

Abstract
Motivation: Deep sequencing of the transcripts of regulatory non-coding RNA generates footprints of post-transcriptional processes. After obtaining sequence reads, the short reads are mapped to a reference genome, and specific mapping patterns can be detected called read mapping profiles, which are distinct from random non-functional degradation patterns. These patterns reflect the maturation processes that lead to the production of shorter RNA sequences. Recent next-generation sequencing studies have revealed not only the typical maturation process of miRNAs but also the various processing mechanisms of small RNAs derived from tRNAs and snoRNAs.
Results: We developed an algorithm termed SHARAKU to align two read mapping profiles of next-generation sequencing outputs for non-coding RNAs. In contrast with previous work, SHARAKU incorporates the primary and secondary sequence structures into an alignment of read mapping profiles to allow for the detection of common processing patterns. Using a benchmark simulated dataset, SHARAKU exhibited superior performance to previous methods for correctly clustering the read mapping profiles with respect to 5′-end processing and 3′-end processing from degradation patterns and in detecting similar processing patterns in deriving the shorter RNAs. Further, using experimental data of small RNA sequencing for the common marmoset brain, SHARAKU succeeded in identifying the significant clusters of read mapping profiles for similar processing patterns of small derived RNA families expressed in the brain.
Availability and Implementation: The source code of our program SHARAKU is available at http://www.dna.bio.keio.ac.jp/sharaku/, and the simulated dataset used in this work is available at the same link. Accession code: The sequence data from the whole RNA transcripts in the hippocampus of the left brain used in this work is available from the DNA DataBank of Japan (DDBJ) Sequence Read Archive (DRA) under the accession number DRA004502.
Contact: yasu@bio.keio.ac.jp
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Since high-throughput sequencing allows for deep sequencing with high sensitivity, RNA sequencing with a next-generation sequencer (RNA-seq) can detect not only the complete expression patterns of transcribed RNAs but also fragments derived due to the splicing, maturation processing, or non-functional degradation of the RNAs. RNA-seq studies targeting microRNAs (miRNAs) have revealed the existence of many different RNA fragments derived from small RNA species other than microRNA, providing further proof that derived RNA fragments are not simply random degradation products but are rather stable entities, which might have functional activity (Martens-Uzunova et al., 2013). The evidence accumulating about shorter sequences or fragments derived from non-coding RNAs indicates that post-transcriptional processes are relatively common mechanisms to derive functional smaller molecules from various RNA families such as tRNAs and snoRNAs. For example, the so-called tRNA-derived RNA fragments (tRFs) are derived from processing at the 5′ or 3′-end of mature or precursor tRNAs (Lee et al., 2009). These sequences constitute a class of short RNAs that are the second most abundant type of RNA after miRNAs. Since the discovery of small RNAs derived from tRNAs, a variety of names have been used to refer to similar entities, such as tRNA-derived RNA fragments (tRFs) (Lee et al., 2009), stress-induced small RNAs (tiRNAs) (Yamasaki et al., 2009) and tRNA-derived small RNAs (tsRNAs) (Haussecker et al., 2010). Their uniform start and stop sites of cleavage in tRNA, together with their nonrandom size property, strongly suggest that tRFs are derived from tRNA cleavage in a specific and regulated manner (Lee et al., 2009). In addition, several studies have identified a class of small fragments derived from snoRNAs (called sdRNAs), some of which may regulate splicing or translation (Chen and Heard, 2013; Taft et al., 2009).
The biological roles of such sno-derived RNA and tRNA-derived small RNAs have primarily been investigated in cancer cells (Martens-Uzunova et al., 2013). tRNA-derived fragments participate in several types of biological processes, including as signal molecules in a stress-induced response and as regulators of gene expression. sdRNAs can function as miRNAs, regulators of alternative splicing, and tumor suppressors and oncogenes.
The reads for such short derived RNAs are relatively abundant, i.e. greater than background levels, in small RNA-seq datasets. Chen and Heard (2013) pointed out that ‘these reads are precisely mapped to specific regions of primary or secondary structures, and might contain special motifs or base biases, reflecting the involvement of special enzymes involved in their generation’. Hence, mapping a large amount of sequence reads onto a reference sequence can reveal specific forms of mapping patterns for the maturation process or random patterns for non-functional degradations (see Fig. 1(a)). These mapping patterns of RNA-seq short reads constitute the so-called read mapping profiles (see Fig. 1(b)) (Pundhir et al., 2015). Chen and Heard (2013) also pointed out that the non-specific degradation products include RNA fragments that are rapidly digested by the surveillance machinery from RNA molecules, which are defective in processing, folding and functions. Thus, it is crucial to reliably distinguish the true shorter RNAs from their non-functional degradation products to clearly identify derived and functional small RNAs and fragments. Therefore, the aim of the present study was to develop a computational tool for the comprehensive analysis and rapid identification of the post-transcriptional processing patterns of non-coding RNAs based on high-throughput RNA-seq data. The algorithm developed in this study was designed to capture specific forms of read mapping patterns mapped to specific regions of primary or secondary structures reflecting the functional activities of enzymes, and to distinguish them from non-functional degradation products.
Fig. 1.
