

PRESENTATION OF CASE
Dr. Eric Hesse (Harvard School of Dental Medicine)
A 56-year-old man was seen in the outpatient endocrinology and oral-surgery clinics of this hospital because of recurrent hypophosphatemia.
The patient had been well until 19 years earlier, when rib pain developed and a left metatarsal stress fracture occurred while he was running. Laboratory tests performed 1 year later at another hospital revealed normal levels of serum creatinine and bicarbonate and normal creatinine clearance, and urine tests for organic or amino acids were negative; other test results are shown in Table 1. Radiographs of the left forefoot showed Looser zones (lucent bands perpendicular to the cortex), and pathological examination of a bone-biopsy specimen reportedly revealed findings that were consistent with severe osteomalacia. Phosphate supplements and calcitriol were administered, after which the patient’s symptoms resolved. An extensive search for a tumor was negative.
Table 1.
Laboratory Data.*
| Variable | Reference Range, Adults† | Other Hospitals | This Hospital | |||||
|---|---|---|---|---|---|---|---|---|
| 18 Yr Earlier | 16 Yr Earlier, while Receiving Treatment | 13 Yr Earlier, after Surgery, while Receiving No Treatment | 9 Yr Earlier, while Receiving Treatment | 6 Yr Earlier | 6 Mo Earlier | On Presentation, while Receiving Treatment | ||
| Serum | ||||||||
|
| ||||||||
| Calcium (mg/dl) | 8.5–10.5 | 8.5–9.1 | 9.36 | 9.2 | 9.1 | 9.5 | ||
|
| ||||||||
| Ionic calcium (mg/dl) | 4.6–5.2 | 4.6 | 5.1 | |||||
|
| ||||||||
| Phosphorus (mg/dl) | 2.6–4.5 | 1.0 (ref 2.5–4.5) | 2.5 | 3.6 | 1.6 (ref 2.2–4.6) | 1.7 (ref 2.5–4.5) | 1.7 (ref 2.5–4.5) | 1.8 |
|
| ||||||||
| Alkaline phosphatase (U/liter) | ||||||||
|
| ||||||||
| Total | 45–115 | 318–391 (ref 100–280) | 256 | 129 (ref 30–110) | 108 (ref 20–125) | 102 | ||
|
| ||||||||
| Bone-specific | 202 (ref <70) | 36.9 μg/liter (ref 5.9–22.9) | ||||||
|
| ||||||||
| Parathyroid hormone (pg/ml) | 10–60 | 34 | 39 (ref 10–65) | 21 (ref 10–65) | 58 | |||
|
| ||||||||
| 25-Hydroxyvitamin D (ng/ml) | 33–100 | 9 (repeated, 19) (ref 3–30) | 55 (ref 20–100) | 39 | ||||
|
| ||||||||
| 1,25-Dihydroxyvitamin D (pg/ml) | 18–64 | 29 (repeated, 39) (ref 25–60) | 31 (ref 15–60) | 39 (ref 15–60) | 29 | |||
|
| ||||||||
| Fibroblast growth factor 23 (C-terminal) (RU/ml) | ≤180 | 91 | 870 | |||||
|
| ||||||||
| Creatinine (mg/dl) | 0.60–1.50 | 1.20 | ||||||
|
| ||||||||
| Urine | ||||||||
|
| ||||||||
| Calcium (mg/24 hr) | 2.0–2.6 (mmol/24 hr) (ref 2.0–9.0) | 149 | 289 (ref 100–300) | |||||
|
| ||||||||
| Calcium (mg/dl) | 15 | |||||||
|
| ||||||||
| Phosphorus (mg/dl) | Not defined | 86 (ref 20–370) | 112.8 | |||||
|
| ||||||||
| Phosphorus (timed) | Diet-dependent | 1596 mg/24 hr (ref 340–1000) | 16.9 mg/15 ml (3.5-hr urine collection) | |||||
|
| ||||||||
| Phosphate excretion index | <0.12 | 0.31–0.74 | ||||||
|
| ||||||||
| Tubular maximum phosphate reabsorption (mg/100 ml glomerular filtrate) | 2.05–4.28 | 1.3 | ||||||
|
| ||||||||
| Creatinine (mg/ml) | 1.22 | |||||||
|
| ||||||||
| Creatinine (g/24 hr) | 0.63–2.50 | 1.66 | 1.72 | |||||
|
| ||||||||
| Ratio of calcium (in mg) to creatinine (in g) | 30–210 | 174 | ||||||
Ref denotes the reference range at another hospital. To convert the values for calcium to millimoles per liter, multiply by 0.250. To convert the values for phosphorus to millimoles per liter, multiply by 0.3229. To convert the values for 25-hydroxyvitamin D to nanomoles per liter, multiply by 2.496. To convert the values for 1,25-dihydroxyvitamin D to picomoles per liter, multiply by 2.6. To convert the values for serum creatinine to micromoles per liter, multiply by 88.4.
Reference values are affected by many variables, including the patient population and the laboratory methods used. The ranges used at Massachusetts General Hospital are for adults who are not pregnant and do not have medical conditions that could affect the results. They may therefore not be appropriate for all patients.
