26 (dd, J 6.5 Hz, J 8.2 Hz, 1 H), 5.20 (d, J 2.9 Hz, 1 H), 5.16 (td, J 4.9 Hz, J 8.1 Hz, 1 H), 5.02 (d, J 7.9 Hz, 1 H), 4.94 (d, J 11.3 Hz, 1 H), 4.73 (d, J 6.2 Hz, 1 H), 4.68 (d, J 11.4 Hz, 1 H), 4.61 (dd, J 3.1 Hz, J 12.1 Hz, 1 H), 4.44 (dd, J 5.2 Hz, J 12.1 Hz, 1 H), 4.20 (bs, 1 H), 4.15–4.09 (m, 3 H), 4.01–3.88 (m, 4 H), 3.34 (dd, J 1.9 Hz, J 10.8 Hz, 1 H), 3.27 (dd, J 5.9 Hz, J 10.9 Hz, 1 H), 3.02 (dd, J 8.3 Hz, J 12.1 Hz, 1 H), 2.44–2.30 (m, 2 H), 1.06 ppm (t, J 7.4 Hz, 1 H); 13C NMR (125 MHz, CDCl3): δ=166.1, 165.9, 165.5, 165.5, 165.3, 165.2, 164.9, 138.6, 135.6, 133.5, 133.4, 133.3, 133.3, 133.3, 133.2, 133.1, 133.0, 130.0, 130.0, 129.9, 129.9, 129.9, 129.8, 129.7, 129.5, 129.4, 129.4, 129.0, 128.9, 128.5, 128.5, 128.5, 128.4, 128.4, 128.2, 128.2, 128.0, 127.8, 127.5, 127.4, 127.2, 126.6, 126.1, 125.9, 100.8, 100.6, 81.3, 77.3, 77.1, 76.2, 73.1, 72.9, 72.6, 72.2, 71.9, 71.5, 71.4 (2C), 69.8, 69.5, 69.3, 63.3, 61.9, 25.2, 14.7 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C86H76O21NaS: 1499.4498, found: 1499.4547; Anal. calcd for C86H76O21S: C 69.91, H 5.18, S 2.17, found: C 69.75, H 5.28, S 2.43.
Ethyl β‐d‐glucopyranosyl‐(1→2)‐[β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (5). A catalytic amount of sodium methoxide (75 mg, 1.38 mmol) was added to a solution of 4 (4.10 g, 2.77 mmol) in dry MeOH (150 mL). The mixture was stirred at 20 °C overnight (TLC, CH2Cl2−MeOH, 9:1). After complete conversion, Dowex (H+) acidic ion‐exchange resin was added for neutralisation, the resin was filtered off, washed with MeOH (30 mL), and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, 600 mL, 7.5 cm, DCM→DCM/MeOH, 95:5→90:10→88:12→86:14→84:16→83:17→82:18) gave 5 (1.68 g, 80 %) as a colourless, amorphous solid: R f=0.13 (CH2Cl2/MeOH, 9:1); [α]D 20+45.6 (c 1.0, MeOH); 1H NMR (600 MHz, DMSO) δ 7.93 (1 H, s), 7.88–7.84 (1 H, m), 7.81 (m, 2 H), 7.56 (dd, 1.6 Hz, J 8.5 Hz, 1 H), 7.48–7.43 (m, 2 H), 7.43–7.28 (m, 4 H), 7.27–7.23 (m, 1 H), 5.40 (d, J 2.0 Hz, 1 H), 5.10 (d, J 4.8 Hz, 1 H), 4.94–4.83 (m, 5 H), 4.69–4.60 (m, 2 H), 4.53 (d, J 11.9 Hz, 1 H), 4.44 (d, J 11.9 Hz, 1 H), 4.39 (t, J 5.6 Hz, 1 H), 4.33 (d, J 7.8 Hz, 1 H), 4.22 (d, J 7.4 Hz, 1 H), 4.19 (t, J 2.8 Hz, 1 H), 4.00–3.90 (m, 2 H), 3.86 (dd, J 4.8 Hz, J 11.1 Hz, 1 H), 3.75 (dd, J 1.6 Hz, J 11.2 Hz, 1 H), 3.73–3.64 (m, 2 H), 3.61 (dd, J 3.4 Hz, J 8.3 Hz, 1 H), 3.42–3.35 (m, 1 H), 3.32–3.27 (m, 1 H), 3.19–3.09 (m, 2 H), 3.09–2.96 (m, 4 H), 2.88 (dd, J 10.2 Hz, J 11.5 Hz, 1 H), 2.64–2.49 (m, 2 H), 1.16 ppm (t, J 7.4 Hz, 3 H); 13C NMR (125 MHz, DMSO): δ=138.6, 136.6, 132.8, 132.4, 128.2, 127.6, 127.5, 127.4, 127.3, 126.2, 126.0, 125.9, 125.6, 103.9, 101.6, 81.8, 77.2, 76.8, 76.7, 76.6, 74.7, 74.1, 73.9, 73.1, 72.2, 71.7, 70.3, 70.3, 69.7, 68.8, 65.9, 61.5, 24.5, 14.9 ppm; HRMS (ESI): [M−H]− m/z calcd for C37H47O14S: 747.2687, found: 747.2702.
Ethyl 6‐O‐tert‐butyldimethylsilyl‐β‐d‐glucopyranosyl‐(1→2)‐[β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (6). tert‐Butyldimethylchlorosilane (82 mg, 0.55 mmol) and a catalytic amount of DMAP (1 mg, 8 μmol) were added to a solution of 5 (273 mg, 0.36 mmol) in dry pyridine (10 mL), and the mixture was stirred at 20 °C for 4 h. The progress of the reaction was followed by TLC (DCM/MeOH, 9:1). The mixture was concentrated in vacuo, and then redissolved and coevaporated with toluene (3×30 mL) (water bath temperature of rotary evaporator: <30 °C). Purification by flash column chromatography (SiO2, DCM→DCM/MeOH, 95:5→94:6→93:7→92:8→91:9→90:10) gave 6 (240 mg, 76 %) as colourless, amorphous solid: R f=0.35 (CH2Cl2/MeOH, 9:1); [α]D 20+31.5 (c 0.89, MeOH); 1H NMR (500 MHz, CDCl3): δ=7.82–7.74 (m, 4 H), 7.53 (d, J 8.5 Hz, 1 H), 7.47–7.39 (m, 2 H), 7.35–7.19 (m, 5 H), 5.32 (s, 1 H), 4.85 (d, J 11.5 Hz, 1 H), 4.78 (d, J 11.5 Hz, 1 H), 4.59 (d, J 12.0 Hz, 1 H), 4.51 (d, J 12.0 Hz, 1 H), 4.43–4.36 (m, 2 H), 4.31–4.19 (m, 3 H), 4.13–4.07 (m, 2 H), 3.93–3.77 (m, 6 H), 3.73–3.64 (m, 3 H), 3.55–3.40 (m, 4 H), 3.35–3.26 (m, 3 H), 3.00 (t, J 10.6 Hz, 1 H), 2.89 (bs, 1 H), 2.61–2.46 (m, 2 H), 1.20 (t, J 7.4 Hz, 1 H), 0.84 (s, 9 H), 0.00 (s, 3 H), 0.00 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=137.6, 134.8, 133.3, 133.2, 128.6, 128.2, 128.2, 128.1, 128.0, 127.8, 127.7, 126.7, 126.2, 126.1, 103.2, 100.7, 82.6, 77.0, 76.3, 76.1, 75.6, 74.2, 74.0, 73.7, 73.5, 72.3, 72.2, 72.0, 71.5, 69.7, 68.7, 65.6, 64.3, 25.9, 25.6, 18.3, 15.0, −5.3, −5.4 ppm; HRMS (ESI): [M−H]− m/z calcd for C43H61O14SSi: 861.3551, found: 861.3558
Ethyl 2,3,4‐tri‐O‐benzyl‐6‐O‐tert‐butyldimethylsilyl‐β‐d‐glucopyranosyl‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (7). NaH (100 mg, 2.49 mmol, 60 % oil dispersion) was washed with pentane (3×10 mL) prior to use. NaH was added portionwise to a solution of 6 (239 mg, 0.27 mmol) in dry DMF (10 mL) at 0 °C in an atmosphere of nitrogen. After 15 min, benzyl bromide (236 μL, 2.00 mmol) was added dropwise at 0 °C under vigorous stirring. The temperature was then allowed to rise to 20 °C overnight (TLC, toluene/EtOAc, 9:1). After complete consumption of the starting material, residual NaH was quenched with MeOH (1 mL), and then with H2O (50 mL). The resulting mixture was extracted once with EtOAc (40 mL), the layers were separated, and the organic layer was washed with brine (1×40 mL), dried over MgSO4, and concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 97:3→94:6→92:8→91:9) gave 7 (300 mg, 77 %) as a colourless syrup. R f=0.61 (toluene/EtOAc, 9:1); [α]D 20+29.3 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.89 (s, 1 H), 7.85–7.74 (m, 3 H), 7.62 (d, J 9.7 Hz, 1 H), 7.49–7.13 (m, 35 H), 7.10–7.02 (m, 2 H), 5.47 (d, J 2.1 Hz, 1 H), 5.14 (d, J 10.4 Hz, 1 H), 5.06 (d, J 12.4 Hz, 1 H), 4.94 (d, J 11.0 Hz, 1 H), 4.87–4.69 (m, 7 H), 4.66 (m, 2 H), 4.57–4.50 (m, 3 H), 4.38–4.22 (m, 5 H), 4.08–4.02 (m, 1 H), 3.92–3.75 (m, 5 H), 3.68–3.51 (m, 5 H), 3.43 (t, J 8.9 Hz, 1 H), 3.37–3.28 (m, 2 H), 2.90 (dd, J 10.1 Hz, J 11.6 Hz, 1 H), 2.68–2.54 (m, 2 H), 1.26 (t, J 7.4 Hz, 3 H), 0.85 (s, 9 H), 0.01 (s, 3 H), 0.00 ppm (s, 3 H); 13C NMR (126 MHz, CDCl3): δ=138.9, 138.8, 138.8, 138.7, 138.4, 138.4, 138.3, 136.3, 133.4, 133.0, 129.1, 129.0, 128.5, 128.4, 128.4, 128.2, 128.2, 128.2, 128.0, 128.0, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 127.6, 127.5, 127.4, 126.8, 126.6, 125.9, 125.6, 103.4, 102.0, 85.0, 84.1, 82.4, 81.8, 78.3, 77.5, 76.4, 76.3, 75.7, 75.5, 75.4, 75.0, 75.0, 74.9, 74.6, 73.0, 72.9, 71.9, 71.1, 69.0, 63.6, 62.7, 26.2, 26.1, 25.5, 18.4, 15.0, −5.1, −5.2 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C85H98O14NaSSi: 1425.6344, found: 1425.6320.
Ethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyl‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (8). TBAF trihydrate (52 mg, 0.16 mmol) was added to a solution of 7 (153 mg, 0.11 mmol) in THF (5 mL), and the mixture was stirred at 20 °C for 2 h (TLC, toluene/EtOAc, 9:1). After complete consumption of the starting material, the reaction mixture was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 93:7→90:10→87:13→84:16→81:19→78:22→75:25) gave 8 (135 mg, 96 %) as a colourless, amorphous solid: R f=0.39 (toluene/EtOAc, 6:1); [α]D 20+34.5 (c 0.42, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.84–7.77 (m, 4 H, Har), 7.56–7.54 (m, 1 H, Har), 7.48–7.44 (m, 2 H, Har), 7.38–7.36 (m, 2 H, Har), 7.32–7.20 (m, 31 H, Har), 7.12–7.11 (m, 2 H, Har), 5.37 (d, 1 H, J 3.4 Hz, H‐1), 5.09 (d, 1 H, J 10.4 Hz, PhCH 2), 4.93 (d, 1 H, J 11.1 Hz, PhCH 2), 4.90–4.84 (ABq, 2 H, PhCH 2), 4.82–4.70 (m, 6 H, PhCH 2), 4.63–4.50 (m, 5 H, H‐1′, PhCH 2), 4.35–4.26 (m, 3 H, H‐1′′, PhCH 2), 4.17 (m, 1 H, H‐4), 4.09 (m, 1 H, H‐2), 4.04–4.02 (m, 1 H, H‐5), 3.92 (m, 1 H, H‐3), 3.78–3.73 (m, 3 H, H‐6a, H‐6′a, H‐5′′a), 3.63–3.49 (m, 5 H, H‐6b, H‐2′, H‐3′, H‐6′b, H‐4′′), 3.43–3.39 (m, 2 H, H‐4′, H‐3′′), 3.33–3.29 (m, 1 H, H‐5′), 3.27 ( t, 1 H, J 8.4 Hz, H‐2′′), 2.86–2.80 (m, 1 H, H‐5′′b), 2.69–2.56 (m, 2 H, SCH 2CH3), 1.26 ppm (t, 3 H, J 7.4 Hz, SCH2CH 3); 13C NMR (125 MHz, CDCl3): δ 138.7, 138.7, 138.5, 138.5, 138.3 (2C), 138.0, 136.1, 133.2, 133.0 (Car,quart), 128.8, 128.44, 128.40, 128.33, 128.28, 128.22, 128.21, 128.17, 128.1, 128.0, 127.9, 127.83, 127.78, 127.76, 127.7, 127.6, 127.51, 127.47, 127.4, 126.7, 126.1, 125.8 (Car), 104.1 (C‐1′′), 103.2 (C‐1′), 84.5 (C‐3′), 84.0 (C‐3′′), 82.2 (2C, C‐1, C‐2′′), 81.7 (C‐2′), 78.0 (C‐4′′), 77.8 (C‐2), 77.6 (C‐4′), 77.0 (C‐3), 75.6 (PhCH2), 75.4 (PhCH2), 75.4 (C‐5′), 75.1 (PhCH2), 75.0 (PhCH2), 74.8 (PhCH2), 73.0 (PhCH2), 73.0 (PhCH2), 72.4 (NapCH2), 72.0 (C‐5), 69.0 (C‐6), 63.6 (C‐5′′), 62.2 (C‐6′), 25.4 (SCH2CH3), 15.0 ppm (SCH2 CH3). The signal of C‐4 could not be observed; HRMS (ESI): [M+Na]+ m/z calcd for C79H84O14NaS: 1311.5479, found: 1311.5487.
Ethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐1‐thio‐α‐d‐mannopyranoside (9). TEMPO (2 mg, 13 μmol) and BAIB (300 mg, 0.93 mmol) were added to a vigorously stirred solution of 8 (116 mg, 90 μmol) in CH2Cl2/H2O (12 mL, 3:1), and the mixture was stirred at 20 °C for 4 h. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 9:1). The reaction was quenched by adding 10 % aq. Na2S2O3 solution (10 mL). The resulting mixture was extracted once with EtOAc (20 mL), the layers were separated, and the organic layer was washed with brine (1×10 mL), dried over MgSO4, and concentrated in vacuo. Cs2CO3 (57 mg, 0.17 mmol) was added to a solution of the crude in dry DMF (3 mL) at 20 °C. After 15 min, benzyl bromide (22 μL, 0.18 mmol) was added dropwise at 0 °C. The temperature was then allowed to rise to 20 °C over 16 h (TLC, toluene/EtOAc 9:1). After complete consumption of the starting material, water (10 mL) was added, and the resulting mixture was extracted with Et2O (2×15 mL), the layers were separated, and the organic layer was dried over MgSO4, and concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene‐EtOAc, 70:30) gave 9 (75 mg, 60 %) as a pale yellow syrup: R f=0.45 (toluene/EtOAc, 9:1); [α]D 20+31.7 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.84–7.77 (m, 3 H), 7.74 (d, J 8.4 Hz, 1 H), 7.59–6.97 (m, 43 H), 5.39 (s, 1 H), 5.11 (d, J 10.1 Hz, 1 H), 5.07–5.00 (m, 2 H), 4.92–4.85 (m, 2 H), 4.84–4.77 (m, 2 H), 4.77–4.69 (m, 4 H), 4.65–4.60 (m, 2 H), 4.55–4.50 (m, 2 H), 4.48–4.44 (m, 2 H), 4.37 (d, J 7.5 Hz, 1 H), 4.31–4.24 (m, 2 H), 4.21 (s, 2 H), 4.05 (dd, J 3.8 Hz, J 9.6 Hz, 1 H), 3.94 (d, J 9.8 Hz, 1 H), 3.88–3.74 (m, 4 H), 3.67–3.53 (m, 4 H), 3.43 (t, J 8.9 Hz, 1 H), 3.33–3.29 (m, 1 H), 2.92–2.87 (m, 1 H), 2.65–2.54 (m, 2 H), 1.25 ppm (t, J 7.4 Hz, 3 H); 13C NMR (125 MHz, CDCl3): δ=168.1, 138.8, 138.8, 138.7, 138.5, 138.4, 138.3, 138.0, 136.2, 135.1, 133.4, 133.1, 129.3, 128.8, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.8, 127.7, 127.7, 127.5, 127.5, 127.4, 126.8, 126.6, 125.9, 125.7, 103.5, 102.8, 84.2, 84.1, 82.5, 82.0, 81.1, 79.0, 78.5, 76.6, 76.3, 75.8, 75.6, 75.2, 75.2, 75.1, 74.9, 74.4, 73.0, 73.0, 72.0, 71.5, 68.9, 67.4, 63.7, 25.7, 15.1 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C86H88O15NaS: 1415.5742, found: 1415.5712.