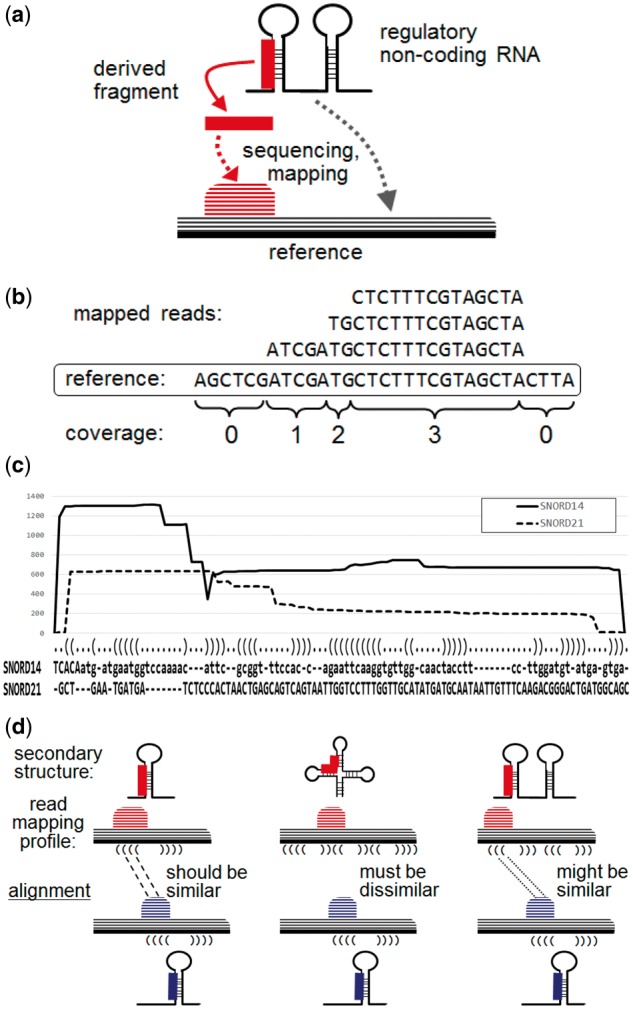
(a) Schematic illustration of a derived RNA fragment and the mapping pattern obtained from sequencing. (b) Read mapping profile and calculation of coverage. (c) An alignment of two read mapping profiles for SNORD14 and SNORD21 output by SHARAKU, using the small RNA-seq data for the common marmoset brain with the annotations of RNA sequence alignment and the predicted secondary structure. The solid line represents the read coverages of SNORD14, and the dashed line represents the read coverages of SNORD21. (d) Schematic illustration of the necessity of incorporating the primary and secondary structures of RNA sequences into an alignment
There are only a few existing tools (Erhard and Zimmer, 2010; Hoogstrate et al., 2015; Langenberger et al., 2012; Videm et al., 2014) to analyze such read mapping profiles of RNA-seq data and detect footprints of the post-transcriptional processes or degradations of RNAs. FlaiMapper (Hoogstrate et al., 2015) predicts and annotates non-coding RNA-derived fragments by determining the start and stop positions from read mapping profiles. FlaiMapper can predict miRNA boundaries with high accuracy. deepBlockAlign (Langenberger et al., 2012), which our present study takes quite a similar approach to, is an alignment-based method for alignments of read mapping profiles to find non-coding derived RNAs that share similar post-transcriptional processing. ALPS (Erhard and Zimmer, 2010) is also alignment-based, but is not designed for the purpose of identification of short derived RNA fragments. BlockClust (Videm et al., 2014) computes a similarity score between read mapping profiles to detect similar processing patterns and cluster read mapping profiles. BlockClust includes a high-dimensional feature representation to encode read mapping profiles and calculates the similarity scores based on a graph kernel. Therefore, BlockClust does not rely on alignment-based techniques and is not designed for the purpose of calculating the alignment of read profiles. However, all of these methods are computed based only on the information of read mapping profiles and do not take the RNA sequence and secondary structure information into account. Further, those methods did not address the reliable distinguishability of processing patterns from the random degradation.
Constructing an alignment for a pair of biological sequences such as DNA, RNA and protein sequences is a fundamental and robust method of sequence analysis (Durbin et al., 1998). The pairwise alignment of biological sequences is achieved according to insertions, deletions and match operations so that the two sequences are aligned with the same column length. The similarity score of an alignment is calculated according to the predefined scores for insertions, deletions and matches. Similarly, one can define the alignment between a pair of read mapping profiles (see Fig. 1(c)).
In previous work with the deepBlockAlign algorithm (Langenberger et al., 2012), the alignment program was designed to compare and align any read mapping profiles regardless of the RNA family to which they belong. The alignment program was also applied to the comparison of read mapping profiles of the same RNA gene across different samples that might be expected to induce different read mapping profiles (Pundhir and Gorodkin, 2015). This method is effective for the differential processing analysis of read mapping profiles of each non-coding RNA gene. In contrast with previous work, our primary goal was to detect specific forms among similar read mapping patterns mapped to specific regions of primary or secondary structures reflecting the functional activities of enzymes by alignments of read mapping profiles, and to distinguish these from non-functional degradation products. We developed a new read mapping profile alignment program named SHARAKU, which incorporates the primary and secondary structures of RNA sequences into an alignment of read mapping profiles. This is accomplished in combination with DAFS, a program for the simultaneous aligning and folding of RNA sequences (Sato et al., 2012). As a result, the most advanced feature of SHARAKU is that it simultaneously aligns the read mapping profiles and RNA sequences with the folding RNA secondary structures. Since each type of derived RNA fragments is cleaved from its precursor with a specific context of primary sequence and secondary structure, we can expect that the simultaneous alignment of read mapping profiles with the primary and secondary structures contributes to precise identifications of the type of derived RNAs. Thus, application of SHARAKU to the mixture of different RNA families would enable the accurate clustering of read mapping profiles with respect to 5′-end processing or 3′-end processing of each RNA family, and facilitate the detection of common processing patterns shared among different RNA families (see Fig. 1(d)). Note that SHARAKU was not designed for classification of RNA sequences into different RNA families. Further, SHARAKU produces an alignment of read mapping profiles at the nucleotide-level resolution, as Figure 1(c) displays. In contrast, deepBlockAlign uses blockbuster (Langenberger et al., 2009) to generate block groups and aligns any read mapping profiles at the level of individual blocks and block groups, including those in unannotated regions or unknown RNA genes. On the other hand, the current version of SHARAKU can only be applied to the annotated non-coding RNA regions.
Several previous studies (Ono et al., 2011; Scott and Ono, 2011) have explored the similarities and evolutionary relationships between snoRNAs and miRNA precursors. These similarity features represent molecules involved in the same processing pathways with a similar set of processing enzymes and the same RNAi targets. These similarity features are often confirmed based on the conservation of their primary and secondary structures, such as structural characteristics of typical H/ACA or C/D boxes. We hypothesized that in addition to sequence structure conservation, determining the similarity among read mapping profiles might help to identify the functional or processing similarities between snoRNAs and miRNA precursors.
2 Methods
When read mapping profiles for a pair of non-coding RNAs are obtained, SHARAKU fundamentally aligns two read mapping profiles by inserting gaps so that the sum of the differences of coverages at all positions between the two profiles is minimized. Simultaneously, SHARAKU takes information on the sequence and secondary structures of RNAs into account when aligning read mapping profiles by integration with DAFS, which calculates reliable structural alignments that maximize the expected accuracy of a predicted common secondary structure and its sequence alignment. The algorithm can be efficiently implemented by using dynamic programming. Second, SHARAKU calculates a similarity score (correlation coefficient) between two read mapping profiles based on the alignment. Third, SHARAKU produces a similarity score matrix for all pairs of read mapping profiles of non-coding RNAs in the target reference genome. Subsequently, the agglomerative hierarchical clustering is constructed based on the similarity score matrix.
2.1 Construction of read mapping profiles
Let denotes the four nucleotides and denotes the set of all finite RNA sequences consisting of bases in Σ.
A set of sequence reads generated by RNA-seq data for non-coding RNAs are mapped against the reference genome using standard mapping tools such as BWA (Li and Durbin, 2009) (i.e. alignment programs) for a huge number of short reads. The read coverage, i.e. the number (count) of mapped reads, at each position in an annotated RNA gene-coding region is calculated from the output of the mapping tool in BAM format. In an RNA gene-coding region, the read coverage ci at each position i is normalized so that every normalized value of read coverage is between 0 and 1. For an RNA sequence, denoted , of an annotated RNA gene-coding sequence, we refer to the sequence of normalized coverages, , as the read mapping profile of the RNA sequence (see Fig. 1(a)). Note that SHARAKU only deals with the read mapping profile within the annotated RNA-gene sequence and calculates the secondary structure within the annotated RNA-gene sequence.
2.2 Alignment algorithm for a pair of read mapping profiles
Let and denote a pair of RNA sequences, and denote the normalized read mapping profiles. The optimal alignment of a pair of two normalized read mapping profiles ca and cb is calculated by the following recursive formula to minimize the sum of the differences of coverages at all positions. The mismatch score at a position of an alignment between ca and cb is simply defined as the absolute difference of the two coverage values and . To define the gap score, first, the coverage value at a gap inserted between j and j + 1 is defined as (i.e. the average of and ). Second, the gap score between position i of ca and the gap inserted between j and j + 1 of cb is defined as the absolute difference of and .
at the end of sequence:
In order to avoid an unnecessary number of gaps inserted, which could cause artificial alignments, the number of gaps inserted into the alignment is restricted by the predefined parameter γ. This can be implemented by dynamic programming to control the number of inserted gaps.
2.3 Alignment of read mapping profiles incorporating sequence structures
Previously, we developed DAFS (Sato et al., 2012), an algorithm that simultaneously aligns and folds RNA sequences on the basis of maximizing the expected accuracy (MEA) of a predicted common secondary structure and its alignment. That is, DAFS calculates the maximization of the expected accuracy over all possible structural alignments of a pair of RNA sequences. Then, DAFS finds one optimal structural alignment, i.e. the most accurate structural alignment with the most plausible secondary structure of each input RNA sequence. Here, we combine the alignment algorithm for read mapping profiles with DAFS according to the same MEA principle in order to incorporate the primary and secondary sequence structures into the alignment of read mapping profiles.
For an RNA sequence , let denote the number of bases appearing in a, which is called the length of a. Given two RNA sequences , let denote the set of all possible alignments of a and b, that is, RNA sequence alignments without considering the secondary structures. Let and denote the set of all possible secondary structures of a and the set of all possible secondary structures of b, respectively. An alignment is represented as a binary-valued matrix , where tik = 1 if and only if the base ai is aligned (matched) with bk. A secondary structure is represented as a binary-valued triangular matrix , where xij1 = if and only if bases ai and aj form a base pair. Let denote a set of all possible structural alignments of a and b, that is, simultaneous alignment of RNA sequences and their secondary structures. We write , which means that a structural alignment consists of an alignment , and two secondary structures and .
First, in DAFS, a gain function of a structural alignment with regard to the correct structural alignment is defined as the weighted sum of gain functions and of the respective secondary structures and a gain function of the alignment:
| (1) |
where is a parameter that controls the weight of the secondary structures and the sequence alignment. Intuitively, the gain function can be regarded as a kind of ‘accuracy’ that represents the weighted sum of the number of true positive predictions and true negative predictions in , which is correlated to balanced accuracy measures such as Matthews correlation coefficient (MCC) and F-measure. In the case of the gain function for the secondary structure, a true prediction in is a pair of bases ai and aj such that . In the case of for the sequence alignment, a true prediction in is a pair of bases ai and bk such that .
Second, when we calculate a structural alignment for a pair of RNA sequences, their correct structural alignment is unknown. In such case, we compute the expectation of a gain function under the distribution over all possible structural alignments of the pair of RNA sequences. The expectation of the gain function under a given probability distribution over the space of structural alignments is maximized to find a structural alignment :
| (2) |
where is a probability distribution of the RNA structural alignments. To reduce the computational complexity of Equation (2), the probability distribution of the structural alignments is factorized as follows by assuming the independence of the structures and alignment:
where and are the probability distributions of RNA secondary structures over and , respectively, and is a probability distribution of alignments over . The expected gain function (2) can then be approximated as follows:
where denotes a base-pairing posterior probability, denotes an alignment-matching posterior probability, σ and τ are balancing parameters between true positives and true negatives, and is a constant independent of (see Sato et al., 2012 and its Supplementary Material for the derivation).
In order to incorporate the DAFS framework of the simultaneous aligning and folding of RNA sequences into the alignment of read mapping profiles on the basis of the MEA principle, we need to define the expectation of the gain function for alignments of read mapping profiles. First, we define the partition function (McCaskill, 1990) for alignments of read mapping profiles. The partition function is equal to the alignment kernel K(a, b) (Morita et al., 2009; Saigo et al., 2004) that measures the similarity between two sequences a and b by summing the scores obtained from all possible alignments with the gaps of the sequences. The partition function for alignments of read mapping profiles ca and cb are defined as follows:
where denotes the set of all possible alignments between a pair of read mapping profiles ca and cb, and denotes the score of an alignment between ca and cb:
The ‘forward’ algorithm to compute the partition function for read mapping profiles can be implemented by the following recursive formula:
where two variables rg and lg are used for controlling the number of inserted gaps. The ‘backward’ algorithm to compute can also be defined in a similar manner.
Now, we can define a posterior probability of positions i and j to be aligned in the alignments of read mapping profiles:
where γr and γl define the maximum number of gaps inserted at a row and column in the alignment, respectively.
The expectation of the gain function for the correct alignment of read mapping profiles, correct sequence alignments and correct secondary structures under a given probability distribution over the space of simultaneous alignments of read mapping profiles, RNA primary sequences and secondary structures is defined and approximated as follows:
where denotes the set of all possible alignments between a pair of read mapping profiles ca and cb, taking the primary and secondary sequence structures between a pair of RNA sequences a and b into account, and is a parameter that controls the weight of the read mapping profile alignment and the sequence alignment.
The ‘optimal’ alignment can be obtained by maximizing the expected gain function on the basis of the MEA principle. Further, the maximization of the expected gain function can be efficiently implemented using the techniques such as ‘dual decomposition’, ‘subgradient optimization’ and ‘dynamic programming’ (see Sato et al., 2012 for more details).
2.4 Similarity score calculation between read mapping profiles
Let be the alignment for a pair of read mapping profiles, ca and cb, obtained by maximizing the expected gain function . We define the similarity score function denoted between ca and cb based on the alignment as follows:
The similarity score is exactly equal to the normalized inner product (cosine coefficient) of two gap-inserted read mapping profiles in the alignment . Therefore, represents the correlation coefficient between two read mapping profiles ca and cb based on the alignment .
3 Results
3.1 Datasets
In order to evaluate the performance of our method, we generated simulated read data in the form of typical next-generation sequencing output (based on an Illumina-type of short sequence reads). To evaluate the clustering ability to detect similar processing patterns, the labeled data of various classes of different processing patterns are required, such as 5′-end processing, 3′-end processing and non-processing. However, comprehensive annotations of such labels for all small non-coding RNAs are not available in existing real data such as the sequence read archive. The only exception is the miRNA family, for which the mature miRNA annotations are accessible in the NCBI Reference Sequence Database.
First, we obtained the small RNA annotation and sequence data for miRNAs (24 genes), snoRNAs (37 genes) and tRNAs (41 genes) with a length less than 120 nt from the Ensembl genome browser for the mouse genome (GRCm38). Second, according to the results of previous studies that analyzed and identified the existence of various types of small RNA-derived fragments (Chen and Heard, 2013; Kawaji et al., 2008; Ono et al., 2011; Scott et al., 2012), we determined the start or end sites of derived fragments that simulated 5′-end processing or 3′-end processing in snoRNAs and tRNAs to be within a few bases from the 5′-end or 3′-end, respectively. Further, we set the length of the derived fragments to 20 nt for snoRNAs and to 30 nt for tRNAs. For miRNAs, we used the annotations for mature miRNAs in the NCBI Reference Sequence Database. Third, we generated 88 324 simulated sequence reads that contain the full-length transcripts of snoRNAs and tRNAs (that is, the complete sequence reads exactly identical to the annotated snoRNA and tRNA sequences), mature miRNAs and their derived fragments. In addition, the set of ‘degradation reads’ was artificially constructed from the simulated complete reads of snoRNAs and tRNAs. In general, there are three major classes of RNA-degrading enzymes (Houseley and Tollervey, 2009): endonucleases that cut RNA internally, 5′ exonucleases that hydrolyze RNA from the 5′-end and 3′ exonucleases that degrade RNA from the 3′-end. In our study, we only considered the degradation of RNA from the 3′-end, which is more reflective of non-functional degradation for unstable RNAs. Thus, the set of degradation reads was obtained by eliminating 40–60% of the nucleotides from the 3′-end in order to simulate the degradation process. The degradation reads constituted 20% of the set of simulated complete reads. In total, the simulated read dataset consisted of the complete reads of snoRNAs and tRNAs, the derived fragment reads for 5′-end processing of miRNAs, snoRNAs and tRNAs, the derived fragment reads for 3′-end processing of miRNAs, snoRNAs and tRNAs, and the degradation reads. Each of the 102 small RNAs used for the simulated data was labeled according to one of the above-mentioned classes as follows: (i) 5′-end processing of miRNAs, (ii) 3′-end processing of miRNAs, (iii) 5′-end processing of snoRNAs, (iv) 3′-end processing of snoRNAs, (v) 5′-end processing of tRNAs, (vi) 3′-end processing of tRNAs, (vii) degradation of snoRNAs and tRNAs. These class labels were used for experimental evaluation of the clustering performance. (See also Supplemental Figure S1 that displays all read mapping profiles obtained by mapping the simulated read data to the 102 annotated small RNA sequences.)