Five years later, during a routine dental evaluation, swelling was noted in the area of the second bicuspid and the first molar on the bottom right (29 and 30, respectively, according to the universal numbering system). Radiographs reportedly showed a slight alteration in the trabecular pattern of the right mandible. Computed tomography (CT) of the head revealed a destructive lesion (1.2 cm in diameter) in the right mandibular body, with marginal trabecular preservation, centered between the second premolar (i.e., the second bicuspid) and the first molar on the right side. The mass was excised, and pathological examination of the specimen reportedly showed an atypical proliferation of mixed epithelial and mesenchymal cells, including fascicles of spindled cells and reactive fragments of trabecular bone; there was insufficient nuclear atypia or mitotic activity to warrant a diagnosis of a malignant tumor. Tumor was focally present at the surgical margin. After the excision, the serum phosphorus level returned to normal and supplementation with phosphate and vitamin D was stopped.
Nine years before this evaluation, back and rib pain, stress fractures, and hypophosphatemia recurred, and phosphate supplementation was resumed. Octreotide scanning (scintigraphy using octreotide labeled with indium-111) was reportedly positive for radionuclide uptake in the right mandible, a feature consistent with persistent or recurrent tumor. During the next 8 years, the patient was seen at several other hospitals. Test results are shown in Table 1. Multiple surgical procedures were performed, including mandibular biopsies and resections and the insertion of a fibular implant, as well as dissection of a cervical lymph node on the right side; pathological examination of the specimens showed a spindle-cell lesion infiltrating bone and soft tissue. Hypophosphatemia and the need for treatment with phosphate and calcitriol persisted.
Five years before this evaluation, the right mandible and the fibular bone implant were resected, and a second bone-replacement graft was inserted. Pathological examination showed reactive changes and a foreign-body–type giant-cell reaction, without the spindle-cell proliferation seen in previous biopsy specimens. Test results are shown in Table 1. Two years later, dental-implant surgery and supplementary bone grafting were performed. Three years later, the patient was referred to this hospital for evaluation and management of persistent hypophosphatemia.
The patient reported bilateral hip pain when walking, fatigue, muscle weakness, intermittent wheezing with exercise, and mild chronic diarrhea that was attributed to phosphate supplements. He had no fever, sweats, weight loss, chest or abdominal pain, new intestinal symptoms, or hematuria. His stools were brown, without visible fat. He had had cutaneous mastocytosis for 30 years, asymptomatic nephrolithiasis noted on CT in the past 5 years, and a tonsillectomy as a child. Medications included calcitriol and a combination of potassium phosphate and sodium phosphate. He had no known allergies. He was married, lived with his wife and children, and worked in journalism. He had traveled to Europe, he drank alcohol in moderation, and he did not smoke or use illicit drugs. His parents were deceased; his mother had had lung cancer, his father had had prostate cancer, a brother had melanoma, and a sister was healthy.
On examination, the blood pressure was 152/84 mm Hg, the pulse 85 beats per minute, the weight 77.1 kg, and the height 170.2 cm. There were brown, infiltrative lesions (5 to 10 mm in diameter) over the trunk and back, consistent with mastocytosis, and a mass on the back of the neck, consistent with a lipoma. The oropharynx was normal. There was enlargement of the lingual mandible anteriorly and on the right, with no dental dysplasia or dysplasia of the palate or jaw bone. There was surgical scarring and fibrosis over the mandible and neck on the right side. The remainder of the examination was normal. Panoramic occlusal radiographs of the teeth and mandible showed a radiolucency associated with the right anterior mandibular implant and a wire in the lingual cortex. The complete blood count and serum levels of electrolytes, total protein, albumin, globulin, magnesium, glucose, and tryptase were normal, as were tests of coagulation and renal and liver function; other test results are shown in Table 1. CT of the hips showed no soft-tissue calcifications and no evidence of stress fractures.
Eight weeks later, samples obtained from multiple sites of venous catheterization revealed elevated levels of fibroblast growth factor 23 (FGF23) at all sites tested, but without a clinically significant gradient in the FGF23 levels in the veins of the neck or extremities. Single-photon-emission CT (SPECT) imaging of the head, chest, and abdomen after the administration of indium-111–labeled pentetreotide revealed high uptake in the lower face that was thought to be in the mandible or reconstructed bone, and subtle pentetreotide localization in the upper abdomen that was thought to be nonspecific.
Additional diagnostic tests were performed.
DIFFERENTIAL DIAGNOSIS
Dr. Clemens Bergwitz
I am aware of the diagnosis in this case. This patient presented initially with stress fractures in the metatarsals and pain in the ribs. Although most stress fractures are of the fatigue type, occurring in persons who engage in repetitive vigorous activity to which they are unaccustomed,1 the presence of rib pain in this patient suggests an underlying disorder affecting the entire skeleton. Bone films of his forefoot showed Looser zones. The affected sites — the ribs, tibial plateau, and metatarsal bones — are typical for osteomalacia (a disorder of bone mineralization that often leads to incomplete or complete fractures) and not typical for osteoporotic fractures, which usually occur in the femoral neck, vertebral bodies, or wrists.
The most common cause of osteomalacia is vitamin D deficiency, which results in secondary hyperparathyroidism. In view of the patient’s history of nephrolithiasis, primary hyperparathyroidism also needs to be considered. However, the serum levels of parathyroid hormone, calcium, and 25-hydroxyvitamin D were normal. It is very important at this point to check for the presence of hypophosphatemia; this is often overlooked, since electrolyte panels do not routinely include a serum phosphorus level. The patient’s serum phosphorus levels were extremely low, suggesting hypophosphatemic osteomalacia as the underlying cause of his bone pain and stress fractures.
HYPOPHOSPHATEMIA
What could cause this patient’s hypophosphatemia (Table 2)? Phosphate is absorbed from the diet in the gut, stored in the skeleton, and excreted by the kidneys (Fig. 1). Phosphate, unlike calcium, is plentiful in our diet and readily absorbed. A common cause of hypophosphatemia is malnutrition. Catabolism may maintain normal serum phosphorus levels during starvation, but it often decreases markedly during refeeding, because total body phosphate is depleted and glucose and insulin drive phosphate into cells. Nothing in this patient’s history suggests phosphate malnutrition.
Table 2.
Causes of Hypophosphatemia.*
| Extrarenal causes |
| Poor intestinal uptake of phosphate (caused by vitamin D deficiency, intestinal phosphate binders, or niacin) |
| Increased sequestration into cells (caused by insulin therapy or proliferative malignant conditions such as leukemia) or bone matrix (caused by bisphosphonates) |
| Renal causes |
| PTH-dependent causes (e.g., hyperparathyroidism) |
| FGF23-dependent causes (e.g., tumor-induced osteomalacia or X-linked, autosomal dominant or autosomal recessive forms of hypophosphatemia) |
| PTH-independent and FGF23-independent causes (e.g., alcoholism, drugs or toxins, renal tubular acidosis, Fanconi’s syndrome, or hereditary hypophosphatemic rickets with hypercalciuria) |
FGF23 denotes fibroblast growth factor 23, and PTH parathyroid hormone.
Figure 1. Regulation of Phosphate Homeostasis by Fibroblast Growth Factor 23, Parathyroid Hormone, and 1,25-Dihydroxyvitamin D.
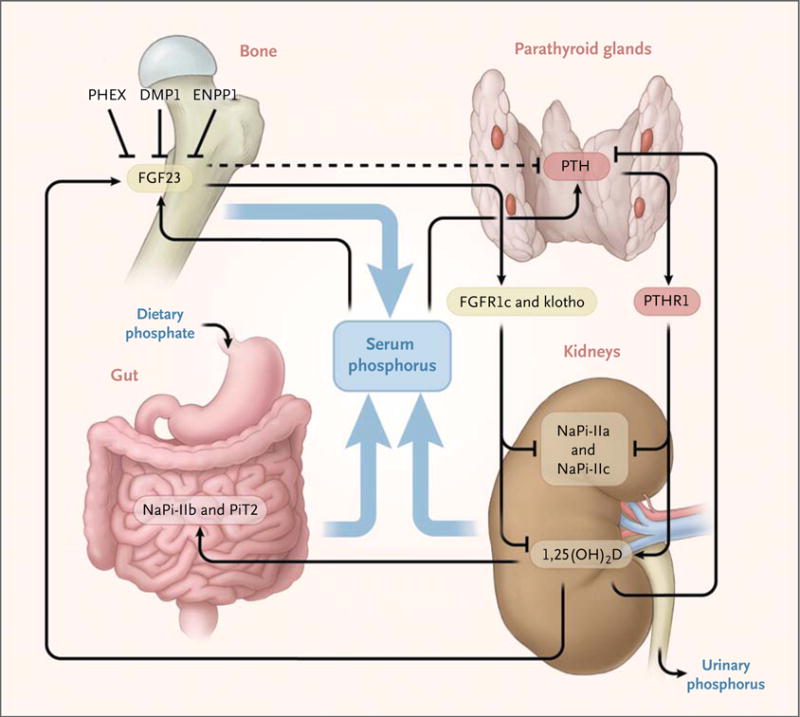
Fibroblast growth factor 23 (FGF23) expression in bone is up-regulated by an increase in dietary phosphate (Pi) intake and by 1,25(OH)2D (1,25-dihydroxyvitamin D), and it is down-regulated, through unknown mechanisms, by PHEX (encoded by PHEX, or phosphate-regulating gene with homologies to endopeptidases on the X chromosome), DMP1 (dentin matrix protein 1), ENPP1 (ectonucleotide pyrophosphatase–phosphodiesterase 1), and probably several additional proteins. FGF23 acts through one or more FGF receptors (e.g., FGFR1c), with the coreceptor klotho, to inhibit renal phosphate reabsorption, to decrease circulating 1,25(OH)2D levels, and possibly to inhibit parathyroid hormone (PTH) secretion by the parathyroid glands (dashed line). The net effect of these PTH-dependent actions is a decrease in serum phosphate levels and an increase in serum 1,25(OH)2D levels. The 1,25(OH)2D levels are probably linked to regulation of FGF23 by phosphate2 but might also be regulated directly by the effects of serum phosphorus levels on 1α-hydroxylase or 24-hydroxylase or both.3 The net effect of hypophosphatemia is up-regulation of 1,25(OH)2D; 1,25(OH)2D is down-regulated by increased serum levels of calcium and phosphorus and by increased FGF23 levels. Vitamin D–receptor and retinoid X–receptor heterodimers facilitate the action of 1,25(OH)2D to enhance the intestinal absorption of phosphate and to stimulate FGF23 synthesis and secretion by osteocytes; furthermore, 1,25(OH)2D inhibits PTH synthesis and secretion by the parathyroid glands. The net 1,25(OH)2D effect is an increase in serum phosphorus levels. NaPi denotes sodium–phosphate cotransporter, PiT2 sodium-dependent phosphate transporter 2, and PTHR1 parathyroid hormone receptor type 1.