2‐Azidoethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranosyl‐(1→3)‐2,4,6‐tri‐O‐benzyl‐α‐d‐mannopyranoside (11). A mixture of 10 (10 mg, 19.3 μmol), 9 (40 mg, 29 μmol), and crushed molecular sieves (4 Å, 20 mg) in dry Et2O (2 mL) was stirred at 20 °C for 30 min. The reaction mixture was cooled to 0 °C, freshly prepared DMTST (15 mg, 60 μmol) was added, and the reaction mixture was stirred at 0 °C for 30 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 9:1). The cooling bath was removed and stirring was continued at 20 °C for 1 h. Et2O (5 mL) was added, and the reaction was quenched with Et3N (50 μL) at 0 °C. The solids were removed by filtration through a pad of Celite, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene/EtOAc, 97:3→70:30) gave 11 (34 mg, 95 %) as a colourless syrup: R f=0.56 (toluene/EtOAc, 9:1); [α]D 20+7.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.77 (s, 1 H), 7.72–7.63 (m, 3 H), 7.53 (d, J 8.4, Hz, 1 H), 7.41–6.98 (m, 57 H), 5.20 (d, J 1.7 Hz, 1 H), 5.08 (d, J 10.5 Hz, 1 H), 5.05–4.99 (m, 2 H), 4.86–4.76 (m, 4 H), 4.76–4.34 (m, 16 H), 4.30–4.15 (m, 4 H), 4.11 (dd, J 9.5, 3.1 Hz, 1 H), 4.06 (d, J 7.8 Hz, 1 H), 4.01–3.89 (m, 3 H), 3.90–3.39 (m, 15 H), 3.34–3.26 (m, 3 H), 3.22 (ddd, J 13.2, 6.1, 3.7 Hz, 1 H), 2.93 ppm (dd, J 11.9, 9.8 Hz, 1 H); 13C NMR (151 MHz, CDCl3): δ=168.2, 138.8, 138.8, 138.7, 138.7, 138.5, 138.4, 138.4, 138.4, 138.4, 138.4, 136.4, 135.2, 133.4, 133.0, 129.2, 128.8, 128.7, 128.6, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 127.6, 127.6, 127.6, 127.5, 127.4, 127.3, 126.9, 126.8, 126.8, 125.9, 125.7, 103.7 (J C−H 161 Hz), 103.2 (J C−H 162.5 Hz), 99.7 (J C−H 172 Hz), 98.0 (J C−H 171.5 Hz), 84.2, 83.6, 82.6, 81.0, 78.8, 78.7, 78.5, 78.4, 75.7, 75.6, 75.6, 75.5, 75.0, 74.9, 74.8, 74.6, 74.6, 74.6, 74.1, 73.5, 73.2, 73.0, 72.8, 72.7, 72.4, 72.0, 69.1, 67.3, 66.7, 63.7, 50.5 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C113H115O21N3Na: 1872.7921, found: 1872.7861.
2‐Azidoethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐2,4,6‐tri‐O‐benzyl‐α‐d‐mannopyranoside (12). DDQ (10 mg, 44 μmol) was added to a vigorously stirred solution of 11 (34 mg, 18 μmol) in CH2Cl2/tBuOH (4.4 mL, 10:1) at 20 °C. The progress of the reaction was monitored by TLC (toluene/EtOAc, 9:1). After 60 min, the reaction was quenched by adding satd. NaHCO3 solution (15 mL). The resulting mixture was extracted once with CH2Cl2 (20 mL), the layers were separated, and the organic layer was washed with, 10 % aq. Na2S2O3 solution (1×15 mL), dried over MgSO4, and concentrated in vacuo to a yellow oil. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 97:3→70:30) gave 12 (23 mg, 74 %) as a colourless syrup: R f=0.24 (toluene/EtOAc, 9:1); [α]D 20+10.2 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.51–6.92 (m, 55 H), 5.22 (d, J 1.6 Hz, 1 H), 5.11 (s, 2H), 5.03 (d, J 10.6 Hz, 1 H), 4.85–4.76 (m, 4 H), 4.74–4.70 (m, 2 H), 4.69–4.56 (m, 8 H), 4.52–4.48 (m, 2 H), 4.44 (d, J 10.6 Hz, 1 H), 4.39 (d, J 11.0 Hz, 1 H), 4.25 (d, J 7.8 Hz,1 H), 4.18 (d, J 11.8 Hz, 1 H), 4.15–4.11 (m, 2 H), 4.06 (dt, J 9.2, 3.8 Hz, 1 H), 4.04–4.00 (m, 2 H), 3.99–3.88 (m, 5 H), 3.83 (ddd, J 10.1, 6.0, 3.6 Hz, 1 H), 3.76 (ddd, J 9.9, 4.9, 1.9 Hz, 1 H), 3.74–3.68 (m, 2 H), 3.68–3.59 (m, 5 H), 3.53 (ddd, J 10.7, 7.0, 3.6 Hz, 1 H), 3.47 (t, J 9.1 Hz, 1 H), 3.44–3.40 (m, 2 H), 3.36–3.28 (m, 3 H), 3.24 (ddd, J 13.2, 6.1, 3.7 Hz, 1 H), 3.13 ppm (t, J 11.1 Hz, 1 H); 13C NMR (151 MHz, CDCl3): δ=168.2, 138.7, 138.6, 138.6, 138.4, 138.4, 138.3, 138.3, 138.2, 138.2, 135.3, 129.0, 128.7, 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.3, 128.2, 128.1, 128.1, 128.0, 128.0, 127.8, 127.8, 127.8, 127.7, 127.7, 127.7, 127.6, 127.6, 127.6, 127.4, 127.1, 126.8, 104.0, 103.1, 100.1, 97.9, 84.1, 83.5, 81.9, a80.9, 79.4, 79.0, 78.3 (2C), 78.0, 77.9, 75.8, 75.7, 75.2, 74.9, 74.8, 74.8, 74.5, 74.4, 73.5, 73.5, 73.1, 72.6, 72.5, 72.0, 69.1, 68.9, 68.5, 67.3, 66.8, 64.3, 50.5 ppm. HRMS (ESI): [M+Na]+ m/z calcd for C102H107O21N3Na: 1732.7205, found: 1732.7264.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐6‐O‐acetyl‐4‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranosyl‐(1→3)‐[benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate‐(1→2)][2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐2,4,6‐tri‐O‐benzyl‐α‐d‐mannopyranoside (14). A mixture of 12 (22 mg, 13 μmol), 13 9 (18 mg, 20 μmol), and crushed molecular sieves (4 Å, 20 mg) in dry Et2O (3 mL) was stirred at 20 °C for 30 min. The reaction mixture was cooled to 0 °C, freshly prepared DMTST (14 mg, 56 μmol) was added, and the reaction mixture was stirred at 0 °C for 30 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 9:1). The cooling bath was removed, and stirring was continued at 20 °C for 1 h. Et2O (6 mL) was added and the reaction was quenched with Et3N (40 μL, 0.20 mmol) at 0 °C. The solids were removed by filtration through a pad of Celite, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 95:5→60:40) gave 14 (26 mg, 79 %) as a colourless syrup: R f=0.47 (toluene/EtOAc, 9:1); [α]D 20 −4.2 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.95 (s, 1 H), 7.82–7.74 (m, 3 H), 7.64 (dd, J 8.3, 1.5 Hz, 1 H), 7.48 −6.91 (m, 78 H), 5.64 (d, J 1.4 Hz, 1 H), 5.25 (s, 1 H), 5.13 (d, J 10.4 Hz, 1 H), 5.08 (m, 2 H), 4.99 (m, 2 H), 4.87 (d, J 10.9 Hz, 1 H), 4.85–4.72 (m, 5 H), 4.72–4.44 (m, 18 H), 4.41–4.34 (m, 4 H), 4.33–4.20 (m, 6 H), 4.20–4.15 (m, 2 H), 4.13–4.08 (m, 1 H), 4.06 (d, J 7.5 Hz, 1 H), 4.03–3.81 (m, 10 H), 3.78–3.71 (m, 2 H), 3.71–3.60 (m, 6 H), 3.59–3.53 (m, 2 H), 3.49 (t, J 8.5 Hz, 1 H), 3.43–3.33 (m, 3 H), 3.28–3.24 (m, 2 H), 3.24–3.15 (m, 2 H), 2.96 (t, J 11.3 Hz, 1 H), 1.63 ppm (s, 3 H); 13C NMR (151 MHz, CDCl3): δ=170.9, 168.2, 139.0, 138.9, 138.8, 138.8, 138.8, 138.7, 138.6, 138.6, 138.4, 138.2, 138.2 (3C), 138.1, 136.7, 135.5, 133.5, 133.1, 129.2, 128.8, 128.8, 128.5, 128.5, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.2, 128.1, 127.9, 127.9, 127.8, 127.8, 127.8, 127.8, 127.7, 127.7, 127.7, 127.6, 127.6, 127.5, 127.4, 127.4, 127.3, 127.3, 127.1, 127.1, 126.7, 125.9, 125.8, 104.3 (J C−H 161 Hz), 103.7 (J C−H 162 Hz), 102.9 (J C−H 159.5 Hz), 100.1 (J C−H 171.6 Hz), 99.6 (J C−H 176.4 Hz), 98.0 (J C−H 169.8 Hz), 84.1, 84.0, 83.5, 82.6, 81.1, 81.0, 79.5, 79.2, 79.2, 79.1, 79.0, 78.4, 78.0, 75.7, 75.6, 75.5, 75.5, 75.4, 75.0, 75.0, 74.9, 74.7, 74.7, 74.6, 74.5, 74.4, 74.3, 73.7, 73.6, 73.3, 73.1, 73.0, 72.5, 72.5, 72.0, 71.7, 70.6, 69.1, 68.2, 67.2, 66.9, 64.3, 64.1, 64.0, 50.5, 20.8 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C154H159O31N3Na: 2569.0855, found: 2569.0840.
