

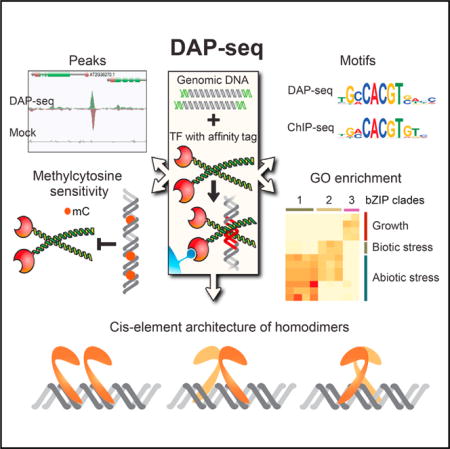
SUMMARY
The cistrome is the complete set of transcription factor (TF) binding sites (cis-elements) in an organism, while an epicistrome incorporates tissue-specific DNA chemical modifications and TF-specific chemical sensitivities into these binding profiles. Robust methods to construct comprehensive cistrome and epicistrome maps are critical for elucidating complex transcriptional networks that underlie growth, behavior, and disease. Here, we describe DNA affinity purification sequencing (DAP-seq), a high-throughput TF binding site discovery method that interrogates genomic DNA with in-vitro-expressed TFs. Using DAP-seq, we defined the Arabidopsis cistrome by resolving motifs and peaks for 529 TFs. Because genomic DNA used in DAP-seq retains 5-methylcytosines, we determined that >75% (248/327) of Arabidopsis TFs surveyed were methylation sensitive, a property that strongly impacts the epicistrome landscape. DAP-seq datasets also yielded insight into the biology and binding site architecture of numerous TFs, demonstrating the value of DAP-seq for cost-effective cistromic and epicistromic annotation in any organism.
Graphical abstract
INTRODUCTION
Comprehensive identification of transcription factor binding sites (TFBS) in a genome, the cistrome, is essential for characterizing regulatory elements and TF function. Chromatin immunoprecipitation sequencing (ChIP-seq) is a powerful approach for TFBS discovery (Kheradpour and Kellis, 2014; Stamatoyannopoulos et al., 2012). However, ChIP-seq experiments have been generally limited in scale as they are difficult to execute, dependent on antibody quality, and challenging for rare or lowly expressed proteins (Kidder et al., 2011). As a result, binding site information is available for relatively few TFs and substantial TFBS coverage is only available for humans and several model organisms. Methods such as DNase hypersensitivity (DHS) assay or ATAC-seq offer more facile approaches for annotating genome-wide regulatory elements across many organisms and cell types (Buenrostro et al., 2015; Sullivan et al., 2014; Thurman et al., 2012). However, without comprehensive knowledge of TF sequence specificity, the targeting TFs of the identified regions cannot be readily verified.
In contrast to ChIP-seq, in vitro mapping of TFBS provides a scalable alternative to rapidly and inexpensively interrogate large numbers of TFs. The two most commonly used in vitro methods are systematic evolution of ligands by exponential enrichment (SELEX) (Jolma et al., 2010) and protein binding microarrays (PBM) (Berger and Bulyk, 2009). In both methods synthetic DNA oligomers are enriched with an affinity-tagged TF and the preferred binding sequences are used to derive binding motifs. Both methods can resolve a large number of TF motifs, which can then be used to predict TFBS genome-wide. However, these assays employ synthetic DNA that lacks genomic DNA properties known to impact TF binding, including primary sequence context and chemical modifications, such as the widespread and tissue-specific 5-methylcytosine found in plants and animals. Efforts have been made to build synthetic oligomer pools that reflect relevant cis-element sequence (Levo and Segal, 2014) or incorporate methylation (Mann et al., 2013), but complex variation in nucleotide sequence and DNA methylation patterns (Schmitz et al., 2013) makes it extremely challenging to fully reproduce native nuclear DNA patterns by synthesis.
Genomic DNA (gDNA) is the native substrate for a TF and therefore ideal for an in vitro TF interaction assay. Unlike synthetic oligomers, gDNA encodes primary sequence and cell-, tissue-, and organism-specific methylation patterns that may impact TF binding. Moreover, as gDNA from different tissue/cell types and species can be easily obtained, the impact of sequence and methylation variation can be experimentally determined. Previous TF:DNA binding assays using naked gDNA were effective in identifying motifs and in vivo binding sites (Guertin et al., 2012; Liu et al., 2005; Rajeev et al., 2014), but this approach has not been applied for global TFBS characterization or to investigate the impact of primary sequence and DNA methylation on in vivo TF binding.
We developed DNA affinity purification sequencing (DAP-seq), a high-throughput assay that uses in-vitro-expressed TF to interrogate naked gDNA fragments to establish binding locations (peaks) and sequence motifs. We demonstrated the ultra-high-throughput capability of the assay by creating a cistrome map for Arabidopsis thaliana, consisting of peaks and motifs for 529 (30%) Arabidopsis TFs. These datasets include 2.7 million experimentally determined genomic-context TFBS covering 11 Mb (9.3%) of the genome, predicting thousands of target genes enriched in known and new functions. Comparison of DAP-seq and ChIP-seq datasets showed that DAP-seq peaks predicted in vivo TF binding better than motif inference. This improved predictive power can be partially explained by the ability of the assay to directly capture the impact of primary sequence and DNA methylation on binding affinities at individual TFBS. Globally, 76% of Arabidopsis TFs surveyed were sensitive to methylation in their motifs. By testing gDNA libraries in which methylcytosines were removed by PCR (ampDAP-seq), we identified ~180,000 TFBS occluded by leaf DNA methylation (the Arabidopsis epicistrome). Finally, we showed that closely spaced motifs significantly affected TF binding by developing a model for cooperative auxin response factor (ARF) homodimer binding to complex motif repeats. In total, ~2,300 individual DAP-seq experiments are reported, with all motifs, peaks, and TF-methylation sensitivities publicly available on our Plant Cistrome Database (http://neomorph.salk.edu/PlantCistromeDB).
RESULTS
DAP-Seq
DAP-seq is an in vitro TF-DNA binding assay that allows low-cost and rapid generation of genome-wide binding site maps for a large number of TFs, while capturing gDNA properties that impact binding in vivo. A DAP-seq gDNA library is prepared by attaching a short DNA sequencing adaptor onto purified and fragmented gDNA (Figure 1A; DAP library). In a separate reaction, an affinity-purified TF is prepared by in vitro expression, bound to ligand-coupled beads, and washed to remove nonspecific cellular components (Figure 1B). The gDNA library is added to the affinity-bound TF and the unbound DNA is washed away (Figure 1C). The bound fraction is eluted, amplified with PCR primers to introduce an indexed adaptor, and the DNA is sequenced. By mapping the reads to a reference genome, enriched loci (peaks) can be used to identify TFBS and motifs. For example, inspection of DAP-seq peaks for the bZIP TF ABI5 revealed enrichment at a known regulatory site that contains two adjacent G-box motifs (CACGTG) (Xu et al., 2014), where a ChIP-seq peak was also found (Figure 1D). The DAP-seq-derived motif matched the motifs derived from both ChIP-seq and PBM (Weirauch et al., 2014), although the DAP- and ChIP-seq motifs shared more sequence similarity at the edges (Figure 1E).
Figure 1. Genome-wide TFBS Discovery by DAP-Seq.
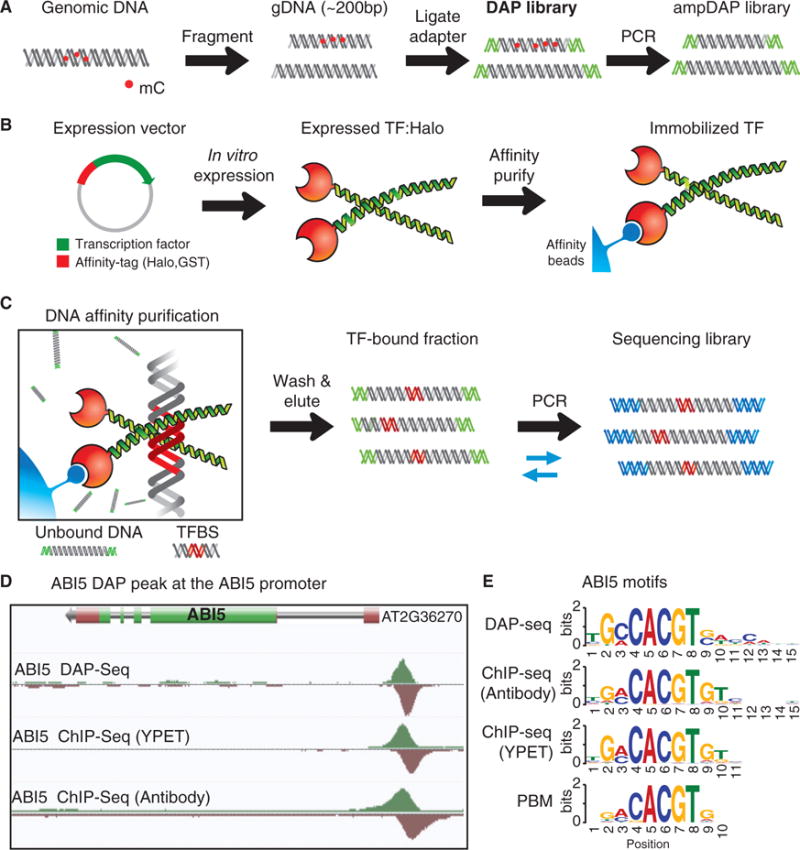
(A) Preparation of DAP- and ampDAP-seq libraries.
(B) Expression and capture of affinity-tagged TFs.
(C) gDNA is bound to immobilized TFs, eluted, and sequenced.
(D) ABI5 DAP- and ChIP-seq peaks at a known regulatory element in the ABI5 promoter.
(E) Motifs derived from DAP- and ChIP-seq match a published ABI5 motif (Weirauch et al., 2014).
See also Table S2.
To measure the impact of DNA modifications on TF binding, we implemented a modified version of DAP-seq, ampDAP-seq, which uses a DNA library in which the DNA modifications are removed by PCR (Figure 1A). Together with DNA chemical modification maps, i.e., base-resolution methylomes (Schmitz et al., 2013), the comparison of DAP-seq and ampDAP-seq data allows for a global assessment of the effects of DNA modifications on TF binding.
The Arabidopsis Cistrome
To create a comprehensive catalog of Arabidopsis motifs and genomic TF binding locations, DAP-seq experiments were carried out on 1,812 TFs comprising 80 families (Pruneda-Paz et al., 2014) (Tables S1A and S1B). Using a computational pipeline that identified highly enriched motifs from the strongest peaks (Supplemental Experimental Procedures; Machanick and Bailey, 2011; Guo et al., 2012), we characterized peaks for 1,055 TFs and derived motifs for 529 TFs. The dataset provided coverage for 52 of the 66 families with more than two members (Figure S1A) and identified a total of ~2.7 million TFBS covering 11 Mb (9%) of the genome. Reproducibility was high, with replicate correlations between 0.71 and 0.99 (Figure S1B). The entire set of motifs (Figure 2A) and peaks (Figure 2B), which we collectively term the Arabidopsis cistrome, can be viewed and downloaded (http://neomorph.salk.edu/PlantCistromeDB).
Figure 2. A Genome-wide Atlas of Arabidopsis TFBS Motifs and Binding Locations.
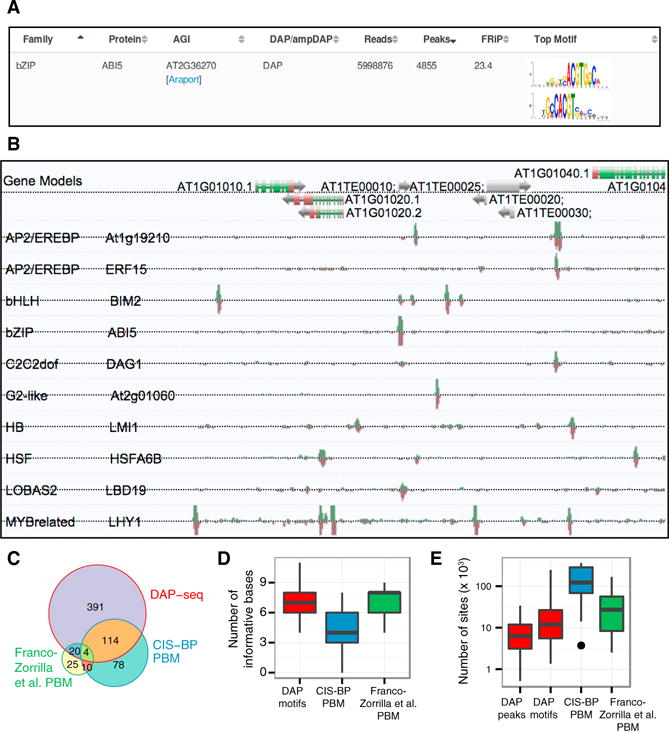
(A) Web portal of TF binding motifs from 529 DAP-seq and 343 ampDAP-seq experiments.
(B) Sample screen shot of genome browser with DAP-seq peaks for selected TFs.
(C) Overlap between TFs from the DAP-seq, CIS-BP PBM (Weirauch et al., 2014), and PBM datasets (Franco-Zorrilla et al., 2014).
(D) Number of informative bases (information content ≥ 0.8 bits) in DAP-seq and PBM motifs.
(E) Number of TFBS predicted by peaks (DAP-seq) or motifs (DAP-seq, CIS-BP, and PBM [Franco-Zorrilla et al., 2014]).
See also Figure S1 and Tables S1 and S2.
We investigated properties of the assay (reproducibility and protein expression levels) and TF family features that may predict the failure or success for a particular TF (Supplemental Experimental Procedures). Overall, technical issues could explain ~10% of failures, and thus some TFs produced peak datasets in retesting (Table S2). Generally, the rescue rate of failed TFs in a retest was related to the overall success rate of the family. For example, retesting 87 failed MADS-box TFs did not produce a single successful DAP-seq dataset, while recovery rates were higher than average in the more successful bZIP and NAC families (Table S2). This suggests that family-specific properties strongly affect the ability to produce a protein with DNA binding activity and may be influenced by protein stability in the assay conditions or a requirement for a protein partner, cofactor, or post-translational modification for activity.
Comparing DAP-seq-derived motifs to curated motif databases (Transfac, JASPAR, and AGRIS), we found most DAP-seq motifs were highly similar to published data (Table S1C). For TFs that were also present in two large-scale Arabidopsis PBM datasets (118 from CIS-BP [Weirauch et al. 2014] and 24 from PBM [Franco-Zorrilla et al. 2014]; Figure 2C; Table S1D), we found quantitative agreement between the DAP-seq and PBM-derived PWM (Figure S1C), although the DAP-seq PWM contained a higher number of informative positions (information content ≥ 0.8 bits; Figure 2D; 4.8 bp for CIS-BP versus 6.8 bp for DAP-seq), and predicted many fewer TFBS (Figure 2E; 122,200 for CIS-BP versus 11,900 for DAP-seq). From DAP-seq and ChIP-seq comparisons of TFs from three different families, the average number of TFBS identified by DAP-seq peaks was similar to the average number of in vivo binding sites recovered (12,352 in DAP-seq versus 8,372 in ChIP-seq; see “DAP-seq Captures TF Binding Sites Identified by ChIP-seq”).
To investigate overall motif relationships, we clustered the PWM of the 529 TFs and observed that related paralogs targeted similar motifs (Figure S2A). Applying a dynamic tree cut (Langfelder et al., 2008) to the clustering dendrogram, we identified 85 motif types (Figure S2A). At the family level, motif clusters from the large and functionally diverse bZIP (Figure 3A) and NAC families (Figure S2B) closely reflected TF phylogeny (Corrêa et al., 2008; Olsen et al., 2005), indicating target sequences are conserved for close paralogs. Binding peaks of these TFs showed a range of enrichment in conserved non-coding regions (Haudry et al., 2013) (Figure S2C). Although the 529 DAP-seq motifs provided a global description of motif types, it was biased toward larger and more tractable families, such as bZIP, NAC, and WRKY, while some families, such as MADS and C3H, were underrepresented (Figure S1A). For a more balanced analysis, a subset of 57 TFs were identified (Figure 3B) that spanned the space of motif diversity (Figure 3C) and captured about 50% of motif types (Figure S2D). They were also selected based on published literature regarding consensus motifs and functions to highlight the known and new properties predicted by DAP-seq (Table S1C).
Figure 3. The Global Diversity of Arabidopsis TF Motifs.
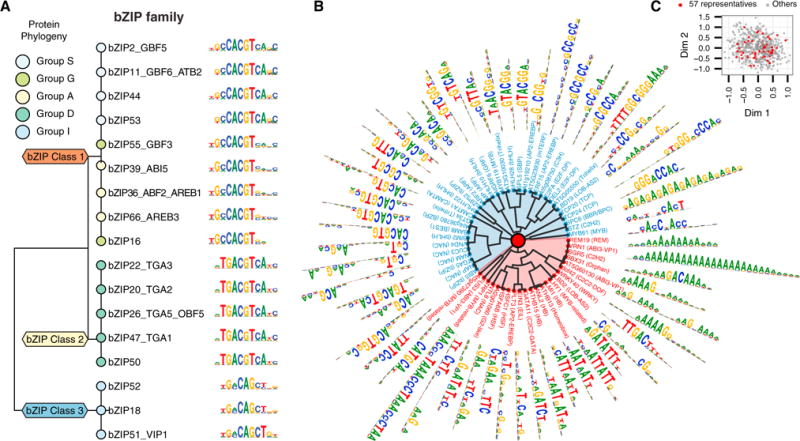
(A) bZIP family motifs from DAP-seq clustered by motif similarity.
(B) 57 TF motifs with GC-rich clusters in blue and AT-rich clusters in red.
(C) Multidimensional scaling plot of the full set of 529 TFs highlighting the 57 representative motifs.
See also Figure S2 and Table S1.
Several new motif types identified in the DAP-seq dataset included members of the C2H2, GRF, and AP2-EREBP family. The discovery of a long poly-A motif for VRN1 and REM19, closely related ABI3-VP1 paralogs, was surprising as previous electrophoretic mobility shift assay experiments found no DNA sequence preference for VRN1, although this was likely because a poly-A oligomer was not tested (Berke and Snel, 2015). Notably, the motif captured for VRN1 (29 bp) was twice as long as REM19 (15 bp), which could be explained by the presence of tandem B3 DNA-binding domains in VRN1 compared to only one copy in REM19. VRN1 and REM19 are master regulators of cold-induced flowering and were recently proposed to be components of the plant Polycomb Repressive Complex PRC1 (Berke and Snel, 2015), suggesting VRN1/REM19 may target the PRC1 to poly-A motifs to repress flowering.
To better understand the genome-wide binding profiles of the different TF families, we computed the enrichment/depletion of binding sites of the 57 representative TFs relative to gene features (Figure S6A) and observed overall distributions similar to those identified by PBM (Weirauch et al., 2014). While substantial positional heterogeneity existed, there was global preference across TF families for enrichment at promoters and 5′ UTR and moderate depletion in coding regions. Enrichment/depletion at long non-coding RNA promoters was weaker and showed patterns different from protein coding genes, suggesting distinct modes of regulation.
Target genes predicted for the 57 representative TFs were strongly enriched (1 × 10−4 < p < 1 × 10−64) in gene ontology (GO) terms that agreed with known functions and indicated potential new functions (Figure S3; Table S1C). By removing generic and redundant GO terms, we could highlight a set of TFs whose target genes predicted functions that were pertinent to all organismal biology (Figure 4, asterisks; related citations in Table S1C). We noted the largest split in TF functions was between those involved in hormone and endogenous response pathways (Figure 4, black bar) and those involved in intrinsic pathways (gray bar). Within this larger division, we observed six specific functional categories: hormone-regulated development (Figure 4, box 1) and growth (Figure 4, box 2), defense (Figure 4, box 3), cell division (Figure 4, box 4), metabolism and nutrition (Figure 4, box 5), and intrinsically regulated growth (Figure 4, box 6).
Figure 4. Critical Biological Processes Are Enriched in DAP-Seq Target Genes.
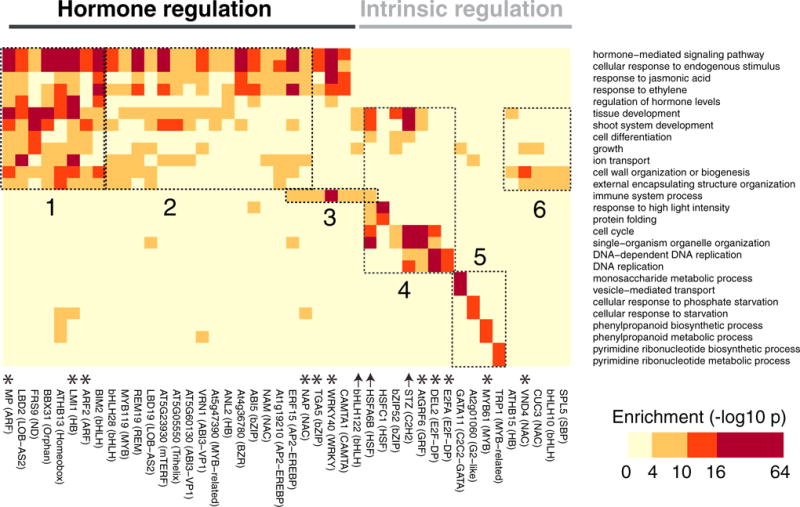
Target genes predicted for 44 diverse TFs (subset of the 57 representatives) are enriched for functional terms associated with basic cellular properties.
See also Figure S3 and Table S1.
TFs enriched for GO terms related to hormone-regulated development (Figure 4, box 1) included two ARFs (ARF2 and MP/ARF5), master regulators of auxin hormone responses, and a Homeobox (HB) family TF (LMI1) also known to play a role in auxin responses. TFs enriched for innate immune response (Figure 4, box 3) included two master regulators of plant defense (WRKY40 and TGA5). Factors enriched in cell-cycle function (Figure 4, box 4) included the E2F family (E2FA and DEL2), direct regulators of DNA replication, and Growth-regulating Factor 6 (AtGRF6), which belongs to a family modulating cell-cycle progression and growth. The metabolism and nutrition category (Figure 4, box 5) contained very specific functions for several TFs that were consistent with the literature, such as the role of MYB61 in phenylpropanoid regulation and lignification. Finally, hormone (Figure 4, box 2) and intrinsic growth (Figure 4, box 6) both contained NAC TFs, important regulators of growth. These functions are consistent with known roles of NAP in hormone-regulated growth and defense, and VND4 in vascularization, an intrinsically regulated process. In summary, many of the predicted functions of the representative TFs are consistent with known functions.
New functions for many TFs were also predicted (Figure 4, arrows; related citations in Table S1C). For the heat-shock factor HSFA6B, we saw enrichment for high heat responses as expected, but also observed enrichment in mitotic functions (cell cycle and DNA replication; Figure 4, box 4). While plant HSFs have not been implicated in mitosis, a recent study of the human HSF1 indicates that this family may directly regulate cell division in proliferating cancer cells (Mendillo et al., 2012). For the C2H2 TF STZ, where mutants have enhanced tolerance to salt and abiotic stresses, we also saw enrichment in mitosis-related functions (Figure 4, box 4). As C2H2 family members from both plants and animals are known to regulate the cell cycle and DNA replication (Staudt et al., 2006; Welch et al., 2007), STZ enrichment for mitotic functions suggests that its stress response phenotype may involve direct regulation of cell division. Finally, bHLH122, known to be important for abiotic drought responses, targeted genes in immune processes (Figure 4, box 3), suggesting that it may also play a role in biotic defense. Overall, GO analysis of DAP-seq-derived target genes revealed TF functions consistent with the literature and identified new possible TF functions.
DAP-Seq Captures TF Binding Sites Identified by ChIP-Seq
To examine the relevance of in-vitro-derived DNA binding profiles compared to those from in vivo experiments, we performed ChIP-seq experiments for three Arabidopsis TFs from unrelated families: ABI5 (bZIP family), ATHB5 (HB family), and ANAC055 (NAC family). The bZIP family is found in all eukaryotes, while the NAC and HB families are plant specific. All three families have functions in plant hormone and growth regulation, although at different stages. The bZIP family in plants includes master regulators of salicylic and abscisic acid (ABA) hormone responses (Finkelstein and Lynch, 2000). ANAC055 is downstream of ABA and jasmonic acid signaling pathways and affects abiotic growth responses (Bu et al., 2008). HB family members play important roles in water stress and interact directly with auxin regulation (Ré et al., 2014). Three independent ChIP-seq experiments were performed on ABI5: two with an anti-ABI5 antibody in dark- and light-grown seedlings (ABI5 Ab etiolated and light) and one with an anti-GFP antibody in light grown seedlings containing a recombineered YPET-tagged ABI5 fusion protein (ABI5 YPET light). ChIP-seq for ANAC055 and ATHB5 used the same YPET-tagging strategy as the ABI5 YPET experiment.
Genome-wide comparison of the three TFs showed that DAP-seq peaks captured significant fractions of ChIP-seq peaks (36% to 81%; p ≤ 1 × 10−5; Figure 5A, blue bars). Ranking ChIP-seq peaks by motif scores in the peak, we observed increased overlap with DAP-seq peaks as motif score increased; 69% to 97% of ChIP-seq peaks that ranked in the top 25% by motif score overlapped with DAP-seq peaks (Figure 5A, red bars). This result suggests that DAP-seq preferentially captures in vivo binding sites associated with high scoring motifs. To confirm this, we compared the motif scores at peaks present in both ABI5 DAP-seq and ChIP-seq (DAP-ChIP) to those unique to one of the datasets (DAP-only and ChIP-only). Overall, we found that DAP-ChIP and DAP-only peaks contained high-scoring motifs, while the motif scores under ChIP-only peaks were only slightly elevated over background (Figure 5B). As a substantial fraction of ChIP-seq peaks do not contain a detectable target motif sequence, it was suggested that only a portion of the ChIP-seq peaks are from direct TF binding (Worsley Hunt and Wasserman, 2014). Our results indicate that DAP-seq may preferentially capture direct in vivo binding targets and thus can provide valuable binding affinity measurements at these sites.
Figure 5. Concordance of In Vitro and In Vivo Binding Sites for Multiple TF Families.
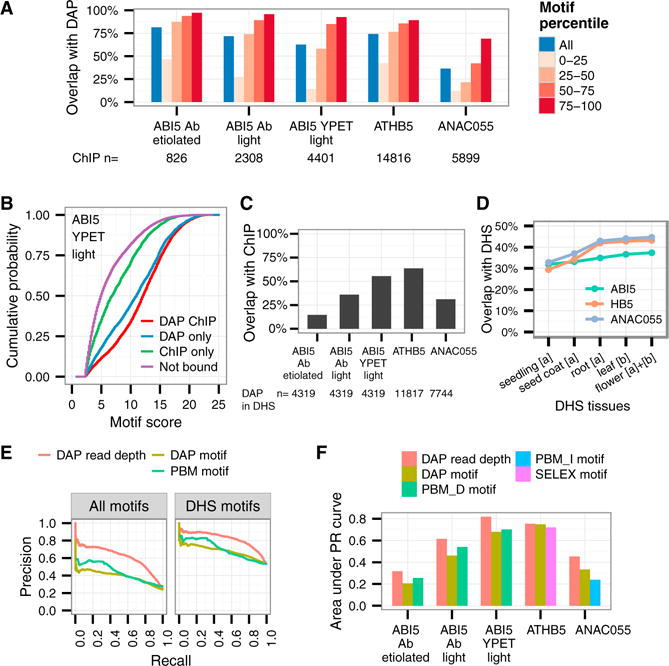
(A) Percent overlap of ChIP-seq peaks with DAP-seq peaks (blue), which increase for peaks associated with higher motif scores (red).
(B) Empirical cumulative distribution of motif scores shows shared ChIP- and DAP-seq peaks (DAP-ChIP) contain higher scoring motifs than do ChIP-only peaks, in which motif scores are similar to the motifs not bound in either assay.
(C) Percent DAP-seq peaks in DHS that overlap with ChIP-seq peaks.
(D) Using DHS data from multiple sources ([a] Sullivan et al., 2014; [b] Zhang et al., 2012) increases coverage of DAP-seq peaks.
(E) Precision (y axis) and recall (x axis) curve shows DAP-seq read depth (signal) predicts in vivo ABI5 binding sites better than mapping DAP-seq and PBM-derived motifs to genome, for all motifs (left) and motifs in DHS (right).
(F) By area under the precision-recall (PR) curve as in (E), all ChIP-seq datasets are most accurately predicted by DAP-seq read depth. PBM_D, motif directly determined by PBM. PBM_I, motif inferred by PBM based on DNA binding domain similarity.
See also Figure S4.
Having identified that indirect binding may explain a large portion of the ChIP-only sites, we investigated whether chromatin properties could explain why certain strong binding sites detected by DAP-seq were not observed in ChIP-seq (DAP-only). Chromatin accessibility is known to influence TF binding affinities in vivo, and although this property cannot be directly measured by DAP-seq, integration with DHS datasets (Sullivan et al., 2014; Zhang et al., 2012) can provide information regarding in vivo site availability (Guertin et al., 2012). Within DHS regions 15% to 64% of DAP-seq peaks overlapped with a ChIP-seq peak (Figure 5C), significantly higher than the 5% to 28% of all DAP-seq peaks overlapping ChIP-seq peaks. As a single tissue captures only a subset of open chromatin states, we sequentially added DHS sites from four tissue types and found each tissue-specific DHS set overlapped with a unique set of DAP-seq peaks (Figure 5D). As these DHS experiments were performed on whole organs, many tissue-, cell-, and condition-specific DHS regions may still be unidentified, and the chromatin-free TF binding profiles from DAP-seq provide a valuable dataset for characterizing open chromatin regions.
We were interested to determine how well in vitro binding captured by DAP-seq peak signal could predict in vivo binding sites compared to conventional motif matching approaches. Using (1) PWM from published PBM, (2) PWM from DAP-seq, and (2) DAP-seq peak signal strength, we established ranked lists of binding sites inside DHS for comparison to the ABI5 YPET ChIP-seq experiment. We computed precision-recall metrics with increasing thresholds on each ranked list: precision is the fraction of predicted sites captured by ChIP-seq, and recall is the fraction of ChIP-seq bound sites captured by predicted sites. We found DAP-seq binding signal achieved 14%–17% higher precision than PWM matching (Figure 5E). For the other four ChIP-seq datasets, DAP-seq binding signal also outperformed PWM predictions, except for ATHB5 (Figure 5F). These results indicated that a direct biochemical interaction assay better predicted in vivo binding compared to motif inference, suggesting that the DAP-seq experiments measure the impact of genomic properties that influence TF binding in vivo.
We examined several primary sequence properties of genomic DNA known to impact in vivo binding, including motif clusters (Pott and Lieb, 2015) and TF sensitivity to DNA methylation in motifs (Domcke et al., 2015), by restricting predictions to peaks containing a single motif with no strong motifs within 100 bp, or to peaks containing motifs with no methylcytosine (Figure S4A). We observed improved performance of motif inference relative to DAP signal for ABI5, but not for ANAC055, suggesting these two TFs have different binding environment requirements and DAP-seq signal may achieve better predictive power by directly capturing environment features other than core recognition sequence.
To more thoroughly investigate this hypothesis, we constructed two random forest (RF) models using both motif and environment features. The first model used DAP signal as the motif feature, and the second used motif match score. Both included the same environment features for motif clusters, cytosine methylation in the motif, and predicted DNA shape parameters for sequences flanking the motifs (Zhou et al., 2015). As expected, adding environment features improved the accuracy for predicting in vivo binding for both types of motif features (Figure S4B), but the importance of the environment features was markedly different for each TF (Figure S4C). The motif score RF model for ANAC055 heavily relied on shape features, while the motif cluster feature and the motif methylation feature were more important for ABI5 YPET ChIP-seq. In contrast, these environment features were less important in DAP-seq signal RF models, suggesting DAP-seq natively captured the TF-specific effects of motif environment.
Cooperative Binding of ARF Homodimers at Phased Motif Repeats
Next, we explored TF-specific effects of motif clustering and how they impact the plant cistrome landscape. Even with the higher resolution of DAP-seq compared to ChIP-seq, for many TFs we observed strong binding at closely spaced motif clusters where multiple binding events were resolved as a single peak (Figures S5A and S5B). Although not surprising for TFs known to target repeat sequences (FRS9 and TRP1), this was also observed for many non-repeat binding TFs (ERF15, BIM2, and ABI5). Several TFs were at the opposite extreme, where strong DAP-seq peaks contained much less than one motif on average (STZ, NAP, ARF2, and MP/ARF5; Figure S5B). Unexpectedly, this group included two ARFs (ARF2 and MP/ARF5) with only 0.1–0.2 motifs per peak despite evidence that they bind to motif repeats in vitro and in vivo (Boer et al., 2014; Ulmasov et al., 1997). This suggests that direct examination is needed to understand the more complex binding site architecture required for strong binding for some families. We explored this hypothesis in more depth focusing on the ARF family.
ARFs are important regulators of many basic plant processes, and multiple lines of evidence indicate that homodimerization, and possibly hetero- and multimerization, are important for ARF binding and function (Farcot et al., 2015). ARF DNA binding is known to strongly prefer direct (DR) and everted repeats (ER) of the well-characterized auxin response element (AuxRE: TGTCTC), although no binding at inverted repeats (IR) was reported (Figure 6A). The spacing between individual AuxREs is important as the DR was bound only when tested with spacing of 10–12 bp (i.e., between the two T’s in bold TGTCTC-N4–6-TGTCTC), and the ER AuxRE with spacing of 15–18 bp (TGTCGG-N6–9-CCGACA) (Ulmasov et al., 1997). A crystal structure of an ARF bound to an ER AuxRE showed that the 15–18 bp spacing allowed the bound ARF homodimer to stabilize through interaction of the ARF dimerization domain (DD) (Boer et al., 2014). Similarly, the DR spacing preference may be explained by stabilizing interactions through a second dimerization domain, the III/IV domain (Figure 6B) (Nanao et al., 2014). Importantly, although substantial evidence supports a role for motif dimers in ARF binding and auxin response regulation, no comprehensive model yet exists to explain or predict ARF binding site preferences (Farcot et al., 2015).
Figure 6. The ARF Family Preferentially Binds to Phased Motif Clusters that Are Enriched in Target Gene Promoters.
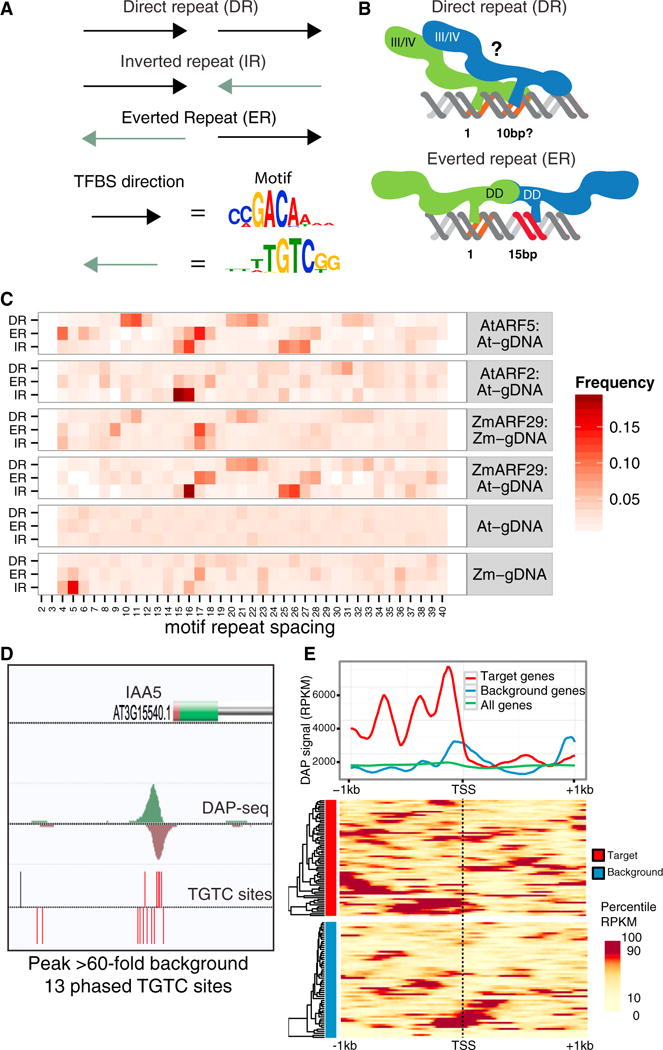
(A) Three possible orientations of an ARF motif repeat.
(B) ARF homodimers could be stabilized at a DR by an interaction of the III/IV domain (top) (Nanao et al., 2014) and at an ER by the dimerization domain (bottom) (Boer et al., 2014).
(C) Relative frequencies of DAP-seq peaks at DR, ER, and IR pairs for Arabidopsis (AtARF5 and AtARF2) and maize (ARF5/ZmARF29) proteins interrogating Arabidopsis (At-gDNA) or maize (Zm-gDNA) DAP-seq libraries.
(D) A cluster of 13 phased TGTC sites (red ticks) in the promoter of the ARF5 target IAA5. Black ticks are non-phased TGTC sites.
(E) DAP-seq signal at the TSS (x axis) of ARF5 direct target genes, non-auxin-responsive background genes, and all genes.
See also Figure S5.
To refine the model of motif repeat orientation and spacing for ARF homodimer binding, we first identified genome-wide tandem motifs in the three repeat types above (Figure 6A). Since the TGTCGG motif reported by both DAP-seq and PBM was present in only ~30% of strong DAP-seq peaks and was distinct from the AuxRE sequence TGTCTC, we used the consensus sequence TGTC as our motif model. We extracted all instances of inverted, everted, and direct TGTC repeats in the genome (IR-TGTC, ER-TGTC, or DR-TGTC), recorded the distance between each pair in the repeat, and tabulated the number of strong DAP-seq peaks found at each repeat type as a function of spacing (Figure 6C). For ARF5/MP, DR-TGTC binding preferentially occurred at three spacing groups: 10–12, 20–23, and 31–34 bp. For ER-TGTC, we observed three spacing groups at 4–8, 15–18, and 25–28 bp. Importantly, our results exactly matched the known spacing of 10–12 bp for DR (Ulmasov et al., 1997) and 15–18 bp for ER (Boer et al., 2014). We also identified novel binding events at IR repeats, which showed similar spacing preferences to those seen for ER-TGTC repeats but had only two spacing groups (15–16 and 25–27 bp). To explain homodimer binding at this third repeat type, we propose a model in which a third isoform of the ARF5 homodimer, with interactions between positively and negatively charged sides of the III/IV dimerization domain (Nanao et al., 2014), stabilizes the complex at specific spacing of the IR-TGTC (Figure S5C). To summarize, we observed three repeat-specific patterns of ARF binding that may be explained by three different ARF dimerization models. The multiple spacing groups for each repeat type and the flexibility within each group suggest that dimers can be stabilized by protein interactions spanning multiple helical turns as long as the interacting protein domains are in phase relative to the DNA helix.
Although the two functionally distinct ARF family members ARF2 and ARF5 had similar binding motifs (Figure 3B), their genome-wide binding correlation was only 0.09, much lower than the typical range of 0.6 to 0.8 for family members with very similar motifs such as those in the bZIP (Figure S5D) and NAC families (Figure S5E). Analysis of repeat spacing preferences for ARF2 revealed a more restricted pattern dominated by the IR-TGTC with a narrower range of flexibility within a spacing group compared to ARF5 (Figure 6C). Therefore, the low genome-wide binding correlation between ARF2 and ARF5 may be explained, in part, by the divergence of preferred spacing groups and motif repeat types, which, in turn, may be due to differences in the protein dimerization properties of the two phylogenetically distinct ARF proteins.
As the ARF family traces its origins back to the first land plants, we investigated whether the maize and Arabidopsis ARF binding properties have diverged in the 140–150 million years since their last common ancestor (Finet et al., 2013). Testing a maize co-ortholog of ARF5 (ZmARF29; Galli et al., 2015) on maize gDNA (Zm-gDNA) by DAP-seq, we observed similar, but not identical, motif spacing preferences with two dominant spacing groups in maize compared to the eight more distributed groups in Arabidopsis (Figure 6C). To determine if the ZmARF29 protein or the maize gDNA influenced the spacing differences, we assayed ZmARF29 using Arabidopsis gDNA (At-gDNA). The resultant ZmARF29:At-gDNA pattern was more similar to maize than to Arabidopsis, indicating that the spacing divergence is primarily due to ARF5 dimerization properties. Together, the ARF2/ARF5 and maize/Arabidopsis comparisons illustrate how natural variation of homodimer interactions can impact TF binding properties and thus the global TFBS landscape. The ZmARF29 experiments also demonstrate that the DAP-seq assay works in a large, repeat-rich genome (~2.5 Gb), similar in size to mammalian genomes.
To evaluate the in vivo relevance of our spacing model, we identified a set of 69 ARF5 target genes that rapidly respond to ARF5-specific repression with IAA19/BODENLOS and auxin treatments (Schlereth et al., 2010). 64% of these ARF5 targets contained a DAP-seq peak in their promoter, 3-fold enrichment over expectation (Figure S5F; p < 1 × 10−10). For example, the promoter of the ARF5 target IAA5 contained 13 phased TGTC sites in a ~400 bp DAP-seq peak that showed 60-fold enrichment over background (Figure 6D). We plotted the average DAP-seq read depth in 2-kb regions centered on the TSS of the 69 target genes and 62 non-auxin-responsive genes (Supplemental Experimental Procedures) and observed very strong binding primarily in the target gene promoters. Strikingly, there was a strong phased signal with a period of ~300 bp beginning ~150 bp upstream of the TSS (Figure 6E). Although DAP-seq was carried out on naked gDNA, the phasing pattern of ARF5 binding in target gene promoters resembled in vivo nucleosome phasing patterns found in active eukaryotic gene promoters (+1, −1 nucleosome, etc., locations) (Struhl and Segal, 2013). This suggests that the rapid responses of these ARF target genes may be due, in part, to high ARF occupancy relative to the preferred nucleosome positions characteristic of an active promoter.
In summary, our results support a model in which three flexible ARF homodimer isoforms bind to three distinct motif-repeat types spanning multiple helical turns, and that spacing preferences affect both ARF paralog and ortholog binding specificity. Moreover, enrichment of phased clustered repeats in the promoters of ARF5 target genes suggests that promoter location of ARF5 regulatory elements may play an important role in regulation of auxin-responsive genes.
The Epicistrome
Arabidopsis thaliana leaf nuclear DNA contains 5-methylcytosine at ~11% of cytosines (Schmitz et al., 2013; see the Supplemental Experimental Procedures), an important epigenomic feature for gene silencing. Several examples demonstrate that TF DNA methylation-sensitivities can impact in vivo TF binding, but the global impact on the cistrome has not yet been established in any organism. To determine how DNA methylation affected binding, we used base-pair methylation maps from Arabidopsis leaf DNA (Schmitz et al., 2013) to quantify DAP-seq and ChIP-seq binding at high-scoring motifs that contained 5-methylcytosine. As plant DNA methylation is equally distributed between two mutually exclusive patterns (Cokus et al., 2008; Lister et al., 2008), we classified these motifs into two categories: (1) motifs in mC-all regions identified by dense methylation in all contexts (CHH, CHG, and CG, where H is A, C, or T) associated with silenced genes and transposons (Figure 7A, inset), and (2) motifs in mCG-only regions exclusively methylated in the CG context, more sparsely distributed, and enriched in expressed genes (Figure 7B, inset). As a control, we identified a set of motifs that neighbored a methylated region (within 200 bp), but themselves did not contain methylation.
Figure 7. Motif Methylation Impacts Binding For 76% of TFs Surveyed.
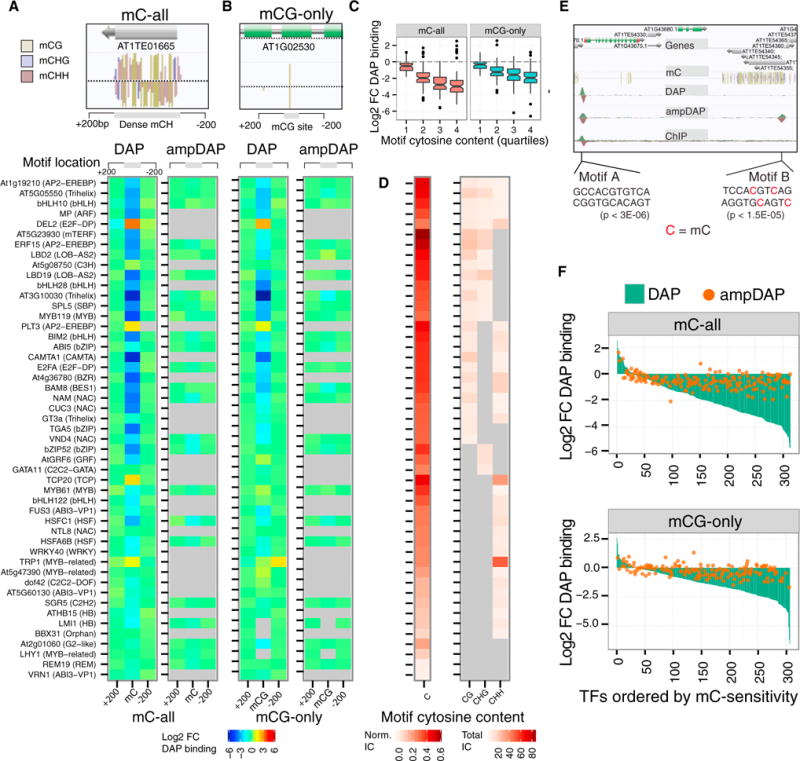
(A) Inset: mC-all regions contain dense methylation in all cytosine contexts. Left: binding fold change (FC) at motifs containing relative to motifs neighboring (within 200 bp) an mC-all site. Right: relative ampDAP-seq binding at the same motifs. Gray boxes indicate TFs with too few (<25) methylated motifs to score or a failed experiment.
(B) Inset: an isolated mCG-only site. Left: binding FC at motifs containing relative to motifs neighboring (within 200 bp) an mCG-only site. Right: relative ampDAP-seq binding at the same motifs.
(C) TF methylation sensitivity is correlated with cytosine content of the motif, defined as the informative content (IC) of cytosines, divided by total IC of the motif.
(D) Cytosine content (left) and informative CG, CHG, and CHH content for each motif (right) of TFs in (A and B).
(E) Genome browser showing DAP-, ampDAP-, and ChIP-seq peaks at methylated and unmethylated ABI5 motifs.
(F) Waterfall plot of log2 relative binding at methylated motifs for 349 TFs in DAP-seq and 219 TFs in ampDAP sets. In total, 248 of 327 TFs (76%) that had sufficient motif instances for quantification were found to be methylation-sensitive.
See also Figure S6 and Table S3.
By calculating the ratio of the ChIP-seq or DAP-seq binding strength (read depth) at methylated and unmethylated motifs, we observed strong binding inhibition for ABI5 both in vitro and in vivo (Figures 7A, 7B, and S6B). Across all TF families both mC-all and mCG-only methylation impacted binding (Figures 7A and 7B), although inhibition by mC-all methylation was more pronounced, possibly due to the higher methylation density in these regions (Figure 7A; inset). For the entire set of 327 TFs that had sufficient motif instances for quantification, mC-all inhibition was seen for 72% (234) of TFs, weak to no binding inhibition for 24% (79), while 4.3% (14) preferentially bound methylated motifs (Figure 7F). Interestingly, E2F family member DEL2, with specific roles in DNA replication, preferentially bound to methylated motifs, suggesting a possible relationship between this epigenetic mark and central regulators of cell division (Harashima et al., 2013).
To independently confirm the effect of methylation on TF binding, we used the modified DAP-seq assay, ampDAP-seq, where PCR replaces the 5-methylcytosines in the gDNA library with un-methylated cytosines (Figure 1A). ampDAP-seq of the 529 TFs resulted in motifs and peaks for 343 TFs. To ensure even comparison, we analyzed 219 TFs that had greater than 5% reads in peaks in both DAP- and ampDAP-seq (Figure S1D). DNA methylation sensitivities detected by DAP-seq were absent in the methylation-free ampDAP-seq datasets (Figure 7F), supporting our conclusion that 5-methylcytosine (or, although less likely, another chemical modification) was responsible for the observed TF binding changes (Figures 7A, 7B, and 7F; Table S3). Importantly, our ampDAP-seq data also provided the methylation-free binding strength of 178,135 TFBS normally occluded by leaf methylation, the Arabidopsis epicistrome.
We found that the cytosine content of a TF’s PWM correlated with its binding sensitivity to 5-methylcytosine (Figures 7C and 7D), with a few exceptions, such as TCP20, MYB61, and bHLH122, suggesting the relationships for some TFs between motif cytosine content and methylation sensitivity are more complex. The Arabidopsis methylome is established by distinct DNA methyltransferases, each with a preference for one of three cytosine contexts CG, CHG, and CHH (Law and Jacobsen, 2010; Zemach et al., 2013). Comparing the CG, CHG, and CHH content in the motifs (Figure 7D) to the methylation sensitivities revealed three general rules: (1) TFs with strong CG or CHG in their motifs were strongly inhibited in both mC-all and mCG-only regions, (2) TFs with only CHH in their PWM were generally insensitive to methylation, and (3) motifs containing multiple cytosine contexts typically showed very high methylation inhibition. These general rules suggest that regulatory relationships could potentially exist between specific DNA methyltransferases and TF families.
One possible mechanism for the role of DNA methylation in gene and transposon silencing is through exclusion of TF binding at regulatory sites. Consistent with this model, the loss of methylation in DNA methyltransferase mutants results in increased expression of thousands of transposons and genes (Zhang et al., 2006). However, since cytosine methylation is also required for targeting of silencing-related chromatin modifications (Law and Jacobsen, 2010), it is difficult to delineate the contributions of individual epigenomic features to gene silencing in vivo. In this regard, the less dense mCG-only methylation provides a valuable complement for analyzing the effects of methylation on binding in vivo since it has not been associated with silencing. We compared the degree of binding reduction between in vitro DAP-seq and in vivo ChIP-seq using the ABI5 datasets. We observed examples of strong ampDAP-seq peaks at high-scoring ABI5 motifs with no equivalent peaks for either DAP-seq or ChIP-seq in both mC-all (Figure 7E) and mCG-only regions (Figure S6C). The extent of reduced binding genome-wide at methylated motifs was similar in the DAP-seq and ChIP-seq datasets at both mC-all and mCG-only sites (Figures 7A, 7B, and S6B). While these observations do not directly demonstrate that motif methylation contributes in vivo to TF exclusion, our findings are consistent with this model.
DISCUSSION
The in vivo protein-DNA interaction landscape is affected by multiple factors including primary sequence, DNA modifications, and chromatin accessibility, along with stabilizing and destabilizing interactions between proteins associated with the DNA (Lelli et al., 2012; Levo and Segal, 2014). Our in vitro DAP-seq assay offers a simple method to examine TF binding to its cognate target (gDNA) in a chromatin-free context, while maintaining important information related to primary genome sequence and DNA methylation. The assay’s high-throughput capability allowed us to create a comprehensive atlas of the Arabidopsis cistrome consisting of 529 TFs targeting 2.7 million binding sites. Furthermore, by integrating DAP-seq TFBS, methylome maps, and direct measurements of binding in the absence of methylation in ampDAP-seq, we have performed the largest analysis to date for evaluating the relationship between TFs and methylated DNA. ampDAP-seq of 219 TFs identified ~180,000 TFBS occluded by leaf DNA methylation, characterizing an Arabidopsis epicistrome atlas. The precise base at which methylation affects binding cannot be easily isolated in mC-all regions as multiple 5-methylcytosines are often found both in and proximal to a motif. However, the same trends of binding changes were observed at the sparsely methylated mCG-only sites, and these binding changes correlated with motif cytosine content and context. Therefore, we propose that DNA methylation at high information positions in the motif may directly affect the interaction between TF and genomic DNA and contribute to the observed TF methylation sensitivity. Finally, by demonstrating the utility of these datasets to generate biological insights (GO enrichment and ARF motif architecture), we believe DAP-seq will be a powerful tool for understanding regulatory DNA functions in eukaryotic genomes. With the availability of hundreds of sequenced genomes and methylomes of wild accessions, these cistrome and epicistrome maps provide a valuable resource to evaluate the impact of natural genetic and epigenomic variation on transcriptional networks controlling plant adaptation.
Our analysis of ARF cis-element architecture shows how genome-wide DAP-seq datasets can be used to characterize regulatory sequence in a native genomic context. Evidence demonstrating preferential binding of ARFs to DR, ER, and IR supports a model in which three distinct ARF homodimer isoforms can form stable protein-protein interactions across multiple turns of the DNA helix. Considering that (1) ARF5 homodimers may be able to associate with DNA in three distinct isoforms, (2) multimeric binding sites are associated with very strong DAP-seq peaks, and (3) ARF5 direct target genes contain multimeric binding sites (e.g., 13 TGTCs in IAA5 promoter), we propose that ARF5 multimerization on genomic DNA could play a functional role in regulating auxin transcriptional response in vivo. Although the experiments presented here did not test nucleosome or TF heterodimer cooperativity, the method may be extended to test cis-element architecture associated with heterodimers and higher-order chromatin complexes. Such assays will be useful for studying heterodimer binding properties important in the biological functions of the ARF and other TF families.
EXPERIMENTAL PROCEDURES
DAP-Seq and ChIP-Seq Experiments
For DAP-seq, gDNA was extracted from young Arabidopsis leaves, fragmented, and ligated with a truncated Illumina TruSeq adaptor. Separately, HALO-tagged TFs were expressed in an in vitro wheat germ system. HALO-TFs were immobilized on Magne HALO-Tag beads, washed, and incubated with the DNA library. After bead washing, DNA was eluted and amplified with indexed TruSeq primers. Sequencing was performed on an Illumina HiSeq 2500 with 100-bp SR reads. For ABI5, ANAC055, and HB5 ChIP-seq, the YPET or wild-type lines were germinated and grown for 36 hr under dark or long day light conditions. ChIP-seq was carried out as previously described with minor modifications (Chang et al., 2013).
The ampDAP-Seq DNA library was prepared by PCR amplification of a standard DAP-seq gDNA library using Phusion High-Fidelity DNA Polymerase (NEB; 15 ng of DNA in a 50 μl reaction) and the A and B adaptor oligos (25 μM each; Supplemental Experimental Procedures) with the cycling conditions below: 2 min at 95°C, 30 s at 98°C, 10 cycles of 15 s at 98°C, 30 s at 60°C, 2 min at 72°C, and a final extension time of 10 min at 72°C, followed by a hold at 4°C. The DNA was purified by Sera-Mag beads (Thermo) and resuspended in 30 μl elution buffer. Following the DAP binding protocol, the recovered DNA was PCR amplified for 20 cycles using the same conditions as DAP-seq using the full-length Illumina primers.
DAP-Seq and ChIP-Seq Data Processing
Reads were mapped to the TAIR10 genome for Arabidopsis and B73_v2 for maize. DAP-seq peaks were called by the GEM peak caller (Guo et al., 2012) and ChIP-seq peaks by MACS2 (Zhang et al., 2008) with the IDR pipeline for replicated samples (Li et al., 2011). Motif discovery was performed using the MEME-ChIP suite (Machanick and Bailey, 2011). Binding signals were calculated by deepTools (Ramírez et al., 2014), and GO enrichment was calculated by g:Profiler (Reimand et al., 2011).
See the Supplemental Experimental Procedures for additional details.
Supplementary Material
Highlights.
2.7 million binding targets for hundreds of TFs define the Arabidopsis cistrome
Methylation sensitivities of 76% of TFs surveyed shape the Arabidopsis epicistrome
Strong enrichment of relevant gene functions is predicted for TF target genes
Auxin response factor motif architecture promotes cooperative binding
Acknowledgments
We thank Andrew Kuruzar for assistance with images (p40design@gmail.com). We thank Chris Benner, Ian Quigley, Debra Fulton, Robert Schmitz, and Yue Zhao for their critical reading of the manuscript. We thank Rosa Castanon for sharing biological materials. A.G. acknowledges funding from NSF (IOS-0820729/IOS-1114484). This work was supported by grants from the NSF (MCB1024999) and the Gordon and Betty Moore Foundation (GBMF3034) (to J.R.E.). J.R.E. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
ACCESSION NUMBERS
The accession number for the raw and processed data of DAP-seq and ChIP-seq reported in this paper has been uploaded to GEO: GSE60143.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.04.038.
AUTHOR CONTRIBUTIONS
R.C.O. and J.R.E. designed the experiments. R.C.O., A.B., S.C.H., L.S., M.G.L., M.G., and A.G. performed the experiments. S.C.H. and R.C.O. established the sample processing and bioinformatics pipelines and carried out the computational analyses. J.R.N. carried out the DNA sequencing. R.C.O., S.C.H., M.G., and J.R.E. prepared the manuscript.
References
- Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke L, Snel B. The plant Polycomb repressive complex 1 (PRC1) existed in the ancestor of seed plants and has a complex duplication history. BMC Evol Biol. 2015;15:44. doi: 10.1186/s12862-015-0319-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer DR, Freire-Rios A, van den Berg WAM, Saaki T, Manfield IW, Kepinski S, López-Vidrieo I, Franco-Zorrilla JM, de Vries SC, Solano R, et al. Structural basis for DNA binding specificity by the auxin-dependent ARF transcription factors. Cell. 2014;156:577–589. doi: 10.1016/j.cell.2013.12.027. [DOI] [PubMed] [Google Scholar]
- Bu Q, Jiang H, Li CB, Zhai Q, Zhang J, Wu X, Sun J, Xie Q, Li C. Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res. 2008;18:756–767. doi: 10.1038/cr.2008.53. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KN, Zhong S, Weirauch MT, Hon G, Pelizzola M, Li H, Huang SSC, Schmitz RJ, Urich MA, Kuo D, et al. Temporal transcriptional response to ethylene gas drives growth hormone cross-regulation in Arabidopsis. eLife. 2013;2:e00675. doi: 10.7554/eLife.00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa LGG, Riaño-Pachón DM, Schrago CG, dos Santos RV, Mueller-Roeber B, Vincentz M. The role of bZIP transcription factors in green plant evolution: adaptive features emerging from four founder genes. PLoS ONE. 2008;3:e2944. doi: 10.1371/journal.pone.0002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–579. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- Farcot E, Lavedrine C, Vernoux T. A modular analysis of the auxin signalling network. PLoS ONE. 2015;10:e0122231. doi: 10.1371/journal.pone.0122231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finet C, Berne-Dedieu A, Scutt CP, Marlétaz F. Evolution of the ARF gene family in land plants: old domains, new tricks. Mol Biol Evol. 2013;30:45–56. doi: 10.1093/molbev/mss220. [DOI] [PubMed] [Google Scholar]
- Finkelstein RR, Lynch TJ. The Arabidopsis abscisic acid response gene ABI5 encodes a basic leucine zipper transcription factor. Plant Cell. 2000;12:599–609. doi: 10.1105/tpc.12.4.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, López-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc Natl Acad Sci USA. 2014;111:2367–2372. doi: 10.1073/pnas.1316278111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli M, Liu Q, Moss BL, Malcomber S, Li W, Gaines C, Federici S, Roshkovan J, Meeley R, Nemhauser JL, Gallavotti A. Auxin signaling modules regulate maize inflorescence architecture. Proc Natl Acad Sci USA. 2015;112:13372–13377. doi: 10.1073/pnas.1516473112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Martins AL, Siepel A, Lis JT. Accurate prediction of inducible transcription factor binding intensities in vivo. PLoS Genet. 2012;8:e1002610. doi: 10.1371/journal.pgen.1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Mahony S, Gifford DK. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 2012;8:e1002638. doi: 10.1371/journal.pcbi.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima H, Dissmeyer N, Schnittger A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013;23:345–356. doi: 10.1016/j.tcb.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Haudry A, Platts AE, Vello E, Hoen DR, Leclercq M, Williamson RJ, Forczek E, Joly-Lopez Z, Steffen JG, Hazzouri KM, et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet. 2013;45:891–898. doi: 10.1038/ng.2684. [DOI] [PubMed] [Google Scholar]
- Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpää MJ, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42:2976–2987. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder BL, Hu G, Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol. 2011;12:918–922. doi: 10.1038/ni.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24:719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68. doi: 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levo M, Segal E. In pursuit of design principles of regulatory sequences. Nat Rev Genet. 2014;15:453–468. doi: 10.1038/nrg3684. [DOI] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5:1752–1779. [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Noll DM, Lieb JD, Clarke ND. DIP-chip: rapid and accurate determination of DNA-binding specificity. Genome Res. 2005;15:421–427. doi: 10.1101/gr.3256505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, Vinson C. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res. 2013;23:988–997. doi: 10.1101/gr.146654.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanao MH, Vinos-Poyo T, Brunoud G, Thévenon E, Mazzoleni M, Mast D, Lainé S, Wang S, Hagen G, Li H, et al. Structural basis for oligomerization of auxin transcriptional regulators. Nat Commun. 2014;5:3617. doi: 10.1038/ncomms4617. [DOI] [PubMed] [Google Scholar]
- Olsen AN, Ernst HA, Leggio LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10:79–87. doi: 10.1016/j.tplants.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8–12. doi: 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- Pruneda-Paz JL, Breton G, Nagel DH, Kang SE, Bonaldi K, Doherty CJ, Ravelo S, Galli M, Ecker JR, Kay SA. A genome-scale resource for the functional characterization of Arabidopsis transcription factors. Cell Rep. 2014;8:622–632. doi: 10.1016/j.celrep.2014.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeev L, Luning EG, Mukhopadhyay A. DNA-affinity-purified chip (DAP-chip) method to determine gene targets for bacterial two component regulatory systems. J Vis Exp. 2014 Jul 21;:89. doi: 10.3791/51715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ré DA, Capella M, Bonaventure G, Chan RL. Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol. 2014;14:150. doi: 10.1186/1471-2229-14-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Arak T, Vilo J. g:Profiler–a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlereth A, Möller B, Liu W, Kientz M, Flipse J, Rademacher EH, Schmid M, Jürgens G, Weijers D. MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature. 2010;464:913–916. doi: 10.1038/nature08836. [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, Ecker JR. Patterns of population epigenomic diversity. Nature. 2013;495:193–198. doi: 10.1038/nature11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. Mouse ENCODE Consortium An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt N, Fellert S, Chung HR, Jäckle H, Vorbrüggen G. Mutations of the Drosophila zinc finger-encoding gene vielfältig impair mitotic cell divisions and cause improper chromosome segregation. Mol Biol Cell. 2006;17:2356–2365. doi: 10.1091/mbc.E05-11-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan AM, Arsovski AA, Lempe J, Bubb KL, Weirauch MT, Sabo PJ, Sandstrom R, Thurman RE, Neph S, Reynolds AP, et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014;8:2015–2030. doi: 10.1016/j.celrep.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmasov T, Hagen G, Guilfoyle TJ. ARF1, a transcription factor that binds to auxin response elements. Science. 1997;276:1865–1868. doi: 10.1126/science.276.5320.1865. [DOI] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch D, Hassan H, Blilou I, Immink R, Heidstra R, Scheres B. Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Genes Dev. 2007;21:2196–2204. doi: 10.1101/gad.440307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley Hunt R, Wasserman WW. Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome Biol. 2014;15:412–428. doi: 10.1186/s13059-014-0412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Li J, Gangappa SN, Hettiarachchi C, Lin F, Andersson MX, Jiang Y, Deng XW, Holm M. Convergence of Light and ABA signaling on the ABI5 promoter. PLoS Genet. 2014;10:e1004197. doi: 10.1371/journal.pgen.1004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SWL, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, Ecker JR. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137.1–R137.9. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang T, Wu Y, Jiang J. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell. 2012;24:2719–2731. doi: 10.1105/tpc.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Shen N, Yang L, Abe N, Horton J, Mann RS, Bussemaker HJ, Gordân R, Rohs R. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc Natl Acad Sci USA. 2015;112:4654–4659. doi: 10.1073/pnas.1422023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.