

Abstract
Molecular surveillance provides a powerful approach to monitoring the resistance status of parasite populations in the field and for understanding resistance evolution. Oxamniquine (OXA) was used to treat Brazilian schistosomiasis patients (mid-1970s to mid-2000s) and several cases of parasite infections resistant to treatment were recorded. The gene underlying resistance (SmSULT-OR) encodes a sulfotransferase required for intracellular drug activation. Resistance has a recessive basis and occurs when both SmSULT-OR alleles encode for defective proteins. Here we examine SmSULT-OR sequence variation in a natural schistosome population in Brazil ~40 years after the first use of this drug. We sequenced SmSULT-OR from 189 individual miracidia (1–11 per patient) recovered from 49 patients, and tested proteins expressed from putative resistance alleles for their ability to activate OXA. We found nine mutations (four non-synonymous single nucleotide polymorphisms (SNPs), three non-coding SNPs and two indels). Both mutations (p.E142del and p.C35R) identified previously were recovered in this field population. We also found two additional mutations (a splice site variant and 1 bp coding insertion) predicted to encode non-functional truncated proteins. Two additional substitutions (p.G206V, p.N215Y) tested had no impact on OXA activation. Three results are of particular interest: (i) we recovered the p.E142del mutation from the field: this same deletion is responsible for resistance in an OXA-selected laboratory parasite population; (ii) frequencies of resistance alleles are extremely low (0.27–0.8%), perhaps due to fitness costs associated with carriage of these alleles; (iii) that four independent resistant alleles were found is consistent with the idea that multiple mutations can generate loss-of-function alleles.
Keywords: Schistosoma mansoni, Oxamniquine resistance, Sulfotransferase, Loss-of-function, Biochemical assay, Soft selective event
Graphical Abstract
1. Introduction
Surveys of drug resistance alleles using molecular markers provide a powerful approach to identify pathogen populations in which resistance is emerging, to map resistance spread, and for evidence-based resistance management. Molecular approaches are now widely used for tracking resistance in malaria (Pearce et al., 2009; Ashley et al., 2014), HIV (Panichsillapakit et al., 2015) and bacterial diseases (Bhembe et al., 2014), and for managing insecticide resistance ( Essandoh et al., 2013; Djègbè et al., 2014). Such methods are also now actively used for monitoring resistance in helminths of veterinary importance such as Haemonchus contortus ( Rufener et al., 2009; Chaudhry et al., 2015; Redman et al., 2015), and have enormous potential for monitoring resistance in helminth parasites infecting humans, because existing phenotypic screening methods based on reductions in production of eggs or larval stages are insensitive for detection of low frequency resistance alleles and cannot detect recessive resistance alleles present in heterozygous worms. However, molecular surveillance is only possible when resistance genes have been identified, which is rarely the case for human helminth infections such as schistosomiasis.
Schistosomiasis, caused by three major species of blood flukes of the genus Schistosoma (Dye et al., 2013; Colley et al., 2014), is the second most important tropical parasitic disease after malaria (Steinmann et al., 2006), affecting an estimated 260 million people across Africa, Asia and South America, and killing over 200,000 people per year. Two drugs are available for treating schistosomiasis. Praziquantel (PZQ) is currently used as a monotherapy in expanding mass drug administration programs in Africa (Dye et al., 2013), making resistance evolution a major concern. A second drug, oxamniquine (OXA), is the focus of this paper. OXA acts specifically against Schistosoma mansoni, which is found in Africa, together with Schistosoma haematobium, and in South America, where it is the only species present. OXA was manufactured in Brazil by Pfizer (Cheetham, 1994) and widely used to treat S. mansoni infections from the mid-1970s to the mid-2000s (Coura and Amaral, 2004), but has now been replaced by PZQ as the first line drug.
Schistosomes resistant to OXA were first identified in Brazil in 1973 (Katz et al., 1973a), around the same time as the first clinical study of OXA treatment (Katz et al., 1973b). This rapid emergence of resistance was most likely due to the previous treatment of the same populations with the related drug hycanthone (Katz et al., 1968; Coura and Conceici¸ão, 2010), as both OXA and hycanthone have the same target (Jansma et al., 1977; Pica-Mattoccia et al., 1993) and the same mechanism of resistance (Pica-Mattoccia et al., 1992a). Later genetic studies showed that resistance is a recessive single locus trait (Cioli and Pica-Mattoccia, 1984; Pica-Mattoccia et al., 1993), most likely involving the absence of a sulfotransferase activity necessary for drug activation in resistant schistosomes (Pica-Mattoccia et al., 2006). The gene encoding the S. mansoni sulfotransferase involved in OXA resistance (SmSULT-OR) was recently identified by classical quantitative trait mapping in concert with crystallographic and functional analyses (Valentim et al., 2013). This work identified an amino acid deletion (p.E142del) in the laboratory-selected resistant parasite (HR), while an independent loss-of-function mutation (p.C35R) was identified in a field-collected resistant parasite line (MAP).
The identification of the gene involved in OXA resistance now allows us to examine distribution of resistance alleles of SmSULT-OR in natural populations. Despite the fact that the Brazilian government has switched from OXA to PZQ during the last decade (Utzinger et al., 2003), the recessive nature of OXA resistance allows the persistence of these alleles, because alleles encoding non-functional enzyme are not counter-selected as long as they segregate with alleles encoding a functional enzyme. We collected miracidia larval stages from a village in Minas Gerais, Brazil, and sequenced the SmSULT-OR gene in these samples. We sought to answer several questions: How common are resistance alleles? How many times have OXA resistance alleles arisen? Are the OXA resistance alleles selected in the laboratory and identified using linkage mapping actually present in nature? More broadly, our goal is to better understand the evolution of drug resistance in schistosomes and to demonstrate the utility of molecular screening approaches in anticipation that the gene(s) underlying PZQ resistance will soon be identified.
2. Materials and methods
2.1. Ethics statement
Stool samples were collected in accordance to the procedures of the Research Ethics Committee of the Universidade Federal de São Paulo, Brazil (process number CAAE: 15567313.8.0000.5091). The purpose of the study and the procedures to be followed were explained and written informed consent was obtained from all participants or their legal guardians prior to any collection.