2‐Aminoethyl β‐d‐xylopyranosyl‐(1→2)‐6‐O‐acetyl‐α‐d‐mannopyranosyl‐(1→3)‐[ β‐d‐glucopyranosyluronate‐(1→2)][ β‐d‐xylopyranosyl‐(1→4)]‐α‐d‐mannopyranosyl‐(1→3)‐α‐d mannopyranoside (15). 10 % w Pd/C (17 mg, 16.1 mmol) was added to a solution of compound 14 (26 mg, 12.4 mmol) in AcOEt/H2O/AcOH (4:2:1, 1.75 mL). The mixture was hydrogenolysed in a high‐pressure reactor (Berghof, Eningen, Germany) at 20 °C (p=30 bar). After 48 h, the solids were removed by filtration using a ‘sandwich filter’ (3 frits stacked on top of each other in the following order: 20 μm, 10 μm, 5 μm), rinsed with H2O (3×2 mL) and EtOH (3×2 mL), and the filtrate was concentrated in vacuo. Purification by reversed‐phase chromatography (C‐18, H2O/MeOH, 9:1→8:2→7:3→6:4→2:8→0:10), followed by freeze‐drying gave 15 (7.0 mg, 67 %) as a colourless, amorphous solid; [α]20 D+6.0 (c 0.40, H2O); 1H NMR (500 MHz, D2O): δ=5.25 (s, 1 H), 5.17 (s, 1 H), 4.90 (s, 1 H), 4.47 (d, J 7.8 Hz, 1 H), 4.40 (d, J 7.8 Hz, 1 H), 4.37–4.33 (m, 2 H), 4.31 (d, J 7.8 Hz, 1 H), 4.26–4.12 (m, 5 H), 4.11–3.86 (m, 10 H), 3.84–3.57 (m, 10 H), 3.54–3.20 (m, 9 H), 2.17 ppm (s, 3 H); 13C anomeric signals taken from Heteronuclear Single Quantum Coherence (HSQC) spectroscopy: 103.7, 102.8, 101.7, 100.9, 99.8, 99.6 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C38H63O31NNa: 1052.3282, found: 1052.3330.
2‐Azidoethyl 4,6‐O‐benzylidene‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (17). A solution 16 (10.0 g, 29.6 mmol) and dibutyltin oxide (9.96 g, 40.0 mmol) in anhydrous toluene (320 mL) was heated at reflux with continuous removal of water (Dean–Stark trap). After 3 h, the mixture was concentrated to half volume, tetrabutylammonium iodide (14.8 g, 40.0 mmol) and 2‐(bromomethyl)naphthalene (7.21 g, 32.6 mmol) were added, and the reaction mixture was brought to reflux for another 3 h. To the reaction mixture, EtOAc (400 mL) was added, and the organic layer was then washed sequentially with water (3×200 mL), 10 % aq. KF‐solution (3×200 mL), and brine (2×200 mL), dried over MgSO4, and concentrated in vacuo to a yellow oily residue. Purification by flash column chromatography (SiO2, pentane→pentane/Et2O, 50:50→35:65→25:75→15:85) gave 17 (12.5 g, 88 %) as a pale yellow syrup: R f=0.33 (toluene/EtOAc, 3:1); [α]D 20+28.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.85–7.36 (m, 12 H), 5.63 (s, 1 H), 4.99 (d, J 12.0 Hz, 1 H), 4.91 (d, J 1.5 Hz,1 H), 4.90 (d, J 12.0 Hz, 1 H), 4.30–4.24 (m, 1 H, H‐6a), 4.16–4.12 (m, 2 H), 3.99 (dd, J 3.0 Hz, J 9.0 Hz, 1 H), 3.89–3.83 (m, 3 H), 3.60 (ddd, J 3.7 Hz, J 6.8 Hz, J 10.6 Hz, 1 H), 3.42–3.32 (m, 2 H,), 2.74 ppm (d, J 1.5 Hz, 1 H); 13C NMR (125 MHz, CDCl3): δ=137.7, 135.5, 133.4, 133.2, 129.1, 128.4, 128.4, 128.1, 127.8, 126.8, 126.3, 126.3, 126.1, 125.9, 101.9, 100.4, 78.8, 75.6, 73.2, 69.9, 69.0, 66.9, 63.9, 50.5 ppm; HRMS (ESI): [M+H]+ m/z calcd for C26H28N3O6: 478.1978, found: 478.1972.
2‐Azidoethyl 2,3,4‐tri‐O‐benzoyl‐β‐d‐xylopyranosyl‐(1→2)‐4,6‐O‐benzylidene‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (18). A catalytic amount of TBDMSOTf (335 μL, 1.46 mmol) was added to a solution of 3 16 (8.89 g, 14.66 mmol) and 17 (7.00 g, 14.66 mmol) in dry CH2Cl2 (125 mL) containing crushed molecular sieves (AW‐300, 180 mg) kept at −78 °C in an atmosphere of nitrogen. The temperature was then allowed to rise to 20 °C overnight (TLC, toluene/EtOAc, 6:1). The reaction mixture was neutralised with Et3N (1.23 mL, 8.79 mmol), the solids were removed by filtration, and the filtrate was concentrated in vacuo to a yellowish foam. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 98:2→96:4→94:6→92:8→90:10→88:12→86:14) gave 18 (11.70 g, 86 %) as a colourless, amorphous solid: R f=0.42 (toluene/EtOAc, 9:1); [α]D 20 −48.8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=8.23–8.22 (m, 2 H), 8.08–8.07 (m, 2 H), 7.97–7.95 (m, 2 H), 7.87 (m, 1 H), 7.81–7.78 (m, 2 H), 7.71–7.69 (m, 1 H), 7.58–7.36 (m, 15 H), 7.27–7.24 (m, 2 H), 5.61 (t, J 4.2 Hz, 1 H), 5.35–5.32 (m, 1 H), 5.15 (d, J 3.0 Hz, 1 H), 5.11 (s, 1 H), 5.10 (d, J 2.4 Hz, 1 H), 5.06–4.98 (ABq, 2 H), 4.95 (dd, J 13.0 Hz, J 1.6 Hz, 1 H), 4.87 (d, J 1.6 Hz, 1 H), 4.30 (dd, J 3.4 Hz, J 1.6 Hz 1 H), 4.10–4.05 (m, 2 H), 3.99 (t, J 9.5 Hz, 1 H), 3.82–3.76 (m, 2 H), 3.75–3.70 (m, 1 H), 3.55–3.50 (m, 1 H), 3.48 (t, 1 H, J 10.5 Hz), 3.37–2.27 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=165.8, 165.5, 165.1, 137.9, 136.1, 133.7, 133.6, 133.2, 130.6, 130.3, 130.2, 129.8, 129.6, 129.5, 129.2, 128.7, 128.6, 128.5, 128.3, 128.2, 127.9, 126.5, 126.4, 126.3, 126.0, 125.9, 101.7, 98.4, 96.8, 78.7, 75.0, 74.7, 73.3, 68.9, 68.5, 68.1, 67.5, 67.0, 64.7, 59.6, 50.6 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C52H47N3O13Na: 944.3007, found: 944.2960.
