

Abstract
Overcoming resistance to chemotherapy is a major and unmet medical challenge in the treatment of pancreatic cancer. Poor drug delivery due to stromal barriers in the tumor microenvironment and aggressive tumor biology are additional impediments toward a more successful treatment of pancreatic cancer. In attempts to address these challenges, we developed IGF1 receptor (IGF1R)-directed, multi-functional theranostic nanoparticles for targeted delivery of therapeutic agents into IGF1R-expressing drug-resistant tumor cells and tumor-associated stromal cells. These nanoparticles were prepared by conjugating recombinant human IGF1 to magnetic iron oxide nanoparticles (IONPs) carrying the anthracycline doxorubicin (Dox) as the chemotherapeutic payload. Intravenously administered IGF1-IONPs exhibited excellent tumor targeting and penetration in an orthotopic patient-derived xenograft (PDX) model of pancreatic cancer featuring enriched tumor stroma and heterogeneous cancer cells. IGF1R-targeted therapy using the theranostic IGF1-IONP-Dox significantly inhibited the growth of pancreatic PDX tumors. The effects of the intratumoral nanoparticle delivery and therapeutic responses in the orthotopic pancreatic PDX tumors could be detected by magnetic resonance imaging (MRI) with IONP-induced contrasts. Histological analysis showed that IGF1R-targeted delivery of Dox significantly inhibited cell proliferation and induced apoptotic cell death of pancreatic cancer cells. Therefore, further development of IGF1R-targeted theranostic IONPs and MRI-guided cancer therapy as a precision nanomedicine may provide the basis for more effective treatment of pancreatic cancer.
Keywords: IGF1R-targeted cancer therapy, theranostic nanoparticles, orthotopic pancreatic cancer patient tissue derived xenografts, MRI, image-guided cancer therapy
Pancreatic cancer has one of the worst outcomes of all cancer types, with a five-year survival of 5%.1 The majority of pancreatic cancer patients are diagnosed at the advanced stage and can only be treated with chemotherapy and radiotherapy. However, resistance to chemotherapy is the most common reason for the failure of pancreatic cancer treatment.2–5 This is because pancreatic cancer has extensive tumor stromal components that consist of over 50% of a tumor mass, as well as disorganized tumor vasculatures, creating physical barriers for drug delivery.6–8 In addition, genetic alterations and dysfunctional signaling pathways in pancreatic tumor cells also lead to intrinsic drug resistance.9 Therefore, the development of new therapeutics for effective treatment of pancreatic cancer requires novel approaches to break drug delivery barriers in the tumor stroma and increase the drug concentration delivered to tumor cells in order to overcome drug-resistant mechanisms.8,10
Targeted delivery of nanoparticles carrying drug payloads has shown the potential to deliver large amounts of therapeutic agents into pancreatic cancer cells to effectively treat drug-resistant tumor cells.11–17 In the delivery of the tumor cell targeted theranostic nanoparticles to pancreatic tumors in vivo, a dense tumor stromal barrier prevents targeted nanoparticle drug carriers from reaching tumor cells by limiting diffusion of nanoparticles in the interstitial space after extravasation from tumor vessels mediated by the enhanced permeability and retention (EPR) effect.16,18,19 Therefore, it is necessary to develop nanoparticle drug carriers targeting both pancreatic tumor and stromal cells. Insulin-like growth factor 1 receptor (IGF1R) is found highly expressed in 40–90% of pancreatic cancer tissues and in both tumor and stromal cells.20,21 In contrast, its expression in normal pancreas is relatively low.22,23 Furthermore, IGF1R is a logical cell surface marker for targeting drug-resistant tumor cells since the level of IGF1R expression is further increased in the drug-resistant tumor cell population.24,25 A high level of IGF1R in tumor cells may facilitate efficient intracellular drug delivery by receptor-mediated endocytosis.26 Thus, IGF1R-targeted theranostic nanoparticles are promising drug delivery carriers for the development of novel targeted therapeutics for the treatment of both drug-sensitive and -resistant pancreatic cancer cells. Insulin-like growth factor 1 (IGF1) is a 70 amino acid protein with three intramolecular disulfide bridges. It binds to its receptor, IGF1R, with high affinity (Kd: 1.6 nM).27 Therefore, it is a natural targeting ligand for IGF1R with high specificity, high affinity, and low immunogenicity, compared to antibody- or antibody fragments-derived ligands.
Magnetic iron oxide nanoparticles (IONPs) are well-suited for the development of theranostic nanoparticles for targeted therapy of pancreatic cancer.13,28–33 IONPs are biocompatible and biodegradable nanomaterials with a low toxicity for the development of therapeutic agents that need to be administrated repeatedly in large doses. More importantly, IONPs offer magnetic resonance imaging (MRI) contrast that enhances the ability for the detection of theranostic nanoparticles in pancreatic tumors located deep in the retroperitoneal cavity, taking advantage of three-dimensional high-resolution imaging and tissue characterization capabilities of MRI.34 Although early studies, including ours, showed in vivo efficacy of tumor cell targeted theranostic nanoparticles in human pancreatic cancer cell line derived xenograft models,12,17,33 those xenograft models lack histological and pathological characteristics of primary human pancreatic cancer tissues and tumor microenvironment, particularly stromal components and heterogeneous presence of tumor cells.35 The results of those in vivo studies could not reffect accurately the efficiency of targeted delivery of theranostic nanoparticles in stroma-rich cancers and responses to therapy in highly heterogeneous tumor cells as well as tumor microenvironment. To address this problem, we have established an orthotopic human pancreatic cancer patient tissue derived xenograft (PDX) model in SCID and nude mice for studying IGF1R-targeted theranostic nanoparticles carrying the chemotherapy drug doxorubicin (Dox) on targeted drug delivery and induction of tumor cell death following treatment. Dox is a potent chemotherapy drug for many cancer types but is not currently used for pancreatic cancer treatment due to its cardiotoxicity. The total Dox dose that can be administrated in a patient's lifetime is limited to <550 mg/m2.36,37 Since pancreatic cancer has low drug delivery efficiency and poor therapeutic response, very high drug doses must be given to pancreatic cancer patients. For example, the therapeutic dose for the first line chemotherapy drug gemcitabine is 1000 mg/m2 weekly for 12 treatments. The maximum tolerated dose of gemcitabine is 2400 mg/m2 weekly for 12 treatments.38 Results of previous clinical studies have shown a significant reduction in Dox-induced cardiotoxicity using liposome-encapsulated Dox (Doxil).39,40 Therefore, targeted delivery of Dox using theranostic nanoparticles developed in this study has the potential to improve the delivery of potent Dox into tumor cells but avoid systemic toxicity. Additionally, demonstration of efficacy of the receptor-targeted theranostic nanoparticles carrying Dox in a human pancreatic cancer PDX model should allow further development of targeted and image-guided therapy for pancreatic cancer patients who have developed drug resistance to the first line chemotherapeutics, such as gemcitabine or the combination of fluorouracil, oxaliplatin, leucovorin, and irinotecan (FOLFIRINOX).41
The early passages of the orthotopic pancreatic cancer PDX xenografts not only retained intratumoral heterogeneity and histological characteristics of the primary human pancreatic cancer tissues but also regenerated tumor microenvironment, such as vasculatures, tumor stromal fibroblasts and macrophages, and extracellular matrix.42–44 Orthotopic human cancer PDX models have been used to study tumor biology and evaluate efficacy of cancer therapeutic agents.45–47 However, the effects of targeted delivery of theranostic nanoparticles and response to the therapy in human pancreatic PDX tumors have not been investigated. Here, we report that IGF1R-targeted nano-particles carrying Dox were delivered into orthotopic pancreatic PDX tumors by efficiently penetrating tumor stroma, leading to significant inhibition of the tumor growth. Targeted delivery of theranostic IONPs and tumor response to therapy could be determined by noninvasive MRI. Our results demonstrated that IGF1-conjugated theranostic IONP is a new and effective nanoparticle drug delivery system for improving targeted therapy of stroma-rich pancreatic cancer.
RESULTS AND DISCUSSION
Orthotopic Human Pancreatic PDX Tumors with Histological and Pathological Characteristics of Primary Human Pancreatic Cancers
Orthotopic human pancreatic PDX tumor models were established by implanting tissue fragments of surgically resected fresh human pancreatic cancer tissues into the pancreas of SCID mice (Figure 1a). Histological analysis of frozen tissue sections of the paired primary human pancreatic cancer and the PDX tumors obtained from patient #1 showed that human pancreatic PDX tumors had infiltrating ductal carcinoma cells surrounded by tumor stromal components (Figure 1b), which closely resembled the primary tumor tissues. On the other hand, orthotopic pancreatic tumor xenografts derived from the human pancreatic cancer MIAPaCa-2 cell line had dense tumor cell clusters with a relatively low level of stromal cells scattered in the tumor. Importantly, immunofluorescence labeling revealed high levels of IGF1R in the primary human pancreatic cancer tissues and PDX tumors in contrast to a low expression level in MIAPaCa-2 cell line derived tumor xenografts (Figure 1b). Picro-Sirius red staining showed extensive stromal collagen surrounding ductal carcinoma cells in both primary human pancreatic cancer tissues and PDX tumors. However, the MIAPaCa-2 cell line derived tumor xenograft had significant differences in the histological and tumor stroma characteristics (Figure 1b). Furthermore, immunofluorescence labeling using antibodies specific for human fibroblast active protein (FAP), which is expressed in active fibroblasts, revealed similar phenotypic features and intratumoral distributions of tumor stromal cell populations in the PDX tumors compared with primary human pancreatic cancer tissues (Figure 1b).
Figure 1.

Establishment and characterization of an orthotopic human pancreatic PDX tumor model. (a) Schematic illustration of the protocol for establishment of an orthotopic pancreatic PDX tumor model. (b) Comparison of histological characteristics and the levels of IGF1R, and stromal fibroblasts and collagen in tissue sections of surgically resected primary human pancreatic cancer (patient #1), the first passage of PDX-tumor derived from patient #1 in SCID mice, and pancreatic tumor xenograft derived from the MIAPaCa-2 cell line. H&E staining and Picro-Sirius red staining showed histological characteristics of primary human pancreatic cancer and PDX tumors. Immunofluorescence labeling using antibodies to IGF1R or human FAP and Alexa Fluor 555 conjugated secondary antibody (red). Quantification of the percentages of FAP or collagen-positive tumor areas in the primary pancreatic and PDX-tumor tissues was performed using ImageJ software. Numbers shown in the figure are the average numbers of six tissue sections. (c) Double immunofluorescence labeling of the primary human pancreatic cancer and the first passage of PDX-tumor tissue sections using anti-IGF1R, FAP, CD68, and CD31 antibodies. White arrows indicate tumor stromal cells, and blue arrows point at ductal tumor cells. Cells coexpressing two biomarkers are in orange-yellow. IGF1R was expressed in both tumor stromal fibroblasts and tumor-associated macrophages. CD31-positive endothelial cells lacked IGF1R expression. Scale bars are 100 μm.
We also examined the IGF1R expression in pancreatic cancer cells and stromal fibroblasts, macrophages, and endothelial cells by double immunofluorescence labeling using antibodies against IGF1R, FAP, CD68 (macrophage biomarker), or CD31 (endothelial cells). First, we found that active stromal fibroblast cells were positive for IGF1R antibody staining in both primary pancreatic cancer and PDX-tumor tissue sections (Figure 1c), although the level of IGF1R was higher in tumor cells compared with stromal fibroblasts. A high level of CD68-positive macrophages was detected in the peripheral areas of the PDX tumors, and those macrophages also expressed IGF1R (Figure 1c). Most small and disorganized tumor vessels identified by anti-CD31 antibody were found in the tumor stromal areas but not in IGF1R-positive tumor cell clusters (Figure 1c). However, IGF1R fluorescence signal was not colocalized with CD31-positive endothelial cells, suggesting a low level of IGF1R expression in tumor endothelial cells. The presence of high levels of IGF1R in pancreatic cancer cells, stromal fibroblasts, and macrophages indicated the feasibility of IGF1R-targeted nanoparticles for drug delivery into pancreatic cancer and tumor stromal cells.
The rationale for targeting IGF1R for the treatment of drug-resistant tumor cells was further supported by the observation that residual human pancreatic PDX tumors following six treatments of 5 mg/kg dose of conventional chemotherapeutics, either cisplatin (Cis) or Dox, further upregulated the levels of IGF1R expression 1.5- (Cis) to 1.6 (Dox)-fold compared with untreated control tumors (Figure S1). In comparison with a high level of IGF1R detected in pancreatic tumors, the levels of IGF1R in major normal organs were relatively low. Immunofluorescence labeling using an anti-IGF1R antibody that reacted with both human and mouse IGF1R detected only a weak fluorescent signal in the liver and muscle (Figure S2).
Previously, we developed urokinase plasminogen activator receptor (uPAR)-targeted theranostic IONPs for targeted drug delivery into pancreatic cancer cells and tumor stromal cells.48 However, the level of uPAR expression in the tumor was heterogeneous with invasive tumor regions expressing a high level of uPAR, while tumor center areas had a relatively low level of uPAR.49,50 Since a high level of IGF1R was uniformly expressed in almost all pancreatic cancer cells and its level was further increased in chemoresistant tumor cells,20,21,24,25 targeting IGF1R has an advantage for efficient drug delivery into tumor cells by receptor-mediated endocytosis over drug delivery using uPAR targeting developed previously.34 Establishment of a human pancreatic PDX tumor model that highly expressed IGF1R mediated determining the effects of IGF1R-targeted nanoparticle–drug delivery, tumor imaging, and therapeutic efficacy.
Preparation and Characterization of IGF1-Conjugated IONPs with Dox Payload
IGF1-conjugated IONPs and theranostic IONPs carrying Dox were developed and characterized. For all in vitro and in vivo studies, IGF1R-targeted IONPs were produced by conjugation of a near-infrared dye (NIR 830)51-labeled human recombinant IGF1 to a 10 nm core size and amphiphilic polymer-coated IONP as shown in Figure 2a and the Materials and Methods section. The molar ratio of IGF1 to IONPs for conjugation is 20:1. The size of an IGF1 (7.6 kDa) was much smaller than an antibody (150 kDa), which allowed conjugation of higher numbers of targeting ligands compared to 2 to 3 antibody molecules that could be conjugated to each nanoparticle. Conjugation efficacy was 88.4% as determined by HPLC analysis of supernatants collected after purification (Figures 2c and S3). Transmission electron microscopy (TEM) showed that IGF1-conjugated IONPs remained as disperse nanoparticles with a 10 nm core size and a thin layer of the polymer coating (Figure 2b). Examination of the hydrodynamic size of IGF1-IONPs by dynamic light scattering (DLS) revealed that a polymer-coated IONP was 14.5 nm and conjugation of IGF1 increased its size to 17.2 nm (Figure 2d).
Figure 2.

Production and characterization of IGF1R-targeted IONP-Dox. (a) Schematic illustration for conjugation of NIR830-IGF1 to amphiphilic polymer-coated IONPs and encapsulation of Dox to IGF1-IONPs. (b) The core size of IGF-IONPs was confirmed using TEM images. (c) Quantification of IGF1 conjugated on each IONP by measuring IGF1 in the supernatant using HPLC. (d) Hydrodynamic sizes of IONPs, IGF1-IONPs, and IGF1-IONP-Dox were determined by the dynamic light scattering (DLS) method.
The chemotherapy drug Dox, a hydrophobic molecule, was encapsulated in the hydrophobic space of the amphiphilic polymer layer on the IONP surface and could be released in a pH-dependent fashion, as the amine group of Dox could be protonated for conversion into a hydrophilic molecule to be released from the nanoparticle.52,53 After loading Dox onto the IGF1-conjugated IONPs in borate buffer (pH 8.5) at a Dox:iron of IONP ratio of 1 mg:2 mg, the amount of Dox encapsulated in the IONPs was determined by subtracting the amount of Dox in the supernatant from the input drug amount. The Dox loading capacity was 0.15 mg of Dox per mg of IGF1-IONPs, which is equivalent to be approximately 980 Dox molecules per IGF1-IONP. The hydrodynamic size of IGF1-IONP-Dox was 20.4 nm (Figure 2d). There was only a slight change in zeta-potentials following IGF1 conjugation and Dox encapsulation. The stability of IGF1-IONP-Dox in water, PBS, and cell culture medium (DMEM with 10% FBS) was evaluated over a 48 h period using DLS (Figure S4). IGF1-IONP-Dox showed a similar size of ~20 nm in water or PBS. When added into the DMEM cell culture medium with FBS, its size increased to 80 nm (Figure S4), which was likely due to nonspecific adsorption of serum proteins and formation of small particle clusters. However, those particle sizes were still within the optimal nanoparticle size for extravasation through tumor vessels (<200 nm) for nanoparticle delivery.
Dox release profiles were then examined under pH 5.5 or 7.4. IGF1-IONP-Dox showed a pH-dependent release profile. Only 6.3% of Dox molecules were released from IGF1-IONP-Dox in a pH 7.4 buffer for 48 h. However, more than 80% of Dox was released from the nanoparticle after 48 h incubation in a pH 5.5 buffer (Figure S5). Therefore, results of this study suggested that Dox payloads would be released from the nanoparticles following receptor-mediated endocytosis into an acidic endosomal environment.
Lack of Cell Proliferation Stimulation in Tumor Cells Following in Vitro and in Vivo Treatment with IGF1-Conjugated IONPs
For the development of a tumor-targeted therapeutic agent that involves a growth factor, it is important to address a concern of its potential growth-stimulating effect. Since the binding of the receptor-targeted nanoparticles to tumor cells will likely lead to receptor-mediated internalization of the nanoparticle-receptor complexes, we hypothesized that conjugation of a growth factor to nanoparticles will attenuate its cell growth promotion effect. We first examined specific binding of IGF1-conjugated nanoparticles to IGF1R-expressing human pancreatic cancer cells. The MIAPaCa-2 human pancreatic cancer cell line was used as a cell model for in vitro targeting and toxicity studies (Figure 3). A bovine serum albumin (BSA)-conjugated nanoparticle was used as a non-IGF1R-targeted nanoparticle control. First, the level of IGF1R expression in MIAPaCa-2 cells was determined by immunofluorescence labeling (Figure 3a). Cells were then incubated with IGF1-IONPs or BSA-IONPs for 4 h. Following washing and fixing cells, Prussian blue staining showed a high level of IONP-bound cells treated with IGF1-IONP but not with BSA-conjugated-IONP (Figure 3b).
Figure 3.

Effects of IGF1-IONPs and IGF1-IONP-Dox on cell proliferation and viability in vitro and in vivo. (a) The level of IGF1R in MIAPaCa-2 cells was examined by immunofluorescence labeling using an anti-IGF1R antibody (red). (b) Prussian blue staining of cells incubated with IONPs, BSA-IONPs, and IGF1-IONPs at 20 μg/mL of iron equivalent dose for 4 h. Cells were also counterstained with nuclear fast red. (c) Effect of IGF1 and IGF1-IONP on cell proliferation in vitro. The percentage of viable MIAPaCa-2 cells 96 h following incubation with IGF1 or IGF1-IONPs for 4 h at equivalent IGF1 concentrations was determined by cell proliferation assay. *p < 0.05; **p < 0.001. (d) Effect of IGF-1-IONPs on tumor cell proliferation in vivo in human pancreatic PDX-tumor xenografts. Ki67-positive cells in tumor tissue sections after two tail vein injections of 20 mg/kg iron dose of IGF1-IONPs were determined by immunofluorescence labeling using an anti-Ki67 antibody. (e) In vitro cytotoxicity of unconjugated Dox, BSA-IONP-Dox, and IGF1-IONP-Dox in MIA PaCa-2 cells. Scale bars are 100 μm.
Next, we tested the effect of IGF1-IONP on cell proliferation in human pancreatic cancer cells in vitro. After incubating MIAPaCa-2 cells with unconjugated human recombinant IGF1 or IGF1-IONPs at 50 to 200 ng/mL of IGF1 equivalent concentrations for 96 h, we found that while unconjugated IGF1 increased cell proliferation as expected, conjugation of the same amount of IGF1 to nanoparticles significantly blocked its effect on stimulating cell proliferation (Figure 3c). To further confirm this observation, we examined cell proliferation status in orthotopic pancreatic PDX tumors derived from patient #1 tissue after two intravenous injections of 20 mg/kg of iron equivalent dose of IGF1-IONPs, which is the IONP dose administrated for targeted therapy. Results of immunofluorescence labeling using an antibody against a cell proliferating biomarker Ki67 in tumor tissue sections showed that repeated treatment of the mice bearing PDX tumors with IGF1-IONPs did not increase the cell proliferation index in tumor tissues in comparison with tumor cells in the control, no treatment tumor tissue sections (Figure 3d). Therefore, conjugation of IGF1 to nanoparticles decreased its growth-stimulating function but retained targeting ability. However, mechanisms of this observation have yet to be fully elucidated.
The cytotoxic effect of IGF1R-targeted theranostic IONPs on tumor cells was also examined in human pancreatic cancer MIAPaCa-2 cells. IONP-Dox was used as a nontargeted nanoparticle control. Results of the cell proliferation assay showed a Dox dose-dependent cytotoxicity in tumor cells treated with conventional Dox, IONP-Dox, and IGF1-IONP-Dox (Figure 3e). In vitro results indicated that Dox and IGF1-IONP-Dox had a comparable cytotoxic effect. Nontargeted IONP-Dox induced tumor cell death, but its effect was weaker than that of Dox and IGF1-IONP-Dox. It should be noted that there was no delivery hindrance issue for free Dox to enter into cancer cells in tissue culture. Therefore, similar cytotoxic effects were seen in cancer cells treated with Dox and IGF1-IONP-Dox.
Selective Accumulation of IGF1-Conjugated IONPs in Human Pancreatic PDX Tumors after Systemic Delivery
Nude mice bearing orthotopic pancreatic PDX tumors with sizes around 5 mm in diameter received a tail vein injection of NIR-830 dye labeled IGF1-IONPs and nontargeted BSA-IONPs at 20 mg/kg of iron equivalent dose. Wholebody NIR optical imaging performed 24 h after the IONP administration showed strong optical signals in the lower abdominal areas that corresponded to the location of orthotopic tumors (Figure 4a). As a representative example shown in Figure 4a, the mean signal intensities of the tumor area in the mice that received IGF1-IONPs were 996 (left side image of the mouse) and 1301 (mouse front image), compared to 319 (left side image) and 371 (front image) in the mouse that received BSA-IONPs. Representative ex vivo images of tumors and normal organs showed the presence of high levels of optical signal in tumors injected with IGF1-IONPs (mean signal intensity: 898) but not BSA-IONPs (mean signal intensity: 398) (Figure 4b). Although a high level of optical signal was detected in the kidney, we did not find iron accumulation in the kidney tissue sections (Figure S6), which supported the notion that optical signal in the kidneys might be due to the renal clearance of free small dye molecules or dye-labeled targeting ligands that were cleaved from NIR-830 dye labeled IGF1-IONPs by macrophages in the liver and spleen.
Figure 4.

Detection of targeted delivery of IGF1-IONPs into orthotopic pancreatic PDX tumors by optical imaging, MRI, and Prussian blue staining. (a) NIR optical imaging of whole body 24 h after IONP administration. Optical images were overlaid with X-ray images of the mice. Red numbers shown are the mean signal intensities of tumor areas. (b) Ex vivo optical imaging of representative tumors and organs following sacrificing the mice. Optical images were overlaid with bright-ffeld images to show location and size of tumors and organs. Numbers shown were the mean optical signal intensities of tumors or organs. (c) Pre and post 24 h T2-weighted MR images. Numbers shown are relative mean MRI signal intensities of the entire tumor. Bar figure shows quantification of MRI signals in the tumors prior to and 24 h after administration of difierent IONPs. Relative MRI signal was defined as the mean intensity of the tumor divided by the mean intensity of the muscle on the same MR image. *p < 0.0001. Pink arrows indicate the location of pancreatic PDX-tumor lesions. (d) Prussian blue staining of frozen tumor sections indicating the presence of IONPs in both tumor edge and tumor center (green arrows) after IGF1-IONP administration via the tail vein. IONPs were not detectable in the tumor treated with nontargeted BSA-IONPs. Blue: IONP-positive cells. Red: Nuclear fast red. (e) IGF1R antibody labeled tumor tissue sections obtained from IGF1-IONP-treated mice dual stained with Prussian blue staining. IONPs (blue) were detected in IGF1R highly expressing tumor cells (white arrows) and intermediate IGF1R expressing tumor stromal cells (light blue arrows). (f) Blue IONP-positive cells detected in CK19-positive ductal tumor cells (white arrows) and CK19 negative tumor stromal cells (light blue arrows). (g) Prussian blue staining positive cells found in FAP-positive stromal fibroblasts (light blue arrows). Yellow arrows indicate tumor cell areas. (h) IGF1-IONP-treated tumor tissue section doubly labeled with IGF1R (red) and CD68 (green) and then stained with Prussian blue (blue). IONP-positive cells were detected in CD68-positive macrophages (white arrows).
Prussian blue staining of frozen tumor tissue sections confirmed the accumulation of IGF1-IONPs in the tumor (Figure 4d). However, IONPs were not detectable in the tumor obtained from the mice that received nontargeted BSA-IONPs (Figure 4d). Importantly, high levels of IGF1-IONPs were detected in both tumor and stromal cells in peripheral tumor areas as well as in the central tumor areas (Figure 4d). To further characterize cell populations responsible for uptake of IONPs, tumor tissue sections were dual labeled with fluorescence using antibodies against IGF1R, CK19 (epithelial tumor cell biomarker), FAP, and CD68 and then stained with Prussian blue for IONPs. We found IONPs in IGF1R-positive pancreatic cancer and stromal cells (Figure 4e). Intratumoral cell delivery of IGF1-IONPs was further confirmed by the observation of blue IONP staining in CK19-positive tumor cells (Figure 4f). IONPs were also detected in FAP-positive fibroblasts (Figure 4g). Finally, IONPs were colocalized with CD68-positive stromal macrophages (Figure 4h). Therefore, results of histological analysis supported the conclusion that IGF1R-targeted IONPs could be delivered into both pancreatic cancer cells and tumor stromal fibroblasts and macrophages.
To determine target specificity and biodistribution of IGF1-IONPs following systemic delivery, tissue lysates of pancreatic tumors and normal organs were examined for the amount of iron using Prussian blue color metric analysis (Figure S6). Orthotopic pancreatic PDX tumors obtained from the mice that received IGF1-IONPs had a 4.2-fold higher iron concentration than that of the tumor treated with BSA-IONPs. As expected, we also found high levels of IONPs in the liver and spleen due to nonspecific uptake by macrophages in the reticuloendothelial system. Since we consistently observed a low level of optical signals in the liver and spleen in mouse whole body and ex vivo optical imaging, it is likely that uptake of IONPs conjugated with NIR830 dye labeled IGF1 into macrophages led to degradation of NIR830-IGF1 on IONPs. NIR830 dye or NIR830-IGF1 conjugates were cleared out through the kidney and bile. Therefore, the liver and spleen had low levels of optical signals but high levels of iron concentration.
A major advantage of IGF1-IONPs as drug carriers is its potential to monitor intratumoral theranostic nanoparticle delivery by MRI. To determine specificity and sensitivity of MRI detection of targeted IONP delivery in orthotopic pancreatic PDX tumors, MRI was performed prior to and after administration of difierent IONPs (Figure 4c). T2-weighted MRI that was sensitive to IONP-induced hypointensity contrast revealed a similar level of MRI contrast in the orthotopic tumors before the IONP injection. However, a significant MRI signal decrease was found in the tumor 24 h after administration of IGF1-IONPs, indicating the accumulation of IONPs in the tumor. There was a 37.3% MRI signal decrease in the tumor of the mice that received IGF-1-IONPs (Figure 4c). On the other hand, tumors in the mice that received nontargeted BSA-IONPs did not show apparent change in MRI contrast (Figure 4c).
Inhibition of Orthotopic Human Pancreatic PDX Tumor Growth Following Systemic Delivery of IGF1-IONP-Dox
The effect of IGF1R-targeted therapy using IGF1-IONP-Dox was examined in nude mice bearing orthotopic pancreatic PDX tumors at the first passage. Intravenous nanoparticle delivery started when the tumors reached sizes about 5 mm in diameter by palpating the upper left of the abdominal cavity of the mice. Tumor-bearing mice received unconjugated Dox, nontargeted IONP-Dox, and IGF1-IONP-Dox at a Dox equivalent dose of 5 mg/kg body weight via tail vein injection once per week for 6 weeks.
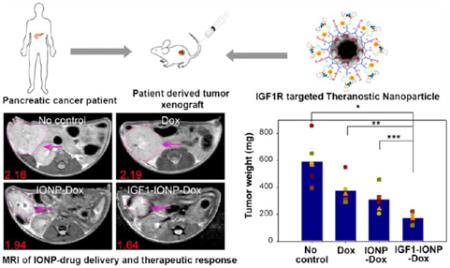
To determine the feasibility of MRI for monitoring nanoparticle-drug delivery, one mouse from each group was chosen for MRI scan 48 h following the last treatment. When comparing T2-weighted MR images of the mice treated with IGF1-IONP-Dox or nontargeted IONP-Dox, a remarkable signal decrease was detected in the tumor area of the mouse that received IGF1-IONP-Dox but not the one that received nontargeted IONP-Dox, suggesting selective accumulation of IGF1R-targeted IONP-Dox in the orthotopic pancreatic PDX tumor (Figure 5a). Additionally, the mice receiving IGF1-IONP-Dox also showed a significant reduction in tumor size compared with tumors in the mice receiving Dox, nontargeted IONP-Dox, or no-treatment control (Figure 5a). These results demonstrated the feasibility of MRI monitoring IONP-drug delivery and tumor responses of the targeted therapy.
Figure 5.

In vivo antitumor effect in an orthotopic pancreatic PDX tumor model. Nude mice bearing orthotopic pancreatic PDX tumors received 5 mg/kg Dox equivalent concentration of difierent IONP-Dox theranostic nanoparticles via tail vein injections once per week for 6 weeks. (a) T2-weighted MRI of the mice from the treatment groups confirmed the accumulation of IGF1-IONP-Dox in the tumor site and tumor growth inhibition compared to both free Dox- and nontargeted IONP-Dox-treated tumors. Pink arrows indicate the locations of pancreatic PDX-tumor lesions. Red numbers show the mean of relative MRI signal intensities of MRI image slices from the entire tumor. A 10.2% MRI signal decrease was detected in nontargeted IONP-Dox-treated tumor, while a 24.1% MRI signal decrease was seen in IGF1-IONP-Dox-treated tumor. (b) Changes in body weight during treatment. (c) Tumor growth inhibition. The mean tumor weight (navy bar) and individual tumor weight distributions as color symbols after the treatment are shown. *p < 0.0001; **p < 0.0006; ***p < 0.005. (d) Representative pancreatic PDX-tumor images with the attached spleens of each treatment group after sacrificing the mice.
The mice were sacrificed 5 days after the final injection, and tumor weights were recorded. Tumors and normal organs were collected for histological analysis. In comparison with the no treatment control group, the PDX tumors collected from the mice in all treated groups showed various degrees of tumor growth inhibition. We found that treatment with unconjugated Dox and nontargeted IONP-Dox led to 33.3% and 49.2% of tumor growth inhibition, respectively, compared to the control group (Figure 5c and d). However, there was no statistically significant difierence between those two groups (Student's t test: Dox vs IONP-Dox: p = 0.168). In contrast, PDX tumors collected from the mice that were treated with IGF1-IONP-Dox were significantly smaller than those from the control groups of no treatment or the Dox- or IONP-Dox-treated group. There was 66.6% tumor growth inhibition in the IGF1-IONP-Dox-treated mouse group. Difierences in tumor weights among groups with difierent treatments were statistically significant (Student's t test: no treatment control vs IGF1-IONP-Dox: p < 0.0001; Dox vs IGF1-IONP-Dox: p < 0.0006; IONP-Dox vs IGF1-IONP-Dox: p < 0.005). Using loss of mouse body weight as an indication of system toxicity, there was no apparent systemic toxicity for all treatment groups following six 5 mg/kg of Dox equivalent doses (Figure 5b). Histological analysis of tissue sections of the major organs after the treatment using hematoxylin and eosin (H&E) staining revealed no apparent tissue damage or morphological changes in the normal organs and tissues for all treatment groups at a Dox equivalent concentration of 5 mg/kg (Figure S7). However, three iv doses of 15 mg/kg of Dox induced severe systemic toxicity, and mice died after the third dose. In contrast, there was no systemic toxicity observed in the mice that received 15 mg/kg of Dox equivalent doses of IGF1-IONP-Dox (Figure S8). Our results demonstrated that in vivo IGF1R-targeted delivery of Dox using IGF1-IONP-Dox is more effective for the treatment of human pancreatic tumors compared to conventional Dox treatment.
Inhibition of Cell Proliferation and Induction of Apoptotic Cell Death by IGF1R-Targeted Therapy Using IGF1-IONP-Dox
Histological analysis was carried out to further evaluate and validate the targeting and antitumor effect. Tumor tissue sections from different treatment groups were examined by H&E staining, Prussian blue staining, and immunofluorescence labeling. In comparison with the morphological characteristics of H&E-stained tissue sections of no-treatment control tumors that contained dense ductal carcinoma cells, tumor cell density in Dox- or IONP-Dox-treated tumors was slightly decreased with scattered necrotic tumor cell areas (Figure 6a). After IGF1-IONP-Dox treatment, tumor cell density was further decreased with tumor cells forming large ductal-like structures that were surrounded by extensive stroma with central necrosis (Figure 6a). Prussian blue staining showed clusters of IONP-positive cells in tumor tissue sections obtained from the mice following treatment with IGF1-IONP-Dox, but not with nontargeted IONP-Dox (Figure 6b). The combination of double immunofluorescence labeling using anti-CD68 and IGF1R antibodies and Prussian blue staining revealed that following 6 weeks of IGF1-IONP-Dox treatment the majority of IONP-containing cells were CD68-positive macrophages in the tumor stroma and necrotic tumor areas, suggesting the possibility of clearance of the dead tumor cells containing IONPs by macrophages in IGF1-IONP-Dox-treated tumors (Figure 6c).
Figure 6.

Histological characterizations of pancreatic PDX tumors following IGF1-IONP-Dox treatment. (a) H&E staining of frozen tumor tissue sections revealed dense ductal carcinoma cells in the no-treatment control or free Dox-treated tumors, while IGF1-IONP-Dox treatment for 6 weeks markedly decreased tumor cell density and formed large ductal-like structures with central necrosis surrounded by extensive stroma. Scale bars are 200 μm. (b) Prussian blue staining showed clusters of IONP-positive cells in PDX-tumor tissue sections from IGF1-IONP-Dox-treated mice. The majority of IONPs were found in cells in stromal areas (blue arrows). IONPs were not detected in the control no-treatment tumor or nontargeted IONP-Dox-treated tumors. (c) Prussian blue staining of IGF1R and CD68 doubly labeled tissue sections from IGF1-IONP-Dox-treated PDX tumors showed the colocalization of IONPs in CD68-positive macrophages. IGF1R-positive tumor cells in residual tumors following 6 weeks of treatments had a low level of IONPs. Scale bars are 100 μm.
To determine the mechanisms mediating tumor growth inhibition in IGF1-IONP-Dox-treated PDX tumors, immunofluorescence labeling was performed using antibodies for cell proliferation marker (Ki67), apoptotic cell death (active caspase 3), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL assay for apoptotic cell death). A marked decrease in the number of Ki-67 positive proliferating cells was observed in the tumor treated with IGF1-IONP-Dox compared with the tumor without treatment. Interestingly, residual tumor tissues obtained from unconjugated Dox or nontargeted IONP-Dox-treated mice retained high levels of Ki-67 positive proliferating cells. These results suggested that targeted therapy using IGF1-IONP-Dox had a stronger inhibitory effect on tumor cell proliferation compared with unconjugated Dox or nontargeted IONP-Dox (Figure 7a and b). Additionally, results from the TUNEL assay indicated that IGF1-IONP-Dox treatment induced 2-fold higher numbers of apoptotic cell death compared with no treatment or unconjugated Dox-treated controls (Figure 7a and c). The presence of the cleaved, active caspase 3 in cells is an indication of activation of apoptotic cell death. We found that the number of active caspase-3-positive cells in IGF1-IONP-Dox-treated tumor tissues was 6.1- to 8.5-fold higher than that in control no-treatment or Dox-treated tumors (Figure 7a and d). Therefore, strong inhibition of tumor cell proliferation and effective induction of the apoptotic cell death observed in IGF1-IONP-Dox-treated tumors likely contributed to the antitumor effect observed in the mice.
Figure 7.

Evaluation of the effects of IGF1-IONP-Dox treatment on cell proliferation and induction of apoptotic cell death in the pancreatic PDX tumors. (a) Immunofluorescence labeling of tumor tissue sections for cell proliferation marker (Ki67, green), TUNEL assay (green), and apoptotic cell death (active caspase-3, red). Hoechst 33342 nuclear stain (blue) was used to identify total cell populations in tumor tissue sections. (b-d) Quantitative analysis for (b) Ki-67-positive cells, (c) apoptotic cells, and (d) cleaved active caspase-3-positive cells from six randomly selected microscopic ffelds of tumor sections by ImageJ. *p < 0.03; **p < 0.007; ***p < 0.0001. Scale bars are 100 μm.
Results of immunofluorescence labeling of active caspase 3 also showed specific induction of apoptotic cell death in pancreatic tumor cells but not in normal tissues (Figure 8a). For example, normal pancreas and spleen adjacent to tumor areas lacked active caspase-3-positive cells, while high levels of caspase-3-positive cells were found in nearby ductal carcinoma cells (Figure 8a). Although nonspecific uptake of IGF1-IONP-Dox was detected in the spleen (Figure S6), active caspase 3 was not found in the spleen tissues, suggesting that nonproliferating macrophages may have a low sensitivity to DNA-damaging drugs.
Figure 8.

Elucidation of the induction effects of the apoptotic cell death in pancreatic PDX-tumor cells following IGF1-IONP-Dox targeted or nontargeted IONP-Dox therapy. (a) Selective induction of apoptotic cell death in pancreatic PDX tumors following systemic delivery of IGF1-IONP-Dox once per week for 6 weeks. Immunofluorescence labeling for active caspase-3-positive cells showed a high level of apoptotic cell death (red) in ductal cancer cells (white arrows) in tumor tissue sections. However, there was no apoptotic cells detected in adjacent normal pancreas and spleen. (b) Phenotypic characterization of the apoptotic cells in IGF1-IONP-Dox-treated tumors by double immunofluorescence labeling of apoptotic cells (active caspase 3, red) and epithelial tumor cells (CK19, green). High levels of the apoptotic cells were detected in ductal-like and CK19-positive tumor cells (white arrows) in the tumor peripheral (upper panel) and central (lower panel) areas. Prussian blue staining shows IONP-positive cells in the tumor area. H&E staining of a similar tumor area shows the presence of tumor cell clusters. (c) Induction of apoptotic cell death in tumor stromal cells after 6 weeks of treatments of nontargeted IONP-Dox. A high level of active caspase-3-positive cells was found in the CK19-negative but CD163- (active macrophages) or FAP-positive cell population at the tumor edge (upper and middle panels, yellow arrows). H&E staining shows that the areas with apoptotic cells had morphological and histological features of tumor stromal cells (yellow arrows). In the tumor center areas, low to intermediate levels of active caspase-3-positive cells were detected in tumor stroma areas (lower panel, yellow arrows). White arrows indicate a low level of CK19-positive tumor cells had active caspase 3 labeling. Prussian blue staining revealed no detectable IONPs in similar areas. Scale bars are 100 μm.
Nontargeted nanoparticle formulated Dox, such as Doxil, have been used for cancer therapy in the clinic through EPR-effect-mediated nanoparticle-drug delivery.40,54,55 Our in vivo study result also showed nearly 50% of tumor growth inhibition in the mice treated with nontargeted IONP-Dox (Figure 5c). To compare cell types that underwent apoptotic cell death in PDX tumors following difierent formulations of Dox treatment, we dual labeled the tissue sections with an epithelial cell marker, CK19, and active caspase 3. In general, unconjugated Dox induced only low levels of apoptotic cells in CK19-positive pancreatic tumor cells following six weekly treatments of 5 mg/kg of Dox in nude mice bearing pancreatic PDX tumors (Figure S9). We observed high levels of active caspase-3-positive apoptotic cells forming clusters in CK19- positive duct-like pancreatic tumor cells in both peripheral and central areas of the tumor tissues obtained from the mice treated with IGF1-IONP-Dox (Figure 8b). IONP-positive cells were also detected in those tumor areas (Figure 8b). In contrast, after treatment with nontargeted IONP-Dox, high levels of active caspase-3-positive cells were detected mostly in the tumor edge, which was enriched in active tumor stromal cells such as active fibroblast (FAP) and active macrophage (CD163), but lacked CK19 expression (Figure 8c). A low level of caspase-3-positive cells was also detected in the central areas of the tumor tissues, and most of the apoptotic cells were in CK19-negative tumor stromal cells (Figure 8c). CK19-positive tumor cells had a low level of active caspase 3 in the tumor tissues treated with nontargeted IONPs (Figure 8c). H&E staining of the tissue section in the same tumor area further showed the location of epithelial ductal cancer cells and tumor stromal cells (Figure 8c). Prussian blue staining failed to detect IONP-positive cells in tumor stromal areas with the apoptotic cells in similar tumor areas (Figure 8c). Therefore, it is likely that the modest antitumor effect observed in the nontargeted IONP-Dox-treated PDX tumors was the result of the EPR effect-mediated delivery into the interstitial space to release drug and induced cell death mostly in stromal cells. Since IONPs were not internalized into cells, Prussian blue staining was not able to detect the IONPs in tumor tissue sections. The ability of IGF1R-targeted drug delivery to reach pancreatic tumor cells and tumor stromal cells led to a stronger anti-tumor effect in the tumor-bearing mice treated with IGF1-IONP-Dox than that of nontargeted IONP-Dox-treated mice.
CONCLUSIONS
We have developed a novel growth factor receptor targeted theranostic nanoparticle (IGF1-IONP-Dox) for targeted drug delivery into IGF1R-overexpressing cancer cells and tumor stromal cells. The efficiency of IGF1R-targeted nanoparticle drug delivery, ability of noninvasive tumor imaging, and effects of targeted therapy were determined in an orthotopic human pancreatic PDX tumor model, which exhibits comparable histological characteristics, phenotypic features, tumor cell heterogeneity, and tumor microenvironment of the primary human pancreatic tumor. It is the most appropriate pancreatic tumor model for the evaluation of the effects of targeted nanoparticle-drug delivery and therapeutic response using theranostic nanoparticles that target both pancreatic ductal carcinoma and tumor-associated stromal cells. Intratumoral nanoparticle delivery of IGF1R-targeted theranostic IONPs was efficient and detectable by noninvasive optical imaging and MRI as well as histological analysis of tumor and normal tissues. IGF1R-targeted therapy by systemic deliveries of IGF1-IONP-Dox significantly inhibited the growth of orthotopic pancreatic PDX tumors by inhibition of cell proliferation and induction of apoptotic cell death in pancreatic cancer cells. However, IGF1-IONPs themselves did not stimulate tumor cell proliferation in vitro and in vivo. There was no systemic toxicity observed following multiple doses of intravenous administrations of IGF1R-targeted theranostic nanoparticles. The results of this study also demonstrated MRI-guided cancer therapy for the evaluation of delivery efficiency of targeted theranostic IONPs and responses to targeted therapy in orthotopic pancreatic PDX tumors. Therefore, IGF1R-targeted theranostic nanoparticles are a promising receptor-targeted drug delivery system for further development of effective approaches for the treatment of stroma-rich pancreatic cancer.
MATERIALS AND METHODS
Establishment of an Orthotopic Human Pancreatic PDX Tumor Model in SCID and Nude Mice
A surgically resected human pancreatic cancer tissue was obtained from pancreatic cancer patient #1 following an approved Emory Institutional Review Board protocol. Fresh tumor tissue fragments were collected at 4 °C in HBSS supplemented with antibiotics. Within 2 h of surgical resection, tumor tissues were cut into 1-2 mm fragments and implanted into the pancreas of immunodeficient SCID mice (8 to 10 weeks old, female) using a surgical procedure approved by Emory Institutional Animal Care and Use Committee. Excess tissues were frozen in liquid nitrogen and stored for further pathological analysis. After surgery, the tumor growth in the mice was monitored by palpating the left upper abdomen twice per week. Orthotopic tumors grew to 5 to 10 mm in diameter in about 8 to 10 weeks. To conduct a large-scale efficacy study, the SCID mice bearing PDX tumors were sacrificed, and tumor fragments at 1 to 2 mm sizes were then implanted into the pancreas of 6- to 8-week-old female nude mice. Over 90% of nude mice had orthotopic tumor growth in about 6 to 7 weeks. All studies shown were conducted using the first passage of the pancreatic PDX tumors derived from patient #1 pancreatic cancer tissue.
Production and Purification of Recombinant Human IGF-1
Recombinant human IGF-1 was expressed in an E. coli strain as a 8.1 kDa precursor protein (pro-IGF1) containing the N-terminal-fused pentapeptide Met(M)-Lys(K)-Lys(K)-Ile (I)-Met (M) as a leader sequence that facilitates the expression and folding of the protein. The expression of pro-IGF-1 was induced by a temperature shift from 30 °C to 42 °C. The inclusion bodies in the insoluble fraction containing the partitioned pro-IGF-1 were collected by continuous flow centrifugation. The pro-IGF1 was refolded and purified by preparative reverse-phase HPLC in an acetonitrile/TFA system. The conversion of pro-IGF-1 to the mature rhIGF-1 was achieved by enzymatic cleavage of the N-terminal pentapeptide using aminopeptidase (1 mg of enzyme for 200 mg of pro-IGF-1). The correctly folded rhIGF-1 protein was further purified by preparative reverse-phase HPLC using the Waters DeltaPrep 4000 system in an acetonitrile/TFA platform to remove the enzyme and any remaining undigested precursor.
Production and Characterization of IGF1R-Targeted Nanoparticles
Amphiphilic polymer-coated IONPs with a core size of 10 nm were obtained from Ocean NanoTech, LLC (San Diageo, CA, USA). NIR 830-labeled human recombinant IGF1 peptide was produced by conjugating active NIR 830-NHS, produced in our group,56 to IGF1 at pH 8.6. NIR 830-labeled human recombinant IGF1 peptides were conjugated to the polymer-coated IONPs mediated by an amide bond between carboxyl groups on the surface of the polymer and amine groups of IGF1 peptides using ethyl-3-dimethylaminopropyl carbodiimide (EDAC, Sigma-Aldrich) and N-hydroxysulfosuccinimide (sulfo-NHS, Sigma-Aldrich) according to the carbodiimide method (Figure 2). Briefly, 109 μg of EDAC and 197 μg of sulfo-NHS were added to the aqueous solution of 1 mg of IONPs in 10 mM borate buffer (pH 4.5), allowing activation of IONPs for 10 min. Activated IONPs were then collected by centrifugation at 3000 rpm for 10 min using a Nanosep 100 K column (Pall Corporation, Port Washington, NY, USA) and then reacted with 420 μg of IGF1 in 10 mM borate buffer (pH 8.5) to produce IGF1-IONP at a molar ratio of IGF1 to IONP of 20:1. The reaction was carried out for 12 h at 4 °C. The final IGF1-IONPs were purified using a Nanosep 100 K column. Conjugation of IGF1 to IONP was confirmed by HPLC analysis of the amount of remaining IGF1 in the supernatants. Size and zeta potential of IGF1-IONP were measured by a Zetasizer Nano (ZS90, Malvern Instruments Ltd., Worcestershire, UK). The core sizes of copolymer-coated and surface-functionalized nanocrystals, IGF1-IONP, and the thickness of polymer layers were viewed and measured by TEM after negative staining (Hitachi H-7500 instrument, 75 kV).
Encapsulation of Dox into IGF1-IONP and Determination of Drug Release Conditions
A 100 μL amount of Dox (500 μg/mL) was mixed with 200 μL of IGF1-IONP (500 μg/mL) in 10 mM borate buffer (pH 8.5). The mixture was shaken at room temperature for 4 h. IGF1-IONP-Dox was then collected by centrifugation at 3000 rpm for 10 min using a Nanosep 100 K column. To determine the amount of encapsulated Dox, unconjugated Dox in the supernatant was collected and analyzed by HPLC. Dox concentration in the supernatant was calculated based on the standard curve obtained from pure Dox. The amount of Dox in the supernatant was subtracted from the input drug amount to calculate the amount of drug encapsulation in IONPs.
The pH-dependent drug release was determined by incubating IGF1-IONP-Dox under pH 5.5 and 7.4 buffer conditions for difierent time points. Dox released from IGF1-IONP-Dox was collected by centrifugation at 3000 rpm for 10 min using a Nanosep 100 K column, and the amount of released Dox in the supernatant was measured by HPLC using the same standard curve as described previously.
Uptake of IGF1-IONP by Pancreatic Cancer Cells
The MIAPaCa-2 human pancreatic cancer cell line (American Type Culture Collection, Manassas, VA, USA) was cultured in a 12-well plate with a density of 20 000 cells/well. Cultures were maintained at 37 °C under a humidified condition with 5% CO2. After 24 h of plating, cells were washed once with PBS, and 20 μg/mL of iron equivalent IONP solution of IGF1-IONPs or nontargeted BSA-IONPs was then added. Cells were incubated with the IONPs for 4 h and then washed three times with cold PBS to remove unbound nanoparticles. Cells were then fixed with 4% paraformaldehyde in PBS, and Prussian blue staining was used to determine the presence of iron in the cells.
Prussian Blue Staining
Fixed cells or frozen tissue sections were incubated with a mixture of 5% potassium ferrocyanide(II) trihydrate and 5% HCl solution for 15 min. After being washed three times with distilled water, cells were counterstained with nuclear fast red solution for 5 min. Following consecutive dehydrations with 70% and 100% EtOH and two rinses in xylene, the slides were mounted. The result of Prussian blue staining was examined by light microscopy.
Cell Proliferation Assay
MIAPaCa-2 cells were cultured in a 96-well plate at a density of 5000 cells/well. After 24 h of plating, cells were washed once with cold PBS. Different IONP-Dox conjugates were diluted in the culture medium at various Dox concentrations and then added into cell cultures. Unconjugated Dox was used as a treatment control. After incubating for 4 h when IONP-Dox bound to and entered into tumor cells, cells were washed three times with cold PBS to remove unbound IONPs. A 100 μL amount of fresh medium was then added to the plate. Cells were incubated for an additional 72 h, and viability of the cells was determined by the Alamar Blue assay (Life Technologies, NY, USA). Cells treated with culture medium alone were used as the no-treatment control. Results shown are the mean value of six repeat studies.
In Vivo Near-Infrared Fluorescence Optical Imaging
Mice were subjected to NIR optical and MR imaging 6 weeks after implanting tumor fragments into nude mice. A 400 picomolar (pmol) amount of NIR-830-labeled BSA-IONPs or IGF1-IONPs was injected via the tail vein into the tumor-bearing mice. NIR optical imaging was conducted using the Kodak in vivo FX imaging system (Carestream Health Inc., Rochester, NY, USA). Mice were sacrificed, and tumors and normal organs were collected for ex vivo optical imaging. All optical images were captured using an 800 nm excitation and 850 nm emission filter set with 3 min exposure time and a gamma value of 0.2. Optical images were analyzed using the software provided by the Kodak imaging system. Regions of interest (ROIs) were selected for measuring the mean fluorescence intensity of tumors and corresponding body background.
In Vivo MRI Scan
MRI scans were carried out on a 4.7 T small-animal MRI scanner (Oxford Magnet Technology, Oxford, UK). For measuring targeted intratumoral delivery of IONPs, mice that received a tail vein injection of 400 pmol of BSA-IONPs or IGF1-IONPs were examined by a T2-weighted MRI scan. MRIs were acquired before and 48 h after IONP injection using a T2-weighted fast spin echo imaging sequence. For monitoring therapeutic responses in PDX tumors, MRI scanning was performed on mice from each treatment group 48 h after the last treatment. MRI contrast in the tumor was quantitatively analyzed using the ROI method and ImageJ software (National Institutes of Health, Bethesda, MD, USA). Averaged signal intensities of the ROI were obtained from the entire tumor area in all MR image slices. MRI contrast in the muscle area was measured as an internal control. MRI signal intensity in the tumor was normalized with the signal of muscle as the intensity of tumor signal/muscle signal.
In Vivo Antitumor Efficacy
Nude mice bearing orthotopic pancreatic PDX tumors derived from patient #1 tumor were randomized into four experimental groups with six mice in each group, including no-treatment control, unconjugated Dox, IONP-Dox, and IGF1-IONP-Dox. Treatment started when the tumor reached sizes of around 5 mm at a Dox equivalent dose of 5 mg/kg body weight via the tail vein once per week for six injections. Mouse body weight was monitored once per week. After the final treatment, a representative mouse from each group was selected for MRI scanning. Mice in all groups were sacrificed 5 days after the final treatment, and tumors and normal tissues were collected for histological, chemical, and immunofluorescence analyses.
Immunofluorescence Labeling
Frozen tissue sections of tumor and normal tissues were used for single or double immunofluorescence labeling. An antibody specific for human IGF1R with a cross reactivity for mouse IGF1R was obtained from Sigma-Aldrich (dilution 1:200). An anti-human fibroblast active protein antibody (Santa Cruz Biotechnology, dilution 1:100) was used for identification of active human fibroblasts in tumor stroma. Anti CD68 antibody (AbD Serotec, dilution 1:100) and CD163 antibody (Santa Cruz Biotechnology, dilution 1:200) were used to identify macrophages, and anti-CD31 antibody (eBioscience, dilution 1:200) was for labeling endothelial cells in tumor blood vessels. Mouse monoclonal anti-CK19 antibody was from Sigma-Aldrich (dilution 1:100). Alexa Fluor 488 dye (green, Invitrogen, dilution 1:500) or Alexa Fluor 555 dye (red, Invitrogen, dilution 1:500) labeled secondary antibodies were used to detect biomarker-positive cells. Images were taken using fluorescence microscopy (Nikon Eclipse E400, Tokyo, Japan).
Picro-Sirius Red Staining of Collagen
Picro-Sirius red staining solution was prepared by dissolving 0.5 g of Sirius red F3B (Sigma-Aldrich) in 500 mL of a saturated aqueous solution of picric acid (Sigma-Aldrich). Picro-Sirius red staining was performed by incubating frozen tissue sections in the staining solution for 1 h, washing twice in acidified water (5% acetic acid in water), dehydrating twice in 100% ethanol, and then clearing in xylene.
Statistical Analysis
One-way ANOVA, the standard Student's t test, and the modified t test were used for statistical analysis of differences among experimental groups in cell viability of tumor cells and tumor weights among different mouse groups. Statistically significant differences were defined when the p value was <0.05 between groups.
Supplementary Material
Acknowledgment
We thank Dr. Erica N. Bozeman for editing the manuscript. This research project was supported by the NIH/NCI grants U01CA151810 (L.Y. and H.M.), R01 CA154129A1 (L.Y.), R01CA154846 (H.M. and L.Y.), NIH/NCI SBIR Contract No. HHSN261201200029C (Y.A.W. and L.Y.), U01-CA-151837 (F.M.U.), and R01-CA-154471 (F.M.U.), and by the Nancy Panoz Endowed Chair Funds (L.Y.).
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b01288.
DLS of IGF1-IONP-Dox, Dox release profiles, and additional immunofluorescence and H&E staining images of tumors and organs from orthotopic pancreatic PDX tumor tissues (PDF)
REFERENCES
- 1.Siegel R, Ma JM, Zou ZH, Jemal A. Cancer Statistics, 2014. Ca-Cancer J. Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Warshaw AL, Fernandez-del castillo C. Pancreatic Carcinoma. N. Engl. J. Med. 1992;326:455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 3.Willett CG, Czito BG, Bendell JC, Ryan DP. Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2005;23:4538–4544. doi: 10.1200/JCO.2005.23.911. [DOI] [PubMed] [Google Scholar]
- 4.Whatcott C, Han H, Posner RG, Von Hoff DD. Tumor-Stromal Interactions in Pancreatic Cancer. Crit. Rev. Oncog. 2013;18:135–51. doi: 10.1615/critrevoncog.v18.i1-2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, Heuchel RL, Haas SL, Lohr JM. Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers. 2014;6:2137–54. doi: 10.3390/cancers6042137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Lohr JM, Friess H. Pancreatic Cancer Microenvironment. Int. J. Cancer. 2007;121:699–705. doi: 10.1002/ijc.22871. [DOI] [PubMed] [Google Scholar]
- 7.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat. Rev. Gastroenterol. Hepatol. 2012;9:454–467. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 9.Bergers G, Hanahan D. Modes of Resistance to Anti-Angiogenic Therapy. Nat. Rev. Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007;99:1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 11.Eck W, Craig G, Sigdel A, Ritter G, Old LJ, Tang L, Brennan MF, Allen PJ, Mason MD. PEGylated Gold Nanoparticles Conjugated to Monoclonal F19 Antibodies as Targeted Labeling Agents for Human Pancreatic Carcinoma Tissue. ACS Nano. 2008;2:2263–2272. doi: 10.1021/nn800429d. [DOI] [PubMed] [Google Scholar]
- 12.Patra CR, Bhattacharya R, Wang EF, Katarya A, Lau JS, Dutta S, Muders M, Wang SF, Buhrow SA, Safgren SL, et al. Targeted Delivery of Gemcitabine to Pancreatic Adenocarcinoma Using Cetuximab as a Targeting Agent. Cancer Res. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 13.Yang LL, Mao H, Wang YA, Cao ZH, Peng XH, Wang XX, Duan HW, Ni CC, Yuan QG, Adams G, et al. Single Chain Epidermal Growth Factor Receptor Antibody Conjugated Nanoparticles for in vivo Tumor Targeting and Imaging. Small. 2009;5:235–243. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanda N, Kattumuri V, Shukla R, Zambre A, Katti K, Upendran A, Kulkarni RR, Kan P, Fent GM, Casteel SW, et al. Bombesin Functionalized Gold Nanoparticles Show in vitro and in vivo Cancer Receptor Specificity. Proc. Natl. Acad. Sci. U. S. A. 2010;107:8760–8765. doi: 10.1073/pnas.1002143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patra CR, Bhattacharya R, Mukhopadhyay D, Mukherjee P. Fabrication of Gold Nanoparticles for Targeted Therapy in Pancreatic Cancer. Adv. Drug Delivery Rev. 2010;62:346–361. doi: 10.1016/j.addr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng H, Xue M, Xia T, Ji ZX, Tarn DY, Zink JI, Nel AE. Use of Size and a Copolymer Design Feature To Improve the Biodistribution and the Enhanced Permeability and Retention Effect of Doxorubicin-Loaded Mesoporous Silica Nanoparticles in a Murine Xenograft Tumor Model. ACS Nano. 2011;5:4131–4144. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng H, Zhao Y, Dong JY, Xue M, Lin YS, Ji ZX, Mai WX, Zhang HY, Chang CH, Brinker CJ, et al. Two-Wave Nanotherapy To Target the Stroma and Optimize Gemcitabine Delivery To a Human Pancreatic Cancer Model in Mice. ACS Nano. 2013;7:10048–10065. doi: 10.1021/nn404083m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain RK, Stylianopoulos T. Delivering Nanomedicine to Solid Tumors. Nat. Rev. Clin. Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouban A, Muraca P, Yeatman T, Coppola D. Expression and Distribution of Insulin-Like Growth Factor-1 Receptor in Human Carcinomas. Hum. Pathol. 2003;34:803–808. doi: 10.1016/s0046-8177(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 21.Valsecchi ME, McDonald M, Brody JR, Hyslop T, Freydin B, Yeo CJ, Solomides C, Peiper SC, Witkiewicz AK. Epidermal Growth Factor Receptor and Insulin like Growth Factor 1 Receptor Expression Predict Poor Survival in Pancreatic Ductal Adenocarcinoma. Cancer. 2012;118:3484–3493. doi: 10.1002/cncr.26661. [DOI] [PubMed] [Google Scholar]
- 22.Bergmann U, Funatomi H, Yokoyama M, Berger HG, Korc M. Insulin-Like Growth Factor-I Overexpression in Human Pancreatic Cancer, Evidence for Autocrine and Paracrine Roles. Cancer Res. 1995;55:2007–2011. [PubMed] [Google Scholar]
- 23.Zheng D, Golubovskaya V, Kurenova E, Wood C, Massoll NA, Ostrov D, Cance WG, Hochwald SN. A Novel Strategy to Inhibit FAK and IGF-1R Decreases Growth of Pancreatic Cancer Xenografts. Mol. Carcinog. 2009;49:200–209. doi: 10.1002/mc.20590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C, et al. Insulin-Like Growth Factor-1 Receptor (IGF-1R) as a Biomarker for Resistance to the Tyrosine Kinase Inhibitor Gefitinib in Non-small Cell Lung Cancer. Cell. Oncol. 2013;36:277–288. doi: 10.1007/s13402-013-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The Insulin-Like Growth Factor 1 Receptor Causes Acquired Resistance to Erlotinib in Lung Cancer Cells with the Wild-Type Epidermal Growth Factor Receptor. Int. J. Cancer. 2014;135:1002–1006. doi: 10.1002/ijc.28737. [DOI] [PubMed] [Google Scholar]
- 26.Shao N, Lu S, Wickstrom E, Panchapakesan B. Integrated Molecular Targeting of IGF1R and HER2 Surface Receptors and Destruction of Breast Cancer Cells using Single Wall Carbon Nanotubes. Nanotechnology. 2007;18:315101. [Google Scholar]
- 27.Kristensen C, Wiberg FC, Andersen AS. Specificity of Insulin and Insulin-Like Growth Factor I Receptors Investigated using Chimeric Mini-Receptors - Role of C-Teriminal of Receptor Alpha Subunit. J. Biol. Chem. 1999;274:37351–37356. doi: 10.1074/jbc.274.52.37351. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Mao H, Cao ZH, Wang YA, Peng XH, Wang XX, Sajja HK, Wang LY, Duan HW, Ni CC, et al. Molecular Imaging of Pancreatic Cancer in an Animal Model Using Targeted Multifunctional Nanoparticles. Gastroenterology. 2009;136:1514–1525. doi: 10.1053/j.gastro.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumagai M, Kano MR, Morishita Y, Ota M, Imai Y, Nishiyama N, Sekino M, Ueno S, Miyazono K, Kataoka K. Enhanced Magnetic Resonance Imaging of Experimental Pancreatic Tumor in vivo by Block Copolymer-Coated Magnetite Nanoparticles with TGF-beta Inhibitor. J. Controlled Release. 2009;140:306–311. doi: 10.1016/j.jconrel.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Loebinger MR, Kyrtatos PG, Turmaine M, Price AN, Pankhurst Q, Lythgoe MF, Janes SM. Magnetic Resonance Imaging of Mesenchymal Stem Cells Homing to Pulmonary Metastases Using Biocompatible Magnetic Nanoparticles. Cancer Res. 2009;69:8862–8867. doi: 10.1158/0008-5472.CAN-09-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olariu CI, Yiu HHP, Bouffier L, Nedjadi T, Costello E, Williams SR, Halloran CM, Rosseinsky MJ. Multi-functional Fe3O4 Nanoparticles for Targeted Bi-Modal Imaging of Pancreatic Cancer. J. Mater. Chem. 2011;21:12650–12659. [Google Scholar]
- 32.Xu Y, Karmakar A, Heberlein WE, Mustafa T, Biris AR, Biris AS. Multifunctional Magnetic Nanoparticles for Synergistic Enhancement of Cancer Treatment by Combinatorial Radio Frequency Thermolysis and Drug Delivery. Adv. Healthcare Mater. 2012;1:493–501. doi: 10.1002/adhm.201200079. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Zhang Q, Luo XF, Li J, He H, Yang F, Di Y, Jin C, Jiang XG, Shen S, et al. Magnetic Graphene-based Nanotheranostic Agent for Dual-modality Mapping Guided Photothermal Therapy in Regional Lymph Nodal Metastasis of Pancreatic Cancer. Biomaterials. 2014;35:9473–9483. doi: 10.1016/j.biomaterials.2014.07.064. [DOI] [PubMed] [Google Scholar]
- 34.Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L. Theranostic Nano-particles with Controlled Release of Gemcitabine for Targeted Therapy and MRI of Pancreatic Cancer. ACS Nano. 2013;7:2078–89. doi: 10.1021/nn3043463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, et al. A Primary Xenograft Model of Small-Cell Lung Cancer Reveals Irreversible Changes in Gene Expression Imposed by Culture In vitro. Cancer Res. 2009;69:3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lefrak EA, Pit'ha J, Rosenheim S, Gottlieb JA. A Clinicopathologic Analysis of Adriamycin Cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Swain SM, Whaley FS, Ewer MS. Congestive Heart Failure in Patients Treated with Doxorubicin - A Retrospective Analysis of Three Trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 38.Fossella FV, Lippman SM, Shin DM, Tarassoff P, CalayagJung M, PerezSoler R, Lee JS, Murphy WK, Glisson B, Rivera E, et al. Maximum-Tolerated Dose Defined for Single-Agent Gemcitabine: A Phase I Dose-Escalation Study in Chemotherapy-Naive Patients with Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 1997;15:310–316. doi: 10.1200/JCO.1997.15.1.310. [DOI] [PubMed] [Google Scholar]
- 39.Batist G, Ramakrishnan G, Rao CS, Chandrasekharan A, Gutheil J, Guthrie T, Shah P, Khojasteh A, Nair MK, Hoelzer K, et al. Reduced Cardiotoxicity and Preserved Antitumor Efficacy of Liposome-Encapsulated Doxorubicin and Cyclophosphamide Compared with Conventional Doxorubicin and Cyclophosphamide in a Randomized, Multicenter Trial of Metastatic Breast Cancer. J. Clin. Oncol. 2001;19:1444–1454. doi: 10.1200/JCO.2001.19.5.1444. [DOI] [PubMed] [Google Scholar]
- 40.Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated Liposomal Doxorubicin (doxil): Reduced Clinical Cardiotoxicity in Patients Reaching or Exceeding Cumulative Doses of 500 mg/m(2) Ann. Oncol. 2000;11:1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 41.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardiere C, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 42.Hidalgo M, Von Hoff DD. Translational Therapeutic Opportunities in Ductal Adenocarcinoma of the Pancreas. Clin. Cancer Res. 2012;18:4249–4256. doi: 10.1158/1078-0432.CCR-12-1327. [DOI] [PubMed] [Google Scholar]
- 43.Kopetz S, Lemos R, Powis G. The Promise of Patient-Derived Xenografts: The Best Laid Plans of Mice and Men. Clin. Cancer Res. 2012;18:5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siolas D, Hannon GJ. Patient-Derived Tumor Xenografts: Transforming Clinical Samples into Mouse Models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hylander BL, Pitoniak R, Penetrante RB, Gibbs JF, Oktay D, Cheng JR, Repasky EA. The Anti-tumor Effect of Apo2L/TRAIL on Patient Pancreatic Adenocarcinomas Grown as Xenografts in SCID Mice. J. Transl. Med. 2005;3:22. doi: 10.1186/1479-5876-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P, et al. Characterization of a Large Panel of Patient-Derived Tumor Xenografts Representing the Clinical Heterogeneity of Human Colorectal Cancer. Clin. Cancer Res. 2012;18:5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 47.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-Derived Tumour Xenografts as Models for Oncology Drug Development. Nat. Rev. Clin. Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L, Sajja HK, Cao Z, Qian W, Bender L, Marcus AI, Lipowska M, Wood WC, Wang YA. uPAR-targeted Optical Imaging Contrasts as Theranostic Agents for Tumor Margin Detection. Theranostics. 2014;4:106–118. doi: 10.7150/thno.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harvey SR, Hurd TC, Markus G, Martinick MI, Penetrante RM, Tan DF, Venkataraman P, DeSouza N, Sait SNJ, Driscoll DL, et al. Evaluation of Urinary Plasminogen Activator, Its Receptor, Matrix Metalloproteinase-9, and Von Willebrand Factor in Pancreatic Cancer. Clin. Cancer Res. 2003;9:4935–4943. [PubMed] [Google Scholar]
- 50.Tan XD, Egami H, Nozawa F, Abe M, Baba H. Analysis of the Invasion-Metastasis Mechanism in Pancreatic Cancer: Involvement of Lasmin(ogen) Cascade Proteins in the Invasion of Pancreatic Cancer Cells. Int. J. Oncol. 2006;28:369–374. [PubMed] [Google Scholar]
- 51.Satpathy M, Wang L, Zielinski R, Qian W, Lipowska M, Capala J, Lee GY, Xu H, Wang YA, Mao H, et al. Active Targeting Using HER-2-Affibody-Conjugated Nanoparticles Enabled Sensitive and Specific Imaging of Orthotopic HER-2 Positive Ovarian Tumors. Small. 2014;10:544–555. doi: 10.1002/smll.201301593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nasongkla N, Bey E, Ren J, Ai H, Khemtong C, Guthi JS, Chin S-F, Sherry AD, Boothman DA, Gao J. Multifunctional Polymeric Micelles as Cancer-Targeted, MRI-Ultrasensitive Drug Delivery Systems. Nano Lett. 2006;6:2427–2430. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 53.Burke TG, Morin MJ, Sartorelli AC, Lane PE, Tritton TR. Function of the Anthracycline Amino Group in Cellular Transport and Cytotoxicity. Mol. Pharmacol. 1987;31:552–556. [PubMed] [Google Scholar]
- 54.Gabizon AA. Pegylated liposomal doxorubicin: Metamorphosis of an Old Drug into a New Form of Chemotherapy. Cancer Invest. 2001;19:424–436. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- 55.Halford S, Yip D, Karapetis CS, Strickland AH, Steger A, Khawaja HT, Harper PG. A Phase II Study Evaluating the Tolerability and Efficacy of CAELYX (Liposomal Doxorubicin, Doxil) in the Treatment of Unresectable Pancreatic Carcinoma. Ann. Oncol. 2001;12:1399–1402. doi: 10.1023/a:1012522120294. [DOI] [PubMed] [Google Scholar]
- 56.Lipowska M, Patonay G, Strekowski L. New Near-Infrared Cyanine Dyes for Labeling of Proteins. Synth. Commun. 1993;23:3087–3094. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.