

Abstract
Excretion of cytoplasmic proteins in pro- and eukaryotes, also referred to as “nonclassical protein export,” is a well-known phenomenon. However, comparatively little is known about the role of the excreted proteins in relation to pathogenicity. Here, the impact of two excreted glycolytic enzymes, aldolase (FbaA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), on pathogenicity was investigated in Staphylococcus aureus. Both enzymes bound to certain host matrix proteins and enhanced adherence of the bacterial cells to host cells but caused a decrease in host cell invasion. FbaA and GAPDH also bound to the cell surfaces of staphylococcal cells by interaction with the major autolysin, Atl, that is involved in host cell internalization. Surprisingly, FbaA showed high cytotoxicity to both MonoMac 6 (MM6) and HaCaT cells, while GAPDH was cytotoxic only for MM6 cells. Finally, the contribution of external FbaA and GAPDH to S. aureus pathogenicity was confirmed in an insect infection model.
INTRODUCTION
Excretion of cytosolic proteins (ECP) has been reported in bacteria and eukaryotes. As none of the classical signal peptide (SP)-dependent or SP-independent pathways could be associated until now with ECP, it has also been referred to as “nonclassical protein export” (1). It is hotly debated whether the release of such proteins is due to cell lysis or whether they are exported by a presently unknown mechanism (2). One of the first reports that typical cytoplasmic proteins are found on the bacterial cell surface came from Pancholi and Fischetti (3). They found that the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is present not only at the cell surface of pathogenic streptococcal groups (3) but also in the supernatant of various bacteria, fungi, and even protozoans (4). GAPDH is a very “sticky” protein as it binds to various human proteins, including plasminogen (5–7), lysozyme, myosin, actin, fibronectin (3), and PAR/CD87 on pharyngeal cells (8). It also stimulates B lymphocytes and induces early interleukin-10 (IL-10) production that facilitates host colonization (9). In group B streptococci (GBS) GAPDH acts as an inducer of apoptosis of murine macrophages (10). Moreover, in enterohemorrhagic and enteropathogenic Escherichia coli, GAPDH was exposed on the surface, where it binds to human plasminogen and fibrinogen, suggesting a role in pathogenesis (11).
For ECP in eukaryotes, the term “moonlighting proteins” has also been coined (12). Moonlighting refers to a single protein that has multiple functions. For example, the mammalian thymidine phosphorylase catalyzes the intracellular dephosphorylation of thymidine but acts outside as a platelet-derived endothelial cell growth factor, which stimulates endothelial cell growth and chemotaxis (12). The glycolytic enzymes GAPDH and enolase and the cell stress proteins chaperonin 60, Hsp70, and peptidyl prolyl isomerase are among the most common of the bacterial moonlighting proteins. They play a role in bacterial virulence since they are involved in adhesion and modulation of cell signaling processes. An overview of moonlighting proteins deriving from bacteria and their role in bacterial virulence is given by Henderson and Martin (13). Such multifunctional proteins are typical for living cells and represent evolutionarily ancient proteins.
Secretome analysis of Staphylococcus aureus showed that there are also typical cytosolic proteins (CPs) in the culture supernatant (14–16). How these CPs are excreted is still not known. Cell lysis has been postulated, and indeed when the cell wall structure is altered in such a way that the cross-linking is affected, a higher release of CPs is observed (17); in a major autolysin (Atl) mutant there is almost no ECP (18). There are, however, good arguments speaking against the hypothesis that ECP is caused only by cell lysis. One of the main arguments is that only certain CPs were excreted, while others, although highly abundant in the cytoplasm, were not found extracellularly, suggesting that there is a selection principle at work (18). Studying the excretion pattern of cytoplasmic proteins using two glycolytic model enzymes, aldolase (FbaA) and enolase (Eno), showed that they are excreted mainly during the exponential growth phase in S. aureus and that the exit site is the septal cleft of dividing cells (19). Another typical excreted CP is the glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is, like FbaA, an essential enzyme in glycolysis of S. aureus (18, 20, 21).
Here, we investigated the role of the excreted glycolytic enzymes FbaA and GAPDH in staphylococci. Application of the two proteins enhanced adherence of S. aureus to host cells but the proteins also adhered to the staphylococcal cell surface via binding to the major autolysin, Atl. However, the most striking activity was their cytotoxicity for monocytes and HaCaT cells and their contribution to the killing of worms in a Galleria mellonella infection model.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in this study are listed in Table S1 in the supplemental material. Most of the studies were carried out with S. aureus USA300 JE2 (22), which is cured from the three plasmids present in the parent strain USA300 LAC (23), a highly characterized community-associated methicillin-resistant S. aureus (MRSA) strain isolated from the Los Angeles County jail. USA300 JE2 and mutants thereof from the Nebraska Transposon Mutant Library (NTML) were obtained from Ken Bayles, Department of Pathology and Microbiology, University of Nebraska Medical Center. Construction of spa (protein A) deletion mutants in S. aureus strains (Δspa strains) was performed using allelic replacement as described by Bae and Schneewind (24). Briefly, ≈2 kb upstream and ≈1 kb downstream of spa were amplified by PCR and ligated to pBASE6 (25) using Gibson assembly (26). The strains of interest were then transformed with the resulting plasmid, pBASE-Δspa. Staphylococcal strains and Escherichia coli strains DC10B and M15 (27) were cultivated at 37°C and with shaking in tryptic soy broth (TSB) and LB, respectively. When appropriate, the medium was supplemented with either 10 μg/ml chloramphenicol for staphylococcal species and 100 μg/ml ampicillin and 25 μg/ml kanamycin for E. coli.
Western blot analysis of cytoplasmic fractions, cell wall-associated proteins, and culture supernatant of staphylococcal strains.
Cell wall-associated proteins were isolated by boiling SA113 Δspa ΔsrtA and SA113 Δspa ΔatlA cells in sample buffer containing β-mercaptoethanol for 7 min. Subsequently cells were pelleted by centrifugation, and supernatant was applied to SDS-PAGE gels. Preparation of cytoplasmic proteins, enrichment of extracellular proteins, and Western blot analyses were performed as described by Ebner et al. (19).
Quantification of FbaA in the cytoplasm and supernatant of S. aureus.
Staphylococcal cultures were grown in basic medium (BM) overnight at 37°C. The cells and supernatants were harvested by applying cells at the same optical densities (optical density at 578 nm [OD578] of 9) and subsequent centrifugation. Supernatant proteins were enriched as described above and analyzed by Western blotting. A standard curve was performed with serial dilutions of purified FbaA, which was used as an internal standard on every blot. Quantification was carried out by comparing the band intensities using ImageJ software.
Purification of FbaA, GAPDH, mCherry, FbaA-mCherry, GAPDH-mCherry, AM, AM-R1/2, and GL.
SA113 Δspa containing the plasmids pCtufamp-fbaA and pCtufamp-gapdh and E. coli DC10B containing the plasmids pCtufamp-mCh (where mCh indicates mCherry), pCtufamp-fbaA-mCh, and pCtufamp-gapdh-mCh were inoculated to an OD578 of ≈0.1 and grown overnight at 37°C. The cells were harvested by centrifugation (7,000 × g for 10 min) and washed twice in phosphate-buffered saline (PBS; Biochrom). Pellets were resuspended in PBS containing complete protease inhibitor cocktail (Roche, Grenzach-Wyhlen, Germany), broken with glass beads (Roth, Karlsruhe, Germany) using a FastPrep FP120 instrument (Thermo Scientific, St. Leon-Rot, Germany), and then subjected to centrifugation (17000 × g for 15 min) to pellet the cell debris. The supernatant containing the cytoplasmic fractions was collected and mixed with preequilibrated Strep-Tactin Superflow resin (IBA, Goettingen, Germany) and incubated for 10 min at 4°C to allow the tagged protein to bind to the resin. The resin-protein mixture was then poured into an empty column and further treated as described by the manufacturer. The His-tagged Atl fragments were composed of the amidase and the two repeats R1 and R2 (AM-R1/2); R1/2 comprises only the repeat sequences R1 and R2, AM comprises only the amidase domain without repeats R1 and R2, and, finally, GL comprises the glucosaminidase domain (28, 29). These proteins were expressed in E. coli M15 using the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible plasmid pEQ30 and purified using Ni-nitrilotriacetic acid (NTA) Superflow resin (Qiagen, Hilden, Germany). Purification of the proteins was performed as described by the manufacturer.
Binding of mCherry, FbaA-mCherry, and GAPDH-mCherry to staphylococcal cells.
To investigate the binding of excreted proteins to staphylococcal cells, 1-ml portions of overnight cultures (16 h) of S. aureus JE2, respective mutants, and Staphylococcus carnosus TM300 were adjusted to the same OD578 values and washed twice with PBS. For each strain, the remaining cell pellet was resuspended in 5 ml of fresh PBS. Subsequently, 0.5 ml of each strain was incubated with 30 μg of either mCherry, FbaA-mCherry, GAPDH-mCherry, or a mixture of 15 μg of FbaA-mCherry and 15 μg of GAPDH-mCherry for 15 min at 37°C. After incubation, the staphylococcal cells were pelleted (17,000 × g for 2 min), and the supernatant was discarded. The pellet was washed twice in 2 ml of PBS and resuspended in 400 μl of PBS. Measurements were carried out using 200 μl in a black F-bottom microtiter plate (Greiner, Frickenhausen, Germany) using a Tecan Infinite 200 (Tecan, Männedorf, Switzerland) plate reader with a constant gain of 193 and with unstained cells as blanks.
Purification of staphylococcal PGN and binding studies of GAPDH to PGN.
The purification of S. aureus peptidoglycan (PGN) was performed essentially as described by de Jonge et al. (30), with modifications according to Ebner et al. (19), resulting in insoluble wall teichoic acid (WTA)-free polymeric PGN. A PGN pulldown assay was performed as described by Terrasse et al. (31), with modifications. Briefly, 20 μg of GAPDH was incubated with PGN for 2 h; subsequently, the unbound protein was removed by centrifugation, and the supernatant was collected. The remaining pellet was mixed with Laemmli buffer and boiled for 10 min to elute bound protein. Subsequently, all samples were loaded on a 12% SDS-PAGE gel and immunoblotted. Detection was carried out using an anti-GAPDH antibody.
Far-Western blot analysis to study interaction of GAPDH and FbaA with extracellular matrix proteins and Atl fragments.
Far-Western blot analysis was performed as described by Wu et al. (32), with modification, using the extracellular matrix proteins plasminogen (Abcam), vitronectin, fibrinogen, fibronectin, human IgG, bovine serum albumin (BSA) (all from Sigma, Munich, Germany) AM-R1/2, AM, GL, and purified GAPDH and FbaA.
Protein digestion for mass analysis.
Protein spots were excised from SDS-PAGE gels and washed with 100 μl of double-distilled H2O (ddH2O) for 5 min (Vibrax VXR; IKA). After the ddH2O was removed, spots were ground with gel-loading pipette tips and destained by the addition of 100 μl of 50% acetonitrile (Merck) and shaking for 5 min. Solvent was removed, and samples were dried in a SpeedVac instrument (Bachofer). A destaining procedure was performed twice or until samples were completely destained. Reduction was carried out with 2.5 μl of dithiothreitol (DTT) solution (45 mM; Merck) in 20 μl of ammonium bicarbonate buffer (25 mM) by shaking for 20 min at 55°C (Thermomixer 5436; Eppendorf). Iodoacetamide (2.5 μl; 100 mM) (Merck) was added, and samples were shaken for 20 min at room temperature. After the supernatant was removed, samples were rinsed with 50 μl of ddH2O and dried in a SpeedVac. Twenty microliters of ammonium bicarbonate buffer (25 mM) and 5 μl of trypsin (0.5 μg, Promega) were added, and samples were digested overnight at 37°C. Supernatants were transferred into protein LoBind tubes (Eppendorf). Peptide extractions were performed twice by adding 15 μl of 50% acetonitrile–1% trifluoroacetic acid (Merck) and transferring the supernatant into the LoBind tubes. Peptide extracts were concentrated down to 30 μl and were further purified and desalted using ZipTip C18 pipette tips (Millipore). Extracted peptides were eluted in 35 μl of 80% acetonitrile–0.1% trifluoroacetic acid, completely dried, and resuspended in 15 μl of 1% acetonitrile–0.05% trifluoroacetic acid. Samples were stored at 4°C until analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Analysis of protein digests by LC-MS/MS.
Peptides were separated by reversed-phase liquid chromatography (UltiMate 3000 RSLCnano nano-ultrahigh-performance liquid chromatograph [UHPLC]) (Dionex) and subsequently analyzed in an online-coupled linear trap quadrupole (LTQ) Orbitrap XL hybrid mass spectrometer (ThermoFisher). Sample volumes of 5 μl were injected onto a 75-μm by 2-cm trapping column (Acclaim PepMap rapid separation LC [RSLC] column; Dionex) at 4 μl/min for 5.75 min. Peptide separation was subsequently performed at 50°C and a flow rate of 175 nl/min on a 50-μm by 25-cm separation column (Acclaim PepMap RSLC; Dionex) applying a 90-min gradient ranging from 2.4 to 32.0% acetonitrile. Eluting peptides were ionized by nanospray ionization and analyzed in the mass spectrometer, implementing a top-five collision-induced dissociation (CID) method which generates fragment spectra for the five most abundant precursor ions in the survey scans. Survey scans were acquired with a resolution of 60,000 and a mass range of 300 to 2,000 m/z, with charge states 2+ and 3+ selected for fragmentation.
Database search and spectral annotation.
Data were processed against the Staphylococcus aureus strain NCTC8325 proteome as contained in the Swiss-Prot database (protein sequences downloaded October 2015; 2,892 reviewed protein sequences [www.uniprot.org]) using the Mascot search engine (Mascot, version 2.2.04; Matrix Science) integrated into the Proteome Discoverer software (version 1.3; ThermoFisher). The search was restricted to tryptic peptides. Precursor mass tolerance was set to 5 ppm, and fragment mass tolerance was set to 0.5 Da. Carbamidomethyl and oxidized methionine were allowed as dynamic modifications. False discovery rates (FDR) were determined by the Percolator algorithm (33) based on processing against a decoy database consisting of the shuffled target database. The FDR was set at a target q value of ≤0.05 (5% FDR). Peptide-spectrum matches (PSMs) with a q value of ≤0.05 were filtered according to additional, orthogonal parameters to ensure spectral quality and validity. Mascot scores were filtered to ≥20.
Adherence to HEK293 and HaCaT cells.
The HaCaT cells, obtained from the Department of Dermatology, University of Tübingen, were cultured in Dulbecco's modified Eagle's medium (DMEM)–high-glucose medium supplied with 10% fetal bovine serum (FBS) and 1% ZellShield at 37°C with 5% CO2. The HEK293 cells, purchased from Invivogen, were cultured in DMEM–high-glucose medium supplemented with 10% FBS (Biochrom GmbH, Berlin, Germany), 1% glutamine, and 1% ZellShield. Prior to stimulation, 5 × 105 cells per well were seeded in a 24-well microtiter plate in 1 ml of culture medium supplemented with antibiotics and grown until confluence at 37°C with 5% CO2. The assay was performed at a multiplicity of infection (MOI) of 30 according to Nguyen et al. (34), with addition of the protein concentrations indicated in the legend for Fig. 3. For invasion, the same experimental procedure was performed, with additional killing of adhered JE2 cells by lysostaphin treatment for 1 h prior to lysis of HaCaT cells. The experiment at an MOI of 30 resulted in too few CFU, so the MOI was increased to 100.
FIG 3.

Impact of FbaA and GAPDH on adherence to and invasion of HaCaT cells. (A) Adherence of JE2 to HaCaT and HEK293 cells normalized to that of H2O. Strep-Tactin elution (Elu) buffer or the addition of BSA (20 μg/ml) made no significant difference in adherence. Only the addition of GAPDH (20 μg/ml), FbaA (20 μg/ml), or GAPDH plus FbaA (each 10 μg/ml) enhanced adherence (*, P < 0.05; **, P < 0.01; Wilcoxon test). (B) Adherence of JE2 to HaCaT cells with increasing concentrations (5, 10, 20, and 40 μg/ml) of GAPDH or FbaA (*, P < 0.05; **, P < 0.001; ns, not significant, by one-way analysis of variance with a Bonferroni posttest). (C) Invasion into HaCaT cells, normalized to that of H2O, in the presence of GAPDH and FbaA (each, 10 μg/ml) (****, P < 0.0001, by t test).
Cytotoxicity assay.
Cytotoxicity assays of JE2-derived FbaA and GAPDH were carried out separately on MonoMac 6 (MM6), a human monocytic leukemia cell line, obtained from the DSMZ (Braunschweig, Germany), and HaCaT cells. Prior to a cytotoxicity assay, 100 μl of HaCaT cells was seeded in a 96-well microtiter plate and incubated for 2 days until the cell number reached 105 cells per well; for MonoMac 6 cells, 105 cells were seeded in a 96-well microtiter plate in 100 μl of culture medium and incubated for 1 h. Both cell lines were incubated at 37°C in 5% CO2. Final concentrations of FbaA and GAPDH (80, 40, 20, 10, 5, and 0 μg/ml) were added to the well, and then cytotoxicity assays were performed using Cell Proliferation Kit I (MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide]) (Roche, Germany). Wavelengths of 570 nm and 690 nm as references were used to measure the absorbance of formed formazan (36).
Galleria mellonella infection model.
Larvae of Galleria mellonella in the final stage (R. J. Mous Live Bait, EX Balk, Netherlands) weighing between 300 and 700 mg were infected with 1 × 106 cells of S. aureus JE2 either untreated or with exogenously applied GAPDH and FbaA at 20 μg/ml each (37, 38). For each experiment, 10 larvae weighing between 300 and 700 mg were infected. This experiment was repeated three times with a total of 30 larvae. Bacteria grown overnight were prepared by being washed twice with PBS (140 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4) and adjusted to an OD578 of 0.1 in PBS. The injection volume for each larva was 10 μl into the last left proleg. A 500-μl Hamilton HPLC syringe and a Hamilton PB600 repeating dispenser were used for injection. After injection, the larvae were incubated at 37°C for 5 days and counted every 24 h.
RESULTS
Excretion of FbaA and GAPDH shows a tendency to correlate with virulence.
In this study, the glycolytic enzymes FbaA and GAPDH as model proteins for excreted CPs were used. We studied the excretion of these proteins in various staphylococcal strains to see whether the amount of excreted proteins correlates with the virulence of the strains. Since ECP occurs mainly during bacterial growth (19), samples were collected after 4 h (mid-exponential phase), and the relative amounts of FbaA and GAPDH in the cytoplasm (control) and culture supernatant were determined by Western blotting. The largest amounts of secreted FbaA and GAPDH were found in the Newman, HG001, HG003, and JE2 strains. FbaA was not detectable in the SA113 and COL strains or in S. carnosus TM300 and S. epidermidis O47 strains. GAPDH was highly excreted in COL and excreted to a lower extent in SA113. Like FbaA, GAPDH was hardly excreted in S. carnosus TM300 and S. epidermidis O47 (Fig. 1). There is a slight tendency that the levels of FbaA and GAPDH released are correlated with virulence. We quantified the amounts of FbaA in the culture supernatant of overnight cultures, which were 0.128 μg/ml in HG003 and 0.294 μg/ml in the Newman strain. However, in lysostaphin-lysed cells, the concentrations increased to 7.299 μg/ml in HG003 and 6.093 μg/ml in Newman (see Fig. S2 in the supplemental material).
FIG 1.
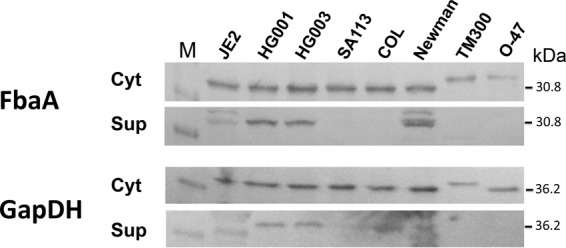
Immunoblotting of FbaA and GAPDH in various staphylococcal strains. Detection of FbaA and GAPDH (theoretical masses, 30.8 and 36.2 kDa, respectively) via specific anti-FbaA and anti-GAPDH antibodies in the cytoplasmic fraction (Cyt) and in the supernatant (Sup). The theoretical masses of FbaA of S. aureus and S. carnosus or S. epidermidis are the same; however, the migration rates are different. The same is true for GAPDH of S. carnosus. Lane M, molecular mass marker.
GAPDH and FbaA bind to host matrix proteins and promote adherence to host cells.
For better understanding of the role of excreted FbaA and GAPDH, we purified the proteins (Fig. 2A) and tested their binding activities to various matrix proteins, like plasminogen, vitronectin, fibrinogen, fibronectin, and BSA (Fig. 2B), by far-Western blot analyses (Fig. 2C). Both proteins bound to plasminogen and a subfragment of vitronectin and to specific fibrinogen chains, but there was no interaction with fibronectin and BSA (Fig. 2C). An overview of the binding specificity is shown in Fig. 2D.
FIG 2.
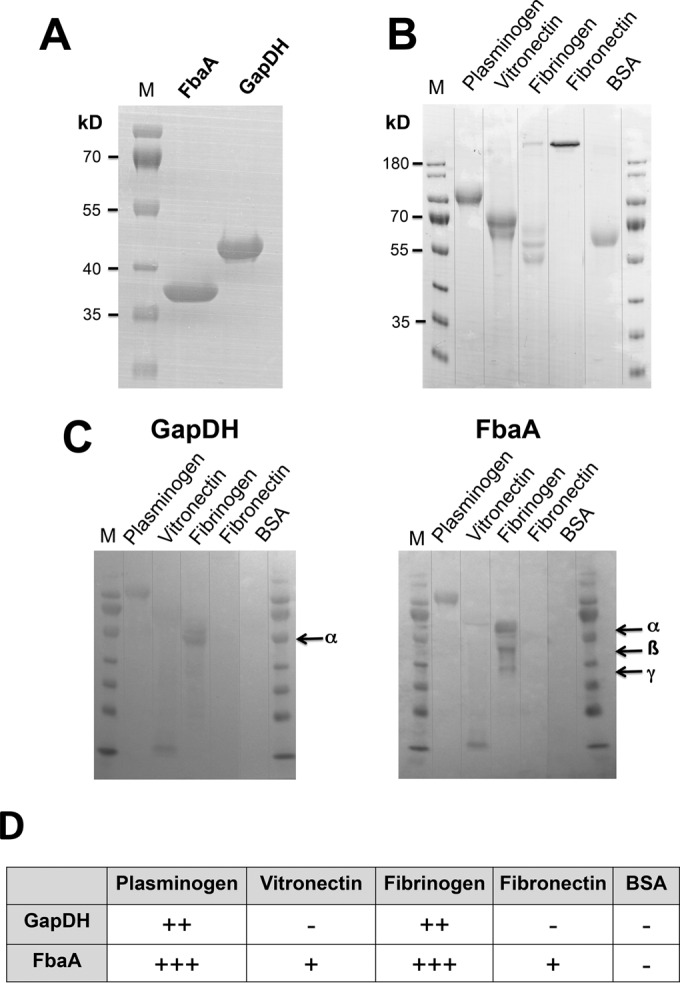
Interaction of FbaA and GAPDH with extracellular matrix proteins. (A) SDS-PAGE of the StrepTag-purified FbaA and GAPDH. (B) SDS-PAGE of plasminogen, vitronectin, fibrinogen, fibronectin, and BSA as a control. (C) Far-Western blot analyses of matrix proteins using purified GAPDH and FbaA as ligands. (D) Summary of the interactions between the matrix proteins and GAPDH and FbaA. The degree of interaction is indicated as follows: +++, strong; ++, intermediate; +, weak; − none. The theoretical masses of FbaA (30.8) and GAPDH (36.2 kDa) were lower than the estimated sizes in SDS-PAGE gels, which were 36 and 42 kDa, respectively. Arrows indicate the alpha, beta, and gamma chains of fibrinogen.
Exogenously applied GAPDH and FbaA promote adherence to HaCaT and HEK293 cells.
To investigate whether GAPDH and FbaA play a role in infection, we performed adherence and invasion assays of JE2 cells. To put the adhesion results on a broader basis, we used two different cell lines: HEK293, a human embryonic kidney cell line, and HaCaT cells, a keratinocyte cell line which has been widely used as a model of keratinocyte function (39). The adhesion assay was performed in the presence and absence of exogenously applied FbaA and GAPDH. As a basis for the adherence assays, we used H2O, Strep-Tactin elution buffer, and BSA. The CFU count with H2O was set to 1.0, and all CFU values were calculated relative to the H2O control. The controls of elution buffer (1.04-fold) and BSA (0.97-fold) did not show any significant difference in adherence levels compared to the level of the H2O control (Fig. 3A). However, in the presence of equal concentrations GAPDH and FbaA, adherence to HaCaT cells was 1.45- and 1.54-fold increased, respectively, and for HEK293 cells adherence was increased 1.23-fold with GAPDH, 1.45-fold with FbaA, and 1.63-fold with both proteins. The strongest increase in adherence, 1.78-fold, was observed in HaCaT cells in the presence of both proteins (Fig. 3A). The adherence of GAPDH and FbaA in a concentration range of 5, 10, 20, and 40 μg/ml was dose dependent (Fig. 3B); saturation was achieved with GAPDH at 40 μg/ml and with FbaA already at 10 μg/ml.
Exogenously applied FbaA and GAPDH decreased invasion into HaCaT cells.
The effect of GAPDH and FbaA on invasion was investigated in HaCaT cells, an aneuploid immortal keratinocyte cell line from adult human skin. HaCaT cells were infected with JE2 (MOI of 100) in the absence and presence of GAPDH and FbaA (each, 10 μg/ml). An MOI of 100 is frequently used for S. aureus invasion into keratinocytes (40). While adherence to HaCaT cells was increased in the presence of both proteins (Fig. 3A), the invasion into HaCaT cells was decreased by more than 50% (Fig. 3C). We wondered why there was such a discrepancy between adherence and invasion and tried to find the reason. The primary mechanism by which S. aureus enters host cells is well characterized: staphylococcal fibronectin binding proteins (FnBPs) interact with cell surface α5β1 integrins via a fibronectin bridge (41). A second internalization mechanism involves the major autolysin, Atl, and heat shock cognate protein Hsc70 as a host cell receptor (42). In the following, we addressed the question of whether GAPDH and FbaA affect these two mechanisms.
Extracellular GAPDH and FbaA bind to staphylococcal cells.
Above, we have shown that GAPDH and FbaA bind to plasminogen and fibrinogen but not to fibronectin and BSA (Fig. 2C), suggesting that the two proteins do not block fibronectin's interaction with the fibronectin binding proteins (FnbA and FnbB) of S. aureus. However, GAPDH and FbaA still might bind to the fibronectin binding proteins at the S. aureus surface. To investigate GAPDH and FbaA binding to the S. aureus surface, we constructed mCherry fusions of both proteins with mCherry as a control. The fluorescent proteins were expressed in E. coli and purified by Strep-Tactin resin (see Fig. S1B in the supplemental material). Their binding to the cell surface was investigated with both S. aureus JE2 and S. carnosus TM300; the two strains showed very similar binding patterns. mCherry as a control showed only low binding to the cells, while binding of the FbaA-mCherry and GAPDH-mCherry fusion proteins was on average increased 10 to 15 times (Fig. 4A). The question now was whether it was a surface protein or a cell wall component like peptidoglycan (PGN) to which FbaA-mCherry and GAPDH-mCherry fusion proteins were bound. To resolve this question, we compared binding of the proteins to JE2 cells with and without trypsin pretreatment for 2 h. Trypsin-treated cells showed a 4- to 5-fold lower binding than that of the untreated cells (Fig. 4C), suggesting that the binding partner is a surface protein.
FIG 4.

FbaA-mCherry and GAPDH-mCherry bind to staphylococci. (A) Binding of mCherry, FbaA-mCherry, and GAPDH-mCherry and the combination of both proteins to JE2 and S. carnosus TM300 cells and measurement of the relative fluorescence units (RFU) (****, P < 0.0001, by t test). (B) Binding of mCherry, FbaA-mCherry, and GAPDH-mCherry to JE2 with and without pretreatment with trypsin for 2 h and measurement of the relative fluorescence units (*, P < 0.05; ****, P < 0.0001, by t test). (C) Pulldown experiments for detection of the interaction between GAPDH and FbaA and staphylococcal PGN. Load, loading control (same amounts of GAPDH and FbaA as used for binding to PGN); Sup, unbound GAPDH and FbaA after incubation with PGN; Elu, GAPDH and FbaA bound to the insoluble PGN, eluted by boiling with Laemmli buffer.
Nevertheless, pulldown experiments with purified insoluble and WTA-free polymeric S. aureus peptidoglycan (PGN) were carried out. Neither FbaA nor GAPDH revealed significant binding to PGN. The amount of applied protein to PGN (Fig. 4B, load) and the amount remaining in the supernatant after 2 h of incubation (Fig. 4B, Sup) were essentially the same. When the proteins were extracted from the PGN pellet (eluate), only tiny amounts of GAPDH and FbaA were eluted (Fig. 4B). These results indicate that PGN is not the binding component, leaving the question open as to which surface protein could be the binding partner.
Extracellular GAPDH and FbaA bind to the major autolysin, Atl.
To identify the staphylococcal surface protein(s) with which GAPDH and FbaA interact, far-Western blot assays were performed. The surface-associated proteins of S. aureus SA113 Δspa ΔsrtA were stripped off by boiling washed whole cells in sample buffer and separating the proteins by SDS-PAGE. The srtA deletion mutant was chosen because the surface proteins of this mutant are not covalently bound and are therefore easier to extract from the cell wall. After the proteins were blotted, one could see in the far-Western blot with both GAPDH and FbaA bands of 65 and 55 kDa, and with GAPDH an additional weak band was visible at 130 kDa (Fig. 5A). The corresponding protein bands were cut from the SDS-PAGE gel and subjected to LC-MS/MS peptide analysis. The predominant protein found in the size range of 55, 65, and 130 kDa belonged to the major autolysin, Atl. Indeed, it has been previously shown that the approximately 130-kDa Atl precursor is processed to 60- and 52-kDa proteins (29).
FIG 5.
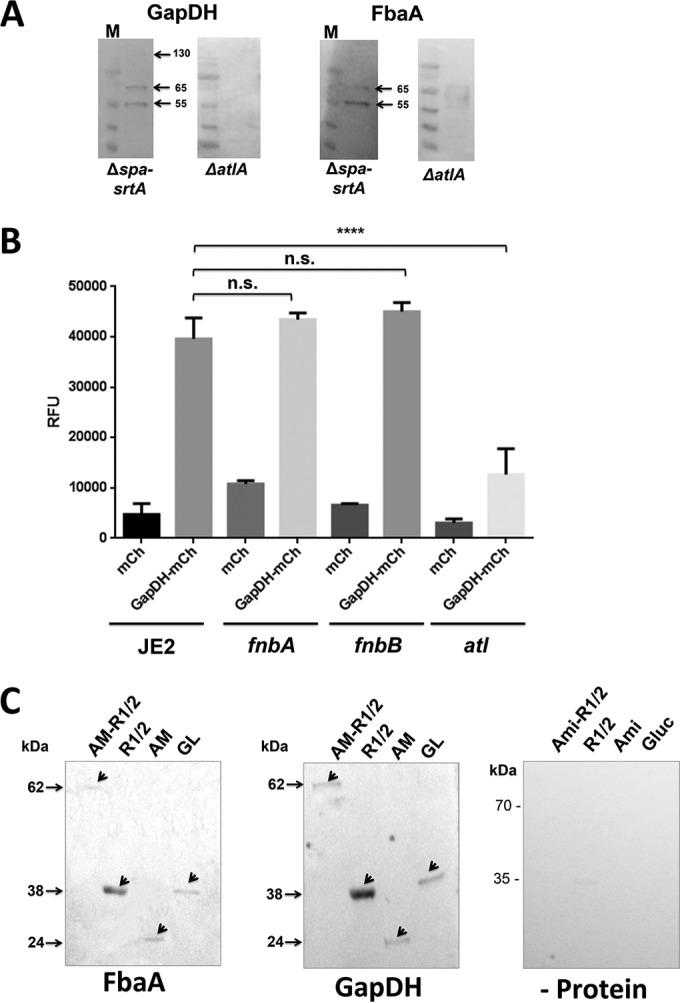
FbaA and GAPDH interact with subfragments of the major amidase (Atl). (A) Far-Western blotting of GAPDH and FbaA with SA113 Δspa ΔsrtA and SA113 ΔatlA cells. (B) Binding of mCherry and GAPDH-mCherry to JE2 as well as to its fnbA, fnbB, and atl transposon mutants; binding was quantified by measurement of relative fluorescence units (****, P < 0.0001, by t test). (C) Far-Western blot analyses of recombinant Atl fragments (AM-R1/2, R1/2, AM [amidase] and GL [glucosaminidase]) purified from E. coli. GAPDH and FbaA were used as ligands; for detection, specific anti-FbaA and anti-GAPDH antibodies were used. As a control, no bait protein was added.
To prove that Atl is the binding partner of GAPDH and FbaA, far-Western blotting was performed with surface proteins derived from the atl deletion mutant, SA113 ΔatlA. In this mutant no interaction was observed (Fig. 5A). Other evidence that Atl is the binding partner was the approximately 4-fold-decreased binding of GAPDH-mCherry to the surface of the atl mutant compared to that of JE2 and its fnbA and fnbB mutants (Fig. 5B).
Finally, we wondered to which Atl domain GAPDH and FbaA are binding. Again, we performed far-Western blot analysis with His-tagged Atl fragments heterologously expressed in E. coli and purified via Ni-NTA. The chosen Atl derivatives were AM-R1/2, composed of the amidase and the two repeats R1 and R2. R1/2 comprises only the repeat sequences R1 and R2, whereas AM is only the amidase domain without the repeats R1 and R2. Finally, GL is only the glucosaminidase domain. The far-Western blotting showed that GAPDH and FbaA bound to all Atl fragments; however, the binding to R1/2 was the strongest (Fig. 5C). The control without ligands showed no signals (see Fig. S1C in the supplemental material). Apparently, the repeats R1/2 are the major target structures of GAPDH and FbaA.
FbaA and GAPDH are cytotoxic.
To investigate whether the binding of FbaA and GAPDH to the matrix proteins has an effect on the host cells, we investigated cytotoxicity with the cell proliferation (MTT) assay in MonoMac 6 (MM6) cells and the keratinocyte cell line HaCaT. With MM6 cells, both proteins exerted a dose-dependent (5 to 80 μg/ml) cytotoxic effect; the toxicity of FbaA was more pronounced than that of GAPDH. At the highest concentrations of GAPDH and FbaA, viability of MM6 cells was decreased by 22% and 52%, respectively (Fig. 6A). With HaCaT cells, GAPDH had no cytotoxic effect; on the contrary, the viability of the cells even appeared increased with increasing concentrations. However, FbaA also showed a high cytotoxic effect with HaCaT cells in a range comparable to that with MM6 cells (Fig. 6B).
FIG 6.
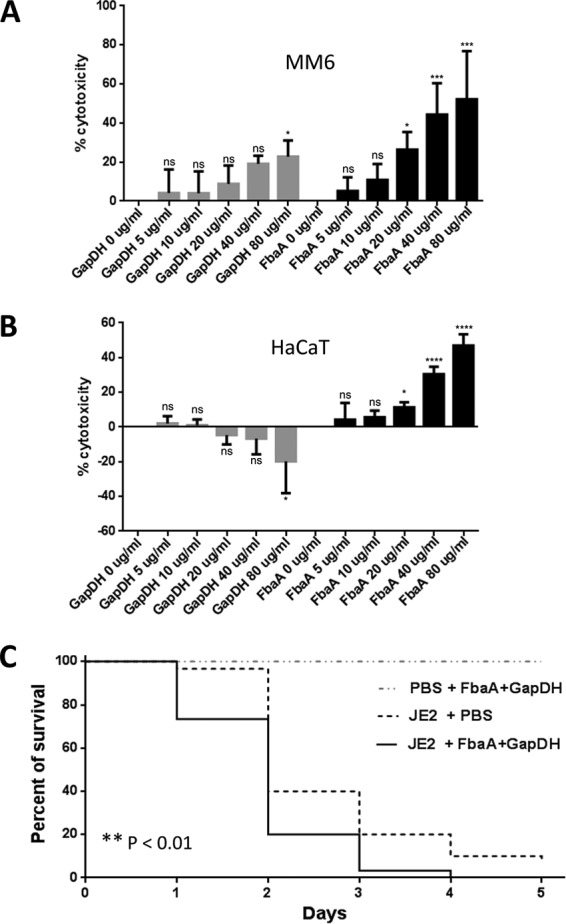
Cytotoxicity test of FbaA and GAPDH in MonoMac 6 and HaCaT cells and infection assay with Galleria mellonella. (A and B) Concentration-dependent cytotoxicity of GAPDH and FbaA in MonoMac 6 cells and HaCaT cells, as indicated. For toxicity testing Cell Proliferation Kit I (MTT) was used. ns, not significant; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001, by one-way analysis of variance with a Bonferroni posttest. (C) Survival of Galleria mellonella treated with JE2 in the presence or absence of FbaA and GAPDH. Survival was followed over 5 days. Statistical significance was measured for JE2 treated with PBS and for JE2 treated with FbaA plus GAPDH (**, P < 0.01, by a Gehan-Breslow-Wilcoxon test; **, P < 0.01, by a Mantel-Cox test).
GAPDH and FbaA enhance virulence of S. aureus USA300 JE2 in a Galleria mellonella infection model.
As external FbaA and GAPDH contribute to host cell adherence and are cytotoxic to host cells, we wondered whether exogenously applied FbaA and GAPDH also have an effect in vivo. To answer this question, we used a Galleria mellonella infection model to compare the virulence of JE2 with and without exogenously applied proteins. The percentage of survival was measured over a 5-day period. In the control group, which was injected with PBS containing FbaA and GAPDH, all larvae survived the 5-day period. The group of larvae infected with only JE2 survived the first day, with a percentage of 97%, followed by a high death rate at the second day, and after 5 days 10% still survived. Larvae infected with JE2 together with FbaA and GAPDH were significantly more rapidly killed. Only 73% survived the first day, and after 4 days 97% were killed; there was no survival after 5 days (Fig. 6C).
DISCUSSION
Excretion of cytoplasmic proteins (ECP) into the culture supernatant is a general phenomenon observed in both pro- and eukaryotes (43, 44). In particular, glycolytic enzymes, chaperones, translation factors, or enzymes involved in detoxification of reactive oxygen species (ROS) were found in the supernatants by secretome analysis (15, 45–48). However, proteome analysis alone does not reveal the potential role of excreted CPs in pathogenicity. Therefore, we concentrated here on two typical cytoplasmic proteins, the aldolase (FbaA) and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Both proteins are glycolytic proteins, and their excretion into the supernatant has been observed in various microorganisms.
One of the first questions we asked was whether the amounts of excreted GAPDH and FbaA differ in various staphylococcal strains and species. For appropriate detection of the proteins in S. aureus by Western blotting, we first had to create protein A (spa) mutants in the corresponding strains, which is the reason why we tested only a limited number of S. aureus strains. However, one tendency can be seen already: the more virulent the strains, the larger the amounts of excreted proteins in the supernatant (Fig. 1). The virulence of some of the strains has been previously investigated in a mouse sepsis model (49). In this study, S. aureus Newman, USA300, HG001, and HG003 showed the highest killing rates. Exactly these strains showed the largest amounts of FbaA and GAPDH in the supernatant; in JE2, a derivative of USA300, there was also a higher migrating band visible that we cannot explain at the moment. In SA113 FbaA and GAPDH were hardly detectable in the supernatant, and in COL only GAPDH was present. In the mouse sepsis model S. aureus COL was almost avirulent; SA113 has not been tested in this study, but it should also be attenuated as it is defective in the global regulators agr and sigB (49), which are involved in virulence. In the biofilm-forming Staphylococcus epidermidis O47 strain, a medium-pathogenic species, and in the nonpathogenic species representative S. carnosus TM300 (50), the two proteins were hardly excreted although the cytoplasmic counterparts were clearly visible. We are aware that this is only a preliminary study, but it suggests a correlation between ECP and virulence.
But how do FbaA and GAPDH contribute to virulence? We showed that they bind to host matrix proteins like plasminogen, vitronectin, and fibrinogen, but they did not bind to fibronectin or BSA. This binding to matrix proteins is not unusual and has been observed in other microorganisms as well, such as the FbaA of mycobacteria or GAPDH and FbaA of Candida albicans that bind to plasminogen (51, 52). In contrast to the FbA of S. aureus, the Neisseria meningitidis FbaA is not essential for growth, but it is required for enhanced adhesion to host cells (21). This is what was also seen with FbaA and GAPDH; they increase adherence of JE2 to HaCaT and HEK293 cells by a factor of 1.5 to 1.8 (Fig. 3A and B). Surprisingly, however, in the presence of FbaA and GAPDH, invasion of JE2 into HaCaT cells was decreased by 50% (Fig. 3C). The mechanism for the enhanced adherence of FbaA and GAPDH to HaCaT and HEK293 cells is unknown. However, it was shown that the Streptococcus pneumoniae Fba directly interacts with the Flamingo cadherin receptor (41); maybe this interaction causes the increased adherence. Since Fba from S. pneumoniae shares 53% identity with the S. aureus FbaA, this interaction might also apply for the S. aureus FbaA.
We also wondered why there was such a discrepancy between adherence and invasion, and we tried to find the reason. The primary mechanism by which S. aureus enters host cells is well characterized: staphylococcal fibronectin binding proteins (FnBPs) interact with the host cell surface α5β1 integrin via a fibronectin bridge (41). A second internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptors (42). In the following, the question of whether FbaA and GAPDH interfere with one of the two mechanisms was addressed. Neither FbaA nor GAPDH binds to fibronectin; therefore, one can rule out that either affects the interaction of the cell surface-bound fibronectin binding proteins (FnbA and FnbB) with fibronectin. However, neither FbaA nor GAPDH could bind to FnbA and FnbB, thus hampering binding to fibronectin. To answer this question, we investigated the binding of fluorescent GAPDH-mCherry and FbaA-mCherry to the cell surface of JE2 cells and of S. carnosus as a control. Both proteins showed strong binding to the cell surfaces of both bacteria. The interaction partner must be a surface protein as trypsin treatment significantly decreased binding, and peptidoglycan as a major cell wall component showed almost no interaction (Fig. 4). At this stage we also could exclude protein A and fibronectin binding proteins as interaction partners as the corresponding genes are absent in the genome of S. carnosus (53). Both far-Western blotting and LC-MS/MS showed that FbaA and GAPDH interact with the major autolysin, Atl, on the surface. Atl appears to be essential as in far-Western blotting with an atl mutant, there was no further binding partner visible. This result suggests that FbaA and GAPDH are partially excreted into the supernatant, but there is also a proportion bound to the cell wall by interaction particularly with the repeat domains of Atl (Fig. 7).
FIG 7.
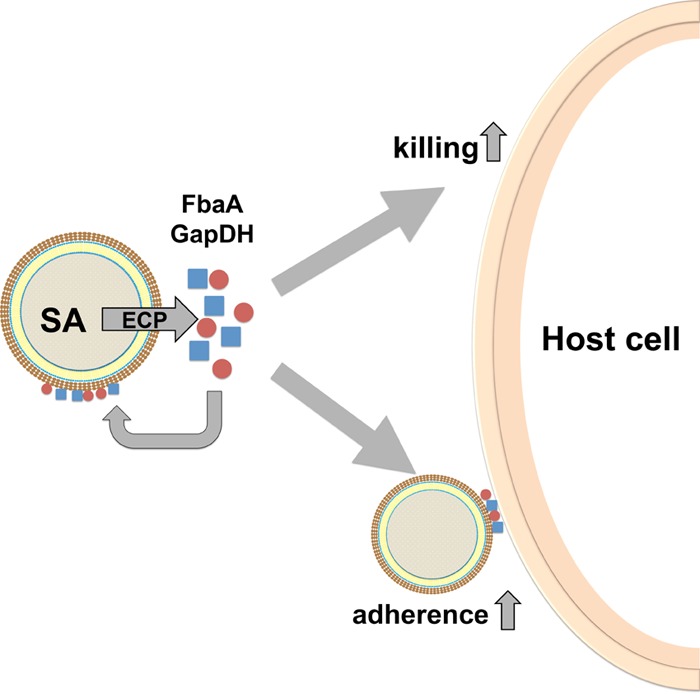
Model for the secondary role of FbaA and GAPDH after excretion. FbaA and GAPDH get excreted and bind to certain extracellular matrix proteins and also bind back to the staphylococcal cells via interaction with Atl and weak interaction with PGN. This affinity to both eucaryotic matrix proteins and staphylococcal surface proteins mediates adherence. SA, S. aureus.
In staphylococci Atl is secreted in a sec-dependent manner as a bifunctional precursor that is composed of two distinct murein hydrolases, an N-acetylmuramyl-l-alanine amidase (AM) and a glucosaminidase (GL) (54, 55). The AM and GL domains are separated by three major repeats, R1 to R3. Following cleavage of the signal peptide, external Atl is proteolytically cleaved at two positions, after the propeptide (PP) and after repeat R2, leading to the formation of processed enzymes AM-R1-R2 and R3-GL (29). Both enzymes bind to the staphylococcal cell wall, particularly to the septum site of dividing cells (56, 57). The repeat sequences are responsible for the targeting of the enzymes to the lipoteichoic acid (LTA) at the septum region, where amidase and a glucosaminidase catalyze the last step in cell division, the cell separation of the daughter cells, by resolving the intertwined murein sacculus (58, 59). By far-Western blotting, we could show that FbaA and GAPDH bound to all Atl fragments; however, the binding to repeat sequences R1/2 was by far the strongest (Fig. 5C). These results suggest that a fraction of FbaA and GAPDH is not excreted but is bound via interaction with Atl, particularly with its repeat domains R1-R2, at the cell surface. Atl is described as a second factor contributing to S. aureus and S. epidermidis internalization by nonprofessional phagocytes via interaction with the host heat shock cognate 71-kDa protein, Hsc70, receptor (42). Which Atl domain is interacting with Hsc70 is unknown; however, the repeat domains are likely candidates. The interaction of FbaA and GAPDH with Atl could occur during the excretion pathway and/or by back-binding of the excreted proteins to Atl. In any case, interference of FbaA and GAPDH with the Atl-Hsc70 internalization pathway is an explanation for the decreased host cell internalization.
Another question was whether excreted FbaA and GAPDH have an effect on host cells. To our great surprise, FbaA showed comparatively high cytotoxicity to both MM6 and HaCaT cells, while GAPDH was cytotoxic only for MM6 cells (Fig. 6A and B). For GAPDH of group B streptococci (GBS) and S. pyogenes, it has been previously described that they induce apoptosis in murine macrophages (10). How the two, seemingly harmless, glycolytic enzymes FbaA and GAPDH affect the viability of host cells is unclear. An explanation for the finding that GAPDH is toxic for MonoMac 6 but not for HaCaT cells could lie in an enhanced internalization (endocytosis-like?) of GAPDH, which might cause apoptosis, as reported for the Streptococcus pyogenes and S. aureus GAPDH proteins (10). However, this cytotoxic activity could contribute to the virulence of S. aureus, maybe not at the same high level as alpha-toxin but to a noticeable extent. It is quite difficult to mimic the in vivo situation exactly since quantification of excreted CPs in an in vivo model is not easily feasible. In a host organism, S. aureus is continuously challenged by the immune system, which makes it very likely that the amount of released CPs in an infection is by far higher than that in an in vitro culture. Subsequently, cell lysis caused by immune cells is expected. The amount of bacteria in an abscess is strongly dependent on the inoculum and strain and was quantified to approximately 6.5 × 106 using 1 × 106 CFU for the Newman strain (40). However, the number of lysed S. aureus cells might be magnitudes higher. In vitro, lysostaphin-lysed S. aureus cells release roughly 7 μg/ml FbaA (see Fig. S2 in the supplemental material), which is already a concentration that exerts cytotoxic activity. Probably, such concentrations may be reached during abscess formation (23). Indeed, it has been shown that a number of cytoplasmic proteins were enriched in neutrophil-depleted abscesses, and among these enzymes was also GAPDH (60). So far, only toxins, binding proteins, cell wall polymers, and defensin or ROS resistance factors have been considered virulence molecules (61). Now we think, however, that the time has come to consider also excreted cytoplasmic proteins as serious virulence factors. Indeed, in a Galleria mellonella infection model, the addition of FbaA and GAPDH aggravated the virulence of JE2 (Fig. 6C). Although Galleria mellonella lacks the adaptive immune system of higher mammals, the results show that in the presence of FbaA and GAPDH, the survival of the larvae was significantly decreased.
Supplementary Material
ACKNOWLEDGMENTS
We thank Regine Stemmler for excellent technical assistance, Cordula Gekeler and Shideh Vatani Shamirzadi for assistance in cell culture, Sebastian Reichert and Lukas Mechler for useful discussion on the topic, and Daniela Eberhart for carefully reading the manuscript. In particular, we thank Kenneth W. Bayles for kindly providing the Nebraska Transposon Mutant Library.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00138-16.
REFERENCES
- 1.Nickel W. 2003. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem 270:2109–2119. [DOI] [PubMed] [Google Scholar]
- 2.Wang G, Chen H, Xia Y, Cui J, Gu Z, Song Y, Chen YQ, Zhang H, Chen W. 2013. How are the non-classically secreted bacterial proteins released into the extracellular milieu? Curr Microbiol 67:688–695. doi: 10.1007/s00284-013-0422-6. [DOI] [PubMed] [Google Scholar]
- 3.Pancholi V, Fischetti VA. 1992. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J Exp Med 176:415–426. doi: 10.1084/jem.176.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pancholi V, Chhatwal GS. 2003. Housekeeping enzymes as virulence factors for pathogens. Int J Med Microbiol 293:391–401. doi: 10.1078/1438-4221-00283. [DOI] [PubMed] [Google Scholar]
- 5.D'Costa SS, Boyle MD. 2000. Interaction of group A streptococci with human plasmin(ogen) under physiological conditions. Methods 21:165–177. doi: 10.1006/meth.2000.0988. [DOI] [PubMed] [Google Scholar]
- 6.Lottenberg R, Broder CC, Boyle MD, Kain SJ, Schroeder BL, Curtiss R III. 1992. Cloning, sequence analysis, and expression in Escherichia coli of a streptococcal plasmin receptor. J Bacteriol 174:5204–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winram SB, Lottenberg R. 1996. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 142:2311–2320. doi: 10.1099/13500872-142-8-2311. [DOI] [PubMed] [Google Scholar]
- 8.Jin H, Song YP, Boel G, Kochar J, Pancholi V. 2005. Group A streptococcal surface GAPDH, SDH, recognizes uPAR/CD87 as its receptor on the human pharyngeal cell and mediates bacterial adherence to host cells. J Mol Biol 350:27–41. doi: 10.1016/j.jmb.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 9.Madureira P, Baptista M, Vieira M, Magalhaes V, Camelo A, Oliveira L, Ribeiro A, Tavares D, Trieu-Cuot P, Vilanova M, Ferreira P. 2007. Streptococcus agalactiae GAPDH is a virulence-associated immunomodulatory protein. J Immunol 178:1379–1387. doi: 10.4049/jimmunol.178.3.1379. [DOI] [PubMed] [Google Scholar]
- 10.Oliveira L, Madureira P, Andrade EB, Bouaboud A, Morello E, Ferreira P, Poyart C, Trieu-Cuot P, Dramsi S. 2012. Group B streptococcus GAPDH is released upon cell lysis, associates with bacterial surface, and induces apoptosis in murine macrophages. PLoS One 7:e29963. doi: 10.1371/journal.pone.0029963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egea L, Aguilera L, Gimenez R, Sorolla MA, Aguilar J, Badia J, Baldoma L. 2007. Role of secreted glyceraldehyde-3-phosphate dehydrogenase in the infection mechanism of enterohemorrhagic and enteropathogenic Escherichia coli: interaction of the extracellular enzyme with human plasminogen and fibrinogen. Int J Biochem Cell Biol 39:1190–1203. doi: 10.1016/j.biocel.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Jeffery CJ. 1999. Moonlighting proteins. Trends Biochem Sci 24:8–11. doi: 10.1016/S0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- 13.Henderson B, Martin A. 2013. Bacterial moonlighting proteins and bacterial virulence. Curr Top Microbiol Immunol 358:155–213. doi: 10.1007/82_2011_188. [DOI] [PubMed] [Google Scholar]
- 14.Sibbald MJ, Winter T, van der Kooi-Pol MM, Buist G, Tsompanidou E, Bosma T, Schafer T, Ohlsen K, Hecker M, Antelmann H, Engelmann S, van Dijl JM. 2010. Synthetic effects of secG and secY2 mutations on exoproteome biogenesis in Staphylococcus aureus. J Bacteriol 192:3788–3800. doi: 10.1128/JB.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM. 2004. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev 68:207–233. doi: 10.1128/MMBR.68.2.207-233.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziebandt AK, Becher D, Ohlsen K, Hacker J, Hecker M, Engelmann S. 2004. The influence of agr and σB in growth phase dependent regulation of virulence factors in Staphylococcus aureus. Proteomics 4:3034–3047. doi: 10.1002/pmic.200400937. [DOI] [PubMed] [Google Scholar]
- 17.Nega M, Dube L, Kull M, Ziebandt AK, Ebner P, Albrecht D, Krismer B, Rosenstein R, Hecker M, Götz F. 2015. Secretome analysis revealed adaptive and non-adaptive responses of the Staphylococcus carnosus femB mutant. Proteomics 15:1268–1279. doi: 10.1002/pmic.201400343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasztor L, Ziebandt AK, Nega M, Schlag M, Haase S, Franz-Wachtel M, Madlung J, Nordheim A, Heinrichs DE, Götz F. 2010. Staphylococcal major autolysin (Atl) is involved in excretion of cytoplasmic proteins. J Biol Chem 285:36794–36803. doi: 10.1074/jbc.M110.167312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebner P, Prax M, Nega M, Koch I, Dube L, Yu W, Rinker J, Popella P, Flötenmeyer M, Götz F. 2015. Excretion of cytoplasmic proteins (ECP) in Staphylococcus aureus. Mol Microbiol 97:775–789. doi: 10.1111/mmi.13065. [DOI] [PubMed] [Google Scholar]
- 20.Ling E, Feldman G, Portnoi M, Dagan R, Overweg K, Mulholland F, Chalifa-Caspi V, Wells J, Mizrachi-Nebenzahl Y. 2004. Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin Exp Immunol 138:290–298. doi: 10.1111/j.1365-2249.2004.02628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tunio SA, Oldfield NJ, Berry A, Ala'Aldeen DA, Wooldridge KG, Turner DP. 2010. The moonlighting protein fructose-1, 6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Mol Microbiol 76:605–615. doi: 10.1111/j.1365-2958.2010.07098.x. [DOI] [PubMed] [Google Scholar]
- 22.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng AG, DeDent AC, Schneewind O, Missiakas D. 2011. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol 19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C. 2012. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog 8:e1003016. doi: 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 27.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 29.Heilmann C, Hussain M, Peters G, Götz F. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol 24:1013–1024. doi: 10.1046/j.1365-2958.1997.4101774.x. [DOI] [PubMed] [Google Scholar]
- 30.de Jonge BL, Chang YS, Gage D, Tomasz A. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J Biol Chem 267:11248–11254. [PubMed] [Google Scholar]
- 31.Terrasse R, Amoroso A, Vernet T, Di Guilmi AM. 2015. Streptococcus pneumoniae GAPDH Is released by cell lysis and interacts with peptidoglycan. PLoS One 10:e0125377. doi: 10.1371/journal.pone.0125377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Li Q, Chen XZ. 2007. Detecting protein-protein interactions by Far Western blotting. Nat Protoc 2:3278–3284. doi: 10.1038/nprot.2007.459. [DOI] [PubMed] [Google Scholar]
- 33.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen MT, Kraft B, Yu W, Demicrioglu DD, Hertlein T, Burian M, Schmaler M, Boller K, Bekeredjian-Ding I, Ohlsen K, Schittek B, Götz F. 2015. The νSaα specific lipoprotein like cluster (lpl) of Staphylococcus aureus USA300 contributes to immune stimulation and invasion in human cells. PLoS Pathog 11:e1004984. doi: 10.1371/journal.ppat.1004984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu W, Götz F. 2012. Cell wall antibiotics provoke accumulation of anchored mCherry in the cross wall of Staphylococcus aureus. PLoS One 7:e30076. doi: 10.1371/journal.pone.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barlow M. 2009. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol Biol 532:397–411. doi: 10.1007/978-1-60327-853-9_23. [DOI] [PubMed] [Google Scholar]
- 37.Mechler L, Herbig A, Paprotka K, Fraunholz M, Nieselt K, Bertram R. 2015. A novel point mutation promotes growth phase-dependent daptomycin tolerance in Staphylococcus aureus. Antimicrob Agents Chemother 59:5366–5376. doi: 10.1128/AAC.00643-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peleg AY, Jara S, Monga D, Eliopoulos GM, Moellering RC Jr, Mylonakis E. 2009. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob Agents Chemother 53:2605–2609. doi: 10.1128/AAC.01533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olaru F, Jensen LE. 2010. Chemokine expression by human keratinocyte cell lines after activation of Toll-like receptors. Exp Dermatol 19:e314–e316. doi: 10.1111/j.1600-0625.2009.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weidenmaier C, McLoughlin RM, Lee JC. 2010. The zwitterionic cell wall teichoic acid of Staphylococcus aureus provokes skin abscesses in mice by a novel CD4+ T-cell-dependent mechanism. PLoS One 5:e13227. doi: 10.1371/journal.pone.0013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blau K, Portnoi M, Shagan M, Kaganovich A, Rom S, Kafka D, Chalifa Caspi V, Porgador A, Givon-Lavi N, Gershoni JM, Dagan R, Mizrachi Nebenzahl Y. 2007. Flamingo cadherin: a putative host receptor for Streptococcus pneumoniae. J Infect Dis 195:1828–1837. doi: 10.1086/518038. [DOI] [PubMed] [Google Scholar]
- 42.Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol 12:1746–1764. doi: 10.1111/j.1462-5822.2010.01506.x. [DOI] [PubMed] [Google Scholar]
- 43.Götz F, Yu W, Dube L, Prax M, Ebner P. 2015. Excretion of cytosolic proteins (ECP) in bacteria. Int J Med Microbiol 305:230–237. doi: 10.1016/j.ijmm.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 44.Jeffery CJ. 2003. Moonlighting proteins: old proteins learning new tricks. Trends Genet 19:415–417. doi: 10.1016/S0168-9525(03)00167-7. [DOI] [PubMed] [Google Scholar]
- 45.Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJ, Raangs GC, Stokroos I, Arends JP, Dubois JY, van Dijl JM. 2006. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol Mol Biol Rev 70:755–788. doi: 10.1128/MMBR.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trost M, Wehmhoner D, Karst U, Dieterich G, Wehland J, Jansch L. 2005. Comparative proteome analysis of secretory proteins from pathogenic and nonpathogenic Listeria species. Proteomics 5:1544–1557. doi: 10.1002/pmic.200401024. [DOI] [PubMed] [Google Scholar]
- 47.Xia XX, Han MJ, Lee SY, Yoo JS. 2008. Comparison of the extracellular proteomes of Escherichia coli B and K-12 strains during high cell density cultivation. Proteomics 8:2089–2103. doi: 10.1002/pmic.200700826. [DOI] [PubMed] [Google Scholar]
- 48.Li M, Rosenshine I, Tung SL, Wang XH, Friedberg D, Hew CL, Leung KY. 2004. Comparative proteomic analysis of extracellular proteins of enterohemorrhagic and enteropathogenic Escherichia coli strains and their ihf and ler mutants. Appl Environ Microbiol 70:5274–5282. doi: 10.1128/AEM.70.9.5274-5282.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbert S, Ziebandt AK, Ohlsen K, Schäfer T, Hecker M, Albrecht D, Novick R, Götz F. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun 78:2877–2889. doi: 10.1128/IAI.00088-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenstein R, Götz F. 2013. What distinguishes highly pathogenic staphylococci from medium- and non-pathogenic? Curr Top Microbiol Immunol 358:33–89. doi: 10.1007/82_2012_286. [DOI] [PubMed] [Google Scholar]
- 51.Crowe JD, Sievwright IK, Auld GC, Moore NR, Gow NA, Booth NA. 2003. Candida albicans binds human plasminogen: identification of eight plasminogen-binding proteins. Mol Microbiol 47:1637–1651. doi: 10.1046/j.1365-2958.2003.03390.x. [DOI] [PubMed] [Google Scholar]
- 52.Puckett S, Trujillo C, Eoh H, Marrero J, Spencer J, Jackson M, Schnappinger D, Rhee K, Ehrt S. 2014. Inactivation of fructose-1,6-bisphosphate aldolase prevents optimal co-catabolism of glycolytic and gluconeogenic carbon substrates in Mycobacterium tuberculosis. PLoS Pathog 10:e1004144. doi: 10.1371/journal.ppat.1004144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenstein R, Nerz C, Biswas L, Resch A, Raddatz G, Schuster SC, Götz F. 2009. Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl Environ Microbiol 75:811–822. doi: 10.1128/AEM.01982-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Albrecht T, Raue S, Rosenstein R, Nieselt K, Götz F. 2012. Phylogeny of the staphylococcal major autolysin and its use in genus and species typing. J Bacteriol 194:2630–2636. doi: 10.1128/JB.06609-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Götz F, Heilmann C, Stehle T. 2014. Functional and structural analysis of the major amidase (Atl) in Staphylococcus. Int J Med Microbiol 304:156–163. doi: 10.1016/j.ijmm.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Komatsuzawa H, Sugai M, Nakashima S, Yamada S, Matsumoto A, Oshida T, Suginaka H. 1997. Subcellular localization of the major autolysin, ATL and its processed proteins in Staphylococcus aureus. Microbiol Immunol 41:469–479. doi: 10.1111/j.1348-0421.1997.tb01880.x. [DOI] [PubMed] [Google Scholar]
- 57.Yamada S, Sugai M, Komatsuzawa H, Nakashima S, Oshida T, Matsumoto A, Suginaka H. 1996. An autolysin ring associated with cell separation of Staphylococcus aureus. J Bacteriol 178:1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schlag M, Biswas R, Krismer B, Köhler T, Zoll S, Yu W, Schwarz H, Peschel A, Götz F. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol Microbiol 75:864–873. doi: 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- 59.Zoll S, Schlag M, Shkumatov AV, Rautenberg M, Svergun DI, Götz F, Stehle T. 2012. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J Bacteriol 194:3789–3802. doi: 10.1128/JB.00331-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Attia AS, Cassat JE, Aranmolate SO, Zimmerman LJ, Boyd KL, Skaar EP. 2013. Analysis of the Staphylococcus aureus abscess proteome identifies antimicrobial host proteins and bacterial stress responses at the host-pathogen interface. Pathog Dis 69:36–48. doi: 10.1111/2049-632X.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi SD, Malachowa N, DeLeo FR. 2015. Pathogenesis of Staphylococcus aureus abscesses. Am J Pathol 185:1518–1527. doi: 10.1016/j.ajpath.2014.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.