2‐Azidoethyl β‐d‐xylopyranosyl‐(1→2)‐4,6‐O‐benzylidene‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (19). A catalytic amount of sodium methoxide (68 mg, 1.27 mmol) was added to a solution of 18 (11.70 g, 12.69 mmol) in dry MeOH (250 mL). The mixture was swirled at 20 °C overnight (TLC, CH2Cl2/MeOH, 9:1). After complete conversion, Dowex (H+) acidic ion exchange resin was added for neutralisation, the resin was filtered off, washed with MeOH (50 mL), and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, DCM→DCM/MeOH, 99:1→98:2→97:3→96:4→95:5) gave 19 (11.70 g, 86 %) as a colourless, amorphous solid: R f=0.39 (DCM/MeOH, 9:1); [α]D 20 −1.8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.82–7.78 (m, 3 H), 7.75–7.73 (m, 1 H), 7.52–7.44 (m, 5 H), 7.40–7.36 (m, 3 H), 5.66 (s, 1 H), 5.05 (d, J 11.7 Hz, 1 H,), 4.88 (d, J 11.7 Hz, 1 H,), 4.85 (d, J 1.5 Hz, 1 H,), 4.53 (d, J 7.0 Hz, 1 H,), 4.27–4.35 (m, 1 H), 4.21–4.17 (m, 2 H), 4.08 (dd, J 3.0 Hz, J 9.0 Hz, 1 H,), 4.05–4.02 (m, 2 H), 3.89–3.81 (m, 3 H), 3.64–3.58 (m, 2 H), 3.49–3.46 (m, 2 H), 3.37 (m, 2 H), 3.26 (dd, J 9.5 Hz. J 11.5 Hz, 1 H,), 3.14 (s br, 1 H), 2.67 ppm (s br, 1 H); 13C NMR (125 MHz, CDCl3): δ=137.4, 134.6, 133.2, 133.1, 129.0, 128.3, 128.0, 127.7, 127.4, 126.1, 101.7, 101.3, 100.0, 79.3, 74.8, 74.5, 74.5, 71.6, 69.8, 69.5, 68.7, 66.7, 65.3, 64.1, 50.4 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C31H35N3O10Na: 632.2220, found: 632.2195.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐4,6‐O‐benzylidene‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (20). NaH (3.81 g, 95.61 mmol, 60 % oil dispersion) was washed with pentane (3×100 mL) prior to use. NaH was added portionwise to a solution of 19 (12.96 g, 21.24 mmol) in dry DMF (200 mL) kept at 0 °C under an atmosphere of nitrogen. After 15 min, benzyl bromide (10.09 mL, 84.96 mmol) was added dropwise at 0 °C under vigorous stirring. The temperature was then allowed to rise to 20 °C over 3 h (TLC, toluene/EtOAc, 6:1). After complete consumption of the starting material, residual NaH was quenched with MeOH, and then with H2O (300 mL). The resulting mixture was extracted once with EtOAc (600 mL), the layers were separated, and the organic layer was washed with brine (1×400 mL), dried over MgSO4, and concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 98:2→96:4→94:6→92:8→90:10→88:12) gave 20 (16.87 g, 90 %) as a colourless syrup: R f=0.55 (toluene/EtOAc, 6:1); [α]D 20 −8.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.86 (m, 1 H), 7.81–7.79 (m, 1 H), 7.76–7.74 (m, 1 H), 7.68–7.66 (m, 1 H), 7.52–7.48 (m, 3 H),7.44–7.42 (m, 2 H), 7.39–7.27 (m, 18 H), 5.61 (s, 1 H), 4.98 (d, J 10.5 Hz, 1 H), 4.95 (d, J 1.6 Hz, 1 H),4.91–4.85 (m, 4 H), 4.73–4.69 (m, 2 H), 4.61 (d, J 11.6 Hz, 1 H), 4.45 (d, J 7.2 Hz, 1 H), 4.28 (m, 1 H), 4.21–4.17 (m, 2 H), 4.03–3.99 (m, 2 H), 3.86–3.78 (m, 3 H), 3.66 (ddd, J 5.2 Hz, J 8.3 Hz, J 9.5 Hz, 1 H), 3.59 (t, J 8.8 Hz, 1 H), 3.56–3.49 (m, 2 H), 3.35 (ddd, J 3.5 Hz, J 7.1 Hz, J 13.3 Hz, 1 H,), 3.30–3.23 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=138.7, 138.3, 138.1, 137.6, 136.0, 133.3, 132.9, 128.8, 128.5, 128.4, 128.3, 128.2, 127.9, 127.8, 127.6, 126.3, 126.1, 125.8, 125.6, 103.9, 101.6, 99.1, 83.8, 81.5, 78.4, 77.8, 76.2, 75.5, 75.1, 74.2, 73.3, 71.9, 68.9, 66.7, 64.6, 64.1, 50.3 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C52H53N3O10Na: 902.3629, found: 902.3613.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐4‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (21). A 1 m solution of BH3 in THF (23.6 mL, 23.6 mmol) was added to a solution of 20 (2.08 g, 2.36 mmol) in dry CH3CN (40 mL) kept at 0 °C under an atmosphere of nitrogen. After 5 min, a 1 m solution of Bu2BOTf (2.36 mL, 2.36 mmol) was added dropwise at 0 °C, and the reaction mixture was stirred for 90 min (TLC, toluene‐EtOAc, 6:1). After complete consumption of the starting material, the reaction was quenched with Et3N (10 mL), followed by dropwise addition of MeOH (10 mL) at 0 °C. The mixture was concentrated in vacuo, and the residue was redissolved and co‐evaporated with MeOH (3×50 mL). The syrupy residue was redissolved in MeOH, the solids were removed by filtration through a short pad of Celite, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 97:3→94:6→91:9→88:12→85:15→82:18→79:21) gave 21 (1.59 g, 76 %) as a colourless syrup: R f=0.16 (toluene/EtOAc, 6:1); [α]D 20+6.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.81–7.80 (m, 2 H), 7.76–7.74 (m, 1 H), 7.71–7.69 (m, 1 H), 7.52–7.50 (m, 1 H), 7.47–7.43 (m, 2 H), 7.37–7.26 (m, 20 H), 5.01–4.98 (m, 2 H), 4.94–4.92 (m, 2 H), 4.86 (ABq, J 12.0 Hz, 2 H), 4.76–4.70 (m, 3 H), 4.62–4.59 (m, 2 H), 4.42 (d, J 7.5 Hz, 1 H), 4.16 (m, 1 H), 4.00–3.97 (m, 2 H), 3.90 (t, J 9.5 Hz, 1 H), 3.82–3.76 (m, 2 H), 3.72–3.63 (m, 3 H), 3.58 (t, J 8.8 Hz, 1 H), 3.54–3.49 (m, 2 H), 3.36–3.32 (ddd, 1 H), 3.29–3.21 (m, 2 H), 1.62 ppm (bs, 1 H); 13C NMR (125 MHz, CDCl3): δ=138.7, 138.6, 138.4, 138.1, 135.6, 133.3, 133.1, 128.5, 128.38, 128.37, 128.3, 127.99, 127.97, 127.94, 127.92, 127.89, 127.87, 127.8, 127.7, 127.6, 127.5, 127.1, 126.5, 125.9, 125.8, 103.8, 98.2, 83.9, 81.2, 78.1, 77.5, 75.5, 75.2, 74.8 (2C), 74.79, 74.4, 73.4, 72.4, 71.5, 66.6, 64.2, 62.5, 50.4 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C52H55N3O10Na: 904.3785, found: 904.3763.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐6‐O‐acetyl‐4‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (22). Acetic anhydride (2.44 mL, 25.76 mmol) was added to a solution of 21 (2.84 g, 1.47 mmol) in dry pyridine (50 mL) at 20 °C, and the mixture was stirred for 3 h (TLC, toluene/EtOAc, 9:1). The reaction mixture was concentrated in vacuo, and then redissolved and co‐evaporated with toluene (3×100 mL). Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 97:3→94:6→91:9→88:12→85:15) gave 22 (2.76 g, 93 %) as a colourless syrup: R f=0.25 (toluene/EtOAc, 9:1); [α]D 20+11.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.83–7.80 (m, 2 H), 7.76 (m, 1 H), 7.72–7.69 (m, 1 H), 7.52 (m, 1 H), 7.48–7.44 (m, 2 H), 7.39–7.25 (m, 20 H), 5.06 (d, J 10.0 Hz, 1 H), 4.99 (d, J 11.5 Hz, 1 H), 4.95–4.93 (m, 2 H), 4.90–4.84 (m, 2 H), 4.75–4.72 (m, 2 H), 4.63–4.61 (m, 2 H), 4.56 (d, J 11.0 Hz, 1 H), 4.40 (d, J 7.0 Hz, 1 H), 4.35–4.29 (m, 2 H), 4.16 (m, 1 H), 4.02–3.98 (m, 2 H), 3.95 (t, J 9.5 Hz), 3.84–3.80 (m, 2 H), 3.70–3.65 (m, 1 H), 3.58–3.51 (m, 3 H), 3.35 (ddd, J 3.5 Hz, J 7.1 Hz, J 13.3 Hz, 1 H), 3.28 (ddd, J 3.0 Hz, J 6.0 Hz, J 9.8 Hz, 1 H), 3.24 (t, J 11.0 Hz, 1 H), 1.75 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.6, 138.8, 138.2, 138.1, 135.5, 133.3, 133.1, 128.8–125.8, 104.1, 98.2, 83.9, 81.4, 78.2, 77.4, 75.6, 75.2, 75.1, 75.0, 73.8, 73.4, 71.5, 70.2, 66.6, 64.2, 63.3, 50.4, 20.5 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C54H57N3O11Na: 946.3891, found: 946.3876.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐4,6‐di‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranoside (23). NaH (155 mg, 3.88 mmol, 60 % oil dispersion) was washed with pentane (3×10 mL) prior to use. NaH was added portionwise to a solution of 21 (0.98 g, 1.11 mmol) in dry DMF (40 mL) kept at 0 °C under an atmosphere of nitrogen. After 15 min, benzyl bromide (395 μL, 3.33 mmol) was added dropwise at 0 °C under vigorous stirring. The temperature was then allowed to rise to 20 °C over 2 h (TLC, toluene/EtOAc, 6:1). After complete consumption of the starting material, residual NaH was quenched with MeOH (2 mL), and then with H2O (80 mL). The resulting mixture was extracted once with EtOAc (160 mL), the layers were separated, and the organic layer was washed with brine (1×100 mL), dried over MgSO4 and concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene→toluene/EtOAc, 98:2→96:4→94:6→92:8→90:10) gave 23 (1.03 g, 95 %) as a colourless syrup: R f=0.38 (toluene/EtOAc, 9:1); [α]D 20+7.8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.82–7.80 (m, 2 H, H), 7.75–7.73 (m, 1 H), 7.70–7.68 (m, 1 H), 7.52–7.50 (m, 1 H), 7.47–7.15 (m, 27 H), 5.06 (d, J 10.5 Hz, 1 H), 4.95–4.92 (m, 3 H), 4.90–4.82 (ABq, 2 H), 4.75–4.71 (m, 2 H), 4.61 (d, J 10.5 Hz, 1 H), 4.55–4.52 (m, 2 H), 4.42–4.40 (m, 3 H), 4.15 (s, 1 H), 4.03–3.96 (m, 3 H), 3.84 (ddd, J 3.5 Hz, J 5.8 Hz, J 10.7 Hz, 1 H), 3.81–3.77 (m, 1 H), 3.75–3.64 (m, 3 H), 3.57–3.49 (m, 3 H), 3.35 (ddd, J 3.5 Hz, J 7.2 Hz, J 13.3 Hz, 1 H), 3.29–3.21 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=138.9, 138.6, 138.6, 138.3, 138.2, 135.7, 133.3, 133.1, 128.9, 128.5, 128.3, 128.2, 128.2, 128.0, 127.95, 127.92, 127.87, 127.64, 127.60, 127.54, 127.51, 127.47, 127.4, 127.1, 126.5, 125.9, 125.7, 104.1, 98.2, 84.0, 81.2, 78.3, 77.4, 75.5, 75.2 (2C), 75.0, 74.4, 73.4, 73.3, 72.1, 71.6, 69.5, 66.5, 64.2, 50.4 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C59H61N3O10Na,: 994.4255, found: 994.4252.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐6‐O‐acetyl‐4‐O‐benzyl‐α‐d‐mannopyranoside (24). DDQ (393 mg, 1.73 mmol) was added to a vigorously stirred solution of 23 (1.00 g, 1.08 mmol) in CH2Cl2/H2O (44 mL, 10:1) at 20 °C. Additional amounts of DDQ (295 mg, 1.30 mmol) were added after 20 min. More DDQ (295 mg, 1.30 mmol) was added after 40 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 6:1). After 60 min, the reaction was quenched by adding 10 % aq. Na2S2O3 solution (200 mL). The resulting mixture was extracted once with CH2Cl2 (200 mL), the layers were separated, and the organic layer was washed sequentially with sat. NaHCO3 solution (3×150 mL), and brine (1×150 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, toluene→toluene/EtOAc, 96:4→93:7→90:10→87:13→84:16→80:20). The first fraction to appear in the eluate contained recovered starting material (51 mg). Solvent removal of the second and main fraction gave 24 (624 mg, 73 %) as a colourless syrup: R f=0.26 (toluene/EtOAc, 6:1); [α]D 20+42.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.36–7.24 (m, 20 H), 4.99 (d, J 11.0 Hz, 1 H), 4.93–4.86 (m, 3 H), 4.74–4.69 (m, 3 H), 4.64–4.61 (m, 2 H), 4.37 (dd, J 2.0 Hz, J 12.0 Hz, 1 H), 4.34 (d, J 8.0 Hz, 1 H), 4.27 (dd, J 4.5 Hz, J 12.0 Hz, 1 H), 4.03 (td, J 3.4 Hz, J 9.2 Hz, 1 H), 3.97–3.91 (m, 2 H), 3.84–3.78 (m, 2 H), 3.66–3.50 (m, 4 H), 3.42–3.36 (m, 2 H), 3.34–3.28 (m, 1 H), 3.23 (dd, J 11.7 Hz, J 10.3 Hz, 1 H,), 3.17 (d, J 9.5 Hz, 1 H), 1.87 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.7, 138.6, 138.3, 138.0, 137.9, 128.6–127.7, 104.4, 98.4, 83.8, 81.5, 80.7, 77.6, 76.3, 75.9, 75.4, 75.0, 73.7, 71.0, 70.0, 66.7, 64.4, 63.6, 50.4, 20.7 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C43H49N3O11Na: 806.3265, found: 806.3230; Anal. calcd for C43H49N3O11: C 65.89, H 6.30, N 5.36, found: C 65.70, H 6.27, N 5.31.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐4,6‐di‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)]‐6‐O‐acetyl‐4‐O‐benzyl‐α‐d‐mannopyranoside (26). A mixture of 24 (66 mg, 84.2 μmol), 25 9 (119 mg, 125.6 μmol), and crushed molecular sieves (4 Å, 20 mg) in dry Et2O (6 mL) was stirred at 20 °C for 30 min. The reaction mixture was cooled to 0 °C, freshly prepared DMTST (64 mg, 248 μmol) was added, and the reaction mixture was stirred at 0 °C for 30 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 9:1). The cooling bath was removed, and stirring was continued at 20 °C for 3 h. Et2O (8 mL) was added and the reaction was quenched with Et3N (200 μL) at 0 °C. The solids were removed by filtration through a pad of Celite, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene/EtOAc, 95:5→7:3) gave 26 (119 mg, 85 %) as a colourless syrup: R f=0.66 (toluene/EtOAc, 9:1); [α]D 20+3.5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.80 (m, 1 H), 7.76–7.74 (m, 1 H), 7.70–7.67 (m, 2 H), 7.50–7.48 (m, 1 H), 7.42–7.14 (m, 47 H), 5.19 (s, 1 H), 5.06 (d, J 11.0 Hz, 1 H), 5.01 (d, J 12.0 Hz, 1 H), 4.97 (d, J 10.5 Hz, 1 H), 4.86–4.76 (m, 7 H), 4.69 (d, J 12.0 Hz, 1 H), 4.64 (d, J 10.0 Hz, 