The reabsorption of phosphate from the urine is tightly regulated in the renal proximal tubules, which are key in maintaining normal phosphate homeostasis. Therefore, we needed to assess the patient’s tubular reabsorption of phosphate to determine whether the cause of his abnormal serum phosphorus level was renal or extrarenal. The clinical assessment of phosphate homeostasis can be challenging, since serum phosphorus levels are influenced by the time of day, relationship to meals, and age of the patient, and none of the methods for determination of tubular reabsorption are entirely satisfying. We generally ask the patient to collect a 3-hour, timed urine sample for analysis of phosphorus and creatinine levels, along with the corresponding serum phosphorus and creatinine levels after an 8-hour fast. The percentage of filtered phosphate that is reabsorbed by the renal tubules is then calculated by subtracting the fractional excretion of phosphorus from 1, according to the following formula: percentage reabsorption = 100 × [1 − (urine phosphorus level × serum creatinine level) ÷ (serum phosphorus level × urine creatinine level)].
This patient’s tubular reabsorption of phosphate was less than 80% on multiple occasions, which establishes a defect in the proximal renal tubules as the underlying cause of his hypophosphatemia. In contrast, in situations involving hypophosphatemia caused by refeeding, we would expect to see tubular reabsorption of nearly 100%, since the cause of hypophosphatemia is extrarenal.
CAUSES OF RENAL PHOSPHORUS WASTING
Causes of generalized dysfunction of the proximal tubules, such as renal Fanconi’s syndrome, can be ruled out by checking for an absence of glucosuria or aminoaciduria; both were absent in this case. Common causes of isolated renal phosphate wasting, which leads to hypophosphatemia, include hyperparathyroidism and renal tubular acidosis, but the patient’s parathyroid function was normal, and the serum bicarbonate level, which would be reduced in renal tubular acidosis, was normal. He had not been taking medications that can cause hypophosphatemia, such as insulin or bisphosphonates. Excess alcohol or certain medications that enhance the action of parathyroid hormone at the proximal tubule (e.g., acetazolamide or theophylline) or chemotherapeutic agents that block renal phosphate transporters (e.g., forscarnet or ifosfamide) can lead to renal phosphate losses.4 In childhood, hypophosphatemia causes rickets, which leads to bowing of the legs and short stature. Since this patient had normal stature and no bowing of the long bones, we concluded that his hypophosphatemia began after growth was completed, which eliminates most inherited forms of renal phosphate wasting.
FGF23-DEPENDENT HYPOPHOSPHATEMIA
In the past few years, the hormone FGF23 has been shown to be an essential regulator of phosphate homeostasis (Fig. 1).5,6 FGF23 binds to fibroblast growth factor receptor 1c (FGFR1c) and its coreceptor klotho,7,8 and activation of this receptor complex inhibits the reabsorption of phosphate by reducing expression of the sodium–phosphate cotransporters NaPi-IIa and NaPi-IIc in the proximal tubule.9 FGF23 also decreases the synthesis of 1,25(OH)2D (1,25-dihydroxyvitamin D) in the renal proximal tubules.10,11 These actions of FGF23 serve to lower blood phosphate levels. Hyperphosphatemia leads to an increase in the secretion of FGF23 by osteocytes, and hypophosphatemia leads to suppression of it. This pattern explains the long-standing observation that hypophosphatemia is normally associated with an increase in 1,25(OH)2D levels.
In this patient with hypophosphatemia, the 1,25(OH)2D level should be elevated because of the hypophosphatemia-driven lowering of FGF23 levels. However, after correcting for vitamin D deficiency and secondary hyperparathyroidism, which sometimes can elevate 1,25(OH)2D levels independent of FGF23 action, we found normal 1,25(OH)2D levels in this patient. This finding suggests an FGF23-dependent cause of his renal phosphate leak. A consequence of increased production of 1,25(OH)2D may be increased absorption of calcium in the gut, resulting in hypercalciuria. Therefore, low excretion of urinary calcium in this patient further supported an FGF23-dependent cause of the renal phosphate leak.
The differential diagnosis of FGF23-dependent hypophosphatemia includes a handful of rare inherited conditions: X-linked hypophosphatemia, autosomal dominant hypophosphatemia, and autosomal recessive hypophosphatemia. Although the onset of some inherited hypophosphatemias, such as autosomal recessive hypophosphatemia, may occur as late as 45 years of age,12 most develop in childhood, adolescence, or young adulthood and are unlikely in this patient, who had no family history of hypophosphatemia and was 38 years of age when symptoms initially developed. At the time of his initial diagnosis, therefore, tumor-induced osteomalacia due to an FGF23-producing tumor was strongly suspected, but no tumor could be found. Several years later, a tumor of the jaw became apparent, and resection led to clinical improvement. Symptoms of hypophosphatemia recurred, however, and multiple local recurrences of the tumor were resected. At the time of the current evaluation, when the patient was 56 years of age, he had severe hypophosphatemia.