2.2. Sampling of Schistosoma mansoni miracidia
We collected stools from school children from Ponto dos Volantes (Minas Gerais, Brazil, GPS coordinates: 16°45′3.301″ S, 41°30′13.755″ W) and shipped these at 4°C by ground transportation overnight to the Universidade Federal de Minas Gerais in Belo Horizonte, Brazil. We processed samples as follows: several grams of stools were filtered through three layers of sieves (mesh size: 250 to 45 μm) to obtain schistosome eggs. Eggs were transferred from the third sieve grid to a Petri dish and exposed under artificial light for at least 1 h. All filtering steps and egg transfer were performed with locally available bottled mineral water.
Washed eggs were observed under a stereomicroscope. For each patient, 1 – 11 living miracidia were sampled individually in ~2 μL of water and spotted onto CloneSaver FTA cards (GE Healthcare Life Sciences, USA). Spotted samples can be easily located on the cards because the pink dye on the cards turns white after water contact. Full cards were allowed to dry for 1 h at room temperature on the bench before being stored in a plastic bag and finally shipped to San Antonio, Texas, USA.
2.3. Preparation of FTA samples for whole genome amplification (WGA)
For each sample, we removed a 2 mm diameter disc from the FTA card using a 2 mm Harris Micro-punch (GE Healthcare Life Sciences). The 2 mm disc corresponds to the entire spot containing the whole miracidium. Each punch was placed individually in a 1.5 mL sterile tube. Punches were washed three times with FTA Purification Reagent (GE Healthcare Life Sciences) then rinsed twice with TE−1 buffer (10 mM Tris, 0.1 mM EDTA, pH 8). Washing and rinsing steps were performed by adding 200 μL of solution to each tube followed by 5 min of incubation on a nutating mixer (24 RPM) at room temperature and then discarding the solution while minimizing contact between the pipette tip and the punch. Punches were finally dried in tubes for 10 min at 56°C on a dry bath incubator.
2.4. WGA
We performed WGA on each punch using the illustra GenomiPhi V2 DNA Amplification kit (GE Healthcare Life Sciences). Punches were transferred in 0.2 mL sterile tubes using a sterile tip. We performed reactions following the manufacturer’s instructions, immersing each punch in 9 μL of sample buffer and keeping tubes on ice at all times after the denaturation step. After amplification, we quantified DNA using the Qubit dsDNA BR assay (Invitrogen, USA).
2.5. Sequencing of SmSULT-OR exons
The two exons of the SmSULT-OR gene were amplified and sequenced independently. Each PCR was performed using the TaKaRa Taq kit (Clontech, USA) and composed of 9.325 μL of sterile water, 1.5 μL of 10× buffer, 1.2 μL of dNTP (2.5 mM each), 0.9 μL of MgCl2 (25 mM), 0.5 μL of each primer (10 μM; Table 1), 0.075 μL of Taq polymerase (5 U.μL−1) and 1 μL of DNA template. Amplifications were done using a GeneAmp PCR system 9700 thermocycler (Applied Biosystems, USA) with the following program: 95°C for 5 min; 95°C for 30 s, 60°C for 30 s, 72°C for 45 s, for 35 cycles; then 72°C for 10 min.
Table 1.
Primer sequences used for PCRs and sequencing of the two exons of the Schistosoma mansoni SmSULT-OR gene in this study.
| Primer type | Primer sequence (5′ to 3′ orientation) | Expected amplicon size (bp) | Usage | |
|---|---|---|---|---|
| Exon 1 | Outer forward primer | GCGAGATTCAAACCCAGGAT | 822 | PCR |
| Outer reverse primer | GCCGTGATATTACTATCAATCCC | PCR | ||
| Nested forward primer | GGGTAAAGGAAGAGGGTTGG | 545 | PCR | |
| Nested reverse primer | TAAGAACAGACATATTAGACGAGT | PCR and sequencing | ||
| Sequencing forward primer | TATATATGAAATATTATAACATTAC | - | sequencing | |
| Exon 2 | Forward primer | ACTTCAACCAATCCACAAATCC | 672 | PCR and sequencing |
| Reverse primer | AGTCCATTCATTCAATGTTTCAA | PCR and sequencing |
Exon 1 required a nested PCR in order to obtain a specific product. Products from the first PCR were cleaned up by adding 4 μL of ExoSAP-IT (Affymetrix USB products, USA). Tubes were then incubated at 37°C for 30 min and at 80°C for 15 min using a thermocycler. Cleaned PCR products were then used as templates for the second PCR following the above protocol.
Sequencing reactions were performed using a BigDye® Terminator v3.1 cycle sequencing kit (Applied Biosystems) on final PCR products. PCR products were cleaned up using ExoSAP-IT as described above. Sequencing reactions were performed using 2.59 μL of sterile water, 1 μL of 5× running buffer, 0.25 μL of BigDye Terminator ready reaction mix (Applied Biosystems), 0.16 μL of forward or reverse primer used in the final PCR step, and 1 μL of final PCR product. Sequencing fragments were generated using a GeneAmp PCR system 9700 thermocycler (Applied Biosystems) with the following program: 96°C for 1 min; 96°C for 10 s, 50°C for 5 s, 60°C for 4 min, for 25 cycles. Sequencing reactions were cleaned up using a BigDye XTerminator® purification kit (Applied Biosystems). In each reaction, 20.45 μL of SAM™ solution and 4.55 μL of XTerminator™ solution were added. Reactions were then vortexed for 30 min and run on a 3730xl DNA Analyzer (Applied Biosystems).
Sequencing files were first screened using FinchTV (v1.4.0; Geospiza Inc.) to identify failed sequencing reactions. In the case of failure, sequencing reactions were performed a second time.
2.6. Variant identification and functional impact evaluation
We scored variants using PolyPhred software (v6.18) (Nickerson et al., 1997) which relies on Phred (v0.020425.c), Phrap (v0.990319), and Consed (v29.0) software, analyzing each exon independently. We identified single nucleotide polymorphisms using a minimum phred quality score (-q) of 40, a minimum genotype score (-score) of 90, and a reference sequence that includes the SmSULT-OR gene and surrounding regions (position 1519500 to 1525200 of chromosome 6 of S. mansoni reference genome v5.0, ftp://ftp.sanger.ac.uk/pub/pathogens/Schistosoma/mansoni/genome/Assemblyv5/sma_v5.0.chr.fa.gz). Variant sites were labeled as non-reference alleles if they differed from the reference sequence. We identified Insertion/deletion (indel) polymorphisms using a minimum phred quality score (-q) of 40, a minimum genotype score (-iscore) of 80. Polymorphisms were visually validated using Consed. All the sequences were submitted to GenBank (GenBank accession no KU951903-KU952091).
Nucleic sequences showing mutations were translated in silico into protein sequences using the Translate tool from ExPASy portal (Artimo et al., 2012).
We evaluated the potential functional impact of identified polymorphisms on RNA features and on protein structure in silico. Modifications of RNA motifs and sites were analyzed using the RegRNA2.0 website (Chang et al., 2013) for all available features with Drosophila melanogaster as the reference species when required. Modifications in protein structure were assessed using the mutagenesis function of PyMol software (v1.7.2.0, Schrödinger, LLC) using the structure of SmSULT-OR determined previously (PDB code 4MUB, Valentim et al., 2013).
2.7. Population genetics analysis
We evaluated population structure by testing for Hardy-Weinberg equilibrium, measuring fixation indices (Fst and the Fis) of parasites using Genepop (Rousset, 2008) with default options and considering schistosomes from a given patient as belonging to the same population. To identify positive selection, we calculated the number of synonymous (s) and non-synonmous (n) sites (Nei and Gojobori, 1986) and compared these values to synonymous (S) and non-synonymous (N) changes. We also performed a McDonald-Kreitman (MKT) test using the MKT server (http://mkt.uab.es; Egea et al., 2008). The SmSULT-OR homolog sequence in Schistosoma rodhaini was used as an outgroup and was obtained using tblastx from the NCBI server (NCBI Resource Coordinators, 2015) on the S. rodhaini genome assembly (GenBank accession no. GCA_000951475.1) using the SmSULT-OR sequence (GenBank accession no. KF733459.1) as a query.
2.8. Recombinant SmSULT-OR protein production
The SmSULT-OR sequence from the reference genome (Smp_089320; GenBank accession no. HE601629.1) was used to create a codon optimized synthetic gene (GenScript) which was subcloned into the pAG8H vector derived from pKM260 (Melcher, 2000). We introduced mutations using a Phusion site-directed mutagenesis protocol (ThermoFisher Scientific, USA). Transformed Escherichia coli strain BL21 pLysS (Promega) were grown at 37°C until the absorbance (λ = 600 nm) reached 0.7. We then decreased the temperature to 18°C and induced expression by the addition of isopropyl-β-D-thiogalactoside (IPTG) at a final concentration of 1 mM. The cells were washed and resuspended in 50 mL of 50 mM Tris pH 8.0, 500 mM NaCl (column buffer) containing 250 μL of Sigma protease inhibitor cocktail per liter of culture and lysed by sonication on ice. The clarified supernatant was loaded onto a Ni2+-NTA affinity chromatography column (GE Healthcare), washed with five volumes of column buffer, and eluted using a 10–500 mM imidazole gradient. We pooled fractions identified as the Smp_089320 via SDS-PAGE and added His-tagged Tobacco etch virus (TEV) protease at a Smp_0893230:protease ratio of 15:1. The resulting solution was dialyzed overnight at 4°C against 50 mM Tris pH 8.0, 100 mM NaCl, 2 mM dithiothreitol (DTT). We passed the dialysate over the Ni2+-NTA column again to remove the His tag and the His-tagged TEV protease, while the cleaved target protein flowed through. The sample was loaded onto a GE-pre-packed Q anion exchange column and eluted with a 0.1–1.0 M NaCl gradient. We pooled fractions containing Smp_089320 as identified by SDS-PAGE and dialyzed overnight at 4°C against 25 mM Tris pH 8.5, 50 mM NaCl, 2 mM reducing agent tris-(carboxyethyl)-phosphine (TCEP) to prevent formation of intermolecular disulfide bonds. The product was ~98% pure as estimated by SDS-PAGE. We concentrated the purified Smp_089320 protein to 10 mg.mL−1 using the calculated extinction coefficient ε = 39,880 mol−1.cm−1.
2.9. OXA activation assay
This assay utilizes the fact that OXA is a prodrug that is enzymatically converted into a highly reactive molecule that covalently binds intracellular components such as DNA (Pica-Mattoccia et al., 1989). Because the original assay employs worm extracts which require time for preparation and introduce unpredictable variations in SmSULT-OR concentration (Pica-Mattoccia et al., 1992a; Valentim et al., 2013), we developed an improved in vitro assay that uses purified recombinant SmSULT-OR enzyme. This in vitro assay measures the enzymatic sulfonation of tritiated OXA molecules which then bind genomic DNA (gDNA). The resultant radioactive DNA-OXA complexes can then be quantified by a scintillation counter. Enzymes produced by non-functional SmSULT-OR alleles cannot sulfonate OXA, so no DNA-OXA complexes are formed.
Recombinant proteins were expressed and purified commercially by GenScript, USA (p.C35R, p.P67L, p.E142del; Valentim et al., 2013) or by the Hart laboratory (University of Texas Health Science Center, USA) (p.G206V and p.N215Y). To determine which recombinant protein has the ability to activate OXA, 1 nM from each recombinant protein was added to 90 μL of a protease inhibitor cocktail (PIC) consisting of 0.1 M HEPES pH 7.4, 0.1mM leupeptin, 2 μM E-64, 2 μM pepstatin A, 0.1 U of aprotinin, and 10 ng.μL−1 sheared S. mansoni gDNA as a final target. For each reaction 100 μCi of 3H-OXA (Pica-Mattoccia et al., 1989) was solubilized in 2 μL of DMSO and added to 10 μL of a mixture containing the enzyme cofactors ATP and MgCl2 at 50 mM each, and 3′-phosphoadenosine-5′-phosphosulfate (PAPS) at 1 mM. The radio labeled OXA and co-factor mix was then added to the PIC mix containing the recombinant protein. The resulting reaction was incubated for 2.5 h at 37°C and stopped by adding three volumes of 0.1% SDS in 0.1 M sodium bicarbonate. The reaction was then extracted three times with two volumes of dichloromethane and the aqueous phase was counted in a liquid scintillation spectrometer (Beckman LS 6500 Scintillation Counter, USA) for 10 min. We also measured a blank solution (water) and the background scintillation. Blank and background values were subtracted from sample values. We performed three independent reactions for each recombinant protein.
2.10. Statistical analysis
Statistical analyses were done using R (v3.1.3) (R Core Team, 2015). For synonymous and non-synonymous changes, data were compared using a Fisher’s exact test. For the OXA activation assay, the data were compared with a Welch t-test after testing for normality (Shapiro test, P > 0.05).
3. Results
3.1. Brazilian samples
We collected 232 FTA preserved miracidium samples from 51 patients (range: 1–12, mean±S.D.: 4.55±3.65). We successfully amplified DNA from 204 samples (87.93%) from 50 patients (range: 1–11, mean±S.D.: 4.08±3.26). Among the amplified samples, 189 from 49 patients (range: 1–11, mean±S.D.: 3.86±3.18) contained schistosome DNA (92.65% of amplified samples, 81.47% of total samples). Among the 189 samples, we sequenced exon 1 from 183 samples and exon 2 from 188 samples. All samples had at least one exon sequenced (Supplementary Table S1).
3.2. SmSULT-OR variants in a Brazilian schistosome population
We scored nine mutations: four non-synonymous single nucleotide polymorphisms (SNPs), one insertion and one deletion in the coding region, and three non-coding SNPs (Table 2, Supplementary Table S1). The number of mutations in each exon (three in exon 1 and three in exon 2) did not differ regarding the length of the exon (327 bp and 447 bp, respectively) (Fisher’s exact test, P = 0.7).
Table 2.
Mutations scored in the exons, intron, and 3′ untranslated regions (UTR) of the Schistosoma mansoni SmSULT-OR gene. For each mutation, the corresponding nucleotide found in Schistosoma rodhaini, the number of homozygous and heterozygous samples carrying the nonreference allele, the number of samples sequenced (sample size), the allele frequency of the non-reference allele, the corresponding amino acid mutation, and the functional impact are shown.
| Nucleic mutation | Schistosoma rodhaini state | No. of homozygous samples for non-reference allele | No. of heterozygous samples for non-reference allele | Sample size | Frequency of the non-reference allele | Amino acid mutation | Functional impact | |
|---|---|---|---|---|---|---|---|---|
| Exon 1 | c.103T>C | T | 0 | 1 | 183 | 0.0027 | p.C35R | Misfolded protein (Valentim et al., 2013) |
| c.200C>T | C | 169 | 12 | 183 | 0.9563 | p.P67L | No effect (Valentim et al., 2013) | |
| c.214_215insA | - | 0 | 1 | 183 | 0.0027 | p.T72NfsX5 | Truncated protein with no active site | |
| Intron | g.328G>A | G | 1 | 0 | 179 | 0.0056 | p.V110IfsX3 | Splicing site disrupted leading to truncated protein with no active site |
| Exon 2 | c.424_426delGAA | - | 1 | 1 | 188 | 0.0080 | p.E142del | Impaired oxamniquine binding (Valentim et al., 2013) |
| c.617G>T | G | 1 | 0 | 188 | 0.0053 | p.G206V | No effect (Fig. 2) | |
| c.643A>T | A | 0 | 1 | 188 | 0.0027 | p.N215Y | No effect (Fig. 2) | |
| 3′ UTR | g.4720C>T | C | 177 | 9 | 188 | 0.9654 | - | - |
| g.4741T>C | T | 2 | 8 | 188 | 0.0319 | - | - |
The code used for nucleic mutations indicates the sequence type (c = coding, g = gene), the position, and the mutation type (X>Y = substitution of X by Y, insN = insertion of N, delN = deletion of N). The code used for protein mutations indicates the sequence type (p = protein), the reference amino acid, the position, and finally the alternative amino acid, and when frame shift (fs) occurs, the position of the stop codon (X) after the mutation. For details about the nomenclature, see Ogino et al., 2007.
Among the seven SNPs, five (71.43%) were transitions and two (28.57%) were transversions. Four of the SNPs were located in the exonic region, two in exon 1 and two in exon 2. Three were present at very low frequency (0.0027–0.0053) while one was present at very high frequency (0.95). One SNP was identified at the first position of the intron at a low frequency (0.0056). The two remaining SNPs were found in the 3′ untranslated region (UTR), one at high frequency (0.96), and one at low frequency (0.03).
One insertion was identified in exon 1 while one deletion was identified in exon 2, both at very low frequency (0.0027 and 0.0080, respectively).
Low frequency mutations were found in heterozygous or in homozygous states among the samples. Observing rare SNPs present as homozygotes was surprising, suggesting population structure. The test for Hardy-Weinberg equilibrium showed a global deficit in heterozygosity (P < 0.0001). This is likely due to a deficit of heterozygous genotypes within host (Fis = 0.3251) rather than due to population differentiation between infections (Fst = 0.0153). Null alleles are not likely to explain the deficit. While we had small numbers of samples for which WGA failed, SmSULT-OR was successfully amplified from all samples for which WGA was successful.
Due to the absence of the synonymous mutations which precludes dN/dS calculation, we compared synonymous (S) and non-synonymous (N) changes with synonymous (s) and non-synonymous (n) sites to evaluate evidence for selection on SmSULT-OR. This comparison reveals no differences (Fisher’s exact test, P = 1) indicating that there was no evidence for selection. We also tested directional evolution by performing a McDonald-Kreitman test using the homolog sequence of SmSULT-OR from S. rodhaini as an outgroup. We found no evidence for directional evolution (χ2= 1.466, P = 0.225).
3.3. Functional impacts of mutations
3.3.1. Non-coding variants
We evaluated the functional impacts of the polymorphisms identified based on RNA features such as binding sites or splicing or regulatory motifs (Table 2). The mutation g.649G>A was predicted to modify the unique splice donor site at the end of exon 1. The disruption of the splicing site leads to translation of the beginning of the intron which ends four codons later due to the introduction of a stop codon. This results in a truncated protein with no active site (Fig. 1, Supplementary Movie S1). Mutations present on the 3′ UTR were not predicted to be in any regulatory sites.
Fig. 1.

Mapping of the mutations on the gene sequence and structure of Schistosoma mansoni SmSUTL-OR sulfotransferase. Exon 1 and exon 2 are represented in orange and beige, respectively. Single nucleotide polymorphisms and insertion/deletion events are represented in cyan and magenta, respectively. Loss-of-function mutations are highlighted in black. (A) Linear representation of the SmSULT-OR gene showing the relative position of the mutations and their translation in amino acid sequences. (B) Positions of mutations on the SmSULT-OR protein. Oxamniquine (OXA) is represented in yellow, 3′-phosphoadenosine-5′-phosphosulfate (PAPS) co-factor is represented in green, and spatial distortions are represented by red discs. For a more detailed view of the mutations on the structure and their functional impact, see Supplementary Move S1.
3.3.2. Coding variants
All SNPs identified in the coding sequence of the gene were non-synonymous, and derived mutations relative to the outgroup S. rodhaini (Table 2). c.103T>C induced a substitution of cysteine to arginine (p.C35R) leading to a misfolded protein as shown previously (Valentim et al., 2013) (Fig. 1). c.200C>T induced a substitution of proline to leucine (p.P67L) and was previously found in the OXA-sensitive strain (Valentim et al., 2013) (Fig. 1); this mutation therefore does not reduce enzyme activity. c.617G>T induced a substitution of glycine to valine (p.G206V) which occurs close the binding site of the PAPS co-factor and was previously observed in field-collected OXA-resistant strain (Valentim et al., 2013) but was never tested. We postulated that this mutation may have a potential detrimental effect on co-factor binding and finally enzyme activity (Fig. 1, Supplementary Movie S1). The mutation c.643A>T induced a substitution of asparagine to tyrosine (p.N215Y) which occurs on a helix connected to a loop involved in co-factor binding. This mutation is predicted to have little impact on protein structure. We therefore postulated that this will not change protein function (Fig. 1, Supplementary Movie S1).
3.3.3. Indels
Both indels have detrimental effects on the enzyme (Table 2). The insertion c.214_125insA induces a frame shift leading to an early stop codon six codons after the mutation. This frame shift is predicted to produce a truncated enzyme with no active site. The deletion c.424_426delGAA is known from previous functional analyses (Valentim et al., 2013) to disrupt OXA binding.
3.4. OXA activation assay
We produced two recombinant SmSULT-OR enzymes carrying the mutations p.G206V and p.N215Y in order to experimentally test whether these two mutations impact OXA activation, resulting in OXA resistance. We also tested known resistance alleles (p.C35R and p.E142del) as controls. The newly identified alleles are able to activate OXA as well as the reference allele (wild type) which does not carry any of these mutations (Welch t-test, t < -1.55, P > 0.18) while the known resistance alleles showed no activation as expected (Welch t-test, t > 5.58, P < 0.021) (Fig. 2). Therefore the two mutations tested (p.G206V and p.N215Y) did not disrupt co-factor binding or otherwise interfere with enzymatic activity.
Fig. 2.
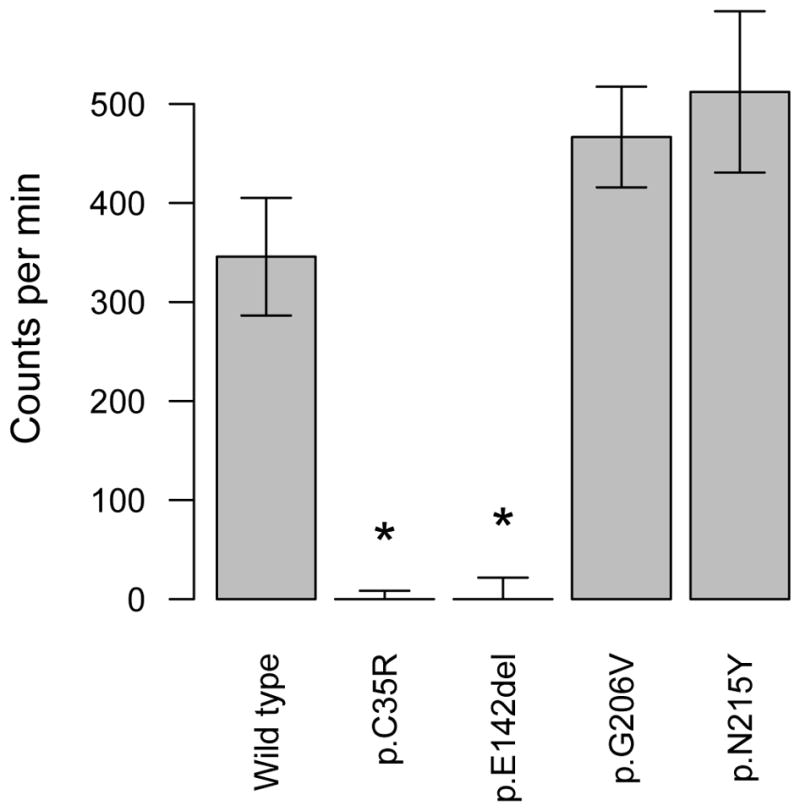
Enzymatic activity of recombinant Schistosoma mansoni SmSULT-OR sulfotransferase expressed from different allelic variants. This in vitro oxamniquine OXA activation assay quantifies DNA-OXA complexes by scintillation (counts per min) (see section 2.9). Bars show the mean of three replicates, while error bars are S.E.M. Enzyme carrying loss-of-function mutations, such as p.C35 or p.E142del, showed no OXA activation, while two newly identified alleles (p.G206V and p.N215Y) did not impair OXA activation. * P < 0.05.
4. Discussion
SmSULT-OR sequences from 189 miracidia collected from 49 Brazilian patients revealed nine mutations, including both mutations previously implicated in OXA resistance (Valentim et al., 2013). We found the p.C35R mutation, previously identified in the MAP strain. MAP was sampled from a patient living in a city of São Paulo state, Brazil (Supplementary Fig. S1) (Pica-Mattoccia et al., 1992b), the neighboring state of that from which our samples were collected. This suggests that the allele has been segregating in Brazilian parasite populations for more than 25 years. The second variant identified in the field, p.E142del, was previously found in the HR laboratory strain, which was initially sampled from a Puerto-Rican patient and subsequently selected with hycanthone in the laboratory (Supplementary Fig. S1) (Cioli and Pica-Mattoccia, 1984). This mutation could have arisen spontaneously in the laboratory. However, given that laboratory schistosome lines are maintained as outbred populations, the simplest explanation is that this mutation was segregating within the parasite population originally established in the laboratory. That we located this same deletion in two miracidia from Brazil and a parasite line collected from Puerto-Rico suggests that this allele may be widespread in schistosome populations from the New World.
It is not clear from our data whether the p.E142del seen in HR and Brazilian field samples arose independently or has a single origin. The HR p.E142del is found together with another mutation (p.L256W) (Valentim et al., 2013), which is absent from the p.E142del allele found in the Brazilian miracidia, providing some evidence for independent origins. Similarly, MAP differs from the Brazilian miracidia carrying the p.C35R mutation; while both also carry p.P67L, MAP carries an additional p.G206V mutation. Additional flanking SNP data will be required to critically test whether the p.E142del and p.C35R OXA resistance mutations have arisen a single time, or have multiple independent origins.
Laboratory selection is commonly used to explore the genetics of pathogen resistance, but a concern with this approach is that the mutations selected in the laboratory may poorly represent those occurring in nature. That we identified the same mutations in laboratory and field selected parasites is extremely encouraging, because ongoing work to identify mutations involved in PZQ resistance also utilizes laboratory selected parasites (Couto et al., 2011). We note that laboratory selection experiments with Plasmodium also tend to identify the same genes and often the same mutations that are observed in the field (Anderson et al., 2011), further validating this approach.
Besides the two known alleles, two of these new mutations, c.214_215insA and g.328G>A, are predicted to confer OXA resistance; both introduce premature insertion of a stop codon producing a protein without an active site. These mutations therefore add two additional loss-of-function mutations that were probably selected during the OXA treatment. That four independent mutations are found in a single sampled parasite population is remarkable. Multiple origins have previously been observed in the evolution of resistance to benzimidazole drugs in the gastrointestinal nematode Haemonchus (Redman et al., 2015). The number of origins of resistance alleles is expected to depend on the size of the parasite population and the rate at which mutation generates resistant alleles (Messer and Petrov, 2013). In the case of Haemonchus contortus, the enormous size of parasite populations is likely to be the main driver, as only several specific mutations within β-tubulin can confer resistance. In the case of OXA resistance, a high mutation rate may be expected as the main driver, because multiple different mutations within SmSULT-OR can generate non-functional proteins. It is also possible that OXA-resistant alleles were present within Brazilian S. mansoni populations prior to hycanthone or OXA treatment. Such standing variation may even have been present prior to the introduction of S. mansoni into South America. Analysis of SmSULT-OR in African populations, where OXA was not used extensively, will help answer this question.
Work on OXA resistance is simplified because we have an effective in vitro functional assay for screening allelic variants. The assay we used is an improvement on those previously described (Pica-Mattoccia et al., 1992a; Valentim et al., 2013), because worm homogenates are replaced by recombinant SmSULT-OR proteins. We identified two additional exon 2 substitutions (p.G206V, p.N215Y): Structural analyses suggested that one of these mutations (p.G206V) may disrupt OXA activation, by interfering with binding of the co-factor (PAPS), while the other (p.N215Y) is likely to have minimal impact on function. We were able to directly test these predictions by performing OXA activation assays. These assays demonstrated that neither mutation prevents OXA activation, allowing us to reject our prediction for p.G206V and confirm our prediction for p.N215Y. Hence, while structural studies are useful for formulating hypotheses about the consequences of mutations, functional assays are essential for critical testing of these hypotheses.
The two last mutations were found in the 3′ UTR of the cDNA. In silico analysis, using predictions based on Drosophila, did not reveal regulatory sites in these regions, suggesting that these mutations do not impact function. However, Drosophila may be a poor model to use. When more information on SmSULT-OR regulatory regions become available, new analyses of these two mutations may reveal potential effects on mRNA stability or translation rate.
The allele frequency of all four OXA resistance alleles combined is 3.8%, and only two parasites of 183 sampled (1%) are homozygous and therefore expected to be phenotypically resistant. The low frequency of the OXA resistance alleles can be explained by two nonexclusive hypotheses. First, resistance alleles may have remained at low levels even when OXA was the first line drug in Brazil. Second, fitness costs associated with these alleles may have driven reductions in allele frequency after the abandonment of OXA treatment. However OXA resistance alleles still persist within the populations in a heterozygous state, the cost being present only when worms are homozygous for the defective alleles.
Whether there is a cost associated with OXA resistance is questionable because miracidia homozygous for resistance alleles were found in the field in the present study. A previous study showed a reduction in infectivity and egg production in OXA-resistant parasites relative to the OXA-susceptible populations from which they were isolated (Cioli et al., 1992). However, this study suffers from a methodological limitation, because resistant and susceptible laboratory populations had different genetic backgrounds, complicating interpretation. Comparison of isogenic wildtype and genetically manipulated resistant parasites, or analysis of fitness in the progeny of a genetic cross between OXA resistant and sensitive parasites would allow measurement of associated fitness costs, while minimizing confounding background effects.
New drugs are urgently needed for schistosome control because treatment currently relies on widespread monotherapy with PZQ. OXA is effective only against S. mansoni, but new OXA derivatives are under active development, with the aim of making compounds that are active against all three major schistosome species infecting humans (Taylor et al., 2015). If such derivatives are to be deployed clinically, understanding the capacity for resistance evolution in schistosome populations is of critical importance. For example, surveys of sequence variation in SmSULT-OR or the S. haematobium homologue would be an important prerequisite for field deployment of an OXA derivative active against both these species in Africa.
Our results have both positive and negative implications for field deployment of OXA derivatives. That multiple resistance alleles are present in a single parasite population suggests that resistance alleles may evolve and spread rapidly. On the positive side, existing resistance alleles are currently at extremely low frequency. It will be essential that OXA derivatives are deployed with appropriate partner drugs to minimize the rate of resistance evolution. We note that resistance to the antimalarial atovaquone was observed in the first clinical trial of this drug (Looareesuwan et al., 1996) and evolved de novo in different treated patients (Musset et al., 2007). Yet this drug is widely and effectively used with proguanil as a combination drug under the trade name Malarone.
Supplementary Material
Impact of mutations on Schistosoma mansoni SmSULT-OR sulfotransferase structure and function. This video details the mutations and potential impact on the protein structure and enzyme activity. OXA: oxamniquine, PAPS: 3′-phosphoadenosine-5′-phosphosulfate
Map of South America showing the places of origin of known and new Schistosoma mansoni oxamniquine (OXA) resistance alleles. Mutation p.C35R was identified in São Paulo state (SP, Brazil) while p.E142del was identified in Puerto Rico (PR). Both locations are distant from our sample site in Minas Gerais state (MG, Brazil; ~500 km and ~3,000 km, respectively).
Highlights.
We surveyed allelic variation in a schistosome drug resistance gene
There were four independent origins of alleles encoding loss-of-function proteins
Resistance mutations are at a very low frequency (< 0.0080)
Loss-of-function mutations were tested using an in vitro oxamniquine activation assay
Acknowledgments
This study was supported by NIH grants [R01-AI097576 (T.J.C.A.), 1R01AI115691 and P50 AI 098507 (P.T.L./P.J.H.)], World Health Organization [HQNTD1206356 (P.T.L.)], the UTHSCSA Presidents Collaborative Research Fund (P.T.L./P.J.H.) and the Robert A. Welch Foundation [AQ-1399 (P.J.H.)]. The molecular work at TBRI was conducted in facilities constructed with support from Research Facilities Improvement Program Grant (C06 RR013556) from the National Center for Research Resources (NIH). The AT&T Genomics Computing Center supercomputing facilities were supported by the AT&T Foundation and the National Center for Research Resources Grant (S10 RR029392). The X-ray Crystallography Core Laboratory is supported by the University of Texas Health Science Center, San Antonio, Office of the Vice President for Research and by the San Antonio Cancer Institute Grant (P30 CA054174). W.L was supported by a Cowles fellowship from Texas Biomedical Research Institute. N.E. was supported by the City of San Antonio Summer Ambassador Program. R.R.A. received support from CNPq (168260/2014-0). G.O. received support from CNPq (309312/2012-4), CAPES (070/13, REDE 21/2015) and FAPEMIG (RED-00014-14, PPM-00439-10). We thank Guilherme Oliveira, Dr. Fernanda Ludolf Ribeiro, Maycon Bruno and Nilvande Ferreira for technical assistance during stool processing and miracidia sampling, Dr. Andrea Gazzinelli and Dr. Leonardo Matoso for stool sampling at Ponto dos Volantes, Dr. Ricardo Toshio Fujiwara at Universidade Federal de Minas Gerais and Dr. Rodrigo Corrêa-Oliveira at Centro de Pesquisas René Rachou for providing laboratory space for stool processing and miracidia sampling, and Joanne Curran for use of a capillary sequencer.
Footnotes
Note: Nucleotide sequence data reported in this paper are available in the GenBank™, EMBL and DDBJ databases under the accession numbers KU951903 - KU952091
Note: Supplementary data associated with this article
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson T, Nkhoma S, Ecker A, Fidock D. How can we identify parasite genes that underlie antimalarial drug resistance? Pharmacogenomics. 2011;12:59–85. doi: 10.2217/pgs.10.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ Tracking Resistance to Artemisinin Collaboration (T. R. A. C) Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhembe NL, Nwodo UU, Govender S, Hayes C, Ndip RN, Okoh AI, Green E. Molecular detection and characterization of resistant genes in Mycobacterium tuberculosis complex from DNA isolated from tuberculosis patients in the Eastern Cape province South Africa. BMC Infect Dis. 2014;14:479. doi: 10.1186/1471-2334-14-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Huang HY, Hsu JBK, Weng SL, Horng JT, Huang HD. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics. 2013;14(Suppl 2):S4. doi: 10.1186/1471-2105-14-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry U, Redman EM, Raman M, Gilleard JS. Genetic evidence for the spread of a benzimidazole resistance mutation across southern India from a single origin in the parasitic nematode Haemonchus contortus. Int J Parasitol. 2015;45:721–728. doi: 10.1016/j.ijpara.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Cheetham PSJ. Case studies in applied biocatalysis from ideas to products. In: Cabral JMS, Best D, Boross L, Tramper J, editors. Applied Biocatalysis. Harwood Academics Publishers GmbH; Chur: 1994. pp. 47–108. [Google Scholar]
- Cioli D, Pica-Mattoccia L. Genetic analysis of hycanthone resistance in Schistosoma mansoni. Am J Trop Med Hyg. 1984;33:80–88. doi: 10.4269/ajtmh.1984.33.80. [DOI] [PubMed] [Google Scholar]
- Cioli D, Pica-Mattoccia L, Moroni R. Schistosoma mansoni: hycanthone/oxamniquine resistance is controlled by a single autosomal recessive gene. Exp Parasitol. 1992;75:425–432. doi: 10.1016/0014-4894(92)90255-9. [DOI] [PubMed] [Google Scholar]
- Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet. 2014;383:2253–2264. doi: 10.1016/S0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coura JR, Amaral RS. Epidemiological and control aspects of schistosomiasis in Brazilian endemic areas. Mem Inst Oswaldo Cruz. 2004;99:13–19. doi: 10.1590/S0074-02762004000900003. [DOI] [PubMed] [Google Scholar]
- Coura JR, Conceici¸ão MJ. Specific schistosomiasis treatment as a strategy for disease control. Mem Inst Oswaldo Cruz. 2010;105:598–603. doi: 10.1590/s0074-02762010000400040. [DOI] [PubMed] [Google Scholar]
- Couto FFB, Coelho PMZ, Araújo N, Kusel JR, Katz N, Jannotti-Passos LK, Mattos ACA. Schistosoma mansoni: a method for inducing resistance to praziquantel using infected Biomphalaria glabrata snails. Mem Inst Oswaldo Cruz. 2011;106:153–157. doi: 10.1590/s0074-02762011000200006. doi: dx.doi.org/10.1590/S0074-02762011000200006. [DOI] [PubMed] [Google Scholar]
- Djègbè I, Agossa FR, Jones CM, Poupardin R, Cornelie S, Akogbéto M, Ranson H, Corbel V. Molecular characterization of DDT resistance in Anopheles gambiae from Benin. Parasit Vectors. 2014;7:409. doi: 10.1186/1756-3305-7-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C, Mertens T, Hirnschall G, Mpanju-Shumbusho W, Newman RD, Raviglione MC, Savioli L, Nakatani H. WHO and the future of disease control programmes. Lancet. 2013;381:413–418. doi: 10.1016/S0140-6736(12)61812-1. [DOI] [PubMed] [Google Scholar]
- Egea R, Casillas S, Barbadilla A. Standard and generalized McDonald-Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008;36:W157–W162. doi: 10.1093/nar/gkn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essandoh J, Yawson AE, Weetman D. Acetylcholinesterase (Ace-1) target site mutation 119S is strongly diagnostic of carbamate and organophosphate resistance in Anopheles gambiae s.s. and Anopheles coluzzii across southern Ghana. Malar J. 2013;12:404. doi: 10.1186/1475-2875-12-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansma WB, Rogers SH, Liu CL, Bueding E. Experimentally produced resistance of Schistosoma mansoni to hycanthone. Am J Trop Med Hyg. 1977;26:926–936. doi: 10.4269/ajtmh.1977.26.926. [DOI] [PubMed] [Google Scholar]
- Katz N, Dias EP, Araújo N, Souza CP. Estudo de uma cepa humana de Schistosoma mansoni resistente a agentes esquistossomicidas. Rev Soc Bras Med Trop. 1973a;7:381–387. doi: 10.1590/S0037-86821973000600008. [DOI] [Google Scholar]
- Katz N, Pellegrino J, Ferreira MT, Oliveira CA, Dias CB. Preliminary clinical trials with hycanthone, a new antischistosomal agent. Am J Trop Med Hyg. 1968;17:743–746. doi: 10.4269/ajtmh.1968.17.743. [DOI] [PubMed] [Google Scholar]
- Katz N, Pellegrino J, Grinbaum E, Chaves A, Zicker F. Preliminary clinical trials with oxamniquine, a new antischistosomal agent. Rev Inst Med Trop Sao Paulo. 1973b;15:25–29. [PubMed] [Google Scholar]
- Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg. 1996;54:62–66. doi: 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- Melcher K. A modular set of prokaryotic and eukaryotic expression vectors. Anal Biochem. 2000;277:109–120. doi: 10.1006/abio.1999.4383. [DOI] [PubMed] [Google Scholar]
- Messer PW, Petrov DA. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol. 2013;28:659–669. doi: 10.1016/j.tree.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musset L, Le Bras J, Clain J. Parallel evolution of adaptive mutations in Plasmodium falciparum mitochondrial DNA during atovaquone-proguanil treatment. Mol Biol Evol. 2007;24:1582–1585. doi: 10.1093/molbev/msm087. [DOI] [PubMed] [Google Scholar]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D6–17. doi: 10.1093/nar/gku1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Gulley ML, den Dunnen JT, Wilson RB Association for Molecular Patholpogy Training Committtee E. Standard mutation nomenclature in molecular diagnostics: practical and educational challenges. J Mol Diagn. 2007;9:1–6. doi: 10.2353/jmoldx.2007.060081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panichsillapakit T, Smith DM, Wertheim JO, Richman DD, Little SJ, Mehta SR. Prevalence of transmitted HIV drug resistance among recently infected persons in San Diego, California 1996–2013. J Acquir Immune Defic Syndr. 2015 doi: 10.1097/QAI.0000000000000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RJ, Pota H, Evehe MSB, Bâ EH, Mombo-Ngoma G, Malisa AL, Ord R, Inojosa W, Matondo A, Diallo DA, Mbacham W, van den Broek IV, Swarthout TD, Getachew A, Dejene S, Grobusch MP, Njie F, Dunyo S, Kweku M, Owusu-Agyei S, Chandramohan D, Bonnet M, Guthmann JP, Clarke S, Barnes KI, Streat E, Katokele ST, Uusiku P, Agboghoroma CO, Elegba OY, Cissé B, A-Elbasit IE, Giha HA, Kachur SP, Lynch C, Rwakimari JB, Chanda P, Hawela M, Sharp B, Naidoo I, Roper C. Multiple origins and regional dispersal of resistant dhps in African Plasmodium falciparum malaria. PLoS Med. 2009;6:e1000055. doi: 10.1371/journal.pmed.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Archer S, Cioli D. Hycanthone resistance in schistosomes correlates with the lack of an enzymatic activity which produces the covalent binding of hycanthone to parasite macromolecules. Mol Biochem Parasitol. 1992a;55:167–175. doi: 10.1016/0166-6851(92)90137-9. [DOI] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Carlini D, Guidi A, Cimica V, Vigorosi F, Cioli D. The schistosome enzyme that activates oxamniquine has the characteristics of a sulfotransferase. Mem Inst Oswaldo Cruz. 2006;101(Suppl 1):307–312. doi: 10.1590/S0074-02762006000900048. [DOI] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Cioli D, Archer S. Binding of oxamniquine to the DNA of schistosomes. Trans R Soc Trop Med Hyg. 1989;83:373–376. doi: 10.1016/0035-9203(89)90508-7. [DOI] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Dias LC, Cioli D. Genetic complementation analysis of two independently isolated hycanthone-resistant strains of Schistosoma mansoni. Mem Inst Oswaldo Cruz. 1992b;87(Suppl 4):211–214. doi: 10.1590/S0074-02761992000800032. [DOI] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Dias LC, Moroni R, Cioli D. Schistosoma mansoni: genetic complementation analysis shows that two independent hycanthone/oxamniquineresistant strains are mutated in the same gene. Exp Parasitol. 1993;77:445–449. doi: 10.1006/expr.1993.1104. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. [Google Scholar]
- Redman E, Whitelaw F, Tait A, Burgess C, Bartley Y, Skuce PJ, Jackson F, Gilleard JS. The emergence of resistance to the benzimidazole anthlemintics in parasitic nematodes of livestock is characterised by multiple independent hard and soft selective sweeps. PLoS Negl Trop Dis. 2015;9:e0003494. doi: 10.1371/journal.pntd.0003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Rufener L, Mäser P, Roditi I, Kaminsky R. Haemonchus contortus acetylcholine receptors of the DEG-3 subfamily and their role in sensitivity to monepantel. PLoS Pathog. 2009;5:e1000380. doi: 10.1371/journal.ppat.1000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- Taylor AB, Pica-Mattoccia L, Polcaro CM, Donati E, Cao X, Basso A, Guidi A, Rugel AR, Holloway SP, Anderson TJC, Hart PJ, Cioli D, LoVerde PT. Structural and functional characterization of the enantiomers of the antischistosomal drug oxamniquine. PLoS Negl Trop Dis. 2015;9:e0004132. doi: 10.1371/journal.pntd.0004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzinger J, Keiser J, Shuhua X, Tanner M, Singer BH. Combination chemotherapy of schistosomiasis in laboratory studies and clinical trials. Antimicrob Agents Chemother. 2003;47:1487–1495. doi: 10.1128/AAC.47.5.1487-1495.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentim CLL, Cioli D, Chevalier FD, Cao X, Taylor AB, Holloway SP, Pica-Mattoccia L, Guidi A, Basso A, Tsai IJ, Berriman M, Carvalho-Queiroz C, Almeida M, Aguilar H, Frantz DE, Hart PJ, LoVerde PT, Anderson TJC. Genetic and molecular basis of drug resistance and species-specific drug action in Schistosome parasites. Science. 2013;342:1385–1389. doi: 10.1126/science.1243106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Impact of mutations on Schistosoma mansoni SmSULT-OR sulfotransferase structure and function. This video details the mutations and potential impact on the protein structure and enzyme activity. OXA: oxamniquine, PAPS: 3′-phosphoadenosine-5′-phosphosulfate
Map of South America showing the places of origin of known and new Schistosoma mansoni oxamniquine (OXA) resistance alleles. Mutation p.C35R was identified in São Paulo state (SP, Brazil) while p.E142del was identified in Puerto Rico (PR). Both locations are distant from our sample site in Minas Gerais state (MG, Brazil; ~500 km and ~3,000 km, respectively).