

Abstract
Rational
SEMA3A-encoded semaphorin is a chemorepellent that disrupts neural patterning in the nervous and cardiac systems. In addition, SEMA3A has an amino acid motif that is analogous to hanatoxin, an inhibitor of voltage-gated K+ channels. SEMA3A knockout mice exhibit an abnormal ECG pattern and are prone to ventricular arrhythmias and sudden cardiac death.
Objective
To determine whether SEMA3A is a naturally occurring protein inhibitor of Kv4.3 (Ito) channels and its potential contribution to Brugada syndrome (BrS).
Methods and Results
Kv4.3, Nav1.5, Cav1.2, or Kv4.2 were co-expressed or perfused with SEMA3A in HEK293 cells and electrophysiological properties were examined via whole-cell patch clamp technique. SEMA3A selectively altered Kv4.3 by significantly reducing peak current density without perturbing Kv4.3 cell-surface protein expression. SEMA3A also reduced Ito current density in cardiomyocytes derived from human induced pluripotent stem cells. Disruption of a putative toxin binding domain on Kv4.3 was used to assess physical interactions between SEMA3A and Kv4.3. These findings in combination with co-immunoprecipitations of SEMA3A and Kv4.3 revealed a potential direct binding interaction between these proteins. Comprehensive mutational analysis of SEMA3A was performed on 198 unrelated SCN5A-genotype negative patients with BrS and two rare SEMA3A missense mutations were identified. The SEMA3A mutations disrupted SEMA3A’s ability to inhibit Kv4.3 channels, resulting in a significant gain of Kv4.3 current compared to WT-SEMA3A.
Conclusions
This study is the first to demonstrate semaphorin3A as a naturally occurring protein that selectively inhibits Kv4.3 and SEMA3A as a possible BrS-susceptibility gene through a Kv4.3 gain-of-function mechanism.
Keywords: Ion channels, potassium channels, Brugada syndrome, genetics, human, SEMA3A, Ito current, Kv4.3 channel blocker
INTRODUCTION
Semaphorins are extracellular and membrane associated proteins involved in many different cellular processes and are well known for their role in nervous system development through neuronal migration and axon guidance.1 Semaphorin 3A (SEMA3A) was the first molecularly characterized neural chemorepellent, that when inactivated disrupts neural patterning and projections.2 Although initial studies focused on SEMA3A’s role in neurodevelopment, SEMA3A is also involved in cardiac innervation patterning.3, 4
In developing murine hearts, SEMA3A is expressed at abundant levels, with expression pattern gradients opposite of sympathetic innervation, emphasizing its role as a chemorepellent essential to neuronal migration.4 Dysregulation of cardiac innervation is associated with an increased risk for ventricular arrhythmias and sudden cardiac death (SCD).5 SEMA3A knockout mice display decreased basal sympathetic activity, SCD during the first postnatal week and electrocardiographic features of sinus bradycardia and mild ST-segment elevation while transgenic SEMA3A overexpressing mice have reduced sympathetic innervation, reduced Ito density, prolonged action potential duration, spontaneous ventricular arrhythmias, and premature SCD.3, 4
The patterning of murine SEMA3A is in a gradient, with greater expression in the endocardium and less expression in the epicardium. This differential gradient is opposite to the expression pattern gradient of the transient outward repolarizing current (epicardium > endocardium; Ito, Kv4.3).4 Interestingly, a portion of the SEMA3A protein is analogous to a tarantula toxin, hanatoxin.6 Hanatoxin and closely related Heteropoda venatoria toxin (HpTx2) block Kv2.1 and Kv4.3 channels, respectively, by modifying energetics of activation via voltage sensor binding.7, 8
Due to its sequence homology with toxins, we hypothesized that SEMA3A may act as a naturally occurring Kv4.3 (Ito) ion channel blocker and the disruption of this interaction would lead to a pathological increase in Ito current density.
Increase in Ito current density is the pathogenic basis for a proportion of Brugada syndrome (BrS), a male predominated disease often presenting in the fourth decade of life, characterized by cardiac conduction abnormalities, ST-segment elevation, and an increased risk for ventricular arrhythmias and SCD.9–11 Sympathetic and parasympathetic patterning may also play an important role in the pathogenesis of BrS12 and arrhythmias in BrS are exacerbated by vagal stimulation.13
Here, we demonstrate that SEMA3A is a naturally occurring protein inhibitor of Kv4.3 (Ito) channels and that SEMA3A is a possible BrS-susceptibility gene.
METHODS
Gene constructs and site-directed mutagenesis
SEMA3A cDNA in the pCR-BluntII-TOPO vector (Open Biosystems, Pittsburgh, PA) was subcloned into the pIRES2-dsRED2 vector (Clontech, Mountain View, CA). The pIreGFP plasmid encoding wild-type (WT) human Kv4.3 (KCND3) and GFP represented Ito current for electrophysiological studies. The Kv4.3-L274A and Kv4.3-V275A mutations were engineered into pIreGFP-KCND3-WT and the SEMA3A-R552C and SEMA3A-R734W mutations were engineered into pIRES2-dsREDs-SEMA3A using primers containing each point mutation (available upon request) in combination with the Quikchange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Constructs for Nav1.5, Cav1.2 and Kv4.2 are included in the supplemental methods section. The integrity of all constructs was verified by DNA sequencing. For perfusion based experiments, human SEMA3A protein (hSEMA3A; R&D Systems, Minneapolis, MN) was dissolved in PBS at a concentration of 1 mM and diluted to work concentrations before experiments.
HEK293 cell culture and transfection
HEK293 cells were cultured in minimum essential medium supplemented with 1% nonessential amino acid solution, 10% horse serum, 1% sodium pyruvate solution, and 1.4% penicillin/streptomycin solution. All cells were plated in T25 flasks and stored in a 5% CO2 incubator at 37°C for 24 hours. Heterologous expression of Kv4.3 and SEMA3A was accomplished by co-transfecting 0.5 μg of pIreGFP-KCND3WT with 1.0 μg pIRES2-dsRed2-SEMA3AWT or pIRES2-dsRed2-SEMA3AR552C or pIRES2-dsRed2-SEMA3AR734W (0.5 μg of WT-SEMA3A + 0.5 μg of mutant-SEMA3A were used for heterozygote co-expression studies) using 5 μL of Lipofectamine transfection reagent (Invitrogen, Carlsbad, CA) in Gibco® OPTI-MEM media (Invitrogen, Carlsbad, CA). Cells fluorescing 48 hours post-transfection were selected for electrophysiological experiments. HEK293 cell culture and transfection procedures for Nav1.5, Cav1.2 and Kv4.2 are included in the online data supplement.
Electrophysiological measurements and data analysis for Kv4.3
Standard whole cell patch clamp technique using an Axopatch 200B amplifier, Digidata 1440A and pClamp version 10.2 software (Axon Instruments, Foster City, CA) was used to measure electrophysiological properties at room temperature (22–24°C). The extracellular (bath) solution contained (mmol/L): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH adjusted to 7.4 with NaOH. The pipette solution contained (mmol/L): 110 KCl, 31 KOH, 10 EDTA, 5.17 CaCl2, 1.42 MgCl2, 4 MgATP and 10 HEPES, pH adjusted to 7.2 with KOH following established protocols.10, 14 Microelectrodes were pulled on a P-97 puller (Sutter Instruments, Novato, CA) and fire polished to a final resistance of 2–3 M′Ω. Series resistance was compensated by 80–85%. Currents were filtered at 5 kHz and digitized at 10 kHz. The voltage-dependence of activation and inactivation was determined using voltage-clamp protocols described in the figure legend. Data was analyzed using Clampfit (Axon Instruments), Excel (Microsoft, Redmond, WA), and fitted with Origin 8 (OriginLab Corporation, Northampton, MA).
Voltage-dependent inactivation curve was fitted with a Boltzmann function: I/I, max= {1+exp [(V–V1/2)/k]}−1, where V1/2 and k are the half-maximal voltage of inactivation and the slope factor respectively. Inactivation time constants for each voltage step were determined by fitting a mono-exponential function to current decay.
Electrophysiological measurements and data analysis for Nav1.5, Cav1.2 and Kv4.2 are included in the online data supplement.
Kv4.3 total cell and cell surface western blot analysis
Cells were transfected in 25 cm2 flasks using 4μg of lipofectamine mixed with 4μg of total plasmids (pBK-CMV encoding KCND3 with td-Tomato co-expressed with or without pBK-CMV encoding KChIP2 and/or pIRES2-dsRED2-SEMA3A). Plasmids were mixed in equal ratios and kept constant at 4μg by the addition of empty (pBK-CMV) vector. Using previously described methods,15, 16 western blot analyses were performed on total protein lysates prepared from transfected HEK293 cells. The primary antibody was anti-Kv4.3 (UC Davis/NIH NeuroMab facility, Davis, CA). The mouse monoclonal anti-transferrin receptor (TransR) antibody (Invitrogen, Carlsbad, CA) was used as a loading control. The rabbit anti-mouse horseradish peroxidase-conjugated secondary antibody (Bethyl Laboratories, Montgomery, TX) followed by SuperSignal West Dura Extended Duration substrate (Pierce, Rockford, IL) was used for signal detection, captured using a Molecular Imager Chemidoc XRS system running Quantity One software version 4.6 (Bio-Rad Laboratories, Hercules, CA). The intensities of the Kv4.3 protein bands were determined using volume analysis from the Quantity One software (Hercules, CA), and were normalized to TransR in the same lane on the same blot. Results are expressed as means ± SEM from 4–6 experiments.
Expanded protocols for the immunoblots of SEMA3A in mouse brain and human heart, as well as the co-immunoprecipitations of SEMA3A and Kv4.3 are available in the online data supplement.
Cell culture, electrophysiological measurements and data analysis for human induced pluripotent stem cell-derived cardiomyocytes
Control human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) were a kind gift from Dr. Timothy Nelson (Mayo Clinic, Rochester, MN). Cardiomyocyte aggregate cultures were maintained in B27/RPMI media (Gibco Invitrogen, Carlsbad, CA). At differentiation days 25–30, the enriched hiPSC-CMs were subjected to enzymatic dissociation using 0.25% Trypsin/EDTA+5% FBS to obtain single cell suspensions of cardiomyocytes. These cells were added to 0.1% gelatin coated glass coverslips maintained in B27/RPMI media and stored in a 5% CO2 incubator at 37°C before use.
Standard whole cell patch clamp technique, as described above, was used to measure Ito currents in hiPSC-CMs at room temperature (22–24°C). Currents were filtered at 1 kHz and digitized at 5 kHz. Data was analyzed as described above.
Study subjects and SEMA3A mutational analysis
Expanded methods regarding the Brugada syndrome study subjects and SEMA3A mutational analysis are available in the online data supplement.
Statistical analysis
All data are expressed as mean ± SEM. One way ANOVA was performed to determine statistical significance among multiple groups and paired t test was used to compare statistical significance before and after SEMA3A perfusion. P<0.05 was considered to be significant.
RESULTS
Kv4.3 current inhibition by SEMA3A in heterologous expression system
Figure 1A shows the representative tracings of Kv4.3-WT, co-expression with SEMA3A-WT, and with paracrine expression of SEMA3A-WT in HEK293 cells. The paracrine expression of SEMA3A-WT represent cells themselves that are not expressing SEMA3A, however they are in the same media as cells expressing SEMA3A (as confirmed by fluorescence). Analysis of the current-voltage relationship indicated that both SEMA3A-WT co-expression and paracrine expression significantly inhibited Kv4.3 current density from −20 mV to +40 mV (n=10 for each group, p<0.05 vs. Kv4.3-WT, Figure 1B). Kv4.3 peak current density at +40 mV (154.7±24.3 pA/pF; n=10) was significantly reduced by 66.3% with SEMA3A-WT co-expression (52.2±12.1 pA/pF; n=10; p<0.05) and 62.2% with paracrine expression of SEMA3A-WT (58.5±14.5 pA/pF; n=10; p<0.05), indicating that SEMA3A-WT is working on the extracellular surface to block Kv4.3 current. We also co-expressed Kv4.3 with KChIP2, a Kv4.3 chaperone, and SEMA3A had a similar marked inhibitory effect as described above (Online Figure I).
Figure 1. SEMA3A-WT inhibits Kv4.3-WT current in heterologous system but does not affect total or cell surface Kv4.3 protein expression.
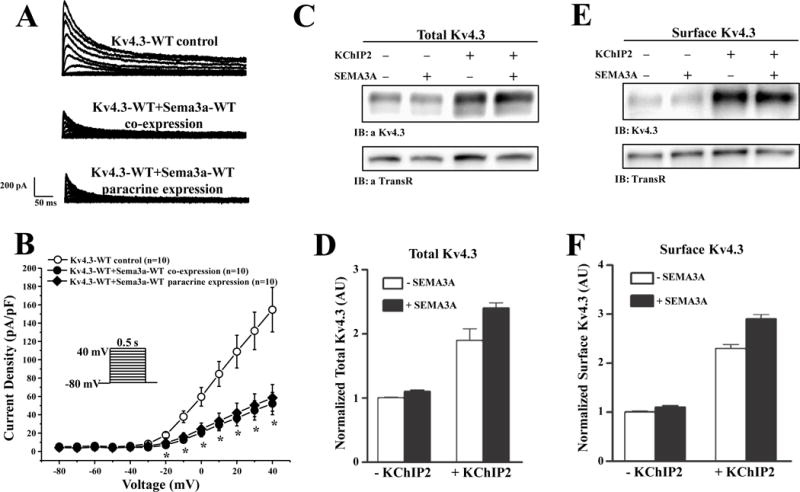
(A) Representative whole-cell tracings of Kv4.3-WT, co-expression with SEMA3A-WT, and paracrine expression of SEMA3A-WT elicited by step depolarization of 500 ms duration from a holding potential of −80 mV to +40 mV in 10 mV increments. (B) The current voltage relationship for Kv4.3-WT (n=10), co-expression with SEMA3A-WT (n=10), and paracrine expression of SEMA3A-WT (n=10). * p<0.05 vs. Kv4.3-WT. Representative western blots of fractionated total (C) and cell surface (E) proteins prepared from HEK293 cells transiently transfected with cDNA constructs encoding td-Tomato and Kv4.3 in the absence (−) and the presence (+) of KChIP2 and/or SEMA3A. The intensities of the total and cell surface Kv4.3 protein bands were measured and normalized to the expression of endogenous transferrin receptor (TransR) in the same lane and subsequently to the total or the surface Kv4.3 in the +/− Sema3A and/or KChIP2 in the same blot. Mean ± SEM (n = 4 blots) total (D) and cell surface (F) Kv4.3 protein expression levels in cells expressing Kv4.3 in the presence and in the absence of KChIP2 and/or SEMA3A are plotted. As reported previously,15, 16 co-expression of Kv4 α-subunits with KChIP2 markedly increased total and cell surface Kv4 protein expression.
SEMA3A’s inhibitory effect on Ito is independent of Kv4.3 expression
To better understand how SEMA3A may be altering the properties of Kv4.3, we first examined the effects of SEMA3A on Kv4.3 protein expression. The overall loss of Kv4.3 current density when co-expressed with SEMA3A is independent of the expression levels of Kv4.3. Specifically, total cell and cell surface Kv4.3 expression is unaffected by SEMA3A in the presence and absence of KChIP2 (Figure 1C–F).
SEMA3A alters the kinetic properties of Kv4.3
Like SEMA3A co-expression, 100 nM human SEMA3A (hSEMA3A) protein perfusion significantly inhibited Kv4.3 current density from −10 mV to +40 mV (n=15, p<0.05 vs. before hSEMA3A perfusion) (Online Figure II). To further determine if hSEMA3A protein could alter Kv4.3-WT current kinetics, we analyzed Kv4.3-WT inactivation time constants and steady-state inactivation parameters before and after perfusion with 100 nM hSEMA3A. 100 nM hSEMA3A protein perfusion significantly decreased Kv4.3 decay time from 0 to 40 mV (n=15, p<0.05). At +40 mV, 100 nM hSEMA3A decreased inactivation time constant by 37.2% from 67.4±3.2 ms to 42.3±4.2 ms (n=15, p<0.05 vs. before hSEMA3A, Figure 2A). Steady-state inactivation was assessed by a standard two-pulse voltage-clamp protocol (Figure 2B) and steady-state inactivation curves were fit using a Boltzmann function.10 100 nM hSEMA3A protein significantly shifted V1/2 of inactivation from −38.9±1.5 mV (before hSEMA3A, n=12) to −51.5±4.7 mV (after hSEMA3A, n=12, p<0.05, Figure 2B). However, recovery from inactivation remained unchanged after perfusion of 100 nM hSEMA3A (Online Figure III).
Figure 2. SEMA3A perfusion alters kinetic properties and inhibits Kv4.3 in a dose-dependent manner.
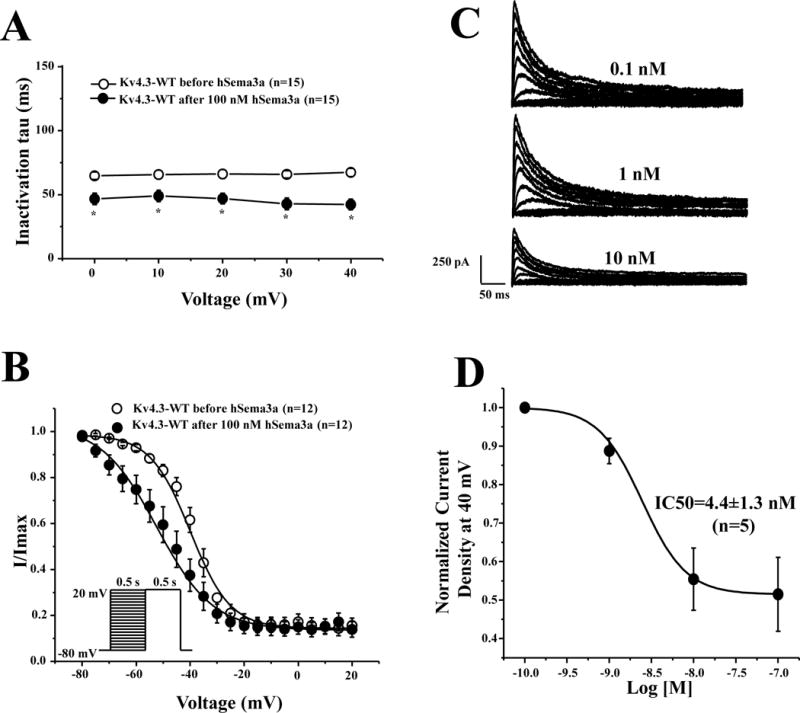
(A) Inactivation time constants (τ) for Kv4.3-WT as a function of voltage before and after 100 nM hSEMA3A perfusion. Inactivation time constants for each voltage step were determined by fitting a mono-exponential function to current decay. * p<0.05 vs. before 100 nM hSEMA3A perfusion. (B) Steady-state inactivation curves of Kv4.3-WT before and after 100 nM hSEMA3A perfusion determined from a holding potential of −80 mV to pre-pulse of +20 mV in 5 mV increments with 0.5 s duration followed by a test pulse of +20 mV with 0.5 s duration and fitted with a Boltzmann function.10 (C) Representative whole-cell Kv4.3-WT tracings with 0.1nM, 1 nM and 10 nM hSEMA3A protein perfusion. (D) Dose-dependent curve with an IC50 of 4.4±1.3 nM (n=5). All values shown represent mean ±SEM.
SEMA3A inhibits Kv4.3 peak current in a dose-dependent manner
In order to explore whether hSEMA3A pharmacologically inhibits Kv4.3 current in a dose-dependent manner, Kv4.3 expressing cells were perfused with 0.1 nM, 1 nM, 10 nM and 100 nM hSEMA3A protein for 5–10 minutes. hSEMA3A protein dose-dependently inhibited Kv4.3 current with an IC50 of 4.4±1.3 nM (n=5, Figure 2C–D).
SEMA3A’s inhibitory effect on Ito in cardiomyocytes derived from human-induced pluripotent stem cells (hiPSC)
In order to explore whether human SEMA3A protein also inhibits Ito channels in human cardiomyocytes, we used control hiPSC-cardiomyocytes at differentiation days of 30–52 (Figure 3A), with average cell capacitance of 34.5±3.9 pF. First, we established that the captured currents elicit a notch in the action potential, which is expected for Ito mediated currents (Online Figure IV). We then examined the effects Ito current density before and after 100 nM hSEMA3A perfusion (Figure 3B), and the current-voltage relationship indicated that 100 nM human SEMA3A protein significantly reduced Ito current density across the voltage from +20 mV to +40 mV (n=5, p<0.05 vs. before hSEMA3A perfusion), (Figure 3C). At +40 mV, Ito peak current density was inhibited by 33.5% from 27.2±7.6 pA/pF (before hSema3a, n=5) to 18.1±5.3 pA/pF (after 100 nM hSema3a, n=5, p<0.05 vs. before hSEMA3A; Figure 3D).
Figure 3. Human SEMA3A protein inhibited Ito channel in hiPSC-CMs.
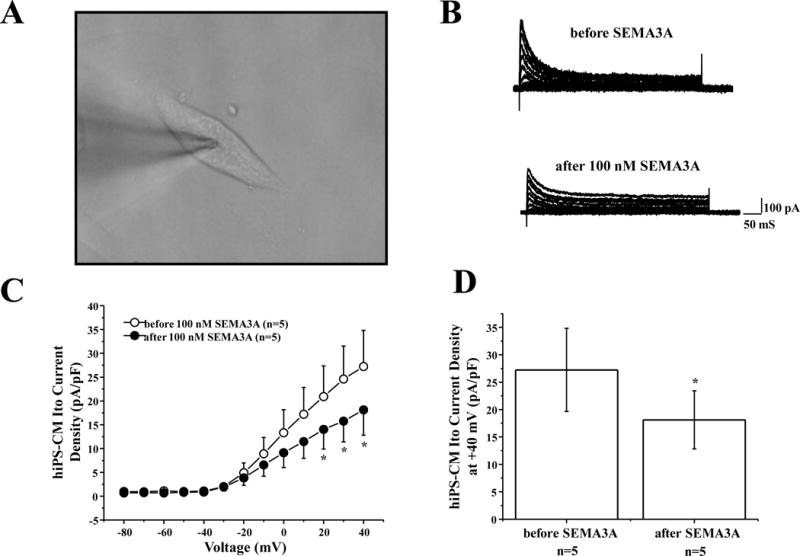
(A) Representative hiPSC-CM cell for Ito recording. (B) Representative whole-cell Ito traces before and after 100 nM human SEMA3A protein perfusion elicited by step depolarization of 500 ms duration to +40 mV from a holding potential of −80 mV in 10 mV increments. (C) The current voltage relationship for Ito before and after 100 nM hSEMA3A perfusion (n=5, * p<0.05 vs. before hSEMA3A). (D) Bar graph showing Ito peak current density at +40 mV before and after 100 nM hSEMA3A perfusion (n=5, *p<0.05 vs. before hSEMA3A). All values represent mean±SEM.
SEMA3A may be a Kv4.3 channel specific blocker
To determine the specificity of SEMA3A’s effects, we incubated hSEMA3A with other cardiac ion channels related to BrS including the sodium channel (SCN5A, INa, Nav1.5), the L-type calcium channel (CACNA1C, ICa,L, Cav1.2), and the Kv4.3 highly homologous voltage-gated potassium channel, Kv4.2 (KCND2). We found that Nav1.5 (Figure 4A–B), Cav1.2 (Figure 4C–D), and Kv4.2 (Figure 4E–F) current densities were not measurably affected by SEMA3A, compared to SEMA3A’s inhibitory effect on Kv4.3 with or without KCHIP2 co-expression.
Figure 4. Channel specificity of SEMA3A.
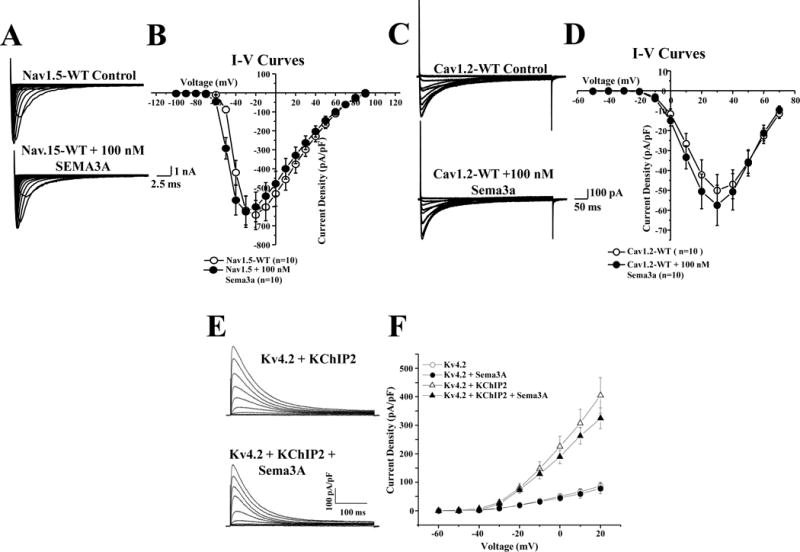
(A) Representative whole-cell voltage-gated inward Na+ currents were recorded from HEK293 cells transiently transfected with cDNA constructs encoding Nav1.5-WT before and after perfusion with 100 nM SEMA3A. Tracings were recorded from holding potential of –100 mV to testing potential of +90 mV in 10 mV increments. (B) The current voltage relationship for Nav1.5 (n=10), perfused with SEMA3A (n=10). (C) Representative whole-cell voltage-gated inward Ca2+ currents were recorded in HEK293 cells transiently transfected with cDNA constructs encoding Cav1.2-WT (α, β, and δ) before and after exposure to 100 nM SEMA3A. Tracings were recorded from holding potential of −90 mV to testing potential of +70 mV in 10 mV increments. (D) The current voltage relationship for Cav1.2 (n=10), exposed to SEMA3A (n=10). (E) Representative whole-cell voltage-gated outward K+ currents were recorded from HEK293 cells transiently transfected with cDNA constructs encoding tdTomato+Kv4.2 in the absence and in the presence of KChIP2 and/or SEMA3A in response to depolarizing voltage steps between −60 and +40 mV in 10 mV increments from a holding potential of −70 mV. Representative Kv4.2+KChIP2 encoded Kv currents, recorded from HEK293 cells transfected in the absence (E upper) or the presence (E lower) of SEMA3A are shown. (F) The current voltage relationship for Kv4.2 (n=8–17) co-expressed with SEMA3A. All values represent mean ±SEM.
Heart expression of SEMA3A and co-immunoprecipitation with Kv4.3
Due to the effects of SEMA3A on Kv4.3, we wanted to confirm whether SEMA3A is expressed in human cardiac tissue. As illustrated in Figure 5, the polyclonal anti-SEMA3A antibody reliably detects native SEMA3A protein in (human) heart and (mouse) brain lysates. Western blot analysis of human ventricular lysates revealed robust expression of SEMA3A in the membrane fraction (Figure 5A). SEMA3A expression in adult mouse brain was particularly robust in the membrane fraction (Figure 5B), therefore, in order to determine if there was a binding interaction between SEMA3A and Kv4.3, we did a co-immunoprecipitation in mouse brain tissue. We found that SEMA3A co-immunoprecipitates with Kv4.3 (Figure 5C). The co-immunoprecipitation of SEMA3A with the anti- Kv4.3 antibody was specific, as evidenced by the absence of signal in the control-IP (Figure 5C). This data suggests a direct binding interaction between these two proteins.
Figure 5. Membrane expression of SEMA3A and association with Kv4.3.
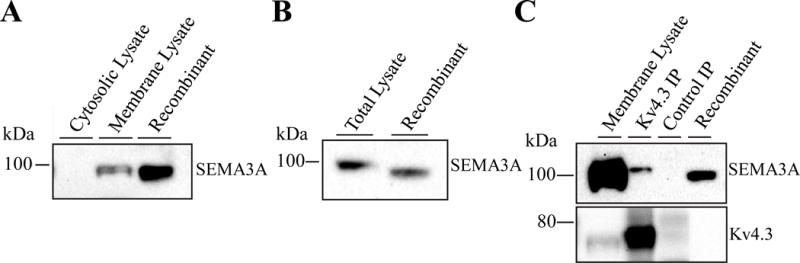
(A) Western blot analysis of human heart membrane and cytosolic lysates revealed robust SEMA3A expression only in the membrane fraction. (B) Western blot analysis of SEMA3A in mouse brain lysates, compared directly with human recombinant SEMA3A. (C) Western blot demonstrating robust expression of SEMA3A in the membrane fraction from mouse brain and co-immunoprecipitation (IP) with Kv4.3 (Kv4.3 IP); no SEMA3A was detected in the control IP.
SEMA3A’s inhibitory effect on Kv4.3 is related to its hanatoxin-like domain
A portion of SEMA3A’s amino acid sequence is analogous to hanatoxin6 (Figure 6A) which is closely related to Heteropoda venatoria toxin 2 (HpTx2). HpTx2 selectively inhibits Kv4.3 through Kv4.3’s voltage sensor, mediated by interactions with two Kv4.3 amino acids (L275 and V276, rat isoform).8 Therefore, to determine whether SEMA3A may be binding to Kv4.3 in a similar location to HpTx2, we mutated the homologous amino acid residues in human Kv4.3, L274 and V275 to alanine. The L274A-Kv4.3 (266.50 pA/pF) and V275A-Kv4.3 (257.02 pA/pF) channels electrophysiologically behave like WT-Kv4.3 (291.85 pA/pF) at +40 mV voltage (Online Figure V). While SEMA3A reduces the current density of WT-Kv4.3 (176.94 pA/pF, 38.44%), L274A-Kv4.3 (215.14 pA/pF, 18.83%), and V275A-Kv4.3 (185.52 pA/pF, 27.93%) channels (Figure 6B, Online Figure V), the overall effect of SEMA3A on peak current density is reduced for L274A. SEMA3A leads to a 38% reduction in WT-Kv4.3 peak current density, however, SEMA3A leads to only an 18% reduction in L274A-Kv4.3 current density (p<0.05; Figure 6C). SEMA3A has a larger effect on V275A-Kv4.3 peak current density, with a 29% reduction (Figure 6C).
Figure 6. SEMA3A Gene Topology and targeted Disruption of Toxin Binding Domain on Kv4.3.
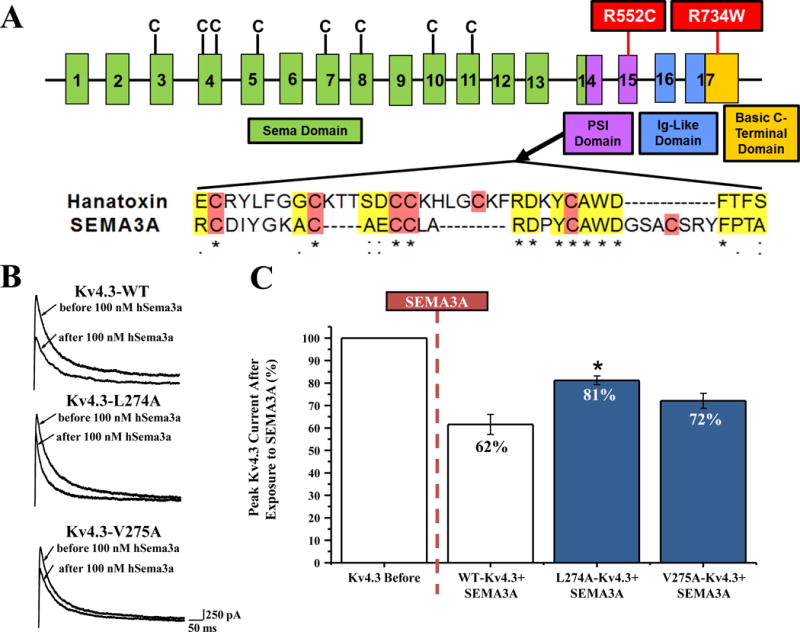
(A) Representation of the linear genetic topology of SEMA3A, exons are labeled numerically. C represents the eight cysteines forming four disulfide brides. Protein domains are represented by colors and boxes. Mutations identified in two BrS patients are highlighted in red boxes. (B) The amino acids encoded within SEMA3A exon 14 share homology with hanatoxin. Cysteines, necessary for toxin folding, are highlighted in red, and homologous amino acids are highlighted in yellow. * represents identical conservation,: represents strongly similar properties scoring > 0.5 and . represents weakly similar properties scoring ≤ 0.5 in the Gonnet PAM 250 matrix. (B) Representative peak current at +40 mV traces before and after perfusion of 100 nM hSEMA3A for Kv4.3-WT, Kv4.3-L274A, and Kv4.3-V275A. (C) Bar graph shown as percent of peak Kv4.3 current density at +40 mV after 100 nM SEMA3A exposure. Kv4.3 before is shown as 100%, and percentages were calculated from Kv4.3-WT (n=10), Kv4.3-L274 (n=10), and Kv4.3-V275A (n=10) before and after 100 nM perfusion of hSEMA3A. p<0.05 vs. WT-Kv4.3+SEMA3A, all values are shown as a mean ± SEM.
Because the effects of SEMA3A are attenuated by L274A and V275A Kv4.3 substitutions, it may be possible that SEMA3A binds to the voltage sensor region on Kv4.3, and these mutations are disrupting this interaction.
SEMA3A Mutations may contribute to the pathogenesis of BrS
Overall, 4 SEMA3A missense mutations were identified in 10 patients (N153S, 2 cases; V435I, 6 cases; R552C, 1 case; R734W, 1 case) within our BrS cohort (Online Table I). However, 2 missense mutations, R552C and R734W (Figure 6A), in 2/198 (1%) unrelated BrS patients (Table 1), were absent in 500 European Caucasian controls, 300 Italian controls, 1094 subjects from the 1000 Genomes Project,17 6503 subjects from the NHLBI GO Exome Sequencing Project,18 and the 12000 Exome Chip19 and were therefore considered as potentially pathogenic missense mutations and investigated functionally.
Table 1.
Demographics of SCN5A-Negative Unrelated BrS Patients
| Brugada Syndrome Patient Demographics | |
|---|---|
| Number of Probands | 198 |
| Age at Diagnosis, years ± SD | 45 ± 13 |
| Range | 9–81 |
| Males (%) | 155 (78) |
| Average QTc, ms ± SD | 406 ± 27 |
| Average PR Interval, ms ± SD | 135 ± 27 |
| Symptomatic Patients (%) | 42 (21) |
| Family History of Cardiac Events/Unexplained Sudden Death (%) | 62 (31) |
| Spontaneous Type 1 Brugada ECG Pattern (%) | 69 (35) |
| Positive Drug Challenge Test (%) | 143 (72) |
R552C-SEMA3A was identified in a 45-year-old male with a history of palpitations at rest. ST-segment elevation was observed in leads V1 and V2 on a Holter ECG, especially after large meals. Flecanide testing revealed a type 1 Brugada ECG pattern (Online Figure VI A–B). Despite a reported negative family history of cardiac events, the patient’s only living family member, a daughter, was clinically evaluated. She had a negative flecanide challenge and was R552C-SEMA3A mutation negative.
R734W-SEMA3A was identified in an asymptomatic 44-year-old male with no family history. An ECG performed for chest pain identified a type 2 Brugada ECG pattern, and a subsequent flecanide test induced a positive type 1 Brugada ECG pattern (Online Figure VI C–D). Ventricular fibrillation was noted during the diagnostic electrophysiology study and an ICD was implanted subsequently. The patient’s son had a negative flecanide test and was R734W-SEMA3A mutation negative.
Both R552C- and R734W-SEMA3A mutations when co-expressed with Kv4.3-WT, resulted in an increased Ito current density from −20 mV to +40 mV (R552C n=23, R734W n=20, p<0.05) compared with Kv4.3-WT+SEMA3A (n=20, Figure 7A–B). SEMA3A mutant co-expression significantly increased Kv4.3 peak current density at +40 mV by 333.5% from 69.3±8.1 pA/pF (WT, n=20) to 300.4±55 pA/pF (R552C, n=23, p<0.05) and by 137.4% to 164.5±31.1 pA/pF (R734W, n=20, p<0.05, Figure 7C). Additionally, R552C and R734W both significantly increased the Ito total charge from −10 to +40 mV (p<0.05 vs. Kv4.3-WT + SEMA3A, Figure 7D). However, neither SEMA3A mutations resulted in significant changes in decay time (Figure 7E) or steady-state inactivation (Figure 7F) when compared to Kv4.3+SEMA3A-WT co-expression.
Figure 7. Possible Brugada syndrome-associated mutations in SEMA3A.
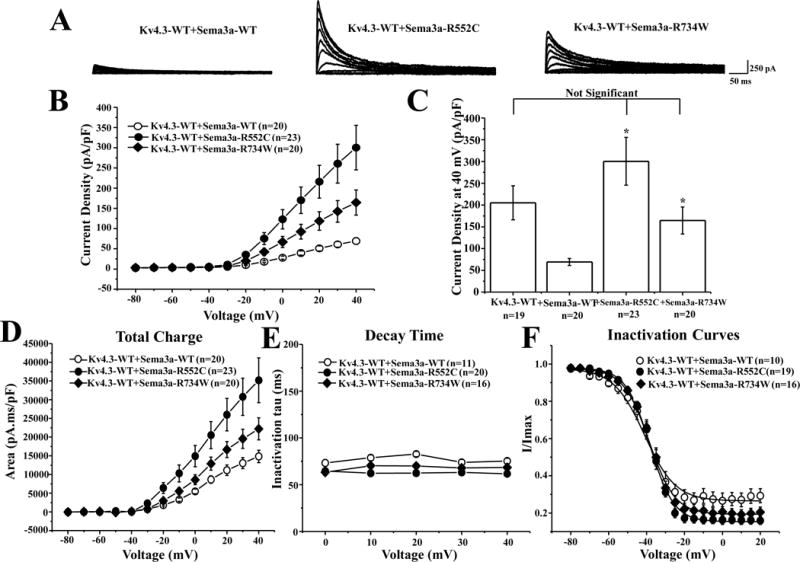
(A) Representative whole-cell Kv4.3-WT co-expressed with SEMA3A-WT, SEMA3A-R552C, or SEMA3A-R734W tracings recorded in HEK293 cells elicited by step depolarization of 500 ms duration from a holding potential of −80 mV to +40 mV in 10 mV increments. (B) The current voltage relationship for Kv4.3-WT co-expressed with SEMA3A-WT (n=20), SEMA3A-R552C (n=23), or SEMA3A-R734W (n=20). * p<0.05 vs. Kv4.3 + SEMA3A-WT. (C) Bar graph showing peak current density at +40 mV for Kv4.3-WT alone or co-expressed with SEMA3A-WT (n=20), SEMA3A-R552C (n=23), or SEMA3A-R734W (n=20). * p<0.05 vs. Kv4.3-WT + SEMA3A-WT. (D) Total charge of Kv4.3-WT co-expressed with SEMA3A-WT, SEMA3A-R552C, or SEMA3A-R734W as a function of voltage obtained by measuring the area under the curve during the first 50 ms of each voltage step. * p<0.05 vs. Kv4.3 + SEMA3A-WT. (E) Inactivation time constants (τ) for Kv4.3-WT with SEMA3A-WT, SEMA3A-R552C, or SEMA3A-R734W as a function of voltage. Inactivation time constants for each voltage step were determined by fitting a mono-exponential function to current decay. (F) Steady-state inactivation curves of Kv4.3-WT with SEMA3A-WT, SEMA3A-R552C, or SEMA3A-R734W determined from a holding potential of −80 mV to pre-pulse of +20 mV in 5 mV increments with 0.5 s duration followed by a test pulse of +20 mV with a 0.5 s duration fitted with a Boltzmann function.10 All values shown represent mean ±SEM.
In addition, electrophysiological analysis was completed in a heterozygous state, with Kv4.3-WT co-expression with SEMA3A-WT and SEMA3A-WT+SEMA3A-R552C or SEMA3A-WT+SEMA3A-R734W (Online Figure VII). With these mutant-WT SEMA3A co-expressions, SEMA3A-WT+SEMA3A-R552C still precipitated a marked increase in Kv4.3 current density from −30 mV to +40 mV (WT+R552C n=15, p<0.05) compared with Kv4.3-WT+SEMA3A-WT (n=15, Online Figure VII C–D). SEMA3A-WT+SEMA3A-R552C significantly increased Kv4.3+SEMA3A-WT peak current density at +40 mV by 220% from 70.9±10.6 pA/pF (SEMA3A-WT, n=15) to 226.9±44.5 pA/pF (SEMA3A-WT+R552C, n=15, p<0.05; Online Figure VII D). In contrast, SEMA3A-WT+SEMA3A-R734W increased the Kv4.3 current density, by only 25.2% compared to SEMA3A-WT (peak current density 88.8±19.9 pA/pF; SEMA3A-WT+R734W, n=14; Online Figure VII C–D).
DISCUSSION
SEMA3A regulates Ito current density and kinetics
SEMA3A has robust expression in human heart tissue, and it has been established previously that Kv4.3 is expressed in the human heart20. In addition, SEMA3A transgenic mice have a reduction of Ito density, reduced sympathetic innervation and have the propensity for spontaneous ventricular arrhythmias.3 In combination with our illustration of SEMA3A’s effect on Kv4.3, this data suggests that SEMA3A not only regulates cardiac innervation patterning but may also regulate Ito current densities in order to maintain a transmural repolarization gradient and prevent potentially lethal cardiac arrhythmias.
Here, we have identified SEMA3A as a novel inhibitory regulator of Kv4.3 current density and kinetics due to direct binding of SEMA3A and Kv4.3 in a manner similar to toxin-channel binding. SEMA3A has several similarities to toxins which are known to physically bind and inhibit voltage gated ion channels. SEMA3A has a 34 amino acid stretch analogous to hanatoxin,6 which contains the 6 stereotypical cysteines (Figure 3) of an “inhibitor cystine knot (ICK)” motif commonly seen in invertebrate toxins.21 This hanatoxin-like sequence in SEMA3A resides within a similar Plexin/Semaphorin/Integrin (PSI) domain in which the structure was described in SEMA4D (a close relative of SEMA3A). Although the function of this domain is unknown, the PSI domain folds using three disulfide bonds akin to the ICK motif.22 Therefore, SEMA3A has protein sequence characteristics of a toxin, which may support its ability to bind and inhibit ion channels.
Toxins are known to bind to the extracellular surface of ion channels. In our study, SEMA3A led to reduced current density of Kv4.3 in HEK293 cells whether co-expressed within the cell (Figure 1A–B), expressed in a paracrine fashion (Figure 1A–B), or with hSEMA3A protein perfusion (Figure 2). SEMA3A perfusion also reduced current density of Ito in cardiomyocytes derived from human induced pluripotent stem cells (Figure 3). In order to establish a binding interaction, we were able to immunoprecipitate SEMA3A with an anti-Kv4.3 antibody in mouse brain (Figure 4B). Altogether, this data supports the conclusion that SEMA3A is binding to Kv4.3, potentially at the extracellular surface; congruent with SEMA3A’s previously established function as a naturally secreted protein binding to cell-surface receptors.2
In an attempt to determine where SEMA3A may be binding to Kv4.3, we focused on what has been previously established for toxin-channel interaction. There are two major mechanisms in which toxins can interact with voltage-gated channels on the extracellular surface, through direct targeting of the ion channel pore or through binding to the channel’s voltage sensor region which influences the stability of closed, open, or inactivated states of the channel.23 Hanatoxin and HpTx2, both fall into the latter category, binding to the voltage sensing domain of Kv2.1 and Kv4.3, respectively.7, 8 When bound, these toxins shift activation to more depolarized voltages, decrease current density, and rapidly inactivate channels,23 all of which are seen when Kv4.3 is exposed to SEMA3A. The binding interaction between toxins and channels is regulated by the overall charge of the voltage sensor paddle region, and mutagenesis of this region can significantly alter the effects of the toxins. Similar to previous studies on Kv4.3 and HpTx2,8 mutagenesis of Kv4.3 at amino acid positions L274 and V275 attenuated SEMA3A’s inhibitory effect on the channel, suggesting a direct interaction between SEMA3A and Kv4.3 voltage sensor.
SEMA3A - A possible Brugada syndrome susceptibility gene
The phenotype of the murine SEMA3A knockout consisting of sinus bradycardia, decreased basal sympathetic activity, ST-segment elevation, and SCD prompted our analysis of our BrS cohort. Here, we identified two ultra-rare SEMA3A mutations, R552C and R734W in two patients diagnosed with BrS. Interestingly, a common I334V-SEMA3A polymorphism was associated recently with a high incidence of unexplained cardiac arrest with ventricular fibrillation among pilsicainide-challenge negative Japanese individuals.24 According to Nakano and colleagues,24 using the 1000 Genomes Project,17 I334V has a prevalence of 2.1% in East Asians, 1.35% among West Africans, a 1.86% among Americans, and 0% among Europeans. This mutation was not identified in our European Caucasian cohort. Functional characterization of this SEMA3A polymorphism identified a loss-of-function of axon collapse and led to disrupted innervation patterning in patient tissues.24 Whether our SEMA3A mutation positive BrS patients have an abnormal cardiac innervation pattern is currently unknown.
Co-expression of Kv4.3 with either SEMA3A mutation in a homozygous fashion led to a significant increase in Ito current compared to Kv4.3 co-expressed with wild-type SEMA3A. We speculate that each mutation may cause misfolding of SEMA3A, thereby either disrupting the hanatoxin-like sequence altering SEMA3A-Kv4.3 binding, or preventing SEMA3A secretion. These effects would presumably disrupt SEMA3A’s normal suppressive effect on Kv4.3 therefore leading to an increase in Ito current. Interestingly, R552C is three amino acids away from the SEMA3A hanatoxin-like sequence. This region, within the PSI domain, is known to fold with three disulfide bonds between 6 cysteine residues (Figure 6A). The addition of a new cysteine as a result of the R552C mutant could lead to the formation of a novel disulfide bond thus altering the folding structure of the PSI domain, directly affecting this toxin-like region, which in turn, could alter binding to Kv4.3. In addition, the R734W mutation within the basic SEMA3A C-terminus substitutes a basic amino acid, arginine, to a hydrophobic uncharged amino acid, tryptophan, which could affect the overall charge of this region and alter its folding structure.
After heterozygote co-expression of mutant and WT-SEMA3A together, R734W no longer accentuates Kv4.3 current density significantly, at least in a HEK293 cell model system during WT + mutant co-expression studies that attempt to mimic the heterozygous state. However, without examination of protein expression in our patient, we do not know if these 50:50 studies are truly reflective of human expression. The mutations themselves could alter the expressivity of the mutant SEMA3A, potentially leading to a more robust phenotype. In addition, growth cone collapse assays for each of the mutants have not been completed. Therefore, it is possible that either of these mutations could still contribute to a BrS-like phenotype, in a nerve growth related manner. These SEMA3A mutants could also alter the normal expression patterning of SEMA3A, disrupting the known SEMA3A and Kv4.3 expression gradients. Additional experimentation in the form of transgenic mice may help elucidate some of the potential developmental innervation changes which may develop due to mutations within SEMA3A.
Altogether, based on our electrophysiological studies, we know that a rare R552C-SEMA3A mutation attenuates SEMA3A’s ability to block Kv4.3, resulting in a substantial accentuation of Kv4.3 current. Previously, we have demonstrated that primary mutations in KCND3-encoded Kv4.3 cause BrS through a marked gain of Kv4.3 current10. Accordingly, in a final common pathway fashion, it is possible that the SEMA3A perturbation underlies their disease. However, since the cases in which these mutations have been identified do not have sufficient pedigree information to test co-segregation, we cannot be certain that the SEMA3A mutation is solely responsible for BrS in these two patients.
The potential for SEMA3A as a therapeutic Ito-specific channel blocker
Gain-of-function in Ito underlies a subset of BrS being first identified in BrS patients harboring mutations in KCNE3.25 Subsequently, we identified two mutations within KCND3-encoded Kv4.3 in patients with BrS.10 Mutations in each of these genes caused a significant gain-of-function in Ito current. One of the current treatment strategies for patients with BrS is quinidine, which blocks a variety of channels. However, quinidine’s Ito blocking activity may underlie its therapeutic efficacy in patients with BrS regardless of the primary pathogenic substrate.26–28 In principle, an Ito-specific blocker might be more effective than quinidine in managing patients with symptomatic BrS. This study provides evidence that SEMA3A may have potential as a novel therapeutic as an Ito-specific blocker. First, SEMA3A has drug-like properties, with a dose dependent response curve as shown in Figure 2C–D. Second, SEMA3A does not alter the current density of other inward cardiac ion channels, such as Nav1.5 or Cav1.2, or another fast transient outward current potassium channel, Kv4.2, lending support that SEMA3A may be a channel specific blocker for Kv4.3. Many of the known Kv4.3 blockers also block other potassium channels or inward currents.26 Therefore, SEMA3A, or, a synthetically derived toxin-like portion of SEMA3A, could potentially be developed into a treatment strategy for patients with symptomatic BrS.
Conclusion
We have identified a novel function for SEMA3A as a potential Kv4.3-specific channel blocker. In addition, with the identification of rare functionally significant mutations, perturbations in SEMA3A may contribute to BrS. The identified effects of SEMA3A on Kv4.3 may be due to a direct binding interaction in a mechanism similar to toxin-channel binding and these findings might stimulate the development of a novel Ito-specific channel blocker for therapeutic intent.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
SEMA3A is a chemorepellent which guides neural patterning and projections, is expressed in murine hearts, and when disrupted through either complete knockout, or transgenic overexpression, leads to sudden cardiac death in mice.
A portion of SEMA3A is analogous to a toxin, known as Hanatoxin, which binds to and inhibits potassium channels.
Gain in function of Kv4.3 potassium channels in the heart can lead to a Brugada syndrome (BrS) phenotype that may be associated with sudden cardiac death.
What New Information Does This Article Contribute?
We have identified a novel function for SEMA3A as a Kv4.3-specific channel blocker, specifically, SEMA3A reduces Kv4.3 channel current density in a dose dependent manner, alters Kv4.3 channel kinetics, yet has no effect on other cardiac ion channels, such as Nav1.5, Cav1.2, and Kv4.2.
SEMA3A co-immunoprecipitated with Kv4.3, suggesting a direct binding interaction between these two proteins.
With the identification of rare SEMA3A mutations, leading to an overall gain-of-function in Kv4.3 current, genetic perturbations in SEMA3A may contribute to Brugada syndrome.
SEMA3A-encoded semaphorin 3A is a chemorepellent that disrupts neural patterning in the nervous and cardiac systems. In addition, SEMA3A has an amino acid motif that is analogous to hanatoxin, an inhibitor of voltage-gated potassium channels. Mice that are lacking SEMA3A (i.e. SEMA3A knockout mice) are prone to ventricular arrhythmias and sudden cardiac death. An increase in voltage-gated Kv4.3 potassium channel current has been recognized as a pathogenic basis for some cases of Brugada syndrome (BrS). In this study, we identified a novel biological role for SEMA3A as a naturally occurring protein inhibitor of Kv4.3 potassium channels. We also showed that SEMA3A gene mutations are a potential contributor to BrS by disrupting SEMA3A’s natural ability to suppress the Kv4.3 channel thus resulting in an increase in potassium channel current. This previously unrecognized interaction between SEMA3A and Kv4.3 could be exploited as a potential drug target, for the regulation of Kv4.3 currents, and have potential therapeutic utility for diseases like BrS.
Acknowledgments
SOURCES OF FUNDING
N.J. Boczek was supported by CTSA Grant (UL1 TR000135) from the National Center for Advancing Translational Science (NCATS), a component of the NIH, and an individual PhD predoctoral fellowship from the American Heart Association (12PRE11340005). L. Crotti and P.J. Schwartz were supported by the Italian Ministry of Education, University and Research (MIUR) FIRB RBFR12I3KA (LC), Italian Ministry of Education, University and Research (MIUR) PRIN 2010BWY8E9 (PJS and LC), Italian Ministry of Health GR-2010-2305717 (LC). M.J. Ackerman was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.
Nonstandard Abbreviations and Acronyms
- BrS
Brugada Syndrome
- SCN5A
Cardiac sodium channel
- INa
Nav1.5
- DHPLC
Denaturing High Performance Liquid Chromatography
- hiPSC
Human induced pluripotent stem cells
- hiPSC-CMs
human induced pluripotent stem cell-derived cardiomyocytes
- HpTx2
Heteropoda venatoria toxin
- IP
Immunoprecipitations
- ICK
Inhibitor Cystine Knot
- CACNA1C
L-type calcium channel
- ICa,L
Cav1.2
- PSI
Plexin/Semaphorin/Integrin
- SEMA3A
Semaphorin 3a
- SDM
Site Directed Mutagenesis
- SCD
Sudden Cardiac Death
- TransR
Transferrin receptor
- KCND3
Transient outward repolarizing current
- Ito
Kv4.3 (or KCND2, Ito, Kv4.2)
- WT
Wild-type
Footnotes
DISCLOSURES
M.J. Ackerman is a consultant for Boston Scientific, Gilead Sciences, Medtronic, and St. Jude Medical and receives royalties from Transgenomic for FAMILION-LQTS and FAMILION-CPVT genetic tests. A.A. Wilde is a member of the scientific discovery board of Sorin. The other authors have no conflicts of interest relevant to this article to disclose.
References
- 1.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiore R, Puschel AW. The function of semaphorins during nervous system development. Front Biosci. 2003;8:484–499. doi: 10.2741/1080. [DOI] [PubMed] [Google Scholar]
- 3.Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Matsumura K, Tomita Y, Makino S, Sano M, Ogawa S, Fukada K. Abstract 1243: Sema3a induces a cardiac ventricular repolarization gradient via sympathetic innervation patterning. Circulation. 2006;114:II_223–II_234. [Google Scholar]
- 4.Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Taniguchi M, Lee J-K, Matsumura K, Tomita Y, Miyoshi S, Shimoda K, Makino S, Sano M, Kodama I, Ogawa S, Fukuda K. Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nature Medicine. 2007;13:604–612. doi: 10.1038/nm1570. [DOI] [PubMed] [Google Scholar]
- 5.Chen LS, Zhou S, Fishbein MC, Chen PS. New perspectives on the role of autonomic nervous system in the genesis of arrhythmias. J Cardiovasc Electrophysiol. 2007;18:123–127. doi: 10.1111/j.1540-8167.2006.00590.x. [DOI] [PubMed] [Google Scholar]
- 6.Behar O, Mizuno K, Badminton M, Woolf CJ. Semaphorin 3a growth cone collapse requires a sequence homologous to tarantula toxin. PNAS. 1999;96:13501–13505. doi: 10.1073/pnas.96.23.13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swartz KJ, MacKinnon R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent k+ channels. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 8.DeSimone C, Lu Y, Bondarenko VE, Morales MJ. S3b amino acid substitutions and ancillary subunits alter the affinity of heteropoda venatoria toxin 2 for Kv4.3. Mol Pharmacol. 2009;76:125–133. doi: 10.1124/mol.109.055657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brugada R, Campuzano O, Brugada P, Brugada J, Hong K. Brugada syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews [Internet] Seattle, WA: University of Washington; 2005. Updated 2012. [Google Scholar]
- 10.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (i(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–1032. doi: 10.1016/j.hrthm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizusawa Y, Wilde AAM. Brugada syndrome. Circ Arrhythm Electrophysiol. 2012;5:606–616. doi: 10.1161/CIRCEP.111.964577. [DOI] [PubMed] [Google Scholar]
- 12.Verrier R, Antzelevitch C. Autonomic aspects of arrhythmogenesis: The enduring and the new. Curr Opion Cardiol. 2004;19:2–11. doi: 10.1097/00001573-200401000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Postema PG. The quest for the identificaion of genetic variants in unexplained cardiac arrest and idiopathic ventricular fibrillation. PLoS Genet. 2013;9:e1003480. doi: 10.1371/journal.pgen.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calloe K, Cordeiro JM, Di Diego JM, Hansen RS, Grunnet M, Olesen SP, Antzelevitch C. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of brugada syndrome. Cardiovasc Res. 2009;81:686–694. doi: 10.1093/cvr/cvn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foeger N, Marinneau C, Nerbonne J. Co-assembly of kv4 alpha subunits with k+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. J Biol Chem. 2010;285:33413–33422. doi: 10.1074/jbc.M110.145185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foeger N, Norris A, Wren L, Nerbonne J. Augmentation of kv4.2-encoded currents by accessory dipeptidyl peptidase 6 and 10 subunits reflects selective cell surface kv4.2 protein stabilization. J Biol Chem. 2012;287 doi: 10.1074/jbc.M111.324574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Exome Variant Server. NESPE; Seattle, WA: (URL: http://evs.gs.washington.edu/EVS/) [accessed September 2013] [Google Scholar]
- 19.Abecasis G, Neale B. Exome chip design. 2011 (URL: Http://genome.Sph.Umich.Edu/wiki/exome_chip_design) [accessed August 2012]
- 20.Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norton RS, Phallaghy PK. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon. 1998;36:1573–1583. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- 22.Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, Esnouf RM. The ligand-binding face of semaphorins revealed by high-resolution crystal structure of sema4d. Nat Struct Biol. 2003;10:843–848. doi: 10.1038/nsb977. [DOI] [PubMed] [Google Scholar]
- 23.Swartz KJ. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49:213–230. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakano Y, Chayama K, Ochi H, Toshishige M, Hayashida Y, Miki D, Hayes CN, Suzuki H, Tokuyama T, Oda N, Suenari K, Uchimura-Makita Y, Kajihara K, Sairaku A, Motoda C, Fujiwara M, Watanabe Y, Yoshida Y, Ohkubo K, Watanabe I, Nogami A, Hasegawa K, Watanabe H, Endo N, Aiba T, Shimizu W, Ohno S, Horie M, Arihiro K, Tashiro S, Makita N, Kihara Y. A nonsynonymous polymorphism in semaphorin 3a as a risk fasctor for human unexplained cardiac arrest with documented ventricular fibrillation. PLoS Genet. 2013;9:e1003364. doi: 10.1371/journal.pgen.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delpon E, Cordeiro JM, Nunez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of kcne3 mutation and its role in the development of brugada syndrome. Circ. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antzelevitch C, Fish JM, DiDiego JM. Cellular mechanisms underlying the brugada syndrome. In: Antzelevitch C, Brugada P, editors. The brugada syndrome: From bench to bedside. Malden, Massachusetts: Blackwell Publishing; 2005. pp. 52–77. [Google Scholar]
- 27.Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with brugada syndrome. Pacing Clin Electrophysiol. 2001;24:1420–1422. doi: 10.1046/j.1460-9592.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- 28.Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with brugada syndrome. Circulation. 2004;110:1731–1737. doi: 10.1161/01.CIR.0000143159.30585.90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.