1 H), 4.58–4.55 (m, 3 H), 4.49 (d, J 11.0 Hz, 1 H), 4.45 (d, J 12.0 Hz, 1 H), 4.39 (d, J 12.0 Hz, 2 H), 4.34–4.20 (m, 6 H), 4.12–4.08 (m, 3 H), 3.98–3.93 (m, 4 H), 3.84–3.68 (m, 6 H), 3.58–3.53 (m, 1 H), 3.50–3.36 (m, 5 H), 3.33–3.25 (m, 2 H), 3.22–3.17 (m, 1 H), 3.06 (t, J 10.8 Hz, 1 H), 2.77 (t, J 10.8 Hz, 1 H), 1.84 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.6, 139.3, 138.95, 138.9, 138.86, 138.6, 138.5, 138.4, 138.1, 138.0, 136.0, 133.3, 133.0, 128.9, 128.7, 128.6, 128.44, 128.37, 128.35, 128.33, 128.30, 128.28, 128.2, 128.1, 128.0, 127.90, 127.88, 127.85, 127.81, 127.79, 127.77, 127.75, 127.70, 127.6, 127.49, 127.47, 127.45, 127.42, 127.35, 127.1, 126.8, 126.6, 126.0, 125.8, 104.3, 103.7, 100.3 (J C−1,H−1 170 Hz), 98.4 (J C−1,H−1 175 Hz), 83.8, 83.4, 81.6, 81.0, 78.5 (2C), 77.7, 77.4, 77.1, 75.8, 75.6, 75.3, 75.12, 75.09, 74.9, 74.7, 74.4, 73.4, 73.2, 72.5 (2C), 72.2, 70.3, 69.9, 66.6, 63.9, 63.5, 63.2, 50.3, 20.7 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C100H105N3O20Na: 1690.7189, found: 1690.7153; Anal. calcd for C100H105N3O20: C 71.97, H 6.34, N 2.52, found: C 71.99, H 6.32, N 2.54.
2‐Azidoethyl 2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)‐4,6‐di‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d xylopyranosyl‐(1→2)]‐6‐O‐acetyl‐4‐O‐benzyl‐α‐d‐mannopyranoside (27). DDQ (22 mg, 99 μmol) was added to a vigorously stirred mixture of compound 26 (104 mg, 62 μmol) in CH2Cl2/H2O (11 mL, 10:1) at 20 °C. Additional amounts of DDQ (17 mg, 75 μmol) were added after 20 min. More DDQ (17 mg, 75 μmol) was added after 40 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 6:1). After 60 min, the reaction was quenched by adding 10 % aq. Na2S2O3 solution (20 mL). The resulting mixture was extracted once with CH2Cl2 (50 mL), the layers were separated, and the organic layer was washed sequentially with sat. NaHCO3 solution (3×30 mL), and brine (1×30 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, toluene→toluene/EtOAc, 96:4→93:7→90:10 →87:13→84:16→81:19→78:22). The first fraction to appear in the eluate contained recovered starting material (13 mg). Solvent removal of the second and main fraction gave 27 (76 mg, 80 %) as a colourless foam: R f=0.25 (toluene/EtOAc, 6:1); [α]D 20+13.6 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.38–7.13 (m, 45 H), 5.19 (s, 1 H), 4.99–4.94 (m, 2 H), 4.94 (d, J 11.0 Hz, 1 H), 4.88–4.80 (m, 6 H), 4.69 (d, J 11.5 Hz, 1 H), 4.66–4.57 (m, 5 H), 4.51 (s, 2 H), 4.36–4.24 (m, 6 H), 4.13–4.06 (m, 3 H), 3.99 (dd, J 5.5 Hz, J 12.0 Hz, 1 H), 3.90 (t, J 9.5 Hz, 1 H), 3.85–3.83 (m, 2 H), 3.80–3.68 (m, 5 H), 3.60 (t, J 9.3 Hz, 1 H), 3.57–3.49 (m, 2 H), 3.46 (t, J 9.3 Hz, 1 H), 3.44–3.39 (m, 2 H), 3.36 (t, J 9.0 Hz, 1 H), 3.30–3.26 (m, 2 H), 3.24–3.19 (m, 1 H), 3.07–3.03 (m, 2 H), 2.85 (t, J 11.3 Hz, 1 H), 1.84 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.6, 139.2, 138.8, 138.7, 138.4, 138.3, 138.20, 138.16, 138.0, 128.7, 128.64, 128.58, 128.4, 128.29, 128.28, 128.23, 128.20, 127.95, 127.85, 127.83, 127.79, 127.77, 127.74, 127.71, 127.64 127.56, 127.54, 127.50, 127.46, 127.44, 127.36, 126.7, 104.4, 104.0, 100.9, 98.3, 83.8, 83.2, 81.4, 80.8, 80.7, 78.8, 77.7, 77.3, 77.1, 75.5, 75.5, 75.1, 74.7, 74.7, 74.6, 74.4, 73.4, 73.3, 72.6, 71.9, 70.8, 70.4, 69.8, 66.5, 63.9, 63.5, 63.1, 50.3, 20.6 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C89H97N3O20Na: 1550.6563, found: 1550.6541.
2‐Azidoethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐3‐O‐(2‐naphthalenylmethyl)‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)]‐4,6‐di‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)]‐6‐O‐acetyl‐4‐O‐benzyl‐α‐d‐mannopyranoside (28). A mixture of 27 (31 mg, 20.3 μmol), 9 (41 mg, 29.4 μmol), and crushed molecular sieves (4 Å, 20 mg) in dry Et2O (3 mL) was stirred at 20 °C for 30 min. The reaction mixture was cooled to 0 °C, freshly prepared DMTST (22 mg, 88 μmol) was added, and the reaction mixture was stirred at 0 °C for 30 min. The progress of the reaction was carefully monitored by TLC (toluene/EtOAc, 9:1). The cooling bath was removed, and stirring was continued at 20 °C for 3 h. Et2O (8 mL) was added and the reaction was quenched with Et3N (200 μL) at 0 °C. The solids were removed by filtration through a pad of Celite, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (SiO2, toluene/EtOAc, 98:2→80:20) gave 28 (48 mg, 83 %) as a colourless syrup: R f=0.44 (toluene/EtOAc, 9:1); [α]D 20 −18.9 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.82–7.78 (m, 2 H), 7.71 (d, J 8.1 Hz, 1 H), 7.67 (d, J 8.4 Hz, 1 H), 7.51 (d, J 8.4 Hz, 1 H), 7.45–6.99 (m, 87 H), 5.25 (s, 1 H), 5.16 (s, 1 H), 5.08–4.90 (m, 5 H), 4.86–4.70 (m, 8 H), 4.69–5.58 (m, 7 H), 4.57–4.39 (m, 9 H), 4.38–4.27 (m, 7 H), 4.27–4.14 (m, 8 H), 4.14–3.98 (m, 8 H), 3.88 (d, J 3.1 Hz, 1 H), 3.84 (d, J 7.7 Hz, 1 H), 3.83–3.58 (m, 9 H), 3.58–3.34 (m, 11 H), 3.32–3.14 (m, 4 H), 3.13–3.04 (m, 2 H), 2.95–2.83 (m, 2 H), 1.78 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.8, 168.1, 139.0, 139.0, 138.9, 138.9, 138.8, 138.7, 138.7, 138.7, 138.6, 138.6, 138.6, 138.5, 138.4, 138.3, 138.2, 138.1, 136.3, 135.2, 133.3, 133.0, 129.2, 128.8, 128.8, 128.8, 128.7, 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.3, 128.3, 128.2, 128.2, 128.1, 1278.0, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 127.5, 127.5, 127.5, 127.4, 127.4, 127.4, 127.4, 127.3, 127.2, 127.2, 126.0, 125.9, 125.8, 104.5 (J C−H 160 Hz), 103.8 (J C−H 161.5 Hz), 103.6 (J C−H 163.5 Hz), 102.8 (J C−H 162.5 Hz), 101.3 (J C−H 172.5 Hz), 99.4 (J C−H 170 Hz), 98.4 (J C−H 171.5 Hz), 84.4, 84.0, 83.4, 83.4, 82.6, 81.7, 81.5, 80.9, 79.5, 79.1, 78.8, 78.8, 78.6, 78.5, 78.2, 76.3 (2C), 75.6 (2C), 75.5 (2C), 75.3 (3C), 75.0 (2C), 74.8, 74.6, 74.5 (2C), 74.3, 73.7 (2C), 73.6, 73.2, 73.1, 72.8, 72.8, 72.6, 72.4, 71.9, 70.5, 69.7 (2C), 67.2, 66.6, 63.7, 63.6, 63.5, 63.4, 50.4, 20.7 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C173H179N3O35Na: 2881.2217, found: 2881.2205.