To demonstrate the practical usefulness of SHARAKU, we used a real dataset obtained from an RNA-seq experiment of the hippocampus of the left brain of a 2-year-old male common marmoset that was being bred at the Central Institute for Experimental Animals (CIEA). The total RNA was extracted and was subject to removal of rRNA and 5′Cap, and then the cDNA library of small RNA was prepared using the TruSeq Small RNA Sample Prep Kit (Illumina). The small RNA transcripts were sequenced with the next-generation sequencer MiSeq (Illumina) for a sequence read length of 270 bp, which enabled generating the complete sequences of most small RNA families. The short reads were subject to cutting adapters and quality filtering. The qualified reads were then mapped to the common marmoset (Callithrix jacchus) draft genome caljac-3.2 (MGSAC, 2014) by BWA (Li and Durbin, 2009). The tRNA annotation was predicted by tRNAscan-SE (Lowe and Eddy, 1997). In total, the annotation and sequence data for 619 non-coding small RNAs were obtained (194 miRNAs, 316 snoRNAs, 109 tRNAs) from the Ensembl genome browser (Ensembl 73 version; ‘Callithrix_jacchus.C_jacchus3.2.1.73.gtf’ file) for the common marmoset genome (C_jacchus3.2.1).
3.2 Validation of clustering accuracy with the simulated dataset
We validated the clustering performance based on our alignment method for all pairs of read mapping profiles using the simulated dataset. First, the similarity score matrix for all pairs of read mapping profiles was calculated by SHARAKU. We used empirically determined parameters: . Second, the (agglomerative) hierarchical clustering method with group averaging was applied based on the similarity matrix. The agglomerative method of hierarchical clustering hclust implemented in the R-package was used for subsequent analysis. We evaluated how well each processing pattern and RNA family in the simulated dataset was separated into a distinct cluster according to the hierarchical clustering. Furthermore, we compared the clustering performance based on SHARAKU with that of the alignment-based method deepBlockAlign (Langenberger et al., 2012) using the default parameters. As the input to deepBlockAlign, the output of blockbuster executed on the input of the simulated dataset was given together with the sequence annotation information. In the case of the simulated dataset, this does not affect the result of performance evaluation, because the sequence reads (except for shorter derived RNAs) in the simulated dataset are identical to the complete RNA sequences annotated in the Ensembl genome browser, so that every block that is output by blockbuster with the input of the simulated dataset is identical to one of the annotated RNA sequences. Note that since another alignment-based scoring system ALPS (Erhard and Zimmer, 2010) was not publicly available, we did not compare the performance with ALPS.
In the validation experiment using the simulated read dataset, the simulated reads were mapped to the mouse genome (GRCm38) by BWA (Li and Durbin, 2009) with the default parameters, except that -a bwtsw was set as the bwa index. From the output in BAM format, the normalized read mapping profiles for non-coding RNAs were obtained and fed to the alignment programs.
We evaluated the overall quality of the clustering tree using the receiver operating characteristic (ROC) analysis proposed in (Will et al., 2007). (Note that we can obtain different resultant clusters from a clustering tree depending on a distance threshold to cut the branches.) Given a distance threshold, the number of true positives (TP) was defined as the number of read mapping profile pairs that have the same class label and are correctly assigned to the same resultant cluster. In the same manner, the numbers of false positives (FP), true negatives (TN) and false negatives (FN) are defined by counting the pairs from different class labels but the same resultant cluster, the pairs from different class labels and different resultant clusters, and the pairs from the same class label but different resultant clusters, respectively. The ROC analysis was performed by plotting the true positive rates TP/(TP + FN) versus the false positive rates FP/(TN + FP) for different distance thresholds. The quality of the clustering was measured by the area under the ROC curve (AUC).
Table 1 shows the results of AUC scores obtained with the three methods, and Figure 2 (left) shows the ROC curves generated by SHARAKU and deepBlockAlign. The AUC scores to indicate the clustering abilities of three methods were calculated for the dataset of all read mapping profiles (the mixture of read mapping profiles of miRNAs, snoRNAs and tRNAs) and for the dataset of read mapping profiles of each family, tRNA, snoRNA and miRNA. For the dataset of all read mapping profiles, SHARAKU achieved an almost perfect AUC score, and hence an almost perfect clustering result, and exhibited higher accuracy than deepBlockAlign. SHARAKU also succeeded in completely discriminating the degradation pattern from post-transcriptional processing such as 5′ or 3′-end processing. The alignment obtained by SHARAKU without considering the primary and secondary sequence structures (‘SHARAKU without DAFS’) presented lower AUC scores, which implied that incorporation of the sequence structure information is required to achieve high clustering accuracy. On the other hand, for the dataset of read mapping profiles of each family, both SHARAKU and deepBlockAlign achieved an AUC score of 1.0, which indicates a perfect clustering result. This result, together with that obtained for all read mapping profiles, clearly indicates that the alignment incorporating the secondary structures is indispensable for the accurate clustering of read mapping profiles generated from the mixture of different RNA families. Figure 2 (right) shows an alignment produced by SHARAKU for a pair of RNAs (Mir92-1 and SNORD63) in the simulated dataset that could be successfully separated into two different clusters by SHARAKU, but were clustered together by deepBlockAlign, thereby highlighting the importance of taking the secondary structures into account for alignments of read mapping profiles. (More alignments are also shown in Supplemental Figs S2 and S3.)
Table 1.
AUC scores of the discrimination accuracy based on clustering trees constructed by three methods
| SHARAKU | deepBlockAlign | SHARAKU without | |
|---|---|---|---|
| DAFS | |||
| ALL | 0.985 | 0.921 | 0.930 |
| tRNA | 1.0 | 1.0 | 1.0 |
| snoRNA | 1.0 | 1.0 | 1.0 |
| miRNA | 1.0 | 1.0 | 0.497 |
Fig. 2.

(Left.) ROC curves representing the true positive rates versus false positive rates based on clustering trees generated by SHARAKU and deepBlockAlign. The solid line represents the ROC curve of SHARAKU, and the dashed line represents the ROC curve of deepBlockAlign. (Right.) An alignment produced by SHARAKU for a pair of RNAs (Mir92-1 and SNORD63) in the simulated dataset with the annotations of RNA sequence alignment and the predicted secondary structure. The solid line represents the read coverages of Mir92-1, and the dashed line represents the read coverages of SNORD63
Table 2 shows the computational times required by the three methods for each simulated dataset. Table 2 indicates that SHARAKU needs a large amount of additional computational time compared with deepBlockAlign and SHARAKU without DAFS to simultaneously align the read mapping profiles and RNA sequences and calculate the secondary structures.
Table 2.
CPU times (seconds) required by the three methods for each simulated dataset. The CPU time was measured per alignment of a pair of read mapping profiles
| SHARAKU | deepBlockAlign | SHARAKU without | |
|---|---|---|---|
| DAFS | |||
| ALL | 0.832 | 0.054 | 0.067 |
| tRNA | 0.495 | 0.036 | 0.036 |
| snoRNA | 0.683 | 0.039 | 0.047 |
| miRNA | 0.554 | 0.029 | 0.051 |
3.3 Clustering a real dataset of RNA transcripts in the marmoset brain
A total of 33.8 million (M) sequence reads with a length of 270 bp for small non-coding RNAs was generated using an Illumina MiSeq sequencer. After quality filtering, 30.5 M reads were mapped to the common marmoset (Callithrix jacchus) draft genome caljac-3.2 using BWA. From the output in BAM format, the normalized read mapping profiles for 619 non-coding RNAs were obtained and fed to the alignment programs.
The dendrogram of clustering tree of hierarchical clustering based on the SHARAKU alignments of read mapping profiles of the 619 non-coding RNAs is shown in Figure 4. Most of the read mapping profiles were clustered and well separated into five major clusters, representing (1) 3′-end processing of miRNAs (containing 80 miRNAs); (2) 5′-end processing of snoRNAs and tRNAs (containing 25 snoRNAs, 62 tRNAs and 5 miRNAs); (3) non-processing and non-degradation of snoRNAs and tRNAs (containing 210 snoRNAs, 10 tRNAs and 1 miRNA); (4) degradation of snoRNAs and tRNAs (containing 42 snoRNAs, 20 tRNAs and 1 miRNA); and (5) 5′-end processing of miRNAs (containing 81 miRNAs and 2 tRNAs). In addition, the read mapping profiles representing 3′-end processing of snoRNAs and tRNAs (containing 26 snoRNAs, 3 tRNAs and 3miRNAs) were scattered into four different clusters (6-1), (6-2), (6-3) and (6-4). These 4 clusters could be defined as the complement of the clusters (3) and (4), which might provide a clear interpretation.
Fig. 4.

Dendrogram of hierarchical clustering based on SHARAKU alignments for the read mapping profiles of 619 non-coding RNAs. Annotations of 5 major clusters plus 4 scattered clusters and a representative read mapping profile in each cluster are shown below the clustering tree
A representative read mapping profile in each cluster is shown below the clustering tree. Interestingly, 60% of the tRNAs were processed for deriving shorter fragments mostly at the 5′-ends, whereas 80% of the snoRNAs were non-processed. A few other interesting small clusters were obtained and will be discussed below.
In addition to confirming the utility of the new algorithm, this experiment revealed the post-transcriptional processing and the expression patterns of small derived RNAs in the marmoset brain. To the best of our knowledge, this represents the first identification of the processing patterns of derived RNAs expressed in the brain. The result of hierarchical clustering based on the deepBlockAlign alignments is shown in Supplemental Figure S4.
3.4 Verification of small derived RNA transcripts by northern blotting
In order to verify that a derived RNA fragment predicted by the clustering method based on SHARAKU is truly derived and expressed and not an experimental artifact, we performed northern blotting analysis with a sample of the marmoset spleen. A small RNA-seq analysis for the total RNAs extracted from the marmoset spleen was executed and northern blotting for Leu-CAA-tRNA was performed. The read mapping profile of this Leu-CAA-tRNA belonged to a cluster representing 5′-end processing of tRNAs in the hierarchical clustering tree generated by SHARAKU. Figure 3 shows the read mapping profile for Leu-CAA-tRNA obtained from the RNA-seq reads (left), as well as the northern blots for Leu-CAA-tRNA (lenght: 105 nt) and the derived fragment (expected length: 35 nt) from the tRNA (right). The results clearly proved the actual presence of the derived RNA fragment.
Fig. 3.

(Left) Read mapping profile of Leu-CAA-tRNA from RNA-seq for the marmoset spleen. (Right) Northern blots for Leu-CAA-tRNA (lenght 105 nt) and the derived fragments. The band indicated with the arrow represents the expected size (35 nt) of the predicted derived RNA fragment. Blots were pre-hybridized, then probes which had been end-labeled with [P] ATP were added to the hybridization chamber and incubated with the blots. The membrane was then exposed to a phosphoimager and scanned. The largest band appearing at a distance just short of the 100-nt marker indicates the expression of the origin tRNA from which the short fragment RNA was derived
4 Discussion
Several previous studies (Ono et al., 2011; Scott and Ono, 2011) have explored the similarities and evolutionary relationships between snoRNAs and miRNA precursors. As a result, various relationships between snoRNAs and miRNAs have been identified, from ‘snoRNAs with miRNA features’ to ‘dual function sno-miRNAs’ and ‘miRNAs with snoRNA features’. These similarity features represent molecules involved in the same processing pathways with a similar set of processing enzymes and the same RNAi targets. These similarity features are often confirmed based on the conservation of their primary and secondary structures, such as structural characteristics of typical H/ACA or C/D boxes. We hypothesized that in addition to sequence structure conservation, determining the similarity among read mapping profiles might help to identify the functional or processing similarities between snoRNAs and miRNA precursors. An interesting small cluster in the clustering tree shown in Figure 4 (highlighted with an arrow and asterisk) consisted of a mixture of snoRNAs, miRNAs and tRNAs. In particular, we found that the snoRNA ‘HBII-52’ and the miRNA ‘let-7a’ have quite similar features in their secondary structures and read mapping profiles, indicating a similar processing mechanism to produce the derived shorter fragments at 3′-ends. Figure 5 shows these features.
Fig. 5.
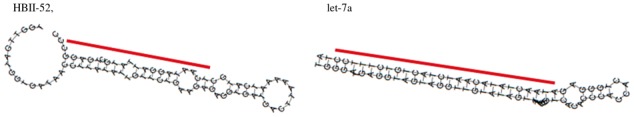
The secondary structures predicted by RNAfold (Hofacker, 2003) and annotations with lines indicating the predicted locations of deriving fragments for a snoRNA ‘HBII-52’ and a miRNA ‘let-7a’ found in the clusters. The line for miRNA secondary structure indicate the predicted locations of the mature miRNA
Indeed, small derived RNAs from HBII-52 were previously reported to resemble miRNAs (Chen and Heard, 2013).
Another interesting cluster is cluster (6) of the 5′-end processing of miRNAs. The cluster contained two tRNAs. ‘Pro-TGG-tRNA’ has the typical miRNA-like secondary structure and miRNA/miRNA* duplex, shown in Figure 6. Supporting this result, previous work has indicated possible cross-talk between tRNA-derived RNA fragments and the canonical pathway of miRNAs (Sobala and Hutvagner, 2011).
Fig. 6.
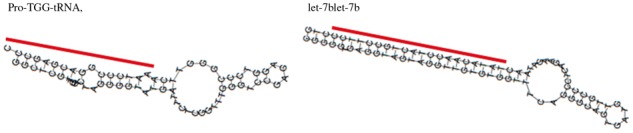
The secondary structures predicted by RNAfold and annotations, with lines indicating the locations of derived fragments for a tRNA ‘Pro-TGG-tRNA’ and an miRNA ‘let-7b’ found in the cluster of the 5′-end processing of miRNAs. The line for miRNA secondary structure indicates the predicted locations of the mature miRNA
Thus, the alignments and clustering of read mapping profiles using SHARAKU can give insight into revealing the common processing patterns in different families of non-coding RNAs and their derived fragments, which could help to clarify the processing pathways and biological functions of derived RNA fragments.
One of the merits of SHARAKU is to calculate the optimal alignment based on not only the pattern of read mapping profiles, but also primary and secondary structures of RNA sequences. Since each type of derived RNA fragments is cleaved from its precursor with a specific context of primary sequence and secondary structure, we can expect that the simultaneous alignment of read mapping profiles with the primary and secondary structures contributes to precise identifications of the type of derived RNAs. To combine the pattern of read mapping profiles and the primary and secondary structures of RNAs, a Sankoff-type algorithm is required to be implemented. We implemented SHARAKU with DAFS framework, which enabled to efficiently combine the pattern of read mapping profiles and the primary and secondary structures of RNAs by the dual decomposition technique.
In the present study, we have only dealt with annotated non-coding RNAs. However, SHARAKU can also be applied to the alignment and clustering of novel and unannotated regions by employing tools such as blockbuster (Langenberger et al., 2009) in order to determine the expressed block regions on the reference genome that are obtained from RNA-seq reads. As non-functional degradation products, we only considered the degradation of RNA from the 3′-end in the simulated dataset. Therefore, performance evaluations of SHARAKU to determine the tolerance for the other two classes of RNA-degradations, endonucleases and 5′ exonucleases, are required. These issues will be addressed in our future work.
5 Conclusion
With the aim of offering computational tools for comprehensively analyzing the post-transcriptional processing patterns of non-coding RNAs and detecting their common processing patterns, based on RNA-seq, we developed a new algorithm called SHARAKU to align two read mapping profiles of next-generation sequencing data for non-coding RNAs. SHARAKU incorporates the primary and secondary structures of RNA sequences into an alignment of read mapping profiles by combining with DAFS, which constructs reliable structural alignments that maximize the expected accuracy of a predicted common secondary structure and its sequence alignment. SHARAKU could simultaneously align the read mapping profiles and RNA sequences with information of the folded RNA secondary structures. In an experiment using a simulated dataset, SHARAKU achieved an almost perfect clustering result, and exhibited higher accuracy than deepBlockAlign. In an experiment with real data of small RNA sequencing for the common marmoset brain, SHARAKU succeeded in identifying the five major clusters plus four scattered clusters representing typical processing patterns. This method also revealed some interesting clusters consisting of mixtures of several RNA families that predicted common processing patterns among different RNA families. These results demonstrate that SHARAKU can be an indispensable tool for analyses of the processing patterns and functions of regulatory non-coding RNAs with deep-sequencing data.
Supplementary Material
Acknowledgements
We thank Dr. Erika Sasaki and Takashi Inoue (CIEA, Japan) for providing the common Marmoset sample.
Funding
This work was supported by a Grant-in-Aid for Scientific Research in Innovative Areas [No. 221S0002] from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) to Y.S., and a Grant-in-Aid for Scientific Research (A) (KAKENHI) [No. 23241066] from the JSPS to Y.S. and K.S., and supported by a Grant-in-Aid for Scientific Research (C) (KAKENHI) [No. 25330348] from the JSPS to K.S. This work was also supported by a MEXT-supported Program for the Strategic Research Foundation at Private Universities, and a grant from the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED) of Japan.
Conflict of Interest: none declared.
References
- Chen C.J., Heard E. (2013) Small RNAs derived from structural non-coding RNAs. Methods, 63, 76–84. [DOI] [PubMed] [Google Scholar]
- Durbin R. et al. (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Erhard F., Zimmer R. (2010) Classification of ncRNAs using position and size information in deep sequencing data. Bioinformatics, 26, i426–i432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D. et al. (2010) Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA, 16, 673–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker I.L. (2003) Vienna RNA secondary structure server. Nucleic Acids Res., 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstrate Y. et al. (2015) FlaiMapper: computational annotation of small ncRNA-derived fragments using RNA-seq high-throughput data. Bioinformatics, 31, 665–673. [DOI] [PubMed] [Google Scholar]
- Houseley J., Tollervey D. (2009) The many pathways of RNA degradation. Cell, 136, 763–776. [DOI] [PubMed] [Google Scholar]
- Kawaji H. et al. (2008) Hidden layers of human small RNAs. BMC Genomics, 9, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberger D. et al. (2009) Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics, 25, 2298–2301. [DOI] [PubMed] [Google Scholar]
- Langenberger D. et al. (2012) deepBlockAlign: a tool for aligning RNA-seq profiles of read block patterns. Bioinformatics, 28, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.S. et al. (2009) A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev., 23, 2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Eddy S.R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res., 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmoset Genome Sequencing and Analysis Consortium. (2014) The common Marmoset genome provides insight into primate biology and evolution. Nat. Genet., 5, 12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens-Uzunova E.S. et al. (2013) Beyond microRNA–novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett., 340, 201–211. [DOI] [PubMed] [Google Scholar]
- McCaskill J.S. (1990) The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers, 29, 1105–1119. [DOI] [PubMed] [Google Scholar]
- Morita K. et al. (2009) Genome-wide searching with base-pairing kernel functions for noncoding RNAs: computational and expression analysis of snoRNA families in Caenorhabditis elegans. Nucleic Acids Res., 37, 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M. et al. (2011) Identification of human miRNA precursors that resemble box C/D snoRNAs. Nucleic Acids Res., 39, 3879–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pundhir S., Gorodkin J. (2015) Differential and coherent processing patterns from small RNAs. Sci. Rep., 5, 12062.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pundhir S. et al. (2015) Emerging applications of read profiles towards the functional annotation of the genome. Front. Genet., 6, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigo H. et al. (2004) Protein homology detection using string alignment kernels. Bioinformatics, 20, 1682–1689. [DOI] [PubMed] [Google Scholar]
- Sato K. et al. (2012) DAFS: simultaneous aligning and folding of RNA sequences via dual decomposition. Bioinformatics, 28, 3218–3224. [DOI] [PubMed] [Google Scholar]
- Scott M.S., Ono M. (2011) From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie, 93, 1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M.S. et al. (2012) Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res., 40, 3676–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A., Hutvagner G. (2011) Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip. Rev. RNA, 93, 1987–1992. [DOI] [PubMed] [Google Scholar]
- Taft R.J. et al. (2009) Small RNAs derived from snoRNAs. RNA, 15, 1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videm P. et al. (2014) BlockClust: efficient clustering and classification of non-coding RNAs from short read RNA-seq profiles. Bioinformatics, 30, i274–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S. et al. (2007) Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol., 3, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S. et al. (2009) Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell. Biol., 185, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.