

Abstract
Despite of the potential implications for cancer immunotherapy, conventional approaches using in vitro expanded CD8+ T cells have suboptimal outcomes, mostly due to loss of functionality from cellular exhaustion. We therefore investigated the phenotypic and functional differences among in vitro activated CD8+ T cells of three different sources, namely naïve (NTeff), memory (MTeff) and tumor-infiltrating lymphocytes (TILeff) from human and mice, to better understand mechanisms behind potent effector functions and potential for overcoming current limitations. In line with the greater proliferation activity and longer telomere lengths of NTeff populations, cells of naïve origin exhibited significantly less amounts of T cell exhaustion markers than those of MTeff and TILeff, and moreover, acquired distinct expression patterns of memory-promoting transcription factors, T-bet and Eomes, induced in a rapid and sustainable manner. NTeff cells appeared to have lower expression of Foxp1 and were refractory to apoptosis upon TGF-β conditioning, implying better survival potential and resistance to tumor-induced immune suppression. Of CD8+ T cell pools activated to tumor-specific CTLs, naïve cell generated effectors possessed the most potent cytotoxic activity, validating implications for use in rational design of adoptive immunotherapy.
Adoptive immunotherapy, or the infusion of ex vivo activated and expanded tumor-specific CD8+ T cells into cancer patients, is a strategy involving removal of CD8+ T cells from the tumor environment and provision of stimulatory conditions necessary for their optimal activation, in attempts to overcome poor T-cell responsiveness to tumors. Adoptive T-cell transfer therapy was first attempted in the late 1980s to early 1990s, following the identification of the first tumor associated antigens and isolation of tumor reactive CD8+ T-cell clones from cancer patients. A sufficient number of activated CD8+ effector T cells were obtained and subsequently transferred intravenously into patients, mediating tumor elimination1. However, current articles have reported that immunotherapy employing the use of CD8+ cytotoxic T lymphocytes (CTLs) is limited by chronic activation and functional impairment of effector cells induced by immunosuppressive factors2,3,4. Investigation of these cells has revealed a so called ‘exhaustion profile’ that includes cell dysfunction, loss of effector function, and progressive increase in the amount and diversity of check point inhibitors such as programmed cell death protein 1 (PD1), cytotoxic T lymphocyte antigen 4 (CTLA4), lymphocyte activation gene 3 protein (LAG3), and killer cell lectin-like receptor G1 (KLRG1)2,3,4. It has also been shown that CTL function is altered by transforming growth factor-β (TGF-β), a lymphocyte inhibitor frequently overexpressed in the tumor mircroenvironment (TME) of multiple tumors5,6. Stephen et al.7 have characterized the mechanism of T cell unresponsiveness in TME driven by Forkhead box protein P1 (Foxp1) upregulation in response to TGF-β, by which antigen-primed CD8+ T cells are prevented from proliferating and promoting cytotoxic function.
The pool of lymphocytes used for adoptive immunotherapy can be derived from any of the CD8+ subsets, including tumor-infiltrating lymphocytes (TILs), naive T cells (TN), and antigen-experienced memory T cells (TM), which can be divided into central memory (TCM) and effector memory (TEM) subsets that differ in phenotype, homing, and function8. Memory CD8+ T-cell subsets are studied more often than naïve cells, because of their tumor specificity and greater ability to rapidly proliferate when exposed to previously encountered antigens. Moreover, Berger et al.9 showed that effector cells derived from TCM rather than TEM possess increased potential to survive and establish immunologic memory after infusion in macaques. These findings have been consistent in mice memory subsets10. However, a more recent study has suggested that TN subsets may serve as a suitable candidate for immunotherapy because of their low level of ‘exhaustion’ in effector cells, sustained replicative potential correlative to telomere length, and minimal effector differentiation translating to greater efficacy after infusion11. Although self or tumor-reactive naïve CD8+ T cells have the capacity to respond to tumor antigens and differentiate into cytotoxic effector cells, some major limitations are the relatively low number of self/tumor-specific lymphocytes that result from negative selection during thymic maturation and inadequate magnitude of stimulation from antigens in the tumor environment12. Studies have shown that CD8+ T cells can be expanded by T cell growth factor interleukin-2 (IL-2) in the absence of T-cell receptor (TCR) signals, and in this situation, IL-2 can induce a unique signaling pathway that is associated with strong lymphocyte-specific protein tyrosine kinase/JAK3-dependent activation of the PI3K/AKT pathway13,14. Such signaling pathways not only induce rapid proliferation of IL-2 activated naïve CD8+ T cells but also upregulate expression of eomesodermin (Eomes), signifying the differentiation fate of primary naïve effectors into long-lived memory cells. Several current studies performed in murine models have demonstrated the role of T-box transcription factors, T-bet and Eomes, in regulation of both effector and memory functions where high expression of T-bet promotes effector function and Eomes is linked to formation of the long-term memory population15,16,17.
Considering some of the current limitations in adoptive T-cell transfer, we sought to evaluate the phenotypic and functional differences of effectors derived from naive, memory, and tumor-infiltrating CD8+ T cells (NTeff, MTeff, and TILeff, respectively), and to further investigate the possibility of generating optimal tumor-reactive CTLs with high-dose IL-2 activation for application in clinical trials. We report that exhaustion phenotypes and expression kinetics of T-box transcription factors for NTeff cell populations show possible advantages over other subgroups for use in cell transