

Abstract
Pseudomonas aeruginosa is an opportunistic Gram‐negative pathogen with high antibiotic resistance. Its lectin LecB was identified as a virulence factor and is relevant in bacterial adhesion and biofilm formation. Inhibition of LecB with carbohydrate‐based ligands results in a decrease in toxicity and biofilm formation. We recently discovered two classes of potent drug‐like glycomimetic inhibitors, that is, sulfonamides and cinnamides of d‐mannose. Here, we describe the chemical synthesis and biochemical evaluation of more than 20 derivatives with increased potency compared to the unsubstituted cinnamide. The structure–activity relationship (SAR) obtained and the extended biophysical characterization allowed the experimental determination of the binding mode of these cinnamides with LecB. The established surface binding mode now allows future rational structure‐based drug design. Importantly, all glycomimetics tested showed extended receptor residence times with half‐lives in the 5–20 min range, a prerequisite for therapeutic application. Thus, the glycomimetics described here provide an excellent basis for future development of anti‐infectives against this multidrug‐resistant pathogen.
Keywords: carbohydrates, glycoconjugates, glycomimetics, LecB/PA-IIL, lectins
Introduction
The Gram‐negative and opportunistic pathogen Pseudomonas aeruginosa is a major threat to hospitalized and immune‐compromised patients.1 Unfortunately, many hospital strains are getting largely unaffected by many antibiotics.2 This resistance results from the presence of drug‐neutralizing tools in P. aeruginosa, for example, beta‐lactamases or efflux pumps, but also from its potential to form biofilms. In these biofilms, bacteria are embedded in a self‐produced matrix, which protects against host immune defense and antibiotic treatment.3, 4
Two lectins produced by P. aeruginosa, LecA (PA‐IL) and LecB (PA‐IIL), play key roles in biofilm formation, and both lectins are virulence factors involved in host damage and bacterial uptake.5, 6, 7, 8, 9, 10 Inhibition of these lectins as an anti‐infective strategy belongs to the new concept of antivirulence therapies.11 In contrast to antibiotics targeting bacterial viability, antivirulence therapeutics solely target the infection without interference on viability and therefore the appearance of resistant mutants is decreased.12 In a proof of concept study for therapeutic suitability of the lectins LecA and LecB as target, Hauber et al. could show a decrease in bacterial load after treatment of pulmonary‐infected patients with an aerosol containing natural ligands of both lectins.13
The carbohydrate‐binding protein LecB was first isolated by Gilboa–Garber et al., and its glycan preference was determined.14, 15 The crystal structure of LecB in complex with its high‐affinity ligand l‐fucose revealed the presence of two calcium ions, which mediate binding between the saccharide and the protein.16 A subsequent study could show that the lower‐affinity ligand d‐mannose binds similarly to LecB as a consequence of three hydroxy groups with identical relative orientation in both l‐fucose and d‐mannose.17 The trisaccharide Lewisa was identified from glycan array analysis and is currently its best known monovalent ligand with a thermodynamic dissociation constant of 210 nm.18, 19 Numerous multivalent inhibitors of LecB based on conjugates of natural l‐fucose have been reported to date in order to further increase binding affinities.20, 21 One multivalent fucosylated peptide dendrimer was shown to be a high‐affinity ligand with the potential to inhibit biofilm formation and disperse established biofilms of P. aeruginosa in a LecB‐dependent fashion.22 In contrast, the development of monovalent inhibitors has been largely neglected due to their intrinsically lower affinities and efforts are summarized in recent reviews.23, 24
We recently reported the discovery of derivatives of d‐mannose with amido‐ and sulfonamido substituents at position 6 (2 and 3, Figure 1).25 Compared with the parent low‐affinity ligand methyl mannoside (1, Figure 1),26 these modifications improved binding affinity between four‐ and 24‐fold. In contrast to α‐l‐fucosides, d‐mannose as scaffold provided the possibility to target an adjacent cleft on the protein in order to increase binding affinity. Because in the crystal structure of LecB with d‐mannose,17 a hydrogen bond between its 6‐OH and Ser23 was observed, particular attention was paid to this hydroxy group in the design of new inhibitors.25, 27, 28 However, we could show through chemical modification that this hydrogen bond does not have a significant influence on binding affinity at ambient conditions in aqueous solution.27, 28 In our previous study, we succeeded in obtaining a co‐crystal structure of sulfonamide 2 in complex with LecB. In the absence of a crystal structure for cinnamide 3 in complex with LecB, molecular dynamics simulations and NMR suggested an intercalation of the cinnamide residue into the beta‐sandwich of the lectin.25
Figure 1.
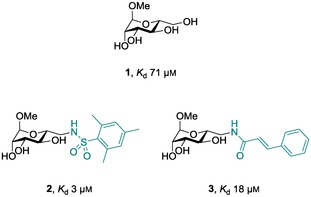
Reported natural and synthetically modified mannose based inhibitors 1, 2, and 3 and their thermodynamic dissociation constants (K d)with the lectin LecB.25, 26
Here, we describe the synthesis of a set of more than 20 substituted cinnamido derivatives to establish the structure–activity relationship (SAR) of the interaction of this class of inhibitors with LecB. Furthermore, we used a diverse set of different biophysical techniques to establish the binding mode of this class of LecB inhibitors.
Results and Discussion
In the predicted binding mode of cinnamide 3, the aromatic moiety opens a cleft in the protein and intercalates into the beta‐sandwich formed by one LecB monomer. In order to get a more detailed picture of the interaction of such cinnamides with LecB, we performed a structure–activity study with a variety of substituents in ortho‐, meta‐, and para‐position of the cinnamic amide residue of 3. The synthesis of the cinnamide derivatives started from methyl α‐d‐mannoside (1). Introduction of an amino functional group allowed amide bond formation with unprotected mannose derivative 4 with a set of selected commercially available cinnamic acid derivatives. Various substituents were introduced in ortho‐, meta‐, or para‐position resulting in the potential LecB inhibitors 5 a–e, 5 g–k, and 5 m–q, respectively, which were obtained in moderate to good yields (Scheme 1). The nitro derivatives 5 e, 5 k, and 5 q were further reduced with iron(II) sulfate following a method by Pribulova et al.29 for aliphatic nitro groups to give the corresponding anilines 5 f, 5 l, and 5 r in high yields. Furthermore, we synthesized the more rigid 2‐naphtoyl derivative 6 as a cyclized analog of 3, as well as the two disubstituted analogs 7 a and 7 b following the same protocol.
Scheme 1.
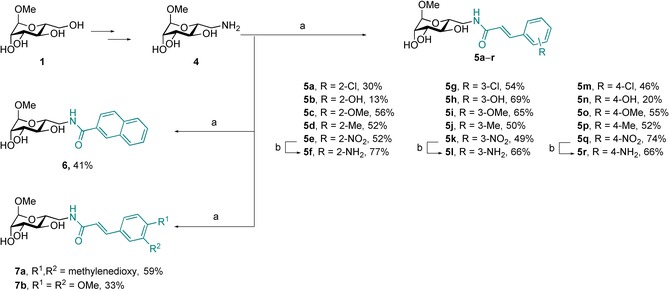
Synthesis of compounds 5 a–r, 6, and 7 a,b. Reagents and conditions: a) carboxylic acids, EDC⋅HCl, Et3N, DMF, 0 °C → rt; b) FeSO4⋅7 H2O, NH4OH, H2O, rt; c) Pd/C, H2, MeOH, rt. Reaction times, yields, and purities are summarized in Table S1 in the Supporting Information.
All synthesized compounds were then biochemically evaluated for inhibition of LecB using a competitive binding assay based on fluorescence polarization readout, recently developed for LecB in our lab.25 Surprisingly, all monosubstituted compounds were within a narrow activity range with IC50 values from 27–73 μm, indicating that the substituents tested had a moderate effect on the activity that remains similar to the parent cinnamide 3 with an IC50 of 37 μm (Figure 2). Introduction of ortho substituents in 5 a–f generally resulted in weaker inhibition of LecB (IC50 49–73 μm), while the same substituents in meta‐ or para‐position (5 g–l and 5 m–r) showed similar or enhanced potency with IC50 values ranging from 27–53 μm. The decreased affinity of ortho‐substituted 5 a–f might result from a steric hindrance with the receptor. Generally, the introduction of polar substituents such as phenolic hydroxy groups or aniline‐NH2 led to a decreased affinity (5 b, 5 f, 5 h, 5 l, 5 n, 5 r), whereas the presence of the lipophilic substituents chloro, methoxy, or methyl resulted in enhanced potencies especially in meta or para position (5 g, 5 i, 5 j, and 5 m, 5 o, 5 p). Methoxy‐substituted cinnamides were superior inhibitors in the ortho, meta, and para series, with highest potencies for meta‐5 i and para‐5 o. We therefore combined both substituents in one molecule as 3,4‐methylenedioxy derivative 7 a or 3,4‐dimethoxycinnamide 7 b to analyze potential synergistic effects. Both compounds showed good inhibition of LecB with dimethoxy‐substituted 7 b as the most potent inhibitor at an IC50 of 19.9 μm. Methylenedioxy derivative 7 a had an IC50 of 29 μm, which was comparable to singly meta‐ or para‐methoxy‐substituted 5 i and 5 o.
Figure 2.
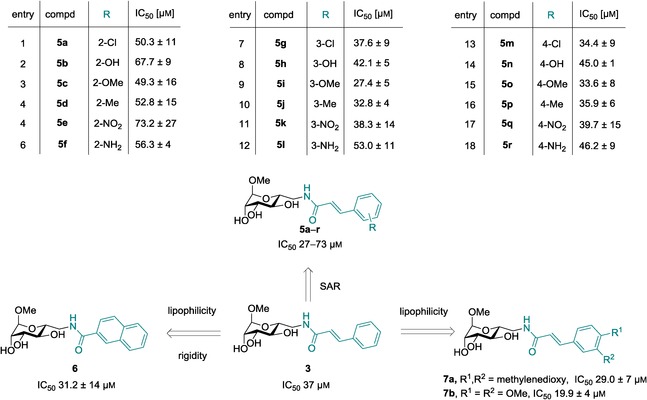
Biochemical evaluation of LecB binding to ligands 5 a–r, 6, and 7 a,b. IC50 values were determined with a competitive fluorescence polarization assay. Means and standard deviations were determined from a minimum of three independent experiments.
In addition, we tested the cyclized analog 6 bearing a 2‐naphtamide in lieu of the cinnamide moiety for LecB inhibition. In accordance with the general trend of higher affinities for more lipophilic substituents among all tested compounds, compound 6 also displayed an increased inhibitory potency (IC50 31 μm). The 2‐naphtamide 6 is structurally similar to ortho‐substituted cinnamides 5 a–f, especially to the 2‐methyl derivative 5 d. Generally, steric hindrance with the receptor, a distorted planarity of the cinnamide moiety, or even a further increase of the already high rotational barrier due to the ortho‐substituents in 5 a–f could account for the decreased affinity in the ortho‐series. Possible reasons for the difference in activity between 6 and 5 a–f cannot be deduced from the present data set. Although a rather flat SAR on the substituents of the cinnamic acid moiety was obtained, a two‐fold increase in potency compared to unsubstituted cinnamide 3 could be achieved through introduction of two methoxy substituents in meta‐ and para position in 7 b.
Can this SAR at the cinnamide residue explain the previously proposed intercalation model of the binding of 3 to LecB? During our previous structural characterization of the cinnamide 3–LecB interaction, we observed global line broadening of the protein signals at a protein–ligand ratio of 2:1; a stoichiometric ratio of 1:1 led to complete vanishing of resonances and visual precipitation of protein after a few hours of recording time. In contrast, the same analysis with sulfonamide 2 yielded distinct shifts of a small set of protein resonances. The protein NMR spectra were interpreted, such that the computationally predicted intercalation of 3 into the beta‐sandwich of LecB leads to a global effect on its structure and, thus, results in changes of most of the protein resonances.
In order to analyze whether the observed effects on LecB induced by cinnamide 3 are carbohydrate‐specific and to exclude simple detergent‐like denaturation of the protein, we designed its glucose‐analog 10. The compound was synthesized in analogy to 3: methyl α‐d‐glucoside (8) was transformed into the tosylate and subsequently the azide 9 was obtained after sodium azide treatment as previously reported by Cramer et al.30 (Scheme 2). The azide was hydrogenolytically reduced using palladium on activated charcoal, and the resulting amine was directly coupled with cinnamic acid to yield glucocinnamide 10 in good yield (46 % over two steps).
Scheme 2.
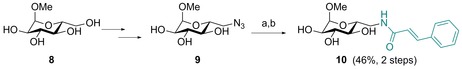
Synthesis of glucose‐derivative 10. Reagents and conditions: a) Pd/C, H2, MeOH, rt; b) cinnamic acid, EDC⋅HCl, Et3N, DMF, 0 °C → rt.
The control compound gluco‐ 10 was then tested in the competitive binding assay to assess the carbohydrate‐based specificity of cinnamide manno‐ 3. No inhibition of LecB function up to 3 mm of gluco‐cinnamide 10 was observed (Figure 3), whereas manno‐ 3 showed inhibition in a dose‐dependent manner as previously reported. This assay relies on the displacement of a fucose‐fluorescein conjugate as detection probe bound to intact LecB. The observed high fluorescence polarization even at high concentrations of gluco‐ 10 therefore indicated persistent binding of the fluorescent probe and thus the absence of any kind of inhibitory influence of gluco‐ 10 on LecB function. In summary, there was no unspecific binding of the cinnamide moiety at elevated ligand concentrations, but the effects observed with manno‐ 3 were instead dependent on the stereochemistry of one single hydroxy group and thus on the specific binding to the carbohydrate recognition site of LecB.
Figure 3.
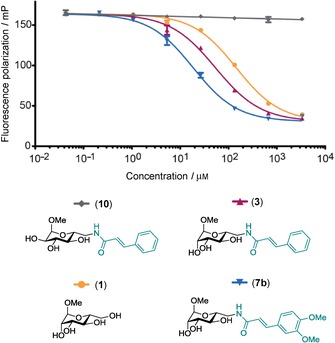
Inhibition of LecB by manno‐ 3 is carbohydrate‐specific, and the control compound gluco‐ 10 has no effect up to a concentration of 3 mm on LecB functional binding to the fucosylated fluorescent probe of the assay system.
To further study the interaction of the most potent cinnamide with LecB, compound 7 b was then titrated to LecB, and the thermodynamics of binding were analyzed by isothermal microcalorimetry (ITC, Figure 4, Table 1). A K d value of 10.9 μm with a stoichiometry of 1 was determined, and binding enthalpy and entropy were obtained. In comparison to sulfonamide 2, both cinnamide 3 and dimethoxycinnamide 7 b showed a reduced binding enthalpy, but in contrast to 2, a beneficial contribution of binding entropy to the Gibbs free energy.
Figure 4.
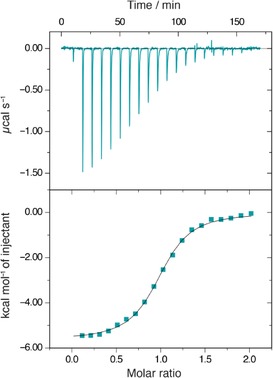
Isothermal titration microcalorimetry (ITC) of LecB with dimethoxycinnamide 7 b. Three independent titrations were performed; one representative example is shown here.
Table 1.
Thermodynamics of ligand binding to LecB by isothermal titration microcalorimetry. Values for 1–3 were taken from the literature.25, 26
| 7 b | 3 | 2 | MeMan [1] | |
|---|---|---|---|---|
| K d [μm] | 10.9±1.80 | 18.5 | 3.3 | 71 |
| ΔG [kcal mol−1] | −6.81±0.16 | −6.6 | −7.5 | −5.4 |
| ΔH [kcal mol−1] | −5.63±0.21 | −4.3 | −7.9 | −4.3 |
| −TΔS [kcal mol−1] | −1.18±0.36 | −2.3 | 0.4 | −1.4 |
| N | 1.03±0.09 | 0.94 | 0.71 | 0.94 |
Subsequently, we performed surface plasmon resonance (SPR) analysis to determine the parameters of binding kinetics of LecB with its ligands. LecB was biotinylated using an activated carboxylic acid derivative of biotin and coupled to the surface of a streptavidin‐coated sensor chip. High surface densities of LecB were obtained, which is necessary for the detection of small molecule binding. Methyl mannoside (1), sulfonamide 2, cinnamide 3, and its derivative 7 b, which showed the highest potency among the cinnamide series in LecB inhibition, were analyzed by SPR (Figure 5, Table 2). Unmodified mannoside 1 showed fast binding kinetics, which are usually observed with lectin–carbohydrate interactions.31 The measured K d of 47 μm was in good agreement with the previously determined thermodynamic value of 71 μm by ITC.26 Sulfonamide 2 showed a two‐fold higher on rate and a very slow off rate of 6.2×10−4 s−1. The half‐life (t1/2=ln2/k off) of the complex of LecB and sulfonamide 2 was 18.6 min, explaining its very high affinity (K d=1.12 μm by SPR, 3.3 μm 25 by ITC) towards LecB compared to a t1/2 of only 45 s for 1. It is important to note, that long drug‐receptor half lives are important for in vivo function and future success for drug development.32, 33 When analyzing cinnamide 3, slow binding kinetics approaching the detection limit of the apparatus were observed with a k on of 100.9 m −1 s−1, which was threefold slower than mannoside 1 and more than fivefold slower than sulfonamide 2. Its dimethoxy derivative 7 b also showed a slow on rate with a k on of 169.6 m −1 s−1, indicating a similar mode of binding to LecB for 3 and 7 b. The dissociation constant of 18.1 μm for 3 was in very good agreement with its previously determined value of 18.5 μm 25 by calorimetry. The most potent cinnamide derivative dimethoxycinnamide 7 b showed a K d by SPR of 7.7 μm and a complex half‐life t1/2 of 8.8 min, demonstrating the high potential of this series for further drug development. The very slow on rates observed for the cinnamides 3 and 7 b could result from a conformational change within the protein upon binding, in favor of the previously proposed intercalation model or from conformational changes within the ligand.
Figure 5.
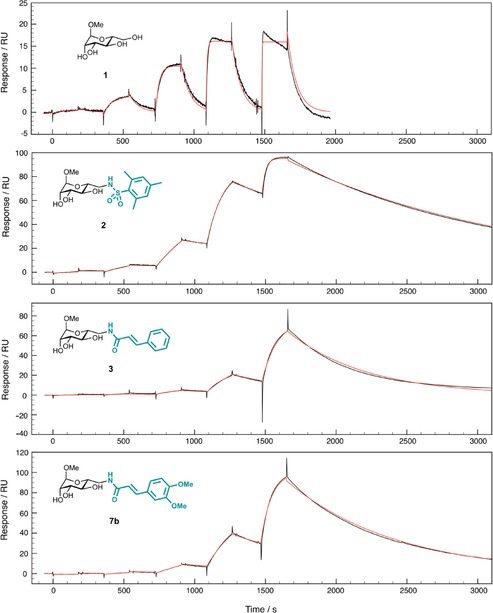
Single cycle kinetics analysis by surface plasmon resonance (SPR) of the direct binding of 1, 2, 3, or 7 b to immobilized LecB. Experimental data are shown in black; calculated fits using a 1:1 binding model with a global fitting on all injected concentrations are shown in red. Ligand concentrations injected were 120 nm, 600 nm, 3 μm, 15 μm, and 75 μm for 2, 3, and 7 b, and 3 μm, 15 μm, 75 μm, 375 μm, and 1.8 mm for 1.
Table 2.
Data obtained from the kinetic fit of a 1:1 binding model to the SPR single cycle kinetics sensograms.
| cmpd | K d [μM] | k on [m −1 s−1] | k off [s−1] | k off [s−1]×103 | t 1/2 [min] | R max |
|---|---|---|---|---|---|---|
| 1 | 47.5 | 326.1 | 0.01548 | 15.48 | 0.75 | 18.8 |
| 2 | 1.12 | 552.6 | 0.00062 | 0.62 | 18.64 | 94.9 |
| 3 | 18.1 | 100.9 | 0.00183 | 1.83 | 6.33 | 93.8 |
| 7 b | 7.67 | 169.6 | 0.00130 | 1.30 | 8.88 | 107.0 |
Analysis of LecB and its complex with cinnamide by circular dichroism (CD) spectroscopy could yield insight on conformational changes induced by ligand binding. Interestingly however, we did not observe significant differences in the CD spectra of LecB with 3 or LecB alone, and both spectra were indicative of beta‐sheets as the predominant secondary structure (Figure 6 A). Then, a thermal denaturation of LecB in presence and absence of cinnamide 3 was performed to assess the influence of 3 on protein stability (Figure 6 B). The CD spectrum of LecB was stable between 37 °C and up to above 90 °C, and no defined melting point (T m) could be determined as shown in Figure 6 B for the ellipticity at 229 nm. In presence of compound 3, only slight differences were detected with somewhat bigger changes without ligand. Further attempts to analyze T m of LecB with a set of compounds by thermal shift analysis using SYPRO® Orange also failed to give signals for unfolded protein (data not shown). The lack of success in both thermal unfolding techniques presumably originates in the extraordinary high thermal stability of LecB.14
Figure 6.
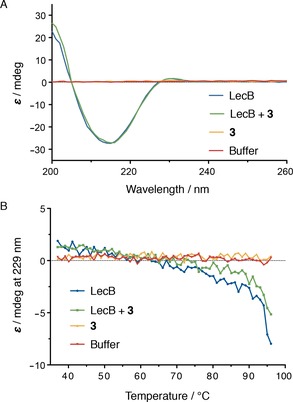
A) CD spectroscopy of LecB (35 μm) in the presence or absence of compound 3 (100 μm) gave identical CD spectra indicative for beta‐sheet secondary structure. B) thermal unfolding followed by CD spectroscopy at 229 nm was not indicative of a destabilization of the protein in presence of 3.
Based on the high degree of structural similarity of cinnamide 3 and dimethoxycinnamide 7 b and their similar binding kinetics, we anticipated comparable binding modes and thus also a similar behavior on protein NMR signals. Surprisingly, the 1H,15N‐TROSY‐HSQC (transverse relaxation‐optimized spectroscopy–heteronuclear single quantum coherence) NMR spectrum of 15N‐labelled LecB showed only distinct shifts of protein resonances in the presence of 7 b, and even saturation of LecB with dimethoxycinnamide 7 b could be achieved at excellent quality of the spectra (Figure 7 B). In contrast to our previously reported data for LecB in complex with the unsubstituted cinnamide 3, no global vanishing of signal intensity was observed with dimethoxycinnamide 7 b. The behavior observed with 7 b rather resembles the spectra obtained with sulfonamide 2 and thus a distinctly defined binding site.
Figure 7.
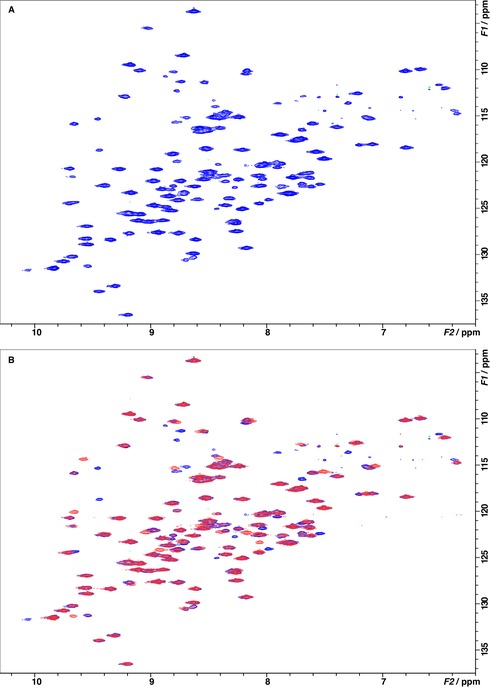
Binding of dimethoxycinnamide 7 b to LecB was analyzed by 1H,15N‐TROSY‐HSQC NMR experiments in the absence (blue) and presence (red) of 7 b. A) Spectrum of 500 μm 15N‐labeled LecB without addition of ligand. B) Spectrum of 500 μm 15N‐labeled LecB in the absence (blue) and in presence (red) of 1 equivalent 7 b. Upon addition of 7 b, the intensities of about 15 peaks decreased, and a number of new crosspeaks appeared. This is indicative of chemical shift perturbations of a defined binding that interacts with 7 b in the slow‐exchange regime.
After an extensive search for crystallization conditions of LecB with cinnamide 3, we finally obtained protein crystals which diffracted to 1.6 Å and allowed structure determination (Figure 8, Table 3). Both, the mannose part and the cinnamide residue were well defined and present in every binding site of the LecB tetramer (Figure 8 A). Interestingly, this crystal structure shows a surface binding of the cinnamide as opposed to the computational model suggesting the intercalation of the aromatic moiety into the protein.25 The asymmetric unit contains one tetramer of LecB with one ligand molecule per monomer. In the overlay of ligand binding sites, a high variability between the ligand conformation in the four binding sites can be observed (Figure 8 A). The mannose residue always adopts the same binding pose, whereas the cinnamide side chain is oriented in three different orientations. After closer examination of the structure, we identified extended π–π stacking contacts between the cinnamide part of two ligands from a neighboring LecB tetramer in the crystal lattice. In only one out of the four binding sites, the cinnamide substituent is in contact with the protein surface, forming van der Waals contacts with Gly97 and Thr98 and a water mediated hydrogen bond with Ser23 (Figure 8 B). In the other three binding sites, the cinnamic acid residues form extended interligand π–π stacking contacts in the crystal lattice. It is well known that crystal lattice contacts can induce certain conformations of ligands or flexible regions in proteins, which sometimes do not occur in solution.34 In solution, however, the three binding modes lacking protein contact of the cinnamide are unlikely to occur. The binding mode observed for the fourth pose with the cinnamide having a lipophilic contact with the protein surface could explain the flat SAR, that is a narrow range of inhibitory activities, obtained.
Figure 8.
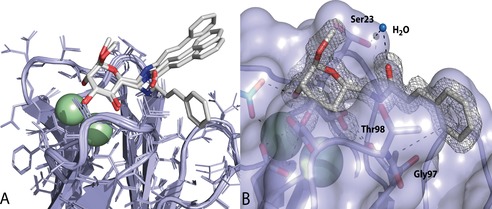
A) The crystal structure of the complex of 3 with LecB at 1.6 Å shows a surface binding of the cinnamide residue to the protein. The superposition of all four binding sites indicates coordination of the cinnamic acid residue to the protein in one monomer and no interaction with LecB in three of the four monomers. The latter results from extended interligand stacking interactions in neighboring binding sites due to the crystal packing. B) Surface for monomer 2 with electron density for 3: the cinnamide substituent forms lipophilic interactions with Gly97 and Thr98 and one water‐mediated hydrogen bond via the carbonyl oxygen with Ser23.
Table 3.
Crystal structure data of the complex of 3 with LecB.
| Data | ||||
|---|---|---|---|---|
| Beamline (wavelength [Å]) | BM30A/0. 97 9618 | |||
| Spacegroup | P1 | |||
| Unit cell dimensions | ||||
| a, b, c [Å] | 44.95, 51.11, 52.35 | |||
| α, β, γ [°] | 101.80, 99.40, 115.95 | |||
| Resolution (outer shell) [Å] | 39.25–1.60 (1.63–1.60) | |||
| Measured/unique reflections | 193 815/50 076 | |||
| Average multiplicity | 3.9 (3.7) | |||
| R merge | 0.035 (0.179) | |||
| Completeness [%] | 96.6 (90.0) | |||
| Mean I/σI | 25.4 (6.1) | |||
| CC1/2 | 0.999 (0.936) | |||
| Refinement | ||||
| R cryst/R free nb reflections/free reflections | 16.7/20.4 47 532/2541 | |||
| R msd bonds [Å] | 0.0144 | |||
| R msd angles [°] | 1.63 | |||
| R msd chiral [Å3] | 0.102 | |||
| Atoms (chain) | A | B | C | D |
| Protein Bfac [Å2] | 851 7.4 | 859 8.3 | 856 7.9 | 838 8.1 |
| Water molecules Bfac, [Å2] | 175 18.7 | 153 19.8 | 165 21.1 | 128 20.2 |
| Ligand Bfac, [Å2] | 23 8.4 | 23 8.3 | 23 9.2 | 23 10.0 |
| Calcium Bfac, [Å2] | 2 6.2 | 2 7.0 | 2 6.9 | 2 6.8 |
| Ramachandran (Molprobity) | ||||
| Allowed Favored Outliers | 100 % 97.3 % 0 % | |||
| PDB code | 5A3O | |||
In order to further understand the distinctly different characteristics of the interaction of LecB with the cinnamide ligands observed by protein NMR spectroscopy, we analyzed inhibitor‐mediated aggregation of LecB. Besides intermediate exchange phenomena or protein unfolding, aggregation also leads to loss in observed signal intensity by NMR spectroscopy. To quantify the compounds’ effect on protein aggregation, we performed titration experiments with a constant concentration of LecB and measured aggregation via light scattering at 600 nm (OD600). Surprisingly, we did not observe measurable light scattering for dimethoxycinnamide 7 b between 14 μm and 1.7 mm, but intense and spontaneous aggregation for cinnamide 3 at 510 μm and above, which could explain the distinct differences observed by NMR spectroscopy (Figure 9). Sulfonamide 2, as well as the gluco‐cinnamide 10, showed no light scattering under identical conditions; all compounds tested in absence of LecB were also translucent across the entire concentration range. We thus concluded that unsubstituted cinnamide 3 dimerizes or even oligomerizes at high concentrations. These oligomers may act as multivalent anchor points that attract individual LecB tetramers and promote aggregation and precipitation. This aggregation can explain the global vanishing of signal intensity in 1H,15N‐TROSY‐HSQC NMR experiments which was observed for 3. Introduction of the two methoxy‐substituents in 7 b prevents aggregation of LecB probably because the methoxy groups prevent strong π–π‐stacking‐mediated oligomerization of compound 7 b itself.
Figure 9.
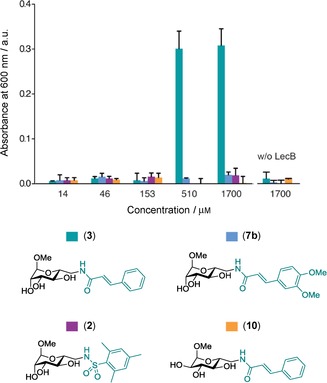
Aggregation assay of LecB (133 μm) with ligands (14 μm to 1.7 mm). Aggregation at OD600 observed for 3 at the two highest concentrations. No aggregation observed for 2, 7 b, and 10.
Conclusions
Cinnamides of d‐mannose, such as 3, were previously identified as potent glycomimetic inhibitors of LecB, a virulence factor and important biofilm component of the opportunistic pathogen P. aeruginosa.25 Based on microcalorimetry data and massive line broadening observed in protein NMR spectroscopy signals in the presence of 3, which may result from global changes in the protein structure and/or aggregation of the protein, we proposed an intercalation binding model. Here, we report an extensive SAR study of substituents at the cinnamide residue of 3 and their influence on binding affinity to LecB. Although a rather flat SAR was obtained, binding affinity of this class of compounds could be further improved for LecB. The dimethoxy‐substituted compound 7 b is the most potent derivative with a seven‐fold higher affinity than 1. The established SAR at the cinnamide residue was, however, not sufficient to explain the previously proposed intercalation model of the binding of 3 to LecB. We therefore further analyzed the binding of 3 to LecB using a number of biophysical methods.
First, the synthesis and biochemical evaluation of the gluco‐analog 10 of manno‐cinnamide 3 showed that the inhibition of LecB by compound 3 is specific and strictly carbohydrate dependent. Furthermore, the data showed that the inhibitory effect on LecB does not result from an unspecific detergent‐like effect of these amphiphilic compounds.
Kinetic analysis by surface plasmon resonance of the interaction of LecB with the two manno‐cinnamides studied here, 3 and 7 b, showed comparable binding kinetics for both compounds. Very slow on rates close to the detection limit of the SPR machine, as well as comparable intermediate off rates were detected for both compounds. This kinetic data suggests a similar mode of action for the structurally highly related compounds cinnamide 3 and dimethoxycinnamide 7 b. However, the interaction study using 1H,15N‐TROSY HSQC NMR spectra of LecB in presence of 7 b showed distinct shifts of about 15 resonances and was therefore in clear contrast to the behavior of its unsubstituted analog 3. After extensive search for crystallization conditions for the complex of LecB and cinnamide 3, we succeeded in determining the crystal structure of its complex. Surprisingly, a surface localization of the cinnamide residue in 3 was detected with defined lipophilic interactions and one directed hydrophilic hydrogen bond with LecB. It was not until we performed aggregation assays with the compounds studied and LecB, that we could fully explain the nature behind the effects observed. Only unsubstituted cinnamide 3 aggregated LecB at high ligand concentrations, similar to those used in NMR experiments. When two methoxy substituents were introduced into the structurally highly similar compound 7 b, aggregation of LecB was no longer observed. Presumably, self‐aggregation of compound 3 at high concentrations in aqueous solution via its lipophilic cinnamide moiety lead to an apparent multivalency, which precipitated tetravalent LecB. Both cinnamides showed comparable very slow on‐rates in the surface plasmon resonance (SPR) experiments with LecB, and therefore similar binding modes of 3 and 7 b are plausible. A possible conformational change of the protein accounting for these slow on rates is unlikely due to identical orientations of the protein in the crystal structures of the apoprotein,17 LecB with 2 25 or LecB with 3 (Figures S1 and S2 in the Supporting Information). The overlay of the crystal structures of LecB and sulfonamide 2 with cinnamide 3 also demonstrates the different areas on the lectin targeted by the sulfonamide and the cinnamide residue, respectively (Figure S2 in the Supporting Information).
Based on these experiments and the crystal structure obtained, we could revise the initially proposed intercalation mode for cinnamide‐based LecB inhibitors. The knowledge gained from this study now allows the structure‐based design of follow‐up derivatives of the highly potent cinnamide‐based class of glycomimetics as LecB inhibitors. Importantly, all glycomimetics tested showed extended receptor residence times with half‐lives in the 5–20 min range. In contrast, the rapid kinetics of natural carbohydrate ligands with half‐lives in the 45 s range hamper their use as therapeutics. Thus, the glycomimetics described here provide an excellent basis for future development of anti‐infectives.
Experimental Section
Chemical synthesis
Thin layer chromatography (TLC) was performed using silica‐gel 60‐coated aluminum sheets containing fluorescence indicator (Merck KGaA, Darmstadt, Germany) using UV light (254 nm) and by charring either in anisaldehyde solution (1 % v/v 4‐methoxybenzaldehyde, 2 % v/v concentrated H2SO4 in EtOH), in aqueous KMnO4 solution or in a molybdate solution (a 0.02 m solution of Ce(NH4)4(SO4)4⋅2H2O and (NH4)6Mo7O24⋅4H2O in aqueous 10 % H2SO4) with heating. Medium pressure liquid chromatography (MPLC) was performed on a Teledyne Isco Combiflash Rf200 system (Lincoln, USA) using pre‐packed silica gel 60 columns from Teledyne Isco, SiliCycle, or Macherey–Nagel. Commercial chemicals and solvents were used without further purification. Deuterated solvents were purchased from Eurisotop (Saarbrücken, Germany). Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III 400 UltraShield spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) at 400 MHz (1H) or 101 MHz (13C). Chemical shifts are given in ppm and were calibrated on residual solvent peaks as internal standard.35 Multiplicities were specified as s (singlet), d (doublet), t (triplet), or m (multiplet). The signals were assigned with the help of 1H, 1H‐COSY, and DEPT‐135‐edited 1H, 13C ‐HSQC experiments. High‐resolution mass spectra (HRMS) were obtained on a Bruker micrOTOF II ESI spectrometer, and the data were analyzed using DataAnalysis from Bruker.
The NMR and MS data of all synthesized compounds as well as reaction times and mass yields (Table S1) can be found in the Supporting Information.
General procedure for amide couplings
Methyl 6‐amino‐6‐deoxy‐α‐d‐mannopyranoside25 (50 mg, max. 0.26 mmol), triethylamine (54 μL, 1.5 equiv), and in case of free acids, N‐(3‐dimethylaminopropyl)‐N′‐ethylcarbodiimide hydrochloride (EDC⋅HCl, 73 mg, 1.5 eq), were dissolved in dry dimethylformamide (DMF, 1.5 mL) and cooled to 0 °C. Then, the carboxylic acid, (0.31 mmol, 1.2 equiv) dissolved in DMF (0.5 mL) was added dropwise under argon. After stirring for 1 h at 0 °C, the reaction was allowed to warm to rt and was further stirred for 1–3 days. The reactions were quenched with saturated aqueous NH4Cl (10 mL) and extracted with EtOAc (1×30 mL, 6×10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by MPLC on silica with CH2Cl2/EtOH or EtOAc/EtOH.
Methyl 6‐(2‐chlorocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 a) was obtained from 2‐chlorocinnamic acid as a white solid (30 % over two steps).
Methyl 6‐(2‐hydroxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 b) was obtained from 2‐hydroxycinnamic acid as a white solid (13 % over two steps).
Methyl 6‐(2‐methoxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 c) was obtained from 2‐methoxycinnamic acid as a white solid (56 % over two steps).
Methyl 6‐(2‐methylcinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 d) was obtained from 2‐methylcinnamic acid as a white solid (52 % over two steps).
Methyl 6‐(2‐nitrocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 e) was obtained from 2‐nitrocinnamic acid as a white solid (52 % over two steps).
Methyl 6‐(2‐aminocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 f). 5 e (64 mg, 0.174 mmol) was dissolved in degassed H2O, and FeSO4⋅7 H2O (7 eq) and NH4OH solution (580 μL, 25 %) were added and stirred under argon atmosphere for 30 min. The suspension was filtered, unsolved residue was washed with NH4OH solution (5×2 mL, 5 %), and solvent was removed in vacuo. After purification by MPLC, 5 f was obtained as a yellowish solid (45 mg, 0.133 mmol, 77 %).
Methyl 6‐(3‐chlorocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 g) was obtained from 3‐chlorocinnamic acid as a white solid (54 % over two steps).
Methyl 6‐(3‐hydroxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 h) was obtained from 3‐hydroxycinnamic acid as a white solid (69 % over two steps).
Methyl 6‐(3‐methoxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 i) was obtained from 3‐methoxycinnamic acid as a white solid (65 % over two steps).
Methyl 6‐(3‐methylcinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 j) was obtained from 3‐methylcinnamic acid as a white solid (50 % over two steps)
Methyl 6‐(3‐nitrocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 k) was obtained from 3‐nitrocinnamic acid as a white solid (49 % over two steps)
Methyl 6‐(3‐aminocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 l). 5 k (64 mg, 0.174 mmol) was dissolved in degassed H2O, and FeSO4⋅7 H2O (7 eq) and NH4OH solution (580 μL, 25 %) were added and stirred under argon atmosphere for 30 min. The suspension was filtrated, unsolved residue was washed with NH4OH solution (5×2 mL, 5 %), and solvent was removed in vacuo. After purification by MPLC, 5 l was obtained as yellowish solid (39 mg, 0.143 mmol, 66 %).
Methyl 6‐(4‐chlorocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 m) was obtained from 4‐chlorocinnamic acid as a white solid (46 % over two steps).
Methyl 6‐(4‐hydroxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 n) was obtained from 4‐hydroxycinnamic acid as a white solid (20 % over two steps).
Methyl 6‐(4‐methoxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 o) was obtained from 4‐methoxycinnamic acid as a white solid (55 % over two steps).
Methyl 6‐(4‐methylcinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 p) was obtained from 4‐methylcinnamic acid as a white solid (52 % over two steps).
Methyl 6‐(4‐nitrocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 q) was obtained from 4‐nitrocinnamic acid as a white solid (74 % over two steps).
Methyl 6‐(4‐aminocinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (5 r). 5 q (48 mg, 0.13 mmol) was dissolved in degassed H2O, and FeSO4⋅7 H2O (7 eq) and NH4OH solution (430 μL, 25 %) were added and stirred under argon atmosphere for 30 min. The suspension was filtered, unsolved residue was washed with NH4OH solution (5×2 mL, 5 %), and solvent was removed in vacuo. After purification by MPLC, 5r was obtained as a yellowish solid (29 mg, 0.087 mmol, 66 %).
Methyl 6‐(2‐naphtoylamido)‐6‐deoxy‐α‐d‐mannopyranoside (6) was obtained from 2‐naphtoic acid as a white solid (41 % over two steps).
Methyl 6‐(3,4‐methylenedioxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (7 a) was obtained from 3,4‐methylenedioxycinnamic acid in as a white solid (59 % over two steps).
Methyl 6‐(3,4‐dimethoxycinnamido)‐6‐deoxy‐α‐d‐mannopyranoside (7 b) was obtained from 3,4‐dimethoxycinnamic acid as a white solid (33 % over two steps).
Methyl 6‐(cinnamido)‐6‐deoxy‐α‐d‐glucopyranoside (10). Methyl 6‐azido‐6‐deoxy‐α‐d‐glucopyranoside30 (9, 270 mg, 1.23 mmol) was dissolved in MeOH (12 mL) and stirred under hydrogen atmosphere (1 bar) with 10 % Pd/C (30 mg, 10 mol %) at rt for 4 h. The mixture was filtered through celite, and the solvent was removed under reduced pressure to give methyl 6‐amino‐6‐deoxy‐α‐d‐glucopyranoside30 (208 mg) as colorless solid that was subsequently coupled under standard conditions with cinnamic acid (43 % over two steps).
Recombinant expression and purification of P. aeruginosa LecB
The protein was expressed and purified as described recently25 using E. coli BL21 (DE3) and plasmid pET25pa2l.36 The purified and lyophilized protein was dissolved in Tris‐buffered saline supplemented with calcium (TBS/Ca: 20 mm Tris, 137 mm NaCl, 2.6 mm KCl at pH 7.4 supplemented with 0.1 or 1 mm CaCl2) before use, and the concentration was determined by UV spectroscopy at 280 nm using a molar extinction coefficient of 6990 m −1 cm−1.37 Recombinant expression and purification of 15N‐labelled LecB was performed using 15NH4Cl as described25 following a procedure by Marley et al.38
Competitive binding assay
The competitive binding assay based on fluorescence polarization was performed as previously described by Hauck et al.25 Briefly, a stock solution of LecB (20 μL of 225 nm) and fluorescent reporter ligand N‐(fluorescein‐5‐yl)‐N′‐(a‐l‐fucopyranosyl ethylen)‐thiocarbamide (15 nm) in TBS/Ca were mixed with serial dilutions (10 μL of 1 mm to 12.8 nm) of testing compounds in TBS/Ca in triplicates. After addition of the reagents, the black 384‐well microtiter plates (Greiner Bio‐One, Germany, cat. no. 781900) were incubated for 8–22 h at rt in a humidity chamber. Fluorescence emission parallel and perpendicular to the excitation plane was measured on a PheraStar FS (BMG Labtech, Ortenberg, Germany) plate reader or on a Tecan INFINITE F500 plate reader (Tecan Group Ltd., Männedorf, Switzerland) with excitation filters at 485 nm and emission filters at 535 nm. For measurements on the Tecan INFINITE F500 plate reader, the G‐factor was set on 0.92154 and the gain to 80. The measured intensities were decreased by buffer values, and fluorescence polarization was calculated. The data were analyzed using BMG Labtech MARS software (v.3.01R2, 2013) and/or with GraphPad Prism (GraphPad Software, Inc., La Jolla, USA) and fitted according to the four‐parameter variable‐slope model. Bottom and top plateaus were defined by the standard compounds l‐fucose and methyl α‐d‐mannoside, respectively, and the data was reanalyzed with these values fixed. A minimum of three independent measurements of triplicates each was performed for every ligand.