Within the past decade, FGF23 assays have become widely available.13 This patient’s FGF23 level rose from 70 reference units (RU) per milliliter (normal, <180) 5 years before our first evaluation to more than 800 RU per milliliter at the time of this evaluation, which confirmed our clinical diagnosis of hypophosphatemic osteomalacia due to FGF23-dependent renal phosphate wasting.
The question was, What was the source of the excess FGF23? Was it the jaw, which on the last evaluation had shown no evidence of tumor? Or had the tumor metastasized? Which imaging studies could help distinguish between local mandibular recurrence and distant metastasis? And finally, how could we manage the patient’s recurrent hypophosphatemia?
Dr. Kamath, may we see the imaging studies?
Dr. Ravi S. Kamath
A panoramic radiograph of the jaws (Fig. 1 in the Supplementary Appendix, available with the full text of this article at NEJM.org) showed postsurgical changes and no evidence of recurrent tumor.
Selective venous sampling was performed at multiple sites in the systemic circulatory system with the use of a transjugular approach and in the portal venous circulatory system with the use of a transportal approach (see Fig. 2 in the Supplementary Appendix). All the samples revealed diffusely elevated levels of FGF23, with no significant difference at any particular anatomical location, including in the venous outflows of the neck or extremities, which might have helped localize the source of the FGF23.
Whole-body imaging and SPECT imaging of the head, chest, and abdomen were performed after the administration of indium-111–labeled pentetreotide (see Fig. 3 in the Supplementary Appendix), which revealed high uptake in the lower face, thought to be in the mandible or in the reconstructed bone. An additional focus of uptake was identified in the right upper abdomen, thought to be in the region of the duodenum or pancreatic head. However, previous and subsequent imaging, including CT and magnetic resonance imaging (MRI), failed to identify a correlate for this abnormal uptake.
CT scans of the chest obtained after the administration of intravenous contrast material (Fig. 2) showed multiple bilateral pulmonary nodules (≤9 mm in diameter), small mediastinal and axillary lymph nodes, nodules in the periscapular subcutaneous fat bilaterally (≤6 mm in diameter), and diffuse sclerosis of the thoracic skeleton (including small lytic lesions in multiple vertebral bodies, the sternum, and the right lateral sixth rib). A CT scan of the abdomen and pelvis showed bilateral punctate, nonobstructing renal calculi and an 8-mm lytic lesion in the fourth lumbar vertebral body, which was unchanged from a previous study and was thought to be benign.
Figure 2. CT Images.
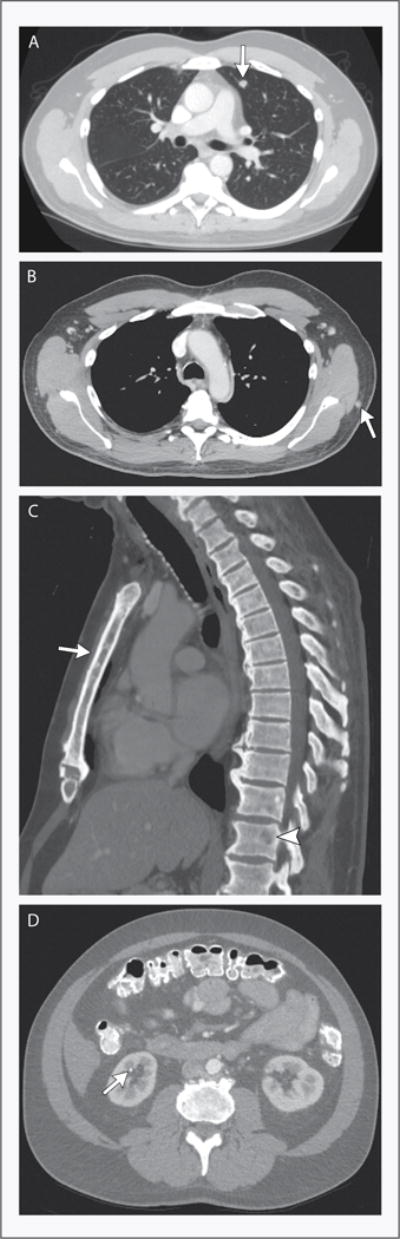
A CT scan of the chest obtained after the administration of intravenous contrast material shows multiple pulmonary nodules. The largest is a 9-mm nodule in the lingula (Panel A, arrow). Panel B shows a soft-tissue nodule in the periscapular regions (arrow), and Panel C shows diffuse bony sclerosis with numerous lytic lesions, including lesions in the sternum (arrow) and vertebral bodies (arrowhead). A CT scan of the abdomen and pelvis obtained after the administration of intravenous and oral contrast material shows punctate, nonobstructing renal calculi (Panel D, arrow).
Dr. Hesse
We asked Dr. John Wain (Surgery) to perform a video-assisted thoracoscopic biopsy of one of the pulmonary nodules.
CLINICAL DIAGNOSIS
Recurrent tumor-induced osteomalacia due to metastatic tumor.