2‐Azidoethyl (benzyl 2,3,4‐tri‐O‐benzyl‐β‐d‐glucopyranosyluronate)‐(1→2)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→4)]‐6‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)]‐4,6‐di‐O‐benzyl‐α‐d‐mannopyranosyl‐(1→3)‐[2,3,4‐tri‐O‐benzyl‐β‐d‐xylopyranosyl‐(1→2)]‐6‐O‐acetyl‐4‐O‐benzyl‐α‐d‐mannopyranoside (29). DDQ (7.2 mg, 32 μmol) was added to a vigorously stirred solution of 27 (45 mg, 15.7 μmol) in CH2Cl2/tBuOH (1.6 mL, 10:1) at 20 °C. The progress of the reaction was monitored by TLC (toluene/EtOAc, 9:1). After 60 min, the reaction was quenched by adding satd. NaHCO3 solution (6 mL). The resulting mixture was extracted once with CH2Cl2 (10 mL), the layers were separated, and the organic layer was washed with 10 % aq. Na2S2O3 solution (6 mL), dried over MgSO4, and concentrated in vacuo to a yellow oil. Purification by flash column chromatography (SiO2, toluene/EtOAc, 94:6→70:30) gave 29 (42.8 mg, 68 %) as a colourless syrup: R f=0.40 (toluene/EtOAc, 9:1); [α]D 20 −9.8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.57–6.86 (m, 85 H), 5.33 (s, 1 H), 5.16 (s, 1 H), 5.14–5.06 (m, 2 H), 5.05–4.98 (m, 3 H), 4.88–4.78 (m, 6 H), 4.76–4.54 (m, 13 H), 4.53–4.40 (m, 6 H), 4.38–4.18 (m, 9 H), 4.17–3.89 (m, 15 H), 3.85–3.75 (m, 2 H), 3.75–3.34 (m, 17 H), 3.33–3.27 (m, 2 H), 3.22 (ddd, J 13.1, 6.4, 3.8 Hz, 1 H), 3.17–3.04 (m, 4 H), 2.93 (dd, J 11.6, 9.3 Hz, 1 H), 1.79 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=170.8, 168.1, 139.0, 139.0, 138.9, 138.8, 138.7, 138.6, 138.6, 138.6, 138.5, 138.5, 138.5, 138.3, 138.3, 138.2, 138.2, 138.0, 135.4, 129.2, 128.8, 128.8, 128.7, 128.7, 128.6, 128.6, 128.5, 128.4, 128.4, 128.4, 128.4, 128.3, 128.3, 128.3, 128.2, 128.1, 128.0, 128.0, 127.8, 127.8, 127.8, 127.7, 127.7, 127.6, 127.6, 127.5, 127.5, 127.5, 127.4, 125.9, 104.5, 104.0, 103.4, 102.7, 101.2, 99.9, 98.3, 84.3, 84.0, 83.4 (2C), 82.0, 81.7, 81.3, 80.5, 79.8, 79.0, 78.9, 78.6, 78.4, 78.2, 78.1, 77.9, 77.9, 77.4, 75.6, 75.6, 75.5, 75.4 (2C), 75.3, 75.2, 74.9, 74.8, 74.6, 74.5, 74.2, 74.0, 73.6 (2C), 73.4, 73.2, 73.2, 73.0, 72.0, 71.9, 70.5, 69.7, 68.9, 68.2, 67.2, 66.7, 64.2, 63.9, 63.7, 63.3, 50.4, 20.8 ppm; HRMS (ESI): [M+Na]+ m/z calcd for C162H171N3O35Na: 2741.1591, found: 2741.1567.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Science Foundation Ireland (Grants 08/IN.1/B2067 and 13/IA/1959) and Marie Curie Intra‐European Fellowships (FP7‐PEOPLE‐2011‐IEF, project number 299710) for financial support.
L. Guazzelli, R. Ulc, S. Oscarson, ChemistryOpen 2015, 4, 729.
This article is part of the Virtual Special Issue “Carbohydrates in the 21st Century: Synthesis and Applications”.
References
- 1. Park B. J., Wannemuehler K. A., Marston B. J., Govender N., Pappas P. G., Chiller T. M., AIDS 2009, 23, 525–530. [DOI] [PubMed] [Google Scholar]
- 2. McFadden D. C., Fries B. C., Wang F., Casadevall A., Eukaryotic Cell 2007, 6, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellerbroek P. M., Lefeber D. J., van Veghel R., Scharringa J., Brouwer E., Gerwig G. J., Janbon G., Hoepelman A. I. M., Coenjaerts F. E. J., J. Immunol. 2004, 173, 7513–7520. [DOI] [PubMed] [Google Scholar]
- 4. Oscarson S., Alpe M., Svahnberg P., Nakouzi A., Casadevall A., Vaccine 2005, 23, 3961–3972. [DOI] [PubMed] [Google Scholar]
- 5. Nakouzi A., Zhang T., Oscarson S., Casadevall A., Vaccine 2009, 27, 3513–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vesely J., Rydner L., Oscarson S., Carbohydr. Res. 2008, 343, 2200–2208. [DOI] [PubMed] [Google Scholar]
- 7. Guazzelli L., Ulc R., Oscarson S., Carbohydr. Res. 2014, 389, 57–65. [DOI] [PubMed] [Google Scholar]
- 8.L. Guazzelli, S. Oscarson, in Proceedings of BeilsteinGlyco-Bioinformatics Symposium, 2013, 149–167.
- 9. Guazzelli L., Ulc R., Rydner L., Oscarson S., Org. Biomol. Chem. 2015, 13, 6598–6610. [DOI] [PubMed] [Google Scholar]
- 10. Alpe M., Oscarson S., Svahnberg P., J. Carbohydr. Chem. 2003, 22, 565–577. [Google Scholar]
- 11. Alpe M., Oscarson S., Svahnberg P., J. Carbohydr. Chem. 2004, 23, 403–416. [Google Scholar]
- 12. Zhao W., Kong F., Carbohydr. Res. 2004, 339, 1779–1786. [DOI] [PubMed] [Google Scholar]
- 13. Zhao W., Kong F., Bioorg. Med. Chem. 2005, 13, 121–130. [DOI] [PubMed] [Google Scholar]
- 14. Zhao W., Kong F., Carbohydr. Res. 2005, 340, 1673–1681. [DOI] [PubMed] [Google Scholar]
- 15. Garegg P. J., Hultberg H., Wallin S., Carbohydr. Res. 1982, 108, 97–101. [Google Scholar]
- 16. Jiang Z.-H., Schmidt R. R., Liebigs Ann. Chem. 1992, 975–982. [Google Scholar]
- 17. Goya P., Martinez A., J. Chem. Soc. Perkin Trans. 2 1990, 783–786. [Google Scholar]
- 18. De Mico A., Margarita R., Parlanti L., Vescovi A., Piancatelli G., J. Org. Chem. 1997, 62, 6974–6977. [Google Scholar]
- 19. Nimrichter L., Frases S., Cinelli L. P., Viana N. B., Nakouzi A., Travassos L. R., Casadevall A., Rodrigues M. L., Eukaryotic Cell 2007, 6, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang L., Chan T.-H., Tetrahedron Lett. 1998, 39, 355–358. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary