

Abstract
Pilus biogenesis on the surface of uropathogenic Escherichia coli requires the chaperone/usher pathway, a terminal branch of the general secretory pathway. In this pathway, periplasmic chaperone-subunit complexes target an outer membrane (OM) usher for subunit assembly into pili and secretion to the cell surface. The molecular mechanisms of protein secretion across the OM are not well understood. Mutagenesis of the P pilus usher PapC and the type 1 pilus usher FimD was undertaken to elucidate the initial stages of pilus biogenesis at the OM. Deletion of residues 2 to 11 of the mature PapC N terminus abolished the targeting of the usher by chaperone-subunit complexes and rendered PapC nonfunctional for pilus biogenesis. Similarly, an intact FimD N terminus was required for chaperone-subunit binding and pilus biogenesis. Analysis of PapC-FimD chimeras and N-terminal fragments of PapC localized the chaperone-subunit targeting domain to the first 124 residues of PapC. Single alanine substitution mutations were made in this domain that blocked pilus biogenesis but did not affect targeting of chaperone-subunit complexes. Thus, the usher N terminus does not function simply as a static binding site for chaperone-subunit complexes but also participates in subsequent pilus assembly events.
Gram-negative bacteria use the chaperone/usher pathway to secrete proteins across their outer membrane (OM) for the formation of complex structures associated with virulence, such as adhesive pili or fimbriae (34). Uropathogenic Escherichia coli uses this pathway to assemble type 1 and P pili, coded for by the fim and pap gene clusters, respectively. Type 1 pili mediate binding to mannosylated glycoproteins present in the bladder and are associated with cystitis (18, 31). P pili are critical for binding to Galα(1-4)Gal moieties present in the kidney and are associated with pyelonephritis (5, 31). These pili are composite structures built from multiple pilus subunits (6). P pili consist of a 6.8-nm-diameter helical rod composed of PapA subunits and a 2-nm-diameter linear tip fibril composed mainly of PapE subunits. The PapG adhesin is located at the distal end of the tip fibril and is joined to PapE by the PapF adaptor subunit. The PapK adaptor subunit joins the tip fibril to the PapA rod. The PapH subunit terminates the rod and is believed to play a role in anchoring the rod to the bacterial surface (1). Type 1 pili have a short tip fibril consisting of the FimH adhesin and the FimF and FimG adaptor subunits. The type 1 pilus rod is composed of FimA subunits.
Assembly of subunits into the complex pilus structure requires the action of two nonstructural secretion components working in concert: the periplasmic chaperone (PapD for P pili, FimC for type 1 pili) and the OM usher (PapC for P pili, FimD for type 1 pili) (34). Each of the structural and assembly components uses the Sec general secretory pathway (17) for translocation across the inner membrane (IM) into the periplasm. Pilus subunits must then form a stable interaction with the periplasmic chaperone. The chaperone allows proper folding of the subunits and prevents premature subunit-subunit interactions (15). Pilus subunits contain an immunoglobulin-like (Ig) fold, except that the subunits lack the seventh (C-terminal) β-strand present in canonical Ig folds (7, 24, 25). The absence of this strand produces a deep groove on the surface of the folded subunit, exposing the subunit's hydrophobic core. The chaperone, which consists of two complete Ig domains (12), functions by donating its G1 β-strand to fill this groove, facilitating subunit folding in a mechanism termed donor strand complementation (3, 7, 24). The subunit groove also comprises an interactive surface involved in subunit-subunit interactions (25, 30, 39). Thus, donor strand complementation couples the folding of subunits with the simultaneous capping of their interactive surfaces.
Chaperone-subunit complexes must next target the OM usher for subunit assembly into pili and secretion to the cell surface. In the absence of the usher, complexes accumulate in the periplasm but no pili are assembled or secreted (16, 21, 37). Pilus assembly is thought to occur at the periplasmic face of the usher, concomitant with secretion of the pilus fiber through the usher to the cell surface (34). This process is “self-energized” and does not require transduction of energy from the IM (14). Pili are assembled in a top-to-bottom fashion; the adhesin is incorporated first, followed by assembly of the tip fibril and finally the rod. The usher facilitates this organization by differentially recognizing chaperone-subunit complexes in accordance with their final position in the pilus (8, 28). Chaperone-adhesin complexes (PapDG or FimCH) have the highest affinity for the usher (PapC or FimD). This ensures incorporation of the adhesin first into the pilus and biogenesis of organelles functional for binding host surfaces.
Interaction with the usher triggers an exchange of chaperone-subunit interactions for subunit-subunit interactions by an unknown mechanism, allowing incorporation of subunits into the pilus fiber. Pilus subunits have a highly conserved N-terminal extension that is exposed in the chaperone-subunit complex (7, 24). This extension has been shown to participate in subunit-subunit interactions (25, 30, 39). At the usher, the G1 β-strand of the chaperone is exchanged for the N-terminal extension of an incoming subunit in a process termed donor strand exchange, which couples chaperone dissociation with pilus assembly (3). Recent crystal structures have captured the donor strand exchange interaction (25, 39). These structures revealed that in the chaperone-subunit complex, the chaperone maintains subunits in an activated, high-energy conformation. Subunit-subunit interactions and donor strand exchange then allow subunits to undergo a topological transition to a more compact, lower-energy state. This topological transition presumably provides the driving force for fiber formation at the OM usher.
The usher is a β-barrel structure predicted to be composed of 24 transmembrane β-strands (36). Electron microscopy has shown that the usher assembles into a ring-shaped structure with a 2- to 3-nm-diameter central pore. This pore size is large enough for a folded pilus subunit to pass through (35). Analysis of PapC C-terminal truncation mutant proteins demonstrated that the usher C terminus is required for pilus biogenesis but not for binding to chaperone-subunit complexes (36). The usher N terminus was proposed to function as the initial targeting site for chaperone-subunit complexes, and the usher C terminus was proposed to function as a distinct domain required for subsequent pilus assembly and secretion events. In the periplasm, a chaperone-adhesin complex would target first the usher N terminus. There is evidence that binding of the chaperone-adhesin complex induces a conformational change in the usher, priming the usher for pilus biogenesis (28). The chaperone-adhesin complex would then shift to the usher C terminus, forming a stable assembly intermediate (28, 36). The N-terminal binding site of the usher would then be available for targeting of the next chaperone-subunit complex. This might position the two chaperone-subunit complexes to drive uncapping of the chaperone from the subunit bound at the C-terminal usher site, followed by donor strand exchange and incorporation of the subunit bound at the N-terminal site into the growing pilus fiber at the C-terminal site. Repetition of this cycle would then result in pilus assembly and secretion to the bacterial surface (36).
In this study, we analyzed the function of the usher N terminus in pilus assembly. Deletion mutagenesis, analysis of chimeric PapC-FimD ushers, and expression of PapC N-terminal fragments demonstrated that the N-terminal region of the usher serves as the initial targeting site for chaperone-subunit complexes. Furthermore, single alanine substitution mutations in the usher N terminus were made that allowed chaperone-adhesin binding to the usher but blocked pilus biogenesis. This indicates that the usher N terminus has functions in addition to serving as the targeting site for chaperone-subunit complexes.
MATERIALS AND METHODS
Strains and plasmids.
The strains and plasmids used in this study are listed in Table 1. The primers used for mutagenesis are listed in Table 2. Plasmid pMJ3 contains the usher-encoding papC gene with a C-terminal hexahistidine tag (His tag), under control of the arabinose (Para) promoter. Plasmid pAP3, coding for PapCΔ2-11, was created from pMJ3 by using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.) to delete residues 2 to 11 of mature PapC. Plasmids encoding alanine-substituted PapC mutant proteins were also derived from pMJ3 with the QuickChange site-directed mutagenesis kit. All of the papC-encoded mutant proteins were fully sequenced to verify the intended mutation and the absence of unintended mutations. Plasmid construction was done with host strain DH5α.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)c | Reference |
|---|---|---|
| Strainsa | ||
| DH5α | hsdR recA endA | 10 |
| SF100 | ΔompT | 2 |
| AAEC185 | Δfim | 4 |
| BL21 | OmpT− Lon− | 23 |
| ORN103 | Δfim | 22 |
| Plasmids | ||
| pMON6235Δcat | Vector; Para Ampr | 15 |
| pMJ3 | PapCHis in pMON6235Δcat | 35 |
| pAP3 | PapCΔ2-11 in pMJ3 | This study |
| pMO1 | PapC E2A in pMJ3 | This study |
| pMO5 | PapC F3A in pMJ3 | This study |
| pMO2 | PapC N4A in pMJ3 | This study |
| pMO6 | PapC T5A in pMJ3 | This study |
| pMO3 | PapC D6A in pMJ3 | This study |
| pMO7 | PapC V7A in pMJ3 | This study |
| pMO8 | PapC L8A in pMJ3 | This study |
| pTN5 | PapC C70A in pMJ3 | This study |
| pTN6 | PapC C97A in pMJ3 | This study |
| pTN7 | PapC C70A + C97A in pMJ3 | This study |
| pHG81 | TnTAP insertion after PapC residue 809 in pMJ3 | —b |
| pHG102 | TnTAP insertion after PapC residue 30 in pMJ3 | —b |
| pHG105 | TnTAP insertion after PapC residue 124 in pMJ3 | —b |
| pHG133 | TnTAP insertion after PapC residue 138 in pMJ3 | —b |
| pHG132 | TnTAP insertion after PapC residue 164 in pMJ3 | —b |
| pHG118 | TnTAP insertion after PapC residue 194 in pMJ3 | —b |
| pHG4 | TnTAP insertion after PapC residue 236 in pMJ3 | —b |
| pTN2 | PapC30NHis in pMON6235Δcat | This study |
| pTN3 | PapC124NHis in pMON6235Δcat | This study |
| pTN20 | PapC138NHis in pMON6235Δcat | This study |
| pTN19 | PapC164NHis in pMON6235Δcat | This study |
| pTN4 | PapC194NHis in pMON6235Δcat | This study |
| pTN18 | PapC236NHis in pMON6235Δcat | This study |
| pMMB91 | Vector; Ptac Kanr | 8 |
| pKD101 | PapC in pMMB91 | 8 |
| pAN2 | FimD in pMMB91 | 28 |
| p10ND | FimDΔ3-12 in pAN2 | This study |
| pNS | PapC10-FimD in pAN2 | This study |
| pNaeIS | PapC25-FimD in pMMB91 | This study |
| pNarIS | PapC180-FimD in pMMB91 | This study |
| pMJ2 | ΔpapC pap operon Ptrc Tetr | 35 |
| pPAP58 | PapDJKEFG Ptac Kanr | 13 |
| pJP1 | PapDG Ptac Kanr | 8 |
| pLS101 | PapD Ptac Kanr | 29 |
| pETS6 | Δfim fim operon natural promoter Clmr | 28 |
| pETS8 | FimC Ptac Ampr | 26 |
| pETS1007 | FimCH Ptac Ampr | 27 |
All strains are E. coli K-12, except BL21, which is E. coli B.
—, C. Martin and D. Thanassi, unpublished data.
Ampr, ampicillin resistance; Kanr, kanamycin resistance; Clmr, chloramphenicol resistance; Tetr, tetracycline resistance; Para, arabinose-inducible promoter, Ptac or Ptrc, IPTG-inducible promoter.
TABLE 2.
Primers used in this study
| Primer | Sequence (forward primer)a |
|---|---|
| papCΔ2-11 | 5′-CGCAGCCAGTGCCGTTGACAAGAAAAATATTGACTTCACC-3′ |
| papCE2A | 5′-GCAGCCAGTGCCGTTGCGTTTAATACAGATGTAC-3′ |
| papCF3A | 5′-GCCAGTGCCGTTGAGGCTAATACAGATGTACTTGACGC-3′ |
| papCN4A | 5′-GCCAGTGCCGTTGAGTTTGCTACAGATGTACTTGACGCAGCGG-3′ |
| papCT5A | 5′-GCCGTTGAGTTTAATGCAGATGTACTTGACGCAGCG-3′ |
| papCD6A | 5′-GCCGTTGAGTTTAATACAGCTGTACTTGACGCAGCGG-3′ |
| papCV7A | 5′-GTTGAGTTTAATACAGATGCACTTGACGCAGCGGAC-3′ |
| papCL8A | 5′-CCGCAGGCCTGTGCGACATCAGATATG-3′ |
| papCD9A | 5′-GAGTTTAATACAGATGTACTTGCCGCAGCGGACAAG-3′ |
| papCC70A | 5′-GCCGCAGGCCGCTCTGACATCAGATATGGTCAG-3′ |
| papCC97A | 5′-CTGGCATGATGGTCAGGCTGCGGATTTTCATGGG-3′ |
| fimDΔ3-12 | 5′-CATCTGCCGACCTCCCCCAGGCTGTGGCC-3′ |
| papC10-fimD | 5′-CATCTGCCGACCTCGAGTTTAATACGGACGTTTTAGACGCTGCTCCCCAGGCTGTGGCC-3′ |
| papC25-fimD | 5′-GGTGCGTGGCGCCTACGCGACAATACCACC-3′ |
| papC180-fimD | 5′-GATTTATCGCGTTTTGAAAATGCCGGCGAATTACCGCCAGGGACG-3′ |
The reverse primer is the reverse complement of the forward primer.
The PapC N-terminal fragments were constructed by taking advantage of a set of TnTAP transposon insertions (9) that had been created in the papC gene on plasmid pMJ3 (C. Martin and D. Thanassi, unpublished data). Each transposon insertion created a unique NotI restriction site in papC. With these NotI sites, we constructed His-tagged N-terminal fragments of different lengths (Fig. 1). Plasmid pHG81 contained a TnTAP insertion between the PapC C terminus (mature residue 809) and the His tag. Plasmids pHG102, pHG105, pHG133, pHG132, pHG118, and pHG4 contained TnTAP insertions in the PapC N terminus following mature residues 30, 124, 138, 164, 194, and 236, respectively. Digestion of pHG81 with NotI and EcoRI generated a fragment including a portion of the TnTAP sequence followed by the C-terminal His tag. Digestion of pHG102, pHG105, pHG133, pHG132, pHG118, and pHG4 with EcoRI and NotI resulted in plasmids carrying the N-terminal region of PapC followed by part of the TnTAP insertion sequence. Ligation of the pHG81 fragment into the EcoRI-NotI-digested plasmids created plasmids encoding N-terminal fragments of PapC, including the signal sequence, followed by 30, 124, 138, 164, 194, or 236 amino acids of the mature N terminus, fused in frame to a 24-amino-acid sequence from the TnTAP transposon (LTLIHKFENLYFQSAAAILVYKSQ) (9) and terminated by the His tag (Fig. 1B).
FIG. 1.

(A) Alignment of the PapC and FimD N termini. The complete usher sequences were aligned by ClustalW with the MacVector software program (Oxford Molecular Ltd., Madison, Wis.). Identical and similar residues are shaded grey, with the conserved cysteines shaded dark grey with white lettering. Predicted transmembrane β-strands of PapC are shown in bold. The PapC and FimD signal sequences are in italics, with the mature proteins beginning at residue number 1. Residues deleted in PapCΔ2-11 and FimDΔ3-12 and exchanged in PapC10-FimD are boxed. The open arrows indicate the positions of the restriction sites in the corresponding DNA sequences used to create the PapC25-FimD and PapC180-FimD chimeras. Introduction of the NaeI site into FimD changed residues 25GQ26 to 25AG26. The filled arrows indicate the C termini of the PapC regions present in the PapC N-terminal fragments. (B) Schematic diagram of PapC N-terminal fragments (not drawn to scale). The fragments contain the signal sequence (SS), 30 to 236 residues of the mature N terminus, a 24-amino-acid sequence from the TnTAP transposon, and a C-terminal His tag. The arrow shows the position of the NotI restriction site in the corresponding DNA sequence that was used for construction of the N-terminal fragments.
FimDΔ3-12 and the PapC-FimD chimeras were constructed as follows. Plasmid pAN2, coding for FimD under control of the Ptac promoter, was cleaved with DraII and EcoRI, and the smaller fragment was subcloned into pPCR-Script (Stratagene) for mutagenesis. The QuickChange site-directed mutagenesis kit was then used to delete residues 3 to 12 of mature FimD or to replace these residues with PapC residues 2 to 11. The mutagenized DraII-EcoRI fragments were then recloned back into pAN2, creating plasmids p10ND (FimDΔ3-12) and pNS (PapC10-FimD). For the larger PapC-FimD chimeras, the QuickChange kit was used to introduce NaeI or NarI sites, as shown in Fig. 1A, into the DraII-EcoRI fragment from pAN2. Introduction of the NarI site did not change the FimD amino acid sequence; introduction of the NaeI site changed FimD residues 25GQ26 to 25AG26, matching the corresponding PapC sequence (Fig. 1A). The mutagenized DraII-EcoRI fragments were then recloned back into pAN2. To create PapC25-FimD, plasmid pKD101, coding for PapC under control of the Ptac promoter (8), was cleaved with ApaI and NaeI and ligated into ApaI-NaeI-digested pAN2, creating plasmid pNaeIS. To create PapC180-FimD, pKD101 was incompletely digested with NarI and a 3-kb fragment was isolated and ligated into a complete NarI digest of pAN2, creating plasmid pNarIS. Sequencing was done to confirm proper construction of each of the chimeras and FimDΔ3-12.
OM isolation and analysis of the ushers in the OM.
Analysis of expression and folding of the ushers in the OM was done with E. coli host strain SF100, which lacks the OmpT OM protease (2). Strains were grown in 10 ml of Luria-Bertani (LB) broth containing appropriate antibiotics at 37°C with aeration. Plasmid-borne genes were induced at an optical density at 600 nm (OD600) of 0.6 for 1 h by addition of 0.2% l-arabinose (for Para) or 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG; for Ptac). Bacteria were harvested, washed, resuspended in 1 ml of 20 mM Tris-HCl (pH 8) containing Complete Protease Inhibitor cocktail (Roche, Indianapolis, Ind.), and lysed by sonication for 2 min (15 s on, 15 s off) in an ice-water bath. Whole bacteria were removed by centrifugation (7,500 × g, 2 min, 4°C). Sarkosyl (sodium-N-lauroylsarcosinate; Fisher, Fairlawn, N.J.) was added to the supernatant fraction to a 0.5% final concentration, and the mixture was incubated for 5 min at 25°C to selectively solubilize the IM (19). The OM was then pelleted by centrifugation (16,100 × g, 30 min, 4°C) and resuspended in 0.1 ml of 20 mM HEPES (pH 7.5)-0.3 M NaCl. An equal volume of 2× sodium dodecyl sulfate (SDS) sample buffer was added, and the sample was incubated for 10 min at 25 or 95°C prior to separation by SDS-polyacrylamide gel electrophoresis (PAGE). For analysis, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Osmonic Inc., Gloucester, Mass.). Following incubation of the membrane with the primary antibody, the appropriate alkaline phosphatase-conjugated secondary antibody was added and the blot was developed with BCIP (5-bromo-4-chloro-3-indolylphosphate)-NBT (nitroblue tetrazolium) substrate (KPL, Gaithersburg, Md.).
HA assays.
Hemagglutination (HA) assays were performed as previously described (36), with E. coli strain AAEC185 (4) as the host. This strain lacks the chromosomal fim gene cluster. All strains were grown in LB broth with aeration at 37°C and induced as described below for 1 h at an OD600 of 0.6. For agglutination of human erythrocytes by P pili, AAEC185 was transformed with pMJ2 carrying a ΔpapC pap operon under control of Ptrc, together with the vector (pMON6235Δcat) alone, pMJ3, or one of the PapC mutant constructs. Plasmid pMJ2 was induced with 0.1 mM IPTG, and the PapC constructs were induced with 0.2% l-arabinose. For analysis of the PapC-FimD chimeras, AAEC185/pMJ2 was transformed with the vector (pMMB91) alone, pKD101, pAN2, or one of the chimeric usher constructs. All plasmids were induced with 50 μM IPTG. The PapC-FimD chimeras and the FimDΔ3-12 deletion construct were also tested for type 1 pilus assembly by agglutination of guinea pig erythrocytes (Colorado Serum Company, Denver). For these assays, AAEC185 was transformed with pETS6, carrying a ΔfimD fim operon under control of its natural promoter, together with the vector (pMMB91) alone, pAN2, pKD101, pND, or one of the PapC-FimD chimeras. Plasmid pETS6 was induced by static passage for 48 h at 37°C, and 50 μM IPTG was added to induce the other plasmids.
Isolation of pili.
Purification of P or type 1 pili from the bacterial surface was done by heat extraction and magnesium precipitation as previously described (36). Bacterial strains, growth, and induction conditions were as described above for the HA experiments. Purified pili were incubated (95°C, 10 min) in SDS sample buffer containing 4 M urea and subjected to SDS-PAGE. The major rod components (PapA and FimA) were detected by Coomassie blue staining. P pilus tip proteins (PapEFG) were detected by blotting with anti-P pilus tip antisera. The FimH adhesin was detected by blotting with anti-FimCH antisera. The blots were developed with the appropriate alkaline phosphatase-conjugated secondary antibody and BCIP-NBT substrate.
EM.
Bacterial strains and growth and induction conditions were as described above for the HA experiments. Cultures were washed and resuspended in phosphate-buffered saline (PBS) and allowed to adhere to Formvar carbon-coated electron microscopy (EM) grids (Ernest F. Fullam, Latham, N.Y.) for 2 min. Bacteria were fixed on the grids with 1% glutaraldehyde in PBS for 1 min, washed twice with PBS briefly, washed twice with water for 1 min, and stained for 20 s with 0.5% phosphotungstic acid. The grids were examined on a JEOL 1200EXII electron microscope at an 80-kV accelerating voltage.
Periplasm preparations.
PapD- and PapDG-containing periplasms were isolated from host strain BL21, and FimC- and FimCH-containing periplasms were isolated from host strain ORN103. Strains were grown in 50 ml of LB broth containing appropriate antibiotics at 37°C with aeration. Bacterial strains harboring pLS101 (PapD), pJP1 (PapDG), pETS8 (FimC), or pETS1007 (FimCH) were grown to an OD600 of 0.6 and induced with 75 μM IPTG for 1 h. Bacteria were harvested, washed, and resuspended in 1 ml of 20 mM Tris-HCl (pH 8)-20% sucrose. Next, 10 μl of 0.5 M EDTA and 5 μl of 15-mg/ml lysozyme were added and the mixture was incubated in ice for 40 min before addition of 10 μl of 1 M MgCl2. The samples were then centrifuged (9,000 × g, 20 min, 4°C). Supernatant fractions (periplasm) containing chaperone or chaperone-adhesin complexes were stored overnight on ice for use the next day in the overlay assay.
Overlay assay.
For analysis of PapC, OM was isolated as described above from SF100 harboring pMON6235Δcat, pMJ3, pAP3, or a plasmid encoding one of the PapC alanine substitution mutant proteins. For analysis of FimD and the PapC-FimD chimeras, OM was similarly isolated from SF100 harboring pMMB91, pAN2, pKD101, p10ND, pNS, pNaeIS, or pNarIS. OM fractions were mixed with an equal volume of 2× SDS sample buffer, incubated at 95°C for 12 min, subjected to SDS-PAGE, and transferred to PVDF membrane. The membrane was blocked overnight with 5% milk in buffer A (10 mM Tris [pH 8.2], 0.5 M NaCl, 0.5% Tween 20). Periplasm preparations containing PapD, PapDG, FimC, or FimCH, isolated as described above, were diluted with an equal volume of buffer A and incubated with the PVDF membrane for 2 h. The membrane was washed three times in buffer A and then probed with anti-PapDG or anti-FimCH antibodies, followed by an alkaline phosphatase-conjugated secondary antibody. The blot was developed with BCIP-NBT substrate. The chaperone alone (PapD or FimC) does not bind to the usher (8, 28); overlay assays with chaperone only were performed as a negative control.
Copurification of chaperone-subunit complexes with PapC.
Strain SF100 harboring pPAP58 (PapDJKEFG under control of Ptrc) was transformed with pMON6235Δcat, pMJ3, pAP3, or pMO5. Strains were grown in 100 ml of LB broth containing appropriate antibiotics at 37°C with aeration. Plasmid-encoded genes were induced at an OD600 of 0.6 for 1 h by addition of 0.2% l-arabinose and 50 μM IPTG, and OM fractions were isolated as described above. The OM pellet was resuspended in 1 ml of 20 mM HEPES (pH 7.5)-0.3 M NaCl and solubilized with 0.5% n-dodecyl-β-d-maltoside (DDM; Anatrace, Maumee, Ohio) by rocking overnight at 4°C. Unsolubilized OM was removed by centrifugation (100,000 × g, 1 h, 4°C). Next, 1 ml of the solubilized OM containing a final concentration of 20 mM imidazole was rocked for 45 min at 25°C with 100 μl of nickel beads (QIAGEN, Hilden, Germany) that had been equilibrated in buffer 1 (20 mM HEPES [pH 7.5], 0.15 M NaCl, 0.05% DDM, 20 mM imidazole). The samples were washed six times with 1 ml of buffer 1, and bound protein was eluted with 50 μl of 1 M imidazole. The elutions were subjected to SDS-PAGE and blotted with anti-P pilus tip antibody. Blots were developed with an alkaline phosphatase-conjugated secondary antibody and BCIP-NBT substrate.
Characterization of the PapC N-terminal fragments.
Expression of PapC N-terminal fragments was done in host strain SF100. Bacteria were grown in 50 ml of LB broth containing appropriate antibiotics at 37°C with aeration. OM and periplasm preparations, isolated as described above, were analyzed to determine the localization of the N-terminal fragments. To purify the N-terminal fragments, periplasm preparations were adjusted to a 20 mM final concentration of imidazole and rocked for 45 min at 25°C with 100 μl of nickel beads (QIAGEN). The beads were washed and eluted as described above. The elutions were subjected to SDS-PAGE and blotted with anti-6-His antibody (Covance, Richmond, Calif.). The blots were developed by chemiluminescence detection with a horseradish peroxidase-conjugated secondary antibody and SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, Ill.). For copurification experiments, the N-terminal fragments were expressed in SF100 together with PapDG (pJP1). Periplasms were isolated, and the N-terminal fragments were purified with nickel beads as described above, except that the blots were probed with anti-PapDG antibody.
N-terminal sequencing of FimD.
OM was isolated as described above from SF100/pAN2. The OM was separated by SDS-PAGE, transferred to PVDF membrane, and stained with Ponceau S. The FimD band was cut out and submitted for N-terminal sequencing by the Proteomics Center at Stony Brook University.
RESULTS
The usher N terminus is the targeting site for chaperone-subunit complexes.
To investigate the function of the usher N terminus, we constructed a deletion mutant form of PapC (PapCΔ2-11) lacking residues 2 to 11 of the mature usher protein (Fig. 1A). The first amino acid was preserved to avoid possible interference with cleavage of the N-terminal signal sequence. The deletion was created in an usher containing a C-terminal His tag for purification and analytical purposes (35). Examination of OM preparations showed that wild-type (WT) PapC and PapCΔ2-11 were similarly expressed and targeted to the OM (Fig. 2A). PapC and other OM β-barrel proteins are resistant to denaturation by SDS, resulting in a characteristic heat-modifiable mobility on SDS-PAGE (32, 35). Compared to protein fully denatured by heating at 95°C in SDS sample buffer, PapC incubated at room temperature produces both a faster-migrating folded monomer form and a slower-migrating oligomeric form (35). PapCΔ2-11 exhibited the same mobility shift as WT PapC (Fig. 2A). This demonstrates that residues 2 to 11 are not required for proper overall folding and oligomerization of PapC.
FIG. 2.

Expression and folding of PapC mutant proteins in the OM. The OM was isolated from strain SF100 expressing WT PapC, PapCΔ2-11 (A), or the indicated alanine substitution PapC mutant protein (B). Duplicate samples were incubated for 10 min in SDS sample buffer at room temperature (RT; 25°C) or 95°C prior to separation by SDS-PAGE. The ushers were detected by blotting with anti-His tag antibody. The presence of the faster-migrating folded monomer form (∼60 kDa) and the slower-migrating oligomeric form (∼200 kDa) in the RT-treated samples indicates proper folding of the usher in the OM. Each of the ushers exhibits heat-modifiable mobility.
The ability of PapCΔ2-11 to complement a ΔpapC pap operon for assembly of adhesive pili was tested by HA assay with human red blood cells. PapCΔ2-11 was not functional for adhesive pilus biogenesis, giving an HA titer of 0 (Fig. 3A). To determine if the deletion mutant protein could assemble any pili on the bacterial surface, bacteria were subjected to heat extraction and magnesium precipitation to isolate pili. In agreement with the HA results, no pilus proteins were purified from bacteria complemented with the PapCΔ2-11 usher (Fig. 3A). Examination of bacteria complemented with PapCΔ2-11 by EM also confirmed a lack of pilus assembly (data not shown). Thus, deletion of PapC residues 2 to 11 caused a complete loss of pilus biogenesis.
FIG. 3.
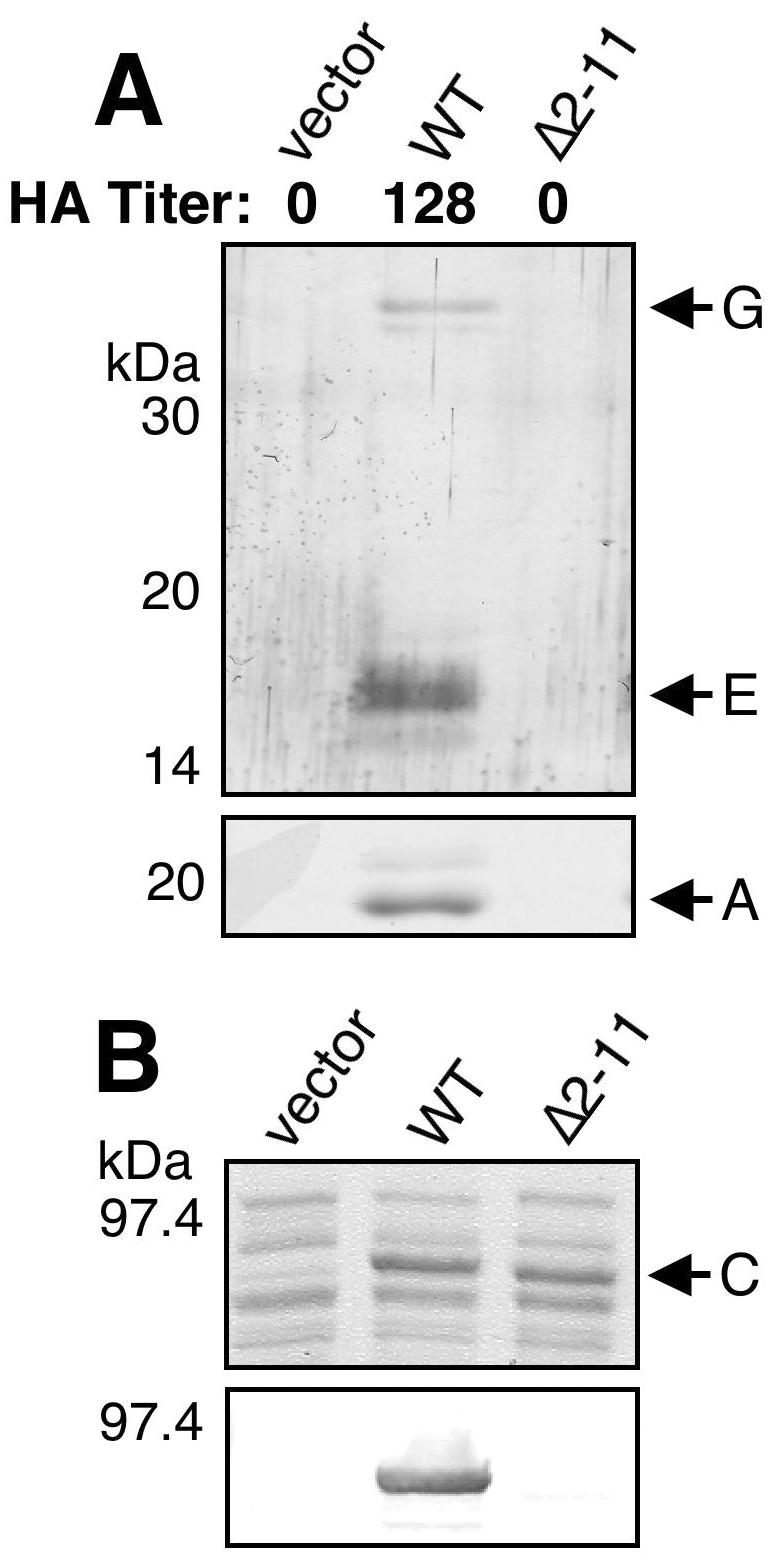
(A) Analysis of pilus biogenesis by PapCΔ2-11. Strain AAEC185/pMJ2 (ΔpapC pap operon) was complemented with the vector alone, WT PapC, or PapCΔ2-11, and assembly of adhesive pili was measured by agglutination of human erythrocytes. The HA titer is the highest fold dilution of bacteria that still provides agglutination. Assembly of pili on the bacterial surface was determined by heat extraction and magnesium precipitation, followed by SDS-PAGE. The rod subunit PapA was detected by Coomassie blue staining (bottom). The tip fibril subunits PapE and PapG were detected by blotting with anti-P pilus tip antibody (top). PapCΔ2-11 was unable to agglutinate erythrocytes or assemble pili. (B) Overlay assay for targeting of PapDG chaperone-adhesin complexes to PapCΔ2-11. OM was isolated from SF100 expressing the vector alone, WT PapC, or PapCΔ2-11. Duplicate samples were separated by SDS-PAGE and either stained with Coomassie blue to show the amount of PapC loaded (top) or transferred to PVDF membrane for the overlay assay (bottom). The PVDF membrane was incubated with PapDG-containing periplasm, and PapDG binding to PapC was detected by blotting with anti-PapDG antibody. The PapCΔ2-11 mutant protein was defective for PapDG binding.
We next examined whether PapCΔ2-11 could interact with chaperone-adhesin complexes by using an in vitro overlay assay (8, 36). Deletion of PapC residues 2 to 11 almost completely abolished PapDG binding to the usher (Fig. 3B), indicating that this region of the PapC N terminus is required for chaperone-adhesin targeting in vitro. To gain in vivo evidence for chaperone-subunit targeting to the usher N terminus, copurification experiments were performed. Previous studies with type 1 pili demonstrated that chaperone-subunit complexes could be purified from the OM as a complex with the FimD usher (27, 28). We coexpressed WT PapC or PapCΔ2-11 with the PapD chaperone and the tip fibril subunits PapK, -E, -F, and -G. OM fractions from these bacteria were isolated and solubilized with the nondenaturing detergent DDM, and PapC was purified by its His tag with nickel beads. As shown in Fig. 4A, PapE, -F, and -G were copurified from the OM with WT PapC. PapD was also copurified with this complex (data not shown). This is consistent with the previous copurification studies done with type 1 pili (27, 28). In contrast, no chaperone-subunit complexes were copurified with PapCΔ2-11 (Fig. 4A). Thus, residues 2 to 11 are essential for targeting of chaperone-subunit complexes to the usher in vivo, explaining why deletion of this region results in loss of pilus biogenesis.
FIG. 4.

Copurification of chaperone-subunit complexes with PapC. OM was isolated from SF100/pPAP58 (PapDJKEFG) expressing the vector alone, WT PapC, PapCΔ2-11 (A), or the F3A PapC mutant protein (B). The OM was solubilized with DDM, and PapC was purified by its His tag with nickel beads. Duplicate samples were separated by SDS-PAGE and either stained with Coomassie blue to show the amount of PapC purified (top) or blotted with anti-P pilus tip antibody to detect pilus subunits that were copurified with the usher (bottom). Pilus tip proteins PapG, F, and E were copurified with WT PapC and the F3A substitution mutant protein, but no pilus subunits were copurified with the PapCΔ2-11 deletion mutant protein.
Analysis of the FimD N terminus and PapC-FimD chimeras.
Each chaperone/usher pathway exhibits specificity for its own subunits. PapDG chaperone-adhesin complexes do not bind to the FimD usher, and FimCH chaperone-adhesin complexes do not bind to the PapC usher (28). The function of the N terminus as the targeting site for chaperone-subunit complexes suggests that this domain may control usher specificity. To test this, and to determine if the FimD N terminus functions in chaperone-subunit targeting as found for PapC, we constructed a FimD N-terminal deletion mutant protein and created PapC-FimD chimeras in which N-terminal regions of FimD were replaced with equivalent regions from PapC. For these studies, we needed to know the processing site for the FimD N-terminal signal sequence. N-terminal sequencing of the mature usher in the OM returned the sequence Asp-Leu-Tyr-Phe, revealing that FimD has a longer-than-usual signal sequence, with cleavage occurring between residues Ala45 and Asp46 (Fig. 1A). Processing of FimD at this site was also reported in a recent publication (20). Hereafter, we will refer to FimD according to its mature sequence, with the Asp following the cleavage site as residue no. 1 (Fig. 1A).
We constructed a FimD N-terminal deletion mutant protein lacking residues 3 to 12 (FimDΔ3-12). These residues correspond to the residues deleted in the PapCΔ2-11 mutant protein (Fig. 1A). We also constructed a PapC-FimD chimera in which FimD residues 3 to 12 were replaced with PapC residues 2 to 11 (PapC10-FimD). In addition, chimeras were constructed in which the FimD signal sequence and residues 1 to 26 or 1 to 174 were replaced with the signal sequence and residues 1 to 25 or 1 to 180 of PapC (PapC25-FimD or PapC180-FimD, respectively). The chimeras were constructed on the basis of amino acid alignments of PapC and FimD (Fig. 1A) to try to match the exchanged regions and avoid disturbing the β-barrel structure of the usher. Each of the constructs was expressed and targeted to the OM (Fig. 5). However, the PapC180-FimD chimera had a lower expression level. Furthermore, PapC180-FimD did not produce a faster-migrating folded monomer species in the heat-modifiable mobility shift assay and thus presumably could not adopt a stable β-barrel fold (Fig. 5). The other chimeras and the N-terminal deletion mutant protein underwent mobility shifts similar to that of WT FimD (Fig. 5).
FIG. 5.

Expression and folding of FimDΔ3-12 and the PapC-FimD chimeras. OM was isolated from SF100 expressing WT FimD, FimDΔ3-12, or the indicated PapC-FimD chimera and incubated for 10 min in SDS sample buffer at room temperature (RT; 25°C) or 95°C prior to separation by SDS-PAGE. The ushers were detected by blotting with anti-FimD antibody. Each of the ushers exhibits heat-modifiable mobility, indicating proper folding in the OM, except for PapC180-FimD. PapC180-FimD also had a lower expression level.
To determine if the FimD constructs were functional, we assayed whether they could complement a ΔfimD fim operon for type 1 pilus biogenesis. By HA of guinea pig erythrocytes, the PapC25-FimD and PapC180-FimD chimeras were not able to assemble adhesive type 1 pili, whereas FimDΔ3-12 and PapC10-FimD assembled adhesive pili, but at reduced levels (Fig. 6). Isolation of type 1 pili from the bacterial surface confirmed these results, with only a very low level of pilus proteins recovered from bacteria complemented with FimDΔ3-12 or PapC10-FimD (Fig. 6). We next tested binding of FimCH chaperone-adhesin complexes to the FimD constructs with the overlay assay. FimCH bound to WT FimD, but no binding to any of the PapC-FimD chimeras or the FimDΔ3-12 deletion mutant protein could be detected (Fig. 7B). FimCH also did not bind to WT PapC, as previously shown (28). Although not detected by the overlay assay, a low level of chaperone-subunit binding to FimDΔ3-12 and PapC10-FimD must occur in vivo, as adhesive pili were assembled by these usher constructs (Fig. 6). These data show that an intact FimD N terminus is required for targeting of chaperone-subunit complexes, as found for PapC, although deletion of a larger region—the first 26 residues—is necessary to disrupt pilus biogenesis completely.
FIG. 6.

Type 1 pilus biogenesis by FimDΔ3-12 and the PapC-FimD chimeras. AAEC185/pETS6 (ΔfimD fim operon) was complemented with the vector alone, WT FimD, WT PapC, FimDΔ3-12, or the indicated PapC-FimD chimera. Assembly of adhesive type 1 pili was measured by agglutination of guinea pig erythrocytes. The HA titer is the highest fold dilution of bacteria that still provides agglutination. Assembly of type 1 pili on the bacterial surface was determined by heat extraction and magnesium precipitation, followed by SDS-PAGE. The rod subunit FimA was detected by Coomassie blue staining (bottom). The FimH adhesin was detected by blotting with anti-FimCH antibody (top). FimDΔ3-12 and the PapC10-FimD chimera had a reduced HA titer and assembled a low level of pili. The other chimeras, as well as WT PapC, were completely defective for type 1 pilus biogenesis and agglutination.
FIG. 7.

Overlay assay for targeting of chaperone-adhesin complexes to FimDΔ3-12 and the PapC-FimD chimeras. OM was isolated from SF100 expressing the vector alone, WT FimD, WT PapC, FimDΔ3-12, or the indicated PapC-FimD chimera, and identical samples were separated by SDS-PAGE. (A) The OM was stained with Coomassie blue to show the amount of usher loaded. The arrowheads indicate the positions of the usher bands. (B and C) The OM was transferred to PVDF membrane and incubated with periplasm containing FimCH (B) or PapDG (C) chaperone-adhesin complexes. Binding of the chaperone-adhesin complexes to the ushers was detected by blotting with anti-FimCH (B) or anti-PapDG (C) antibodies. Only WT PapC and the PapC180-FimD chimera were able to bind PapDG. Only binding of FimCH to WT FimD was detected.
We next performed overlay assays with PapDG to examine whether the PapC regions present in the PapC-FimD chimeras would alter the specificity of the usher and allow targeting of P pilus proteins. WT FimD, FimDΔ3-12, PapC10-FimD, and PapC25-FimD did not bind PapDG (Fig. 7C). However, PapDG bound to the PapC180-FimD chimera (Fig. 7C). This shows that the usher N terminus is able to confer specificity on the usher. Furthermore, this demonstrates that although PapC residues 2 to 11 are required for chaperone-subunit targeting, only a larger N-terminal region encompassing the first 180 residues is sufficient to act as a targeting site. Since PapC180-FimD could bind PapDG in vitro, we asked whether the PapC-FimD chimeras were functional for P pilus biogenesis in vivo. By HA of human erythrocytes, none of the PapC-FimD chimeras was able to complement a ΔpapC pap operon for assembly of adhesive P pili. WT FimD and FimDΔ3-12 were also not functional for adhesive P pilus biogenesis. In agreement with this, P pili could not be isolated from any of these strains by heat extraction and magnesium precipitation or detected by EM (data not shown). The failure of PapC180-FimD to assemble P pili, despite its ability to target PapDG, may be due to the structural defect present in this construct, as shown by its inability to adopt an SDS-resistant fold in the OM (Fig. 5).
PapC N-terminal fragments interact with chaperone-adhesin complexes.
Analysis of the PapC-FimD chimeras localized the chaperone-subunit targeting domain to the first 180 residues of PapC. To further define the targeting domain, we expressed His-tagged PapC fragments containing different lengths of the mature N terminus: 30 (PapC30N), 124 (PapC124N), 138 (PapC138N), 164 (PapC164N), 194 (PapC194N), and 236 (PapC236N) amino acids (Fig. 1). These fragments also contained the N-terminal signal sequence, as well as sequence from a transposon insertion (Fig. 1B; see Materials and Methods). The N-terminal fragments were not able to insert themselves into the OM; instead, they remained soluble in the periplasm. Although expressed at low levels, each of the fragments except PapC30N could be purified from periplasm preparations with nickel beads (Fig. 8A). Note that degradation products were also recovered for each of the fragments except PapC236N (Fig. 8A).
FIG. 8.

Copurification of chaperone-adhesin complexes with the PapC N-terminal fragments. Periplasm was isolated from SF100 expressing the vector alone or the indicated PapC N-terminal fragment, and the fragments were then purified by their His tags with nickel beads. Duplicate samples were subjected to SDS-PAGE and transferred to PVDF membrane. (A) The amount of N-terminal fragments purified was detected by blotting with anti-His tag antibody. Protein was recovered for each of the fragments except PapC30N. (B) PapDG chaperone-adhesin complexes that were copurified with the N-terminal fragments were detected by blotting with anti-PapDG antibody. PapDG was copurified with PapC fragments containing 124, 138, 164, and 194 residues of the mature N terminus.
We next tested the ability of the PapC N-terminal fragments to interact with chaperone-subunit complexes in vivo with the copurification assay. PapC fragments were coexpressed with PapDG, periplasms were isolated, and the PapC fragments were purified by their His tags with nickel beads. As shown in Fig. 8B, PapDG was copurified with PapC194N, PapC164N, PapC138N, and PapC124N. Thus, 124 residues of the mature N terminus are sufficient to form a stable interaction with PapDG in vivo. This is in agreement with a recent study showing that a 139-residue FimD N-terminal fragment bound Fim chaperone-subunit complexes in vitro (20). Surprisingly, PapDG was not copurified with the PapC236N fragment. This larger fragment, which appeared to be the most stable construct (no degradation band), might adopt a conformation in the periplasm that obscures the chaperone-subunit targeting site.
The usher N terminus has additional functions in pilus biogenesis.
The minimal N-terminal binding domain of PapC identified above includes a large periplasmic loop containing a highly conserved cysteine pair (Cys70 and Cys97, Fig. 1A) (36). To identify functionally important residues within the minimal binding domain, we changed each cysteine residue to alanine, individually and as a pair, and we also performed alanine-scanning mutagenesis of the region deleted in the PapCΔ2-11 mutant protein. Each of these mutant proteins was expressed, targeted to the OM, and able to fold properly as evaluated by heat-modifiable mobility on SDS-PAGE (Fig. 2B). The cysteine mutant proteins appeared to be somewhat more susceptible to degradation, as they were expressed at slightly lower levels than the other ushers and degradation products were visible (Fig. 2B). This suggests that although the cysteine pair is not required for correct global folding of the usher (as judged by heat-modifiable mobility), these residues may act to properly structure the large periplasmic loop.
Each of the alanine substitution mutant proteins was tested for the ability to complement a ΔpapC pap operon to assemble P pili. HA of human erythrocytes showed that four of the PapC substitution mutant proteins (F3A, C70A, C97A, and C70A+C97A) did not assemble functional pili (Fig. 9A). The E2A mutant protein had a moderate decrease in the HA titer compared to WT PapC, the N4A mutant protein exhibited no defect, and the other PapC mutant proteins had only a slight decrease in the HA titer (Fig. 9A). Pilus extraction experiments confirmed a lack of pilus assembly by the F3A and cysteine substitution mutant proteins, whereas pili could be isolated for each of the other PapC mutant proteins (Fig. 9A). Examination of the F3A mutant protein by EM also revealed a lack of pilus expression (data not shown). Thus, residues F3, C70, and C97 are required for pilus biogenesis.
FIG. 9.

(A) Analysis of pilus biogenesis by the alanine substitution PapC mutant proteins. AAEC185/pMJ2 (ΔpapC pap operon) was complemented with the indicated PapC mutant protein, and assembly of adhesive pili was measured by agglutination of human erythrocytes. The HA titer is the highest fold dilution of bacteria that still provides agglutination. The HA titer of WT PapC was 128. Assembly of pili on the bacterial surface was determined by heat extraction and magnesium precipitation, followed by SDS-PAGE. The rod subunit PapA was detected by Coomassie blue staining (bottom). The tip fibril subunits PapE and PapG were detected by blotting with anti-P pilus tip antibody (top). The F3A, C70A, C97A, and C70A+C97A PapC mutant proteins were nonfunctional for pilus biogenesis and agglutination. These samples were analyzed together with the WT PapC and vector controls shown in Fig. 3A. (B) Overlay assay for targeting of PapDG chaperone-adhesin complexes to the alanine substitution PapC mutant proteins. The OM was isolated from SF100 expressing the indicated PapC mutant protein. Duplicate samples were separated by SDS-PAGE and either stained with Coomassie blue to show the amount of PapC loaded (top) or transferred to PVDF membrane for the overlay assay (bottom). The PVDF membrane was incubated with PapDG-containing periplasm, and PapDG binding to PapC was detected by blotting with anti-PapDG antibody. All of the PapC mutant proteins were able to bind PapDG complexes. These samples were analyzed together with the WT PapC and vector controls shown in Fig. 3B.
We next determined if the alanine substitution mutant ushers could interact with chaperone-adhesin complexes by the overlay assay. For the F3, C70, and C97 mutant proteins, we expected results similar to those obtained with the PapCΔ2-11 mutant protein, i.e., loss of chaperone-subunit binding. Surprisingly, each of the alanine substitution mutant proteins, including F3A, C70A, C97A, and C70A+C97A, was able to bind PapDG complexes (Fig. 9B). Thus, residues F3, C70, and C97 are essential for pilus biogenesis but not for chaperone-subunit targeting. The overlay assay appears to show differences in the level of PapDG binding to the substitution mutant proteins (Fig. 9B). However, these differences were not consistent. Analysis of PapDG binding to the F3A, N4A, and T5A substitution mutant proteins by enzyme-linked immunosorbent assay showed a twofold reduction for each of these mutant proteins compared to WT PapC (T. Ng and D. Thanassi, unpublished data). As the N4A and T5A mutant forms were functional for pilus biogenesis, this indicates that the defect in the F3A mutant protein is not due to a weaker affinity for chaperone-subunit complexes. We also tested the ability of the F3A mutant form to interact with chaperone-subunit complexes in vivo with the copurification assay. The F3A PapC mutant protein was coexpressed with the PapD chaperone and tip fibril subunits (PapKEFG), and the usher was purified from the OM by its His tag. As shown in Fig. 4B, PapE, -F, and -G were copurified with the F3A mutant form. The levels of the copurified subunits were indistinguishable from that of WT PapC. PapD was also copurified with the F3A mutant form (data not shown). Significantly, these results show that the usher N terminus is required for pilus biogenesis events in addition to the initial targeting and stable association of chaperone-subunit complexes.
DISCUSSION
In this study, we have used pilus biogenesis by the chaperone/usher pathway to elucidate the initial stages of organelle assembly and secretion at the bacterial OM. To probe the function of the usher N terminus, we constructed a PapC deletion mutant protein (PapCΔ2-11) lacking only 10 out of 809 amino acids. This mutant usher was stably expressed in the OM and able to adopt an SDS-resistant β-barrel fold. However, PapCΔ2-11 was unable to complement a ΔpapC pap operon for assembly of P pili. The basis for this defect was revealed by experiments showing that PapCΔ2-11 could not bind to chaperone-adhesin complexes in vitro or form stable interactions with chaperone-subunit complexes in vivo. Similarly, an intact FimD N terminus was required to bind FimCH chaperone-adhesin complexes in vitro and for type 1 pilus biogenesis in vivo, although removal of FimD residues 1 to 26 was required for complete loss of pilus expression. The mature usher N terminus is predicted to reside in the periplasm, prior to the first transmembrane β-strand (Fig. 1A) (36). The results described above demonstrate that this first periplasmic region is critical for the initial targeting of chaperone-subunit complexes to the usher and thus for pilus biogenesis.
The usher is expected to play a central role in determining the specificity of each chaperone/usher pathway for its own subunits. In previous experiments, coexpression of PapC mutant proteins containing C-terminal deletions with fim genes allowed the otherwise defective PapC mutant proteins to complement a ΔpapC pap operon for P pilus assembly (36). These results suggested that the PapC C-terminal truncation mutant proteins were able to interact with FimD, altering the specificity of the FimD usher to allow P pilus biogenesis. This further suggested that specificity might be determined at the level of chaperone-subunit targeting to the usher N terminus. To test this, we constructed PapC-FimD chimeras in which N-terminal regions of FimD were replaced with equivalent N-terminal regions of PapC. Chimeras containing 10 or 25 PapC residues were unable to bind PapDG chaperone-adhesin complexes, but a chimera containing 180 PapC residues (PapC180-FimD) was able to interact with PapDG. However, the PapC180-FimD chimera was not functional for P pilus biogenesis. This may be due to the inability of PapC180-FimD to fold correctly in the OM, as evidenced by its low expression level and lack of SDS resistance. Nevertheless, these results show that the N-terminal targeting region provides specificity to the usher, since the PapC180-FimD chimera failed to interact with Fim chaperone-subunit complexes and instead targeted Pap complexes.
The experiments with the PapC-FimD chimeras localized the chaperone-subunit targeting domain to the first 180 amino acids of PapC. To map the minimal binding domain more precisely, we expressed PapC fragments containing 30 to 236 residues of the mature N terminus. The smallest fragment (PapC30N) was not stably expressed, but each of the other fragments could be isolated from the periplasm. Fragments containing 124 to 194 residues formed stable interactions with PapDG complexes in vivo, narrowing the minimal targeting domain to the first 124 PapC residues. None of the PapC N-terminal fragments was able to insert itself into the OM. Similarly, the 139-residue FimD N-terminal fragment that was shown to bind Fim chaperone-subunit complexes was also found to remain soluble in the periplasm (20). This was interpreted to indicate the absence of transmembrane strands in this region of the usher, as predicted for the K88 pilus usher FaeD (11). In contrast, our analysis of PapC indicates that transmembrane strands are present in the N terminus (N. Henderson and D. Thanassi, unpublished data). We believe that the fragments remain soluble in the periplasm owing to the overall moderate hydrophobicity of β-barrel membrane proteins and the fact that the fragments would not be able to adopt the complete β-barrel fold necessary for membrane insertion (33, 38).
Previous copurification studies with the intact FimD usher demonstrated that chaperone-subunit complexes form stable interactions with a C-terminal region of the usher in vivo (28). The results described above clearly show that chaperone-subunit complexes are also able to form stable interactions with the usher N terminus. Furthermore, our results establish that targeting to the N terminus must precede interaction with the usher C terminus, as chaperone-subunit complexes were not copurified with the PapCΔ2-11 mutant protein.
We performed alanine substitution mutagenesis to identify functionally important residues within the minimal chaperone-subunit targeting region, focusing on residues deleted in the PapCΔ2-11 mutant protein, as well as a highly conserved cysteine pair located within a large periplasmic loop (Fig. 1A) (36). Mutation of F3, C70, and C97 rendered the usher unable to complement a ΔpapC pap operon for pilus biogenesis, as found for the PapCΔ2-11 deletion mutant protein. However, in contrast to PapCΔ2-11, the alanine substitution mutant proteins remained competent for binding chaperone-adhesin complexes in vitro by the overlay assay. The F3A mutant protein was also able to form stable interactions with chaperone-subunit complexes in vivo, as demonstrated by the copurification assay. This indicates that these residues, and thus the usher N terminus, play an important role(s) in pilus biogenesis that is separate from the initial targeting and stable association of chaperone-subunit complexes. Disulfide bonding by the cysteine pair may act to structure the periplasmic loop properly for interaction with chaperone-subunit complexes (36). We consider it likely that the pilus assembly defect of the cysteine mutant proteins is an indirect effect due to loss of the disulfide bond and destabilization of the periplasmic loop.
This study supports our previously proposed model for pilus biogenesis at the OM in which chaperone-subunit complexes are initially targeted to an N-terminal region of the usher (36). We show here that the targeting domain is located within the first 124 residues of the usher and that small deletions in the usher N terminus disrupt chaperone-subunit binding and pilus biogenesis. Targeting of a chaperone-adhesin complex to the usher may prime the usher for pilus assembly, leading to a shift of the chaperone-adhesin complex to the usher C terminus to form an assembly intermediate (36). Subsequent targeting of additional chaperone-subunit complexes to the usher N terminus would then allow donor strand exchange between pilus subunits to build the pilus fiber, concomitant with secretion of the fiber to the cell surface. Significantly, we also show here that the usher N terminus is required for stages of pilus assembly in addition to the initial targeting of chaperone-subunit complexes. The F3A, C70A, and C97A mutations appear to have trapped the usher at one or more of these stages, allowing chaperone-subunit binding but preventing further events necessary for pilus biogenesis. The additional functions of the usher N terminus could include discrimination among different chaperone-subunit complexes, priming the usher for pilus biogenesis, shifting chaperone-subunit complexes to the usher C terminus, and/or preparing subunits for donor strand exchange.
Acknowledgments
We thank the Hultgren laboratory for providing antibodies, Keith Studholme for assistance with EM, Amelie Peryea for work on construction of pAP3, and Stephane Shu Kin So for assistance with overlay assays. We thank Jorge L. Benach for critical reading of the manuscript.
This work was supported by Public Health Service grant GM62987 from the National Institute of General Medical Sciences.
REFERENCES
- 1.Baga, M., M. Norgren, and S. Normark. 1987. Biogenesis of E. coli Pap pili: PapH, a minor pilin subunit involved in cell anchoring and length modulation. Cell 49:241-251. [DOI] [PubMed] [Google Scholar]
- 2.Baneyx, F., and G. Georgiou. 1990. In vivo degradation of secreted fusion proteins by the Escherichia coli outer membrane protease OmpT. J. Bacteriol. 172:491-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnhart, M. M., J. S. Pinkner, G. E. Soto, F. G. Sauer, S. Langermann, G. Waksman, C. Frieden, and S. J. Hultgren. 2000. PapD-like chaperones provide the missing information for folding of pilin proteins. Proc. Natl. Acad. Sci. USA 97:7709-7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blomfield, I. C., M. S. McClain, and B. I. Eisenstein. 1991. Type 1 fimbriae mutants of Escherichia coli K12: characterization of recognized afimbriate strains and construction of new fim deletion mutants. Mol. Microbiol. 5:1439-1445. [DOI] [PubMed] [Google Scholar]
- 5.Bock, K., M. E. Breimer, A. Brignole, G. C. Hansson, K.-A. Karlsson, G. Larson, H. Leffler, B. E. Samuelsson, N. Strömberg, C. Svanborg-Edén, and J. Thurin. 1985. Specificity of binding of a strain of uropathogenic Escherichia coli to Galα(1-4)Gal-containing glycosphingolipids. J. Biol. Chem. 260:8545-8551. [PubMed] [Google Scholar]
- 6.Bullitt, E., and L. Makowski. 1995. Structural polymorphism of bacterial adhesion pili. Nature 373:164-167. [DOI] [PubMed] [Google Scholar]
- 7.Choudhury, D., A. Thompson, V. Stojanoff, S. Langermann, J. Pinkner, S. J. Hultgren, and S. D. Knight. 1999. X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science 285:1061-1066. [DOI] [PubMed] [Google Scholar]
- 8.Dodson, K. W., F. Jacob-Dubuisson, R. T. Striker, and S. J. Hultgren. 1993. Outer membrane PapC usher discriminately recognizes periplasmic chaperone-pilus subunit complexes. Proc. Natl. Acad. Sci. USA 90:3670-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrmann, M., P. Bolek, M. Mondigler, D. Boyd, and R. Lange. 1997. TnTIN and TnTAP: mini-transposons for site-specific proteolysis in vivo. Proc. Natl. Acad. Sci. USA 64:13111-13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grant, S. G., J. Jessee, F. R. Bloom, and D. Hanahan. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 87:4645-4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harms, N., W. C. Oudhuis, E. A. Eppens, Q. A. Valent, M. Koster, J. Luirink, and B. Oudega. 1999. Epitope tagging analysis of the outer membrane folding of the molecular usher FaeD involved in K88 fimbriae biosynthesis in Escherichia coli. J. Mol. Microbiol. Biotechnol. 1:319-325. [PubMed] [Google Scholar]
- 12.Holmgren, A., and C. Brändén. 1989. Crystal structure of chaperone protein PapD reveals an immunoglobulin fold. Nature 342:248-251. [DOI] [PubMed] [Google Scholar]
- 13.Hultgren, S. J., F. Lindberg, G. Magnusson, J. Kihlberg, J. M. Tennent, and S. Normark. 1989. The PapG adhesin of uropathogenic Escherichia coli contains separate regions for receptor binding and for the incorporation into the pilus. Proc. Natl. Acad. Sci. USA 86:4357-4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacob-Dubuisson, F., R. Striker, and S. J. Hultgren. 1994. Chaperone-assisted self-assembly of pili independent of cellular energy. J. Biol. Chem. 269:12447-12455. [PubMed] [Google Scholar]
- 15.Jones, C. H., P. N. Danese, J. S. Pinkner, T. J. Silhavy, and S. J. Hultgren. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 16:6394-6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemm, P., and G. Christiansen. 1990. The fimD gene required for cell surface localization of Escherichia coli type 1 fimbriae. Mol. Gen. Genet. 220:334-338. [DOI] [PubMed] [Google Scholar]
- 17.Mori, H., and K. Ito. 2001. The Sec protein-translocation pathway. Trends Microbiol. 9:494-500. [DOI] [PubMed] [Google Scholar]
- 18.Mulvey, M. A., Y. S. Lopez-Boado, C. L. Wilson, R. Roth, W. C. Parks, J. Heuser, and S. J. Hultgren. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494-1497. [DOI] [PubMed] [Google Scholar]
- 19.Nikaido, H. 1994. Isolation of outer membranes. Methods Enzymol. 235:225-234. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama, M., M. Vetsch, C. Puorger, I. Jelesarov, and R. Glockshuber. 2003. Identification and characterization of the chaperone-subunit complex-binding domain from the type 1 pilus assembly platform FimD. J. Mol. Biol. 330:513-525. [DOI] [PubMed] [Google Scholar]
- 21.Norgren, M., M. Baga, J. M. Tennent, and S. Normark. 1987. Nucleotide sequence, regulation and functional analysis of the papC gene required for cell surface localization of Pap pili of uropathogenic Escherichia coli. Mol. Microbiol. 1:169-178. [DOI] [PubMed] [Google Scholar]
- 22.Orndorff, P. E., P. A. Spears, D. Schauer, and S. Falkow. 1985. Two modes of control of pilA, the gene encoding type 1 pilin in Escherichia coli. J. Bacteriol. 164:321-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg, A. H., B. N. Lade, D. S. Chui, S. W. Lin, J. J. Dunn, and F. W. Studier. 1987. Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56:125-135. [DOI] [PubMed] [Google Scholar]
- 24.Sauer, F. G., K. Fütterer, J. S. Pinkner, K. W. Dodson, S. J. Hultgren, and G. Waksman. 1999. Structural basis of chaperone function and pilus biogenesis. Science 285:1058-1061. [DOI] [PubMed] [Google Scholar]
- 25.Sauer, F. G., J. S. Pinkner, G. Waksman, and S. J. Hultgren. 2002. Chaperone priming of pilus subunits facilitates a topological transition that drives fiber formation. Cell 111:543-551. [DOI] [PubMed] [Google Scholar]
- 26.Saulino, E. T. 2000. Ph.D. thesis. Washington University, St. Louis, Mo.
- 27.Saulino, E. T., E. Bullitt, and S. J. Hultgren. 2000. Snapshots of usher-mediated protein secretion and ordered pilus assembly. Proc. Natl. Acad. Sci. USA 97:9240-9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saulino, E. T., D. G. Thanassi, J. S. Pinkner, and S. J. Hultgren. 1998. Ramifications of kinetic partitioning on usher-mediated pilus biogenesis. EMBO J. 17:2177-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slonim, L. N., J. S. Pinkner, C. I. Branden, and S. J. Hultgren. 1992. Interactive surface in the PapD chaperone cleft is conserved in pilus chaperone superfamily and essential in subunit recognition and assembly. EMBO J. 11:4747-4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soto, G. E., K. W. Dodson, D. Ogg, C. Liu, J. Heuser, S. Knight, J. Kihlberg, C. H. Jones, and S. J. Hultgren. 1998. Periplasmic chaperone recognition motif of subunits mediates quaternary interactions in the pilus. EMBO J. 17:6155-6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soto, G. E., and S. J. Hultgren. 1999. Bacterial adhesins: common themes and variations in architecture and assembly. J. Bacteriol. 181:1059-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugawara, E., M. Steiert, S. Rouhani, and H. Nikaido. 1996. Secondary structure of the outer membrane proteins OmpA of Escherichia coli and OprF of Pseudomonas aeruginosa. J. Bacteriol. 178:6067-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamm, L. K., A. Arora, and J. H. Kleinschmidt. 2001. Structure and assembly of beta-barrel membrane proteins. J. Biol. Chem. 276:32399-32402. [DOI] [PubMed] [Google Scholar]
- 34.Thanassi, D. G., E. T. Saulino, and S. J. Hultgren. 1998. The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr. Opin. Microbiol. 1:223-231. [DOI] [PubMed] [Google Scholar]
- 35.Thanassi, D. G., E. T. Saulino, M.-J. Lombardo, R. Roth, J. Heuser, and S. J. Hultgren. 1998. The PapC usher forms an oligomeric channel: implications for pilus biogenesis across the outer membrane. Proc. Natl. Acad. Sci. USA 95:3146-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thanassi, D. G., C. Stathopoulos, K. W. Dodson, D. Geiger, and S. J. Hultgren. 2002. Bacterial outer membrane ushers contain distinct targeting and assembly domains for pilus biogenesis. J. Bacteriol. 184:6260-6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valent, Q. A., J. Zaal, F. K. de Graaf, and B. Oudega. 1995. Subcellular localization and topology of the K88 usher FaeD in Escherichia coli. Mol. Microbiol. 16:1243-1257. [DOI] [PubMed] [Google Scholar]
- 38.Wimley, W. C. 2003. The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 13:404-411. [DOI] [PubMed] [Google Scholar]
- 39.Zavialov, A. V., J. Berglund, A. F. Pudney, L. J. Fooks, T. M. Ibrahim, S. MacIntyre, and S. D. Knight. 2003. Structure and biogenesis of the capsular F1 antigen from Yersinia pestis: preserved folding energy drives fiber formation. Cell 113:587-596. [DOI] [PubMed] [Google Scholar]