PATHOLOGICAL DISCUSSION
Dr. Andrew E. Rosenberg
Curettage of the right mandible, performed 13 years earlier, yielded fragments of tumor and pieces of cortical and trabecular bone (Fig. 3A). The infiltrative tumor filled the medullary cavity and encompassed preexisting cortical and cancellous bone. The neoplasm was composed of aggregates of epithelium that were ameloblastic in appearance (Fig. 3B) and that were surrounded by fascicles of spindle cells (Fig. 3C). Both epithelial and spindle-cell components had minimal cytologic atypia, few mitotic figures, and no necrosis. Admixed with the spindle cells was hemorrhage, scattered osteoclast-like giant cells, and an extracellular matrix with features suggestive of bone. The large fragments of preexisting bone contained prominent seams of unmineralized bone, which is the histologic correlate to the clinical and radiographic findings of osteomalacia (Fig. 3D).
Figure 3. Pathological Findings (Hematoxylin and Eosin).

The initial biopsy specimen from the right mandible shows an admixture of epithelial and spindle-cell components of tumor infiltrating bone (Panel A), a nest of neoplastic epithelial cells with an odontogenic appearance (Panel B), and intersecting fascicles of spindle cells (Panel C). Panel D shows osteomalacic bone with thickened light-pink osteoid seams (arrows). Panel E shows a recurrent mandibular spindle-cell tumor containing irregularly mineralized woven bone. The metastasis in the lung is composed entirely of the spindle-cell component (Panel F).
The recurrent right mandibular tumors resected 8 and 6 years before this evaluation were similar morphologically to the original tumor. However, in those specimens of recurrent tumors, there was a diminution in the quantity of the epithelial component, such that 6 years before this evaluation, the tumor was composed solely of the neoplastic spindle cells. The tumor focally destroyed the cortex and extended into the soft tissues. In the second recurrence, an extracellular bonelike matrix that was focally mineralized was present (Fig. 3E).
The pulmonary lesion resected during the patient’s evaluation at this hospital was composed of fascicles of spindle cells that were identical morphologically to those present in the primary and recurrent mandibular tumors (Fig. 3F). Also present was a focally mineralized bonelike matrix.
This neoplasm metastasized after a clinical course of 19 years, indicating that biologically it is a low-grade malignant tumor. The morphologic features are most consistent with an odontogenic tumor known as ameloblastic fibrosarcoma, in which the epithelial component resembles ameloblastic epithelium and the spindle-cell element is similar to that present in low-grade fibrosarcoma.14,15
Oncogenic osteomalacia has been associated with many types of carcinomas, hematologic cancers, and mesenchymal tumors. The vast majority of the mesenchymal tumors are benign, and more than 75% are known as phosphaturic mesenchymal tumor of the mixed connective-tissue type.16 This term indicates that the neoplasm is composed of mesenchymal elements that have various lines of differentiation, all of which are considered to be types of connective tissue, including the production of bonelike matrix. Odontogenic tumors, such as the one in this patient, have only occasionally been associated with oncogenic osteomalacia.17
Dr. Hesse
In the operating room, the specimen was rinsed in saline, and the fluid was tested for FGF23. We found high levels of FGF23, which, in the absence of albumin, was most likely derived from the tumor and was probably not due to contamination with peripheral blood. Dr. Collins will discuss the further care of this patient.
DISCUSSION OF MANAGEMENT
Dr. Michael T. Collins
Tumor-induced osteomalacia, also known as oncogenic osteomalacia, is a rare paraneoplastic syndrome that results from the production of the phosphaturic factor FGF23, typically from small, benign mesenchymal tumors. The clinical findings at presentation and at the recurrences in this case are typical — years of diffuse musculoskeletal pain, pathological fractures, hypophosphatemia due to renal phosphate wasting, low-normal or low serum 1,25(OH)2D3, and elevated FGF23 levels in the blood. This patient presented with recurrent hypophosphatemia after what appeared to be a successful excision of a phosphaturic mesenchymal tumor of the mandible. One of the most difficult features of the management of osteogenic osteomalacia can be localizing the offending tumor. The general approach to localization involves functional imaging (combined 18F-fluorodeoxyglucose–positron-emission tomography and CT18 or indium-111–labeled pentetreotide19 or both20), followed by anatomical imaging (CT or MRI or both) of suspicious areas that have been identified on functional imaging, as was done in this case. Selective venous sampling, also performed in this case, may be an important tool in cases in which more certainty is needed.21 In our experience, selective venous sampling has proved useful only in cases in which it is necessary to discriminate between multiple suspected sites on functional imaging, or when greater certainty is needed to show that the suspected lesion is the FGF23-secreting tumor.22 As was the case here, we have not had success with venous-sampling studies in which a suspicious lesion was not identified on imaging studies.
Wide excision of the primary lesion is important, as illustrated by this case. The cellular features are almost always benign,5 but if wide excisions are not performed, tumor margins may be positive, as they were in this case, and positive tumor margins may lead to persistent or recurrent disease. Radiofrequency ablation has been advocated as a treatment option,23 but caution should be exercised to ablate considerably beyond the radiographic margin to prevent recurrence.
Medical treatment of tumor-induced osteomalacia consists of phosphate and calcitriol supplementation, as was administered in this case. Unfortunately, oral phosphate supplementation is inconvenient and poorly tolerated; typically it is taken four times per day and, at the doses needed to heal bone, often causes diarrhea, as occurred in this patient. Phosphate supplementation is adjusted to keep the serum phosphorus level near the lower limit of the normal range. Because of the combined effect of repeat phosphate dosing and the 1,25(OH)2D3-lowering effect of FGF23, secondary hyperparathyroidism often develops; the calcitriol dose is adjusted to keep the serum parathyroid hormone level within the normal range. The dosing regimens for phosphate and calcitriol are 15 to 60 mg of elemental phosphate per kilogram of body weight per day, divided into four or five doses, and 15 to 60 ng of calcitriol per kilogram per day, divided into two doses. The most concentrated and best-tolerated sources of phosphorus are liquid preparations of sodium biphosphate–sodium phosphate, which contain approximately 128 mg of elemental phosphate per milliliter. Medical treatment can result in hypercalciuria, nephrocalcinosis, and nephrolithiasis. Therefore, urine calcium levels should be monitored.
A potentially useful adjunct to conventional therapy is the medicinal induction of hypoparathyroidism by cinacalcet, a calcium-sensing receptor agonist.24 The physiological basis for this therapy is the finding that the full phosphaturic effect of FGF23 is a parathyroid hormone–dependent process.25 As with conventional therapy, hypercalciuria can occur; therefore, it is important to monitor urine calcium levels. The administration of cinacalcet is a reasonable option for this patient.
A pressing issue for this patient is the treatment of the metastatic disease. Folpe et al.16 have reported on the only four known cases of malignant transformation of phosphaturic mesenchymal tumors. In addition, we have encountered one case with multiple miliary pulmonary metastases. Also, there is a report of a patient who survived for 30 years with pulmonary metastases.26 The histologic features of this patient’s tumor appear somewhat different from the typical phosphaturic mesenchymal tumor associated with this syndrome, further complicating his prognosis.
FOLLOW-UP
Dr. Hesse
We obtained another CT scan of the chest 8 months after the first one, and the pulmonary metastases were unchanged. Since the patient’s parathyroid hormone level was in the high-normal range, we added cinacalcet to his regimen at a dose of 30 mg twice a day. We continued the administration of calcitriol at 0.5 μg twice a day and phosphate at 250 mg four times a day. With the patient on this regimen, the calcium level dropped slightly, the serum phosphorus level rose to 1.5 to 2.1 mg per deciliter, and the bone pain resolved. The urinary calcium excretion was 200 mg per gram of creatinine, in an acceptable range, which is important to monitor because of the risk of kidney-stone formation and nephrocalcinosis. The patient improved clinically and was able to return to work.
Dr. Bergwitz
Dr. Edwin Choy (Medical Oncology) followed up with the patient, but no cytotoxic treatment for the metastatic sarcoma was required at the time, since repeat imaging studies showed no change in the lung nodules. Eighteen months after the patient’s evaluation at this hospital, tumor recurred in the mandible, confirmed by biopsy. The FGF23 level was 2010 RU per milliliter. Recently, the tumor was resected at another facility; reportedly, the level fell to 755 RU per milliliter immediately after the surgery. The patient continues to take calcitriol and oral phosphate supplements at unchanged doses, but he has stopped taking cinacalcet.
Dr. Nancy Lee Harris (Pathology)
Are there questions or comments for any of our speakers?
A Physician
Was immunohistochemical analysis for FGF23 performed on the lung-biopsy specimen?
Dr. Rosenberg
There are no good commercially available antibodies currently, so that was not performed.
Dr. John T. Potts, Jr. (Endocrine Unit)
Antibodies against FGF23 are being developed for use in humans to treat X-linked hypophosphatemia. Could FGF23 antibodies be considered as therapy in the present case?
Dr. Collins
Antibody treatment has been reported to be effective in the animal model for X-linked hypophosphatemia.27 There is an ongoing trial involving persons with X-linked hypophosphatemia (ClinicalTrials.gov numbers, NCT00830674 and NCT01340482), and the hope is that antibody treatment will be a viable option in the future for patients such as this one.
A Physician
This patient has two rare problems, the FGF23-producing tumor and cutaneous mastocytosis. Would you comment on whether this could be systemic mastocytosis and whether mast cells were involved in his syndrome?
Dr. Bergwitz
The mastocytosis preceded the diagnosis of osteomalacia, but since hypophosphatemic osteomalacia can sometimes take a few years to become symptomatic, it is possible that the two syndromes developed at the same time. In this patient, we looked for evidence of systemic mastocytosis; a bone marrow–biopsy specimen was normal, as were serum levels of total and mature tryptase. We have found no reports in the literature of tumor-induced osteomalacia occurring in either cutaneous or systemic mastocytosis. Finally, the cutaneous mast-cell lesions were stable over a period of two decades, whereas the FGF23 levels increased by an order of magnitude.
ANATOMICAL DIAGNOSIS
Tumor-induced osteomalacia due to metastatic ameloblastic fibrosarcoma.
Supplementary Material
Acknowledgments
We thank Dr. Lloyd Axelrod for assistance in organizing the conference and for reviewing an earlier draft of the manuscript.
Footnotes
Discussed at the Medicine Grand Rounds.
Dr. Rosenberg reports receiving consulting fees from Genzyme. No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Daffner RH, Pavlov H. Stress fractures: current concepts. AJR Am J Roentgenol. 1992;159:245–52. doi: 10.2214/ajr.159.2.1632335. [DOI] [PubMed] [Google Scholar]
- 2.Stubbs J, Liu S, Quarles LD. Role of fibroblast growth factor 23 in phosphate homeostasis and pathogenesis of disordered mineral metabolism in chronic kidney disease. Semin Dial. 2007;20:302–8. doi: 10.1111/j.1525-139X.2007.00308.x. [DOI] [PubMed] [Google Scholar]
- 3.Segawa H, Onitsuka A, Kuwahata M, et al. Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol. 2009;20:104–13. doi: 10.1681/ASN.2008020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bringhurst FR, Leder BZ. Regulation of calcium and phosphate homeostasis. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 5th. Philadelphia: W.B. Saunders; 2006. pp. 805–43. [Google Scholar]
- 5.Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. doi: 10.1146/annurev.med.051308.111339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–5. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–41. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 8.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 9.Yan X, Yokote H, Jing X, et al. Fibroblast growth factor 23 reduces expression of type IIa Na+/Pi co-transporter by signaling through a receptor functionally distinct from the known FGFRs in opossum kidney cells. Genes Cells. 2005;10:489–502. doi: 10.1111/j.1365-2443.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- 10.Perwad F, Zhang MY, Tenenhouse HS, Portale AA. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol. 2007;293:F1577–F1583. doi: 10.1152/ajprenal.00463.2006. [DOI] [PubMed] [Google Scholar]
- 11.Razzaque MS. Klotho and Na+,K+-ATPase activity: solving the calcium metabolism dilemma? Nephrol Dial Transplant. 2008;23:459–61. doi: 10.1093/ndt/gfm702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674–81. doi: 10.1210/jcem.82.2.3765. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 14.Dallera P, Bertoni F, Marchetti C, Bacchini P, Campobassi A. Ameloblastic fibrosarcoma of the jaw: report of five cases. J Craniomaxillofac Surg. 1994;22:349–54. doi: 10.1016/s1010-5182(05)80116-7. [DOI] [PubMed] [Google Scholar]
- 15.Muller S, Parker DC, Kapadia SB, Budnick SD, Barnes EL. Ameloblastic fibrosarcoma of the jaws: a clinicopathologic and DNA analysis of five cases and review of the literature with discussion of its relationship to ameloblastic fibroma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;79:469–77. doi: 10.1016/s1079-2104(05)80130-1. [DOI] [PubMed] [Google Scholar]
- 16.Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30. doi: 10.1097/00000478-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Jefferis AF, Taylor PC, Walsh-Waring GP. Tumour-associated hypophosphataemic osteomalacia occurring in a patient with an odontogenic tumour of the maxilla. J Laryngol Otol. 1985;99:1011–7. doi: 10.1017/s0022215100098091. [DOI] [PubMed] [Google Scholar]
- 18.Roarke MC, Nguyen BD. PET/CT localization of phosphaturic mesenchymal neoplasm causing tumor-induced osteomalacia. Clin Nucl Med. 2007;32:300–1. doi: 10.1097/01.rlu.0000257180.03964.51. [DOI] [PubMed] [Google Scholar]
- 19.Jan de Beur SM, Streeten EA, Civelek AC, et al. Localisation of mesenchymal tumours by somatostatin receptor imaging. Lancet. 2002;359:761–3. doi: 10.1016/s0140-6736(02)07846-7. [DOI] [PubMed] [Google Scholar]
- 20.Hesse E, Moessinger E, Rosenthal H, et al. Oncogenic osteomalacia: exact tumor localization by co-registration of positron emission and computed tomography. J Bone Miner Res. 2007;22:158–62. doi: 10.1359/jbmr.060909. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi Y, Suzuki H, Ogura S, et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab. 2004;89:3979–82. doi: 10.1210/jc.2004-0406. [DOI] [PubMed] [Google Scholar]
- 22.Andreopoulou P, Dumitrescu CE, Kelly MH, et al. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. J Bone Miner Res. 2011;26:1295–302. doi: 10.1002/jbmr.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hesse E, Rosenthal H, Bastian L. Radiofrequency ablation of a tumor causing oncogenic osteomalacia. N Engl J Med. 2007;357:422–4. doi: 10.1056/NEJMc070347. [DOI] [PubMed] [Google Scholar]
- 24.Geller JL, Khosravi A, Kelly MH, Riminucci M, Adams JS, Collins MT. Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res. 2007;22:931–7. doi: 10.1359/jbmr.070304. [DOI] [PubMed] [Google Scholar]
- 25.Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT. FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–92. doi: 10.1210/jc.2004-0724. [DOI] [PubMed] [Google Scholar]
- 26.Harvey JN, Gray C, Belchetz PE. Oncogenous osteomalacia and malignancy. Clin Endocrinol (Oxf) 1992;37:379–82. doi: 10.1111/j.1365-2265.1992.tb02342.x. [DOI] [PubMed] [Google Scholar]
- 27.Aono Y, Yamazaki Y, Yasutake J, et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879–88. doi: 10.1359/jbmr.090509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.