

Abstract
Long-non-coding RNAs (lncRNAs) have an undefined role in the pathobiology of glioblastoma multiforme (GBM). These tumors are genetically and phenotypically heterogeneous with transcriptome subtype-specific GBM stem-like cells (GSCs) that adapt to the brain tumor microenvironment, including hypoxic niches. We identified hypoxia inducible factor 1 alpha-antisense RNA 2 (HIF1A-AS2) as a subtype-specific hypoxia inducible lncRNA, up-regulated in mesenchymal GSCs. Its deregulation affects GSC growth, self-renewal and hypoxia-dependent molecular reprogramming. Amongst the HIF1A-AS2 interactome, IGF2BP2 and DHX9 were identified as direct partners. This association was needed for maintenance of expression of their target gene, HMGA1. Down-regulation of HIF1A-AS2 led to delayed growth of mesenchymal GSC tumors, survival benefits, and impaired expression of HMGA1 in vivo. Our data demonstrate that HIF1A-AS2 contributes to GSCs’ speciation and adaptation to hypoxia within the tumor microenvironment, acting directly through its interactome/targets and indirectly by modulating responses to hypoxic stress depending on the subtype-specific genetic context.
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common and aggressive primary brain tumor in adults, with a median survival of 14.2 months (Johnson and O’Neill, 2012). One of hallmarks of GBM is its high-level of heterogeneity with cells exhibiting varying degrees of polymorphism, both phenotypically and molecularly (Soeda et al., 2015). A sub-population of GBM cells has been identified that retains stem cell characteristics, including self-renewal and undifferentiated status and these cells are described as GBM stem-like cells (GSCs) (Singh et al., 2004). In fact, characterization of the GBM genome (Parsons et al., 2008) and transcriptome (Phillips et al., 2006; Verhaak et al., 2010) has revealed the existence of several distinct cellular subtypes among GBM patients, known as Mesenchymal (M), Proneural (P), Neural (N) and Classical (C). Cellular heterogeneity was also demonstrated for pure populations of GSC in culture, based on protein-coding gene expression (Mao et al., 2013) and recently, single-cell RNA sequencing revealed co-existence of different GSC subtypes within individual tumors (Patel et al., 2014).
The complexity of solid tumors, including GBM, and their distinct pathophysiology relies on anatomic niches that transmit and receive signals through cellular and acellular mediators (Jones and Wagers, 2008). The GBM microenvironment is a complex “ecosystem” composed of distinct phenotypic cell components (Patel et al., 2014) including heterogeneous tumor cells (both GSCs and more differentiated progenitor cells), associated astrocytes, infiltrating immune cells and microglia, abnormal vasculature (Meacham and Morrison, 2013) and extensive hypoxic and necrotic zones (Li et al., 2009; Mathew et al., 2014). These components are highly reliant on each other and undergo constant architectural, phenotypic and transcriptomic re-arrangements depending on fluctuating microenvironmental contexts as the disease progresses (Godlewski et al., 2015).
In recent years unprecedented progress has been made toward understanding the function of non-coding RNAs (ncRNAs) which constitute a vast majority of the human transcriptome (Marx, 2014). Among numerous subclasses of ncRNA a large category is known as long-non-coding RNAs (lncRNA). Although poorly described, it is recognized that they are capable of tasks such as post-transcriptional regulation, cell-cell signaling, organization of protein complexes, and their allosteric regulation. They are involved in physiological (development and differentiation (Fatica and Bozzoni, 2014)), as well as pathological processes such as carcinogenesis (Huarte, 2015). Several lncRNAs have been described in hypoxia-associated cancer processes, implying a potential role in maintaining cellular homeostasis and enabling adaptive survival during hypoxia (Chang et al., 2016; Takahashi et al., 2014). LncRNAs are involved in numerous brain functions (Qureshi and Mehler, 2012) and have been increasingly implicated in the pathobiology of GBM (Pastori et al., 2015; Vassallo et al., 2015; Zhang et al., 2013). LncRNAs that are associated with GBM subtypes and clinical prognosis have been identified through integrative analysis of their expression profiles and clinical outcome (Du et al., 2013). Although this analysis predicted lncRNAs that could be potential drivers of cancer progression, it lacked functional validation.
Elucidating the biological mechanisms whereby hypoxic tumor cells can adapt and survive under severe conditions is of significant clinical importance. While it has been widely accepted that proteins and microRNAs take part in hypoxic cancer progression, it is not known if and how lncRNAs participate. Here we report that the lncRNA –HIF1A-AS2 is highly expressed in mesenchymal (M) GSCs and in GBM but is largely absent in adjacent brain. HIF1A-AS2 interacts with proteins such as insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) and ATP-dependent RNA helicase A (DHX9), enhancing the expression of several of their targets (e.g. high mobility group AT-hook 1 (HMGA1)), and further downstream leads to changes in Endothelial PAS domain-containing protein 1 (EPAS1, also known as HIF2A) expression and the molecular response to hypoxic stress. Remarkably, HIF1A-AS2 regulates the growth and self-renewal of M GSCs and this phenotype is reflected by gene expression rearrangements that are associated with clinical outcome. Finally, we demonstrate that HIF1A-AS2 is essential for tumorigenicity of M GSC-originated intracranial xenografts and that its expression is stimulated in vivo by hypoxic stress. These results highlight a critical role for HIF1A-AS2 in the maintenance of mesenchymal GSC function, and suggest that this lncRNA in GBM mediate the adaptation of GSCs to hypoxic stress.
RESULTS
LncRNA signature reflects intratumoral heterogeneity of GBM
Recognizing novel molecular determinants, such as lncRNAs, which act in GSC subtypes would allow identification of functional targets and provide much needed insight into the contribution of lncRNA to GBM pathophysiology. To analyze the expression of cancer-related lncRNAs in GBM we designed a platform (Table S1 and supplementary References) to detect 73 cancer-related transcripts. We used our collection of GBM specimens to screen lncRNAs, expressed in tumor tissue and in adjacent, matched (i.e. harvested from the same individual) brain tissue. In parallel, we isolated GSCs from GBM specimens and cultured them in serum-free conditions as described before (Peruzzi et al., 2013) (Figure 1A). The analysis of lncRNA in GBM tissue revealed a tumor-specific pattern of expression: 8 lncRNAs were specifically down-regulated, while 7 were specifically up-regulated in tumor, when compared to adjacent tissue (Figure 1B, left). The GSC collection was characterized using a gene signature which assigns a GSC culture to either proneural (P), mesenchymal (M), or “other” subtype (Mao et al., 2013) (Figure S1A). The analysis of lncRNA expression in those GSCs also uncovered a subtype-specific pattern of expression; 20 of 64 detectable lncRNA transcripts (Figure S1B) were significantly enriched in proneural GSCs, and 7 were up-regulated in mesenchymal GSCs (Figure 1B, right). The lncRNA HIF1A-AS2 was one of the most differentially expressed in both tissues and cells (Figure 1C, top). In fact there was significant enrichment of HIF1A-AS2 in each GBM compared to its matched brain tissue (Figure 1C, bottom, left), as well as in M compared to proneural GSCs (Figure 1C, bottom, right). To validate the platform results, we analyzed the expression of three other lncRNAs that were expressed in GSCs, P-specific MEG3, M-specific WT1-AS and a non-subtype specific MALAT1 (Figure S1C). These results show that there was GBM-specific and GBM stem cell subtype-specific pattern of lncRNA expression, and that HIF1A-AS2 was one of the most tumor-, and subtype-specific lncRNAs.
Figure 1. LncRNA signature reflects intratumoral heterogeneity of GBM.

(A) Workflow depicting isolation of tissue and GSCs from GBM patients for lncRNA analysis.
(B) GBM and GSC lncRNA profile distinguishes tumor (Tumor Tissue - TT) from normal tissue (matched Tissue Adjacent to brain Tumor - TAT) and proneural (blue) from mesenchymal (red) GSC subtypes. LncRNA sets that vary coherently between tissues (left) and GSCs (right) were identified by supervised clustering (fold >2, P value < 0.05).
(C) HIF1A-AS2 is tumor (left) and mesenchymal (M) GSCs (right) enriched lncRNA. Relative expression of all lncRNAs (top) as Nanostring reads (NS) and HIF1A-AS2 (bottom) is shown. Data shown as mean ± SD, * P value < 0.05, ** P value <0.01.
HIF1A-AS2 controls cellular fate and molecular landscape of mesenchymal GSCs
We hypothesized that HIF1A-AS2 de-regulation may have important implications for the pathobiology of GBM. Lentiviral shRNA-mediated knockdown of HIF1A-AS2 resulted in its significant depletion in mesenchymal GSCs (Figure 2A) and significant impairment of growth with a concomitant decrease in cell viability (Figure 2B, Figure S2A). Moreover, HIF1A-AS2 knockdown led to diminished neurosphere-forming capacity and reduced neurosphere size (Figure 2C–D). However targeting of HIF1A-AS2 had little effect on growth or viability of proneural GSCs (Figure S2B). In order to delineate the extent of the HIF1A-AS2-dependent molecular footprint, we used the Nanostring nCounter® PanCancer Pathway Panel that detects transcripts of cancer-related genes. We observed significant de-regulation of 47/730 transcripts (Figure 2E, Figure S2C, top panel). Interestingly, the majority of upregulated genes were not proneural or mesenchymal while genes downregulated by HIF1A-AS2-knockdown were expressed in P or mesenchymal GSCs. (Figure S2C bottom panel). The in silico analysis revealed marked down-regulation of pro-proliferative traits concomitant with up-regulation of cell death-related processes in HIF1A-AS2 knockdown M GSCs (Figure S2D). This prompted us to test whether genes deregulated by HIF1A-AS2 were associated with GBM patient outcome. Despite using pre-selected (biased) list of genes, we were able to detect significant association of genes down-regulated in knockdown cells with poorer outcome (Figure S2E). The physical proximity of the HIF1A-AS2 to the hypoxia inducible factor 1 alpha (HIF1A) genomic locus, prompted us to test the effect of low oxygen tension on HIF1A-AS2 transcription. This revealed that in M GSCs, HIF1A-AS2 was not only the most significantly up-regulated lncRNA despite its high basal (normoxic) levels, but also one of the very few lncRNAs whose levels were affected by hypoxic stress in GSCs (Figure 2F, middle; Table S2), while in proneural GSCs the levels of HIF1A-AS2 remained low regardless of oxygen concentration. This result was confirmed by qPCR analysis, showing consistent up-regulation across all tested mesenchymal GSCs but not proneural GSCs (Figure 2F, bottom). Expectedly, levels of HIF1A and EPAS1 mRNA remained stable upon exposure to hypoxic stress in mesenchymal GSCs (with no apparent pattern in proneural GSCs), while the respective encoded proteins were consistently induced in both subtypes of GSC (Figure S2F). Thus the observed unaltered proliferation and viability in both P and mesenchymal GSC under hypoxic stress (Figure S2G) underline the mesenchymal GSC-specific HIF1A-AS2 driven program. This effect was not limited to the stem-like cells as differentiation-promoting conditions did not abolish HIF1A-AS2 expression or its upregulation in hypoxia (Figure S2H) and also led to maintenance of the phenotypic effect of HIF1A-AS2 knockdown (Figure S2I). To assess HIF1A-AS2-dependent signaling during hypoxia, we measured the dynamics of activation of several genes that were shown to be hypoxia-dependent in GBM (Patel et al., 2014). The activation of these genes either lagged or did not occur in mesenchymal GSCs where HIF1A-AS2 was knocked-down (Figure 2G). Moreover, knockdown of HIF1A-AS2 did not alter the induction of HIF1A upon exposure to hypoxia; while induction of EPAS1 was significantly impaired upon HIF1A-AS2 knockdown (Figure 2H). The hypoxic stress caused swift and robust up-regulation of HIF1A-AS2 in mesenchymal GSCs, and similar but weaker effect was observed in knockdown cells, suggesting that hypoxia-dependent induction of the endogenous transcript overcame the shRNA effects (Figure S2J). Similarly to previous findings, HIF1A-AS2 in mesenchymal GSCs is predominantly nuclear and this distribution was not affected by hypoxic stress (Figure S2K). Knockdown of HIF1A-AS2 resulted in an altered mesenchymal GSCs phenotype concomitant with hypoxia-dependent molecular rearrangements and de-regulation of genes associated with worse patients’ outcome underlining the clinical relevance of this lncRNA in GBM.
Figure 2. HIF1A-AS2 controls cellular fate and molecular landscape of mesenchymal GSCs.

(A) Down-regulation of HIF1A-AS2 in mesenchymal GSCs. Short hairpin (sh) RNA strategy (left) and qPCR analysis is shown (right). Data shown as mean ± SD, ** P value < 0.01.
(B) Knockdown of HIF1A-AS2 reduces mesenchymal GSC proliferation (left) and viability (right). Cell number and percentage of dead cells are shown. Data shown as mean ± SD, ** P value < 0.01.
(C) Knockdown of HIF1A-AS2 inhibits sphere formation. Sphere frequency using linear regression plot is shown.
(D) Knockdown of HIF1A-AS2 reduces sphere growth. Representative microphotographs of GSC spheroids (left) and quantification of sphere volume (right) are shown. Data shown as mean ± SD, ** P value < 0.01. Scale bar 50μm.
(E) Knockdown of HIF1A-AS2 results in gene expression rearrangement. Gene sets that vary coherently between control and HIF1A-AS2 knockdown mesenchymal GSC (two single cell clones #1 and #2 were analyzed) were identified by supervised clustering (fold >2, P value < 0.05).
(F) Hypoxic stress up-regulates HIF1A-AS2 in mesenchymal GSC. Workflow depicting hypoxic stress strategy (top). Global expression of lncRNAs in normoxic vs. hypoxic conditions (middle), dashed lines indicates 2-fold deregulation, qPCR validation of HIF1A-AS2 levels in mesenchymal (M) and proneural (P) GSC upon exposure to hypoxia (bottom) are shown. Data shown as mean ± SD, ** P value < 0.01.
(G) Knockdown of HIF1A-AS2 altered response to hypoxia. QPCR based expression signature is shown.
(H) Knockdown of HIF1A-AS2 suppresses EPAS1 activation. Representative Western blot analysis is shown.
HIF1A-AS2 drives tumor progression in hypoxic environment
GBM patients with the aggressive and predominantly mesenchymal subtype exhibit a particularly high degree of tumor necrosis (Verhaak et al., 2010), and conversely highly necrotic tumors are significantly enriched for the mesenchymal transcriptional gene signature (Cooper et al., 2012). To validate the impact of HIF1A-AS2 on tumorigenicity of GSCs, we implanted highly aggressive mesenchymal GSCs that expressed either control shRNA or HIF1A-AS2 shRNA as intracranial xenografts (Figure 3A). We sacrificed one group after 10 days, while the second group was observed for survival analysis. We observed strikingly smaller tumors in the knockdown group after 10 days (Figure 3B, Figure S3A). The survival benefits in knockdown group were also significant, although ultimately these mice also perished (Figure 3C). We hypothesized that as tumor growth progresses and hypoxia increases in the tumor core, the effect of shRNA was overridden by increased HIF1A-AS2 expression driven by the hypoxic microenvironment in vivo. This resulted in ultimate tumor progression and only modest survival benefits. Thus, we analyzed the expression of HIF1A-AS2, HIF1A and EPAS1 in vivo. As expected, the expression of both HIF1A and EPAS1 was much weaker in the HIF1A-AS2 knockdown vs. control tumors (Figure 3D, left). However, de-repression of the expression of HIF1A-AS2 in knockdown cells occurred in a time-dependent fashion (in agreement with a similar finding in vitro (Figure S2G)) and in terminal tumors it reached levels that were comparable to those in control cells (Figure 3D, right). These findings implied that HIF1A-AS2 knockdown tumors delayed their initiation phase (resulting in overall survival benefits), albeit only transiently due to ultimate increase of hypoxic stress that de-repressed HIF1A-AS2 expression.
Figure 3. HIF1A-AS2 drives tumor progression in hypoxic environment.
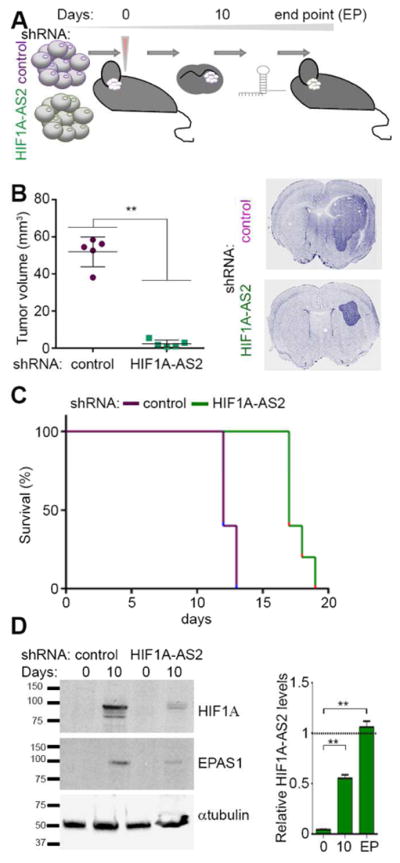
(A) Workflow depicting in vivo experimental design.
(B) HIF1A-AS2 knockdown reduces tumor volume of mesenchymal GSC-originated intracranial xenografts. Quantification of tumor volume and representative DAPI staining of brain sections 10 days post implantation are shown. Data shown as mean ± SD, ** P value < 0.01.
(C) HIF1A-AS2 knockdown in mesenchymal GSC-originated tumors is associated with prolonged survival. Kaplan-Meier curves are shown. N = 5; P = 0.0023.
(D) Tumor microenvironment effect on HIF1A and EPAS1 expression depends on HIF1A-AS2 status. Representative Western blot (left) and qPCR (right) analyses are shown. Data shown as mean ± SD, ** P value < 0.01.
HIF1A-AS2 interactome targets belong predominantly to a class of RNA processing proteins
To identify the HIF1A-AS2 interactome in mesenchymal GSCs, we utilized an in vitro transcription assay coupled with transcript biotinylation to allow pulldown of putative targets followed by mass spectrometry (MS) with additional controls of either no RNA probe or other lncRNA (Figure 4A). We identified a number of putative interacting partners (Table S3), with posttranscriptional regulation of gene expression and mRNA stabilization being the most predominant biological processes (Figure S4A). To validate MS results, we performed Western blot analysis on RNA pulldown material (Figure 4B, left). Identification of direct binding partners of HIF1A-AS2 was achieved by UV-mediated cross-linking followed by RNA pulldown in high stringency conditions (Figure 4B, right). We identified two proteins, DHX9 and IGF2BP2, which directly interact with HIF1A-AS2 (Figure 4B, right) but not with another lncRNA, MEG3 (Figure 4B, bottom panel). The specificity of the HIF1A-AS2-DHX9/IGF2BP2 interactions was additionally validated by protein–RNA immunoprecipitation assays using a panel of lncRNAs (proneural-specific: DLEU2, MEG3, mesenchymal-specific: WT1AS, NEAT1, PRNCR1, GAS5, RMRP and GBM-specific: MALAT1): this showed that only HIF1A-AS2 physically interacted with these two proteins, and that DLEU2, MALAT1, WT1AS were found to interact only with IGF2BP2 although with lower affinity (Figure S4B). Interestingly, DHX9 and IGF2BP2 have been shown to interact with each other (Chatel-Chaix et al., 2013). To select potential downstream effectors of HIF1A-AS2 we first determined if IGF2BP2 targets (Janiszewska et al., 2012) were subtype-specific. As IGF2BP2 is a protein abundantly expressed in both GSC subtypes, it was not surprising that its target genes were not proneural or mesenchymal. Interestingly proneural GSC upregulated targets of IGF2BP2 were not subtype-specific, in contrast to mesenchymal GSC genes which overlapped with mesenchymal signature transcripts (Figure S4C). Among IGF2BP2 target genes, 49 correlated with mesenchymal GSC specific expression (Figure S4D, left panel). Moreover, among IGF2BP2 target genes, there were two genes (HMGA1 and FOS-like antigen 1(FOSL1)) that were down-regulated in both DHX9 knockdown (Figure S4D, right panel) (Lee et al., 2014) and HIF1A-AS2 knockdown cells (Figure S4D). All nine genes that were targets of IGF2BP2 and HIF1A-AS2 were more abundant in mesenchymal GSCs (Figure S4E left panel). Their downregulation by HIF1A-AS2 knockdown was validated by qPCR (Figure S4E, right panel). Next, we tested whether binding of HIF1A-AS2 to its protein partners led to functional consequences for their downstream RNA targets (Figure 4C). As expected, there was an interaction between HIF1A-AS2 and DHX9/IGF2BP2 in mesenchymal GSCs but not in proneural GSCs (Figure 4C, left graphs). In addition, we found that in mesenchymal GSCs there was strong binding between IGF2BP2 and HMGA1, as expected (Janiszewska et al., 2012), and between DHX9 and HMGA1, which has not been previously shown (Figure 4C, middle graphs). Conversely, knockdown of HIF1A-AS2 resulted in significantly diminished interactions between RNA-binding proteins and their mRNA targets (Figure 4C, right graphs). Importantly, knockdown of HIF1A-AS2 did not alter levels of either mRNA or protein of its interacting partners, while it significantly suppressed expression of HMGA1 (Figure 4D, Figure S4F). While the protein levels of DHX9 and IGF2BP2 did not show significant difference between P and mesenchymal GSCs, HMGA1 was strongly enriched in mesenchymal GSCs (Figure S4G). Finally, we analyzed expression of HMGA1 in tumors formed by HIF1A-AS2 knockdown cells and found that its mRNA and protein levels were significantly elevated in the course of tumor progression (Figure 4E), corresponding with up-regulation of HIF1A-AS2 (Figure 3D). Therefore, we conclude that direct interaction of HIF1A-AS2 with DHX9 and IGF2BP2 mRNA binding complexes drives expression of their downstream mRNA targets with pro-oncogenic functions, such as HMGA1, thus explaining the lncRNA-driven tumorigenic phenotype. As only a certain sub-population of tumor cells express HIF1A-AS2 and activates it during hypoxia, we attempted to link the transcriptome profiles of GSC subtypes, HIF1A-AS2 knockdown and hypoxia to the intra-tumoral architecture defined by predominant characteristic phenotypes of tumors (infiltration, proliferation and necrotic zones). As expected, the hypoxic signature was found primarily in necrotic areas, which significantly overlapped with the mesenchymal signature, but not with the proneural signature which was detected mainly in infiltration and proliferation zones. Interestingly, genes down-regulated in HIF1A-AS2 knockdown cells were detected predominantly in the necrotic zone (Figure 4F, Table S4). These data implicate HIF1A-AS2 as a lncRNA contributing to GSCs’ speciation and adaptation to dynamic oxygen fluctuations in the tumor microenvironment.
Figure 4. HIF1A-AS2 interactome partners belong to the class of RNA processing proteins.

(A) Workflow depicting approach to identify HIF1A-AS2 interactome in mesenchymal GSCs by pulldown of biotinylated transcript followed by mass spectroscopy (MS)(top). Coomassie Blue staining of biotinylated HIF1A-AS2-associated proteins is shown (bottom). * indicate DHX9 and ** indicate IGF2BP2 band. Binding to other RNA (MEG3) serves as a control of binding specificity.
(B) DHX9 and IGF2BP2 are direct binding partners of HIF1A-AS2. Western blot analysis of a set of proteins identified by MS (left) and UV-crosslinked pulldown of biotinylated HIF1A-AS2 and control RNA (MEG3) are shown (right and bottom).
(C) Binding between HIF1A-AS2 and its interacting partners affects expression of their downstream target – HMGA1. Workflow depicting RNA immunoprecipitation (RIP) strategy (left). Analysis of UV-crosslinked α-DHX9 RIP (top) and α-IGF2BP2 RIP (bottom). QPCR in proneural (P) and mesenchymal (M) GSCs on HIF1A-AS2 (left) and on HMGA1 (middle) and in control and HIF1A-AS2 knockdown mesenchymal GSCs (right). Data shown as mean ± SD, ** P value < 0.01.
(D) Knockdown of HIF1A-AS2 in mesenchymal GSCs reduces levels of HMGA1 protein. QPCR (left) and Western blotting analysis (right) of selected genes in mesenchymal GSCs upon HIF1A-AS2 knockdown are shown. Expression is relative to control mesenchymal GSCs. Data shown as mean ± SD, ** P value < 0.01.
(E) HIF1A-AS2 knockdown-dependent suppression of HMGA1 in mesenchymal GSCs is maintained at the early stage of tumor progression in vivo. QPCR (left) and Western blot (right) analyses of HMGA1 10 days post implantation in HIF1A-AS2 knockdown M GSCs in vivo are shown. Expression is relative to control M GSCs. Data shown as mean ± SD, ** P value < 0.01.
(F) Expression of mesenchymal (M) or proneural (P)-specific, hypoxia-dependent and HIF1A-AS2-dependent genes is prevalent in necrotic niche of GBM. Ivy GAP database-based expression signature in different areas of GBM for top-30 genes is shown.
(G) A proposed HIF1A-AS2-dependent signaling in a normoxic and hypoxic microenvironment of GBM.
DISCUSSION
GSCs are highly tumorigenic and resistant to conventional radio- and chemo-therapy; therefore, they constitute the primary target for the development of anti-GBM therapies (Lathia et al., 2015; Sorensen et al., 2015). To date, analyses of molecular diversity have focused mostly on protein-coding genes (Patel et al., 2014), but the engagement and contribution of non-coding RNAs remains insufficiently characterized. Although thousands of lncRNAs have been discovered to date, very little is known about their mode of action and possible role in the regulation of cancer-related processes. The positive correlation of bidirectional sense/antisense transcription is in accord with numerous studies showing that antisense RNAs can regulate their neighboring genes in a cis- mode (Kunej et al., 2014). Here we have shown that although HIF1A-AS2 expression responds to hypoxic stress, it had no effect on the expression of its neighboring gene in sense orientation – HIF1A. In fact, the HIF1A locus is ubiquitously transcribed and its protein levels are regulated by rapid degradation under normoxic conditions (Semenza, 2013; Wang et al., 1995). This highly conserved mechanism is unlikely to be controlled by a cell type-specific (Thrash-Bingham and Tartof, 1999) and not conserved (even in mammals) antisense transcript. One of the prominent modes of action for lncRNA is to interact with other cellular factors including proteins, DNA, and other RNA molecules (Minajigi et al., 2015; Ulitsky and Bartel, 2013). The HIF1A-AS2 protein interactome at first indicated a multifunctional role for this lncRNA. We found that identified direct and indirect protein targets were engaged in mRNA metabolism. Interestingly, we did not find that the HIF1A-AS2 lncRNA interacts with transcriptional machinery and protein chromatin remodeling complexes in GSCs, unlike what has been reported for other lncRNAs (Flynn and Chang, 2012). It remains to be investigated whether a direct interaction of HIF1A-AS2 with DNA or pairing with other RNA molecules can occur. IGF2BP2, the direct binding partner of HIF1A-AS2, has been shown to drive a cancer stem cell phenotype in GBM by binding and stabilizing mRNA (including HMGA1) that is enhanced by hypoxic conditions (Janiszewska et al., 2012), suggesting a possible mechanism for GSC adaptation to low-oxygen environments. In fact, IGF2BP2 mRNA and protein are not deregulated in the various GBM subtypes or by hypoxia, suggesting that its function is regulated by co-interaction with other proteins such as DHX9. Such interaction has been already shown to be dependent on the presence of RNA (Chatel-Chaix et al., 2013). Our results suggest that HIF1A-AS2 acts by interacting with RNA binding proteins (IGF2BP2 and DHX9) to stimulate expression of their target mRNAs such as HMGA1, resulting in an increase in protein levels. Importantly, in addition to HMGA1’s function during development (Chiappetta et al., 1996), it is over-expressed in virtually every cancer (Fusco and Fedele, 2007), and its expression levels correlate with the degree of malignancy. In fact, GBM patients with higher levels of HMGA1 exhibit a significantly shorter progression-free survival time (Liu et al., 2015). Our findings that HIF1A-AS2: i) is significantly over-expressed in GBM tumors and in mesenchymal GSCs, ii) promotes stem cell-like and tumorigenic behaviors, and iii) its expression is associated with GBM outcome, suggests that HIF1AS-AS2 is a non-protein coding oncogene. However, it needs to be noted that HIF1AS-AS2 targets genes are highly or even exclusively expressed in mesenchymal GSC, indicating that HIF1A-AS2 tumorigenic function may be cell specific. It suggests that HIF1A-AS2 may be a contextual oncogene, engaged in physiological processes (such as maintenance of homeostasis) in other tissues/organs.
Naturally occurring oxygen gradients serve as morphogenic signals in rapidly growing embryonic tissues (Simon and Keith, 2008), but become extreme in pathophysiological conditions such as ischemia or the rise of solid tumors. Thus chronic exposure to severe oxygen deprivation frequently produces necrotic zones surrounded by densely packed hypoxic tumor cells. This stimulates the development of a tumoral architecture with hierarchical cellular organization/speciation. In fact, our recent study has shown that such cellular organization may be recapitulated in vitro and in vivo (Ricklefs et al., 2016). Interestingly, the gene analysis of GBM tissues harvested by laser microdissection showed GSC-specific signature associated with tumor anatomic sites (glioblastoma.alleninstitute.org.) (Figure 4F). Several prominent reports (Bhat et al., 2013; Bozdag et al., 2014; Halliday et al., 2014; Joseph et al., 2015; Nakano, 2014; Piao et al., 2013) have suggested that certain microenvironmental (such as hypoxia) and therapy-inflicted stressors (such as bevacizumab, irradiation) and/or pathway instabilities (VEGF, NFkB, TNF) may have cause a transition between GBM subtypes. All these reports however provided strong indication of proneural-to-mesenchymal transition, while the evidence for the transition occurring in the opposite direction is still lacking. The subtype-specific expression of HIF1A-AS2 that is induced exclusively in mesenchymal GSC, suggests that this lncRNA drives rather adaptation of mesenchymal GSC to their anatomic hypoxic niche than promotes transition shift from one subtype to the other. The fact that although proneural GSC do not express HIF1A-AS2 but respond to hypoxia and survive this stress, suggest that diverse programs of hypoxic response exist in these cells. The analysis of genes deregulated by HIF1A-AS2 knockdown in mesenchymal GSC has shown extensive deregulation, however a shift toward other subtypes was not observed. In fact, the M-specific signature was downregulated while genes upregulated by HIF1A-AS2 knockdown did not cluster with either subtype. These data, along with the fact that HIF1A-AS2 is not expressed in proneural GSC even upon hypoxic stress, indicate that this lncRNA does not take part in a subtype switch, suggesting that proneural GSCs use a different mechanism for such transition. Characterizing the epigenetic states of phenotypically distinct cells and identifying transcription factors, that are sufficient to reprogram differentiated cells into a tumorigenic stem-like state suggest a plastic developmental hierarchy in GBM cell populations (Carro et al., 2010; Suva et al., 2014). The recent observation that individual tumors contain a spectrum of GBM subtypes and hybrid cellular states (Patel et al., 2014), which is reflected in the diverse expression of ncRNAs (Du et al., 2013) and range of environmental influences (Godlewski et al., 2015), adds further complexity to the pathobiology of GBM. To fully reconstruct a network model that highlights the critical machinery sufficient to fully reprogram differentiated GBM cells, a comprehensive analysis of transcription factors and their downstream effectors (both protein-coding and non-coding RNAs) in the context of the tumor microenvironment is needed. This study shows significant deregulation of lncRNA expression in GBM using highly clinically relevant samples The clinical significance of our findings was underlined by linking HIF1A-AS2 downstream effectors with patient survival outcomes based on GBM subtypes. It is increasingly evident that GBM/GSC subtypes use different signaling and transcriptional networks (e.g. proneural GSC-specific Sox2, Olig2, Notch and PDGFRA and mesenchymal GSC-specific WT1, c-Met and EGFR) (Frattini et al., 2013; Mao et al., 2013; Patel et al., 2014). Moreover GSC subtypes are characterized by divergent epigenetic footprints, as for example the activity of Polycomb Repressor Complexes (one of most important lncRNA effectors) differs significantly between the subtypes (Zheng et al., 2011). Our results clearly indicate that HIF1A-AS2 is selectively important in mesenchymal GSC as its downstream effectors (e.g. HMGA1, FOSL1) are expressed exclusively in M but not in proneural GSCs, providing plausible explanation for its subtype specificity. Importantly, HIF1A-AS2 selectively response to hypoxia in mesenchymal GSC only, as these cells are found predominantly in hypoxic zones, suggesting that microenvironmental adaptation may be one of important drivers of GSC speciation.
The association between necrosis and the mesenchymal transcriptional class in GBM highlights the important contribution of the tumor microenvironment in implementation of hierarchical organization of the tumor (Carro et al., 2010; Orr and Eberhart, 2012). Up-regulation of HIF1A-AS2 by hypoxia in M but not in proneural GSCs suggest that HIF1A-AS2 acting in GBM in tumor anatomic site-dependent fashion may control adaptation of specific set of cells to hypoxic stress. Some experimental evidence suggests that tumor cells may cope with hypoxia by turning on the migratory phenotype to escape from metabolically stressful events/locations (Brat et al., 2004). Here we argue that adaptation rather than behavioral transition drives the survival and proliferation of GBM cells in hypoxic zones (Figure 4G).
GBM is recognized as a complex “ecosystem” composed of cells with distinct phenotypes, genotypes, and epigenetic landscapes. It becomes increasingly clear that the resistance to adverse environmental conditions, such as hypoxia, contributes to the tumor progression and reduced efficacy of anticancer therapies. The mechanisms by which tumor cells respond and adapt to hypoxic stress are crucial in the pathobiology of solid tumors such as GBM. Based on our data, we propose a model depicting an important role for HIF1A-AS2 in the regulation of hypoxic adaptation in tumor cells in tumor anatomic site dependent context which can have important clinical implications, and serve as a proof-of-concept for the development of personalized GBM therapy (Reardon et al., 2015).
EXPERIMENTAL PROCEDURES
Human specimens
Tumor and tumor adjacent tissue samples were obtained as approved by the Institutional Review Board at The Ohio State University and The Harvard Medical School. Surgery was conducted by E.A. Chiocca or I. Nakano. Patient samples were processed for extraction of total RNA or establishment of patient-derived neurospheres.
Cell culture
Primary human GSCs (G2, G6, G33, G34, G35, G44, G62, G88, G146, G157, G91) were isolated by dissociation of gross tumor samples. The unique identity of cultured patient-derived cells was confirmed by short tandem repeats analysis (Kim et al., 2016). Cells where cultured as neurospheres in stem cells enriched condition using Neurobasal (Gibco) supplemented with 1% Glutamine (Gibco), 2% B27 (Gibco) and 20 ng/mL EGF and FGF–2 (PeproTech) or in differentiation-promoting condition using DMEM (Gibco) supplemented with 10% FBS (Sigma). For the differentiation effect, cells cultured in stem cells enriching conditions were transferred to differentiation promoting conditions. Unless otherwise specified, hypoxia experiments were performed at 1% O2 for 24 h. G88, G33, G816, G44 cell lines were infected with lentiviral psi-LVRU6GP shCTR001 vector or psi-LVRU6GP sh217J6/J8 vectors (GeneCopoeia).
Nanostring assay
Nanostring nCounter custom made lncRNA assay and nCounter PanCancer Pathways assay were performed according to manufactures’ instructions (NanoString technologies) and as previously described (Peruzzi et al., 2013).
Immunoblot analysis and antibodies
Immunoblotting was performed as previously described (Mineo et al., 2012). Following antibodies were used: anti-HIF-1A (#610958, BD Biosciences); anti-EPAS1, anti-HMGA1 (#7096 and #7777 respectively, Cell Signaling); anti-DHX9, anti-IGF2BP2 (A300-855A and A303-316A respectively, Bethyl laboratories); anti-NCL, anti-hnRNPA1, anti-G3BP1, anti-HuR (ELAVL1), anti-Fus/TLS (sc9893, sc10030, sc365338, sc5261 and sc25540 respectively, Santa Cruz); anti-Caprin1 (15112-1AP, Protein Tech Group).
RNA pulldown assay
Full length HIF1A-AS2 was cloned into pCI-neo vector (Promega). Biotin-labelled HIF1A-AS2 was transcribed in vitro with Biotin RNA labeling mix (Roche) and T7 polymerase (Roche) and purified using PureLink RNA Mini kit (Ambion). RNA was heated at 65°C for 5 min and then cooled slowly- for 20min to allow secondary structure formation. GSCs were lysed in lysis buffer (50mMTris, 100mM NaCl, 1% Triton, 1 mM EDTA, 1 mM EGTA, 50 mM beta-glycerophosphate, 1 mM DTT, 1mM PMSF, and protease inhibitor cocktail). For pulldown assay, 3 μg of biotin-labeled HIF1A-AS2 RNA was mixed with total cell lysate (500 μg of protein) and incubated at RT for 2 h in the presence of RNasin (100U/ml, Promega). 40 μl of washed streptavidin beads (Invitrogen) were added to the binding reaction and further incubated for 1 h at RT. Beads were washed five times in lysis buffer and bound proteins were analyzed by mass spectrometry as previously described (Bronisz et al., 2014). For UV-crosslink RNA pulldown assays, the binding reaction was UV-irradiated at 400 mJ/cm2 and then incubated with streptavidin beads at RT. After incubation, beads were washed three times with high-stringency wash buffer, three times in high-salt wash buffer, three times in low-salt wash buffer, three times in PXL buffer (Moore et al., 2014), and three times in lysis buffer. RNA was then digested using RNase A and bound proteins were analyzed by immunoblotting.
UV-crosslink RIP
GSCs were UV-irradiated at 400 mJ/cm2 and lysed in modified RIPA buffer (50mM Tris, 150 mM NaCl, 4 mM EDTA, 1% NP-40, 0.1% Na-deoxycholate, 0.5 mM DTT, 100U/ml RNasin, protease and phosphatase inhibitors (Roche)). Cell lysates were precleared with Protein A/G Plus Agarose beads (Pierce) for 1 h at 4°C and incubated with primary antibodies (either IGF2BP2 or DHX9) or rabbit IgG control (Santa Cruz) overnight at 4°C. Protein/RNA complexes were precipitated with Protein A/G Plus Agarose beads, washed three times with modified RIPA buffer, three times with high-salt buffer (1M NaCl modified RIPA buffer), and then three times with modified RIPA buffer. Samples were then treated with Proteinase K (Invitrogen) and RNA was extracted using Trizol. QPCR was performed as described above.
In vivo studies
Female athymic nude mice were purchased from Envigo. For all studies mice were housed at Harvard Medical School (HMS) animal facility in accordance with all NIH regulations. For intracranial tumor injection, cells were analyzed for viability using the Muse Count & Viability Reagent on the Muse Cell Analyzer (Millipore) following the manufactures’ instructions to normalize number of viable cell prior to the transplantation of 5,000 viable GSCs transduced with either control or HIF1A-AS2 shRNA vector and stereotactically injected (2mm right lateral, 0.5mm frontal to the bregma and 4mm deep) into the brain of 6–8 week old mouse. Animals were sacrificed as per protocol and brain tissue was processed as described (Bronisz et al., 2014). Brain sections were imaged using a confocal microscope Zeiss LSM710.
Data and Statistical Analysis
Functional bioinformatic analyses were performed using David Functional Annotation tool (http://david.abcc.ncifcrf.gov/), and STRING v10 protein-protein interaction networks software (Szklarczyk et al., 2015). Experimental and clinical data were analyzed using the GBM-BioDP (URL: http://gbm-biodp.nci.nih.gov) as described (Celiku et al., 2014). Clinical data were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/) as described in The Cancer Genome Atlas research Network (Network, 2013). Gene expression data included data from three platforms HT_HG-U133A (488 patient samples×12042 features), HuEx-1_0-st-v2 (437 patient samples×18631 features), AgilentG4502A_07_1/2 (101+396 patient sample×17813 features). The data from the three platforms were aggregated (Verhaak et al., 2010). GSCs microarray data (Mao et al., 2013) were queried for cluster analysis with PAN Cancer platform data. Clinical data included partial clinical information on 564 patients. The experimental data were already pre-processed as a part of the TCGA data. The genes down-regulated by HIF1A-AS2 knockdown were used to predict patients’ outcome. Genes identified as a IGF2BP2 targets (Janiszewska et al., 2012) were queried with genes that vary coherently between proneural and mesenchymal GSCs (Mao et al., 2013) (Mao et al., 2013). Gene expression in the various anatomical regions of glioblastoma was analyzed using the Ivy Glioblastoma Atlas Project (http://glioblastoma.alleninstitute.org/). Data are expressed as mean ± SD. Statistical analyses were performed using the unpaired two-tailed Student’s t-test from GraphPad Prism software. Differences were considered statistically significant at P<0.05.
Supplementary Material
Acknowledgments
This research was supported by NCI P01 CA69246 (EAC) and NCI 1R01 CA176203-01A1 (JG)
Footnotes
Authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors assisted in drafting and revising the work, approved the final version for publication, and agree to be accountable for all aspects of the work. M.M. conceived overall work and developed the methodology. F.R. performed in vitro and in vivo experiments. A.K.R., S.M.L. performed in vitro experiments. K.I.A. designed the NS platform. P. I. assisted with writing the manuscript, analysis and interpretation of data. I.N. acquired patient’s specimens and assisted with interpretation of data. E.A.C. acquired patient’s specimens and assisted with writing the manuscript, J.G., A.B. conceived and designed overall work and analyzed and interpreted data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdag S, Li A, Baysan M, Fine HA. Master regulators, regulatory networks, and pathways of glioblastoma subtypes. Cancer informatics. 2014;13:33–44. doi: 10.4137/CIN.S14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer research. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- Bronisz A, Wang Y, Nowicki MO, Peruzzi P, Ansari KI, Ogawa D, Balaj L, De Rienzo G, Mineo M, Nakano I, et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer research. 2014;74:738–750. doi: 10.1158/0008-5472.CAN-13-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celiku O, Johnson S, Zhao S, Camphausen K, Shankavaram U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS One. 2014;9:e101239. doi: 10.1371/journal.pone.0101239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YN, Zhang K, Hu ZM, Qi HX, Shi ZM, Han XH, Han YW, Hong W. Hypoxia-regulated lncRNAs in cancer. Gene. 2016;575:1–8. doi: 10.1016/j.gene.2015.08.049. [DOI] [PubMed] [Google Scholar]
- Chatel-Chaix L, Germain MA, Motorina A, Bonneil E, Thibault P, Baril M, Lamarre D. A host YB-1 ribonucleoprotein complex is hijacked by hepatitis C virus for the control of NS3-dependent particle production. J Virol. 2013;87:11704–11720. doi: 10.1128/JVI.01474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M, et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene. 1996;13:2439–2446. [PubMed] [Google Scholar]
- Cooper LA, Gutman DA, Chisolm C, Appin C, Kong J, Rong Y, Kurc T, Van Meir EG, Saltz JH, Moreno CS, et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol. 2012;180:2108–2119. doi: 10.1016/j.ajpath.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, Liu XS. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nature structural & molecular biology. 2013;20:908–913. doi: 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Flynn RA, Chang HY. Active chromatin and noncoding RNAs: an intimate relationship. Current opinion in genetics & development. 2012;22:172–178. doi: 10.1016/j.gde.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nature genetics. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nature reviews Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- Godlewski J, Krichevsky AM, Johnson MD, Chiocca EA, Bronisz A. Belonging to a network--microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro-oncology. 2015;17:652–662. doi: 10.1093/neuonc/nou292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, Holland EC. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5248–5253. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M. The emerging role of lncRNAs in cancer. Nature medicine. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes & development. 2012;26:1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E, Balasubramaniyan V, Wagemakers M, den Dunnen WF, Kruyt FA. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer letters. 2015;359:107–116. doi: 10.1016/j.canlet.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H, Chhipa R, et al. Serine/Threonine Kinase MLK4 Determines Mesenchymal Identity in Glioma Stem Cells in an NF-kappaB-dependent Manner. Cancer cell. 2016;29:201–213. doi: 10.1016/j.ccell.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunej T, Obsteter J, Pogacar Z, Horvat S, Calin GA. The decalog of long non-coding RNA involvement in cancer diagnosis and monitoring. Crit Rev Clin Lab Sci. 2014;51:344–357. doi: 10.3109/10408363.2014.944299. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes & development. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Di Paola D, Malina A, Mills JR, Kreps A, Grosse F, Tang H, Zannis-Hadjopoulos M, Larsson O, Pelletier J. Suppression of the DHX9 helicase induces premature senescence in human diploid fibroblasts in a p53-dependent manner. The Journal of biological chemistry. 2014;289:22798–22814. doi: 10.1074/jbc.M114.568535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pang B, Liu H, Arakawa Y, Zhang R, Feng B, Zhong P, Murata D, Fan H, Xin T, et al. High mobility group A1 expression shows negative correlation with recurrence time in patients with glioblastoma multiforme. Pathology, research and practice. 2015;211:596–600. doi: 10.1016/j.prp.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. A blooming genomic desert. Nat Methods. 2014;11:135–138. doi: 10.1038/nmeth.2817. [DOI] [PubMed] [Google Scholar]
- Mathew LK, Skuli N, Mucaj V, Lee SS, Zinn PO, Sathyan P, Imtiyaz HZ, Zhang Z, Davuluri RV, Rao S, et al. miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:291–296. doi: 10.1073/pnas.1314341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015;349 doi: 10.1126/science.aab2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineo M, Garfield SH, Taverna S, Flugy A, De Leo G, Alessandro R, Kohn EC. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis. 2012;15:33–45. doi: 10.1007/s10456-011-9241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc. 2014;9:263–293. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano I. Proneural-mesenchymal transformation of glioma stem cells: do therapies cause evolution of target in glioblastoma? Future oncology. 2014;10:1527–1530. doi: 10.2217/fon.14.86. [DOI] [PubMed] [Google Scholar]
- Network TC. Corrigendum: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2013;494:506. doi: 10.1038/nature11903. [DOI] [PubMed] [Google Scholar]
- Orr BA, Eberhart CG. Nature versus nurture in glioblastoma: microenvironment and genetics can both drive mesenchymal transcriptional signature. Am J Pathol. 2012;180:1768–1771. doi: 10.1016/j.ajpath.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, Bregy A, Komotar R, St Laurent G, Ayad NG, et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8326–8331. doi: 10.1073/pnas.1424220112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi P, Bronisz A, Nowicki MO, Wang Y, Ogawa D, Price R, Nakano I, Kwon CH, Hayes J, Lawler SE, et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro-oncology. 2013;15:1212–1224. doi: 10.1093/neuonc/not055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:4392–4403. doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13:528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Ligon KL, Chiocca EA, Wen PY. One size should not fit all: advancing toward personalized glioblastoma therapy. Discovery medicine. 2015;19:471–477. [PubMed] [Google Scholar]
- Ricklefs F, Mineo M, Rooj AK, Nakano I, Charest A, Weissleder R, Breakefield XO, Chiocca EA, Godlewski J, Bronisz A. Extracellular vesicles from high grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-15-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM. The evidence of glioblastoma heterogeneity. Sci Rep. 2015;5:7979. doi: 10.1038/srep07979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MD, Fosmark S, Hellwege S, Beier D, Kristensen BW, Beier CP. Chemoresistance and chemotherapy targeting stem-like cells in malignant glioma. Adv Exp Med Biol. 2015;853:111–138. doi: 10.1007/978-3-319-16537-0_7. [DOI] [PubMed] [Google Scholar]
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yan IK, Haga H, Patel T. Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J Cell Sci. 2014;127:1585–1594. doi: 10.1242/jcs.141069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash-Bingham CA, Tartof KD. aHIF: a natural antisense transcript overexpressed in human renal cancer and during hypoxia. J Natl Cancer Inst. 1999;91:143–151. doi: 10.1093/jnci/91.2.143. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassallo I, Zinn P, Lai M, Rajakannu P, Hamou MF, Hegi ME. WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene. 2015 doi: 10.1038/onc.2015.61. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Han L, Bao ZS, Wang YY, Chen LY, Yan W, Yu SZ, Pu PY, Liu N, You YP, et al. HOTAIR, a cell cycle-associated long noncoding RNA and a strong predictor of survival, is preferentially expressed in classical and mesenchymal glioma. Neuro-oncology. 2013;15:1595–1603. doi: 10.1093/neuonc/not131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Houseman EA, Morrison Z, Wrensch MR, Patoka JS, Ramos C, Haas-Kogan DA, McBride S, Marsit CJ, Christensen BC, et al. DNA hypermethylation profiles associated with glioma subtypes and EZH2 and IGFBP2 mRNA expression. Neuro-oncology. 2011;13:280–289. doi: 10.1093/neuonc/noq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.