Microcalorimetry
Isothermal titration calorimetry (ITC) was performed on a MicroCal ITC200 (GE Healthcare, Freiburg, Germany), and the data was analyzed using the MicroCal Origin software (v.7.0, 2009). LecB (300–360 μm) was dissolved in TBS/Ca (the concentration was verified by UV spectroscopy, see above) and placed in the sample cell at 25 °C. The titration was performed with a solution of 7 b (3.0–3.5 mm) in the same buffer. After one preinjection (0.4 μL), 19 injections of 2 μL and 4 s each with a spacing of 460 or 480 s were performed. Three independent titrations were run.
Surface plasmon resonance (SPR) measurements
SPR binding experiments were performed on a Biacore X100 instrument (GE Healthcare, Freiburg, Germany) at 25 °C in Hepesbuffered saline (HBS: 10 mm Hepes/NaOH, pH 7.4, 150 mm NaCl, 0.05 % Tween 20) at a flow rate of 30 μL min−1. LecB was biotinylated using EZ‐Link NHS‐LC‐LC‐Biotin (Pierce, cat. no. 21343) following the manufacturer′s conditions. Streptavidin (7000 RU) was immobilized on a research‐grade CM5 chip using standard procedures, and 4200 RU of biotinylated LecB were captured on channel 2. Binding experiments were performed with the LecB‐captured channel 2, and plots represent the subtracted data (channel 2– channel 1). Binding studies consisted of the injection (association 180 s, dissociation 180 s) of various concentrations of inhibitor (120 nm–75 μm for compounds 2, 3, and 7 b, or 3 μm–1.8 mm for 1) in the single cycle mode. Binding was measured as resonance units over time after blank subtraction, and data were then evaluated by using the Biacore X100 evaluation software (version 2.0).
Biomolecular NMR spectroscopy
For NMR experiments, 15N‐LecB (500 μm) in TBS/Ca was supplemented with 5 % D2O (Eurisotop, Germany). NMR spectra were measured in standard NMR tubes (5 mm) in the presence and absence of ligand (250 or 500 μm) at 300 K. NMR experiments were performed with a Bruker Avance III 600 MHz spectrometer equipped with an inverse H/C/N‐TCI‐cryoprobe (5 mm) with actively shielded z‐gradient. 1H,15N‐TROSY‐HSQC spectra were acquired with 256 increments in the indirect dimension with 16 scans per increment. All NMR spectra were processed and analyzed with Bruker's TopSpin software (version 3.0).
Circular dichroism (CD) spectroscopy
Protein samples for CD measurements and melting assays were purified as described above. CD spectra were measured on a JASCO‐J1500 spectropolarimeter (Gross‐Umstadt, Germany) equipped with a PTC‐510 Peltier Thermostatted Single Cell Holder using a 1 mm optical path. Protein samples were prepared as a 34.8 μm solution in TBS buffer supplemented with 1 mm CaCl2 in a reaction volume of 300 μL with or without 100 μm compound 3. Scans were performed at 37 °C over a wavelength range of 200–280 nm (three accumulations) with a scanning speed of 100 nm min−1, 1 nm data pitch, and 1 nm bandwidth. For thermal denaturation, samples were prepared as described above. Folded samples were heated from 37 °C to 96 °C with a heating rate of 2 °C min−1. The CD signal at 229 nm was recorded every 1 °C.
Crystallization and structure determination
Crystals of recombinant LecB from P. aeruginosa were obtained by the hanging drop vapor diffusion method using 2 μL of drops containing a 50:50 (v/v) mix of protein and reservoir solution at 19 °C. The protein at 10 mg mL−1 was incubated with 15.3 mm of 3 in H2O and 100 μm CaCl2 during 1 h at rt prior to co‐crystallization. Clusters were obtained in a solution containing 25 % PEG6K, 1 m LiCl, and 0.1 m citric acid at pH 3.8. A broken tip was directly mounted in a cryoloop and flash‐frozen in liquid nitrogen. Diffraction data were collected at 100 K at the European Synchrotron Radiation Facility (Grenoble, France) on beamline BM30 A using a MARCCD detector. The data were processed using XDS.39 All further computing was performed using the CCP4 suite.40 Data quality statistics are summarized in Table 3. The structure was solved by molecular replacement using PHASER and the tetramer coordinates of 3ZDV as search model.25, 41 Five percent of the observations were set aside for cross‐validation analysis, and hydrogen atoms were added in their riding positions and used for geometry and structure‐factor calculations. The structure was refined using restrained maximum likelihood refinement in REFMAC 5.842 iterated with manual rebuilding in Coot.43 Incorporation of the ligand was performed after inspection of the 2Fo‐DFc weighted maps. Water molecules, introduced automatically using Coot, were inspected manually. The stereochemical quality of the models was assessed with the program Molprobity,44 and coordinates were deposited in the Protein Data Bank under code 5A3O.
Aggregation assay
A stock solution of LecB (40 μL, 200 μm in TBS/Ca) was mixed with serial dilutions of testing compounds (20 μL of final test concentrations of 1.7 mm to 13.5 μm) into a transparent F‐bottom 384‐well microtiter plate (Greiner Bio‐One, Germany, cat. no. 781901) in duplicates. The highest concentration of each inhibitor was included without LecB in duplicates as control. Absorbance was measured on a PheraStar FS (BMG Labtech, Germany) plate reader at 600 nm after 30 min and 4 h of incubation at rt and did not show differences between the two time points. The data were analyzed using BMG Labtech MARS software and GraphPad Prism.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to Holger Bußkamp (University of Konstanz) and Michael Hofmann (Helmholtz Institute for Pharmaceutical Research Saarland, HIPS) for HRMS measurements, to Dr. Sascha Baumann (HIPS) for fruitful discussions, and Emilie Gillon (Centre de Recherches sur les Macromolécules Végétales, CERMAV, Grenoble) for support in crystallization experiments. Crystal data were collected at the European Synchrotron Radiation Facility on beamline FIP‐BM30A (proposal 20120846), and the authors thank David Cobessi for assistance in using the beamline. The authors also thank the Helmholtz Association (A. T., grant no. VH‐NG‐934), the Konstanz Research School Chemical Biology (R. S. and A. T.), the Zukunftskolleg (A. T.), and the Deutsche Forschungsgemeinschaft (A. T., grant no. Ti756/2‐1) for financial support. A. I. acknowledges support from GDR Pseudomonas and Labex ARCANE (ANR‐11‐LABX‐OO3). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty, A. Titz, ChemistryOpen 2015, 4, 756.
This article is part of the Virtual Special Issue “Carbohydrates in the 21st Century: Synthesis and Applications”.
References
- 1. Peleg A. Y., Hooper D. C., New Engl. J. Med. 2010, 362, 1804–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poole K., Front. Microbiol. 2011, 2, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bjarnsholt T., Ciofu O., Molin S., Givskov M., Høiby N., Nat. Rev. Drug Discovery 2013, 12, 791–808. [DOI] [PubMed] [Google Scholar]
- 4. Davies D., Nat. Rev. Drug Discovery 2003, 2, 114–122. [DOI] [PubMed] [Google Scholar]
- 5. Adam E. C., Mitchell B. S., Schumacher D. U., Grant G., Schumacher U., Am. J. Respir. Crit. Care Med. 1997, 155, 2102–2104. [DOI] [PubMed] [Google Scholar]
- 6. Bajolet-Laudinat O., Girod-de Bentzmann S., Tournier J. M., Madoulet C., Plotkowski M. C., Chippaux C., Puchelle E., Infect Immun. 1994, 62, 4481–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chemani C., Imberty A., de Bentzmann S., Pierre M., Wimmerová M., Guery B. P., Faure K., Infect. Immun. 2009, 77, 2065–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diggle S. P., Stacey R. E., Dodd C., Cámara M., Williams P., Winzer K., Environ. Microbiol. 2006, 8, 1095–1104. [DOI] [PubMed] [Google Scholar]
- 9. Tielker D., Hacker S., Loris R., Strathmann M., Wingender J., Wilhelm S., Rosenau F., Jaeger K.-E., Microbiology 2005, 151, 1313–1323. [DOI] [PubMed] [Google Scholar]
- 10. Eierhoff T., Bastian B., Thuenauer R., Madl J., Audfray A., Aigal S., Juillot S., Rydell G. E., Müller S., de Bentzmann S., Imberty A., Fleck C., Römer W., Proc. Natl. Acad. Sci. USA 2014, 111, 12895–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sommer R., Joachim I., Wagner S., Titz A., CHIMIA 2013, 67, 286–290. [DOI] [PubMed] [Google Scholar]
- 12. Clatworthy A. E., Pierson E., Hung D. T., Nat. Chem. Biol. 2007, 3, 541–548. [DOI] [PubMed] [Google Scholar]
- 13. Hauber H.-P., Schulz M., Pforte A., Mack D., Zabel P., Schumacher U., Int. J. Med. Sci. 2008, 5, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilboa-Garber N., Mizrahi L., Garber N., Can. J. Biochem. 1977, 55, 975–981. [DOI] [PubMed] [Google Scholar]
- 15. Gilboa-Garber N., Methods Enzymol. 1982, 83, 378–385. [DOI] [PubMed] [Google Scholar]
- 16. Mitchell E., Houles C., Sudakevitz D., Wimmerova M., Gautier C., Pérez S., Wu A. M., Gilboa-Garber N., Imberty A., Nat. Struct. Biol. 2002, 9, 918–921. [DOI] [PubMed] [Google Scholar]
- 17. Loris R., Tielker D., Jaeger K.-E., Wyns L., J. Mol. Biol. 2003, 331, 861–870. [DOI] [PubMed] [Google Scholar]
- 18. Marotte K., Sabin C., Préville C., Moumé-Pymbock M., Wimmerová M., Mitchell E. P., Imberty A., Roy R., ChemMedChem 2007, 2, 1328–1338. [DOI] [PubMed] [Google Scholar]
- 19. Perret S., Sabin C., Dumon C., Pokorná M., Gautier C., Galanina O., Ilia S., Bovin N., Nicaise M., Desmadril M., Gilboa-Garber N., Wimmerová M., Mitchell E. P., Imberty A., Biochem. J. 2005, 389, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernardi A., Jiménez-Barbero J., Casnati A., De Castro C., Darbre T., Fieschi F., Finne J., Funken H., Jaeger K.-E., Lahmann M., Lindhorst T. K., Marradi M., Messner P., Molinaro A., Murphy P. V., Nativi C., Oscarson S., Penadés S., Peri F., Pieters R. J., Renaudet O., Reymond J.-L., Richichi B., Rojo J., Sansone F., Schäffer C., Turnbull W. B., Velasco-Torrijos T., Vidal S., Vincent S., Wennekes T., Zuilhof H., Imberty A., Chem. Soc. Rev. 2013, 42, 4709–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Imberty A., Chabre Y. M., Roy R., Chem. Eur. J. 2008, 14, 7490–7499. [DOI] [PubMed] [Google Scholar]
- 22. Johansson E. M. V., Crusz S. A., Kolomiets E., Buts L., Kadam R. U., Cacciarini M., Bartels K.-M., Diggle S. P., Cámara M., Williams P., Loris R., Nativi C., Rosenau F., Jaeger K.-E., Darbre T., Reymond J.-L., Chem. Biol. 2008, 15, 1249–1257. [DOI] [PubMed] [Google Scholar]
- 23. Cecioni S., Imberty A., Vidal S., Chem. Rev. 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- 24. Titz A. in Carbohydrate-Based Anti-Virulence Compounds Against Chronic Pseudomonas aeruginosa Infections with a Focus on Small Molecules in: Topics in Medicinal Chemistry, Vol. 12 (Carbohydrates as Drugs) (Eds.: P. H. Seeberger, C. Rademacher), Springer, Berlin, Heidelberg, 2014, p. 169–186. [Google Scholar]
- 25. Hauck D., Joachim I., Frommeyer B., Varrot A., Philipp B., Möller H. M., Imberty A., Exner T. E., Titz A., ACS Chem. Biol. 2013, 8, 1775–1784. [DOI] [PubMed] [Google Scholar]
- 26. Sabin C., Mitchell E. P., Pokorná M., Gautier C., Utille J.-P., Wimmerová M., Imberty A., FEBS Lett. 2006, 580, 982–987. [DOI] [PubMed] [Google Scholar]
- 27. Sommer R., Exner T. E., Titz A., PLoS One 2014, 9, e112822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hofmann A., Sommer R., Hauck D., Stifel J., Göttker-Schnetmann I., Titz A., Carbohydr. Res. 2015, 412, 34–42. [DOI] [PubMed] [Google Scholar]
- 29. Pribulová B., Petrušová M., Pätoprstý V., Petruš L., Chem. Pap. 2003, 57, 287–291. [Google Scholar]
- 30. Cramer F., Otterbach H., Springmann H., Chem. Ber. 1959, 92, 384–391. [Google Scholar]
- 31. Scharenberg M., Jiang X., Pang L., Navarra G., Rabbani S., Binder F., Schwardt O., Ernst B., ChemMedChem 2014, 9, 78–83, and references therein. [DOI] [PubMed] [Google Scholar]
- 32. Copeland R. A., Pompliano D. L., Meek T. D., Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- 33. Pan A. C., Borhani D. W., Dror R. O., Shaw D. E., Drug Discovery Today 2013, 18, 667–673. [DOI] [PubMed] [Google Scholar]
- 34. Søndergaard C. R., Garrett A. E., Carstensen T., Pollastri G., Nielsen J. E., J. Med. Chem. 2009, 52, 5673–5684. [DOI] [PubMed] [Google Scholar]
- 35. Gottlieb H. E., Kotlyar V., Nudelman A., J. Org. Chem. 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- 36. Mitchell E. P., Sabin C., Snajdrová L., Pokorná M., Perret S., Gautier C., Hofr C., Gilboa-Garber N., Koca J., Wimmerová M., Imberty A., Proteins 2005, 58, 735–746. [DOI] [PubMed] [Google Scholar]
- 37. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F., Methods Mol. Biol. 1999, 112, 531–552. [DOI] [PubMed] [Google Scholar]
- 38. Marley J., Lu M., Bracken C., J. Biomol. NMR 2001, 20, 71–75. [DOI] [PubMed] [Google Scholar]
- 39. Kabsch W., Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4, Acta Crystallogr. Sect. D 1994, 50, 760–763.
- 41. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murshudov G. N., Skubak P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., Acta Crystallogr. Sect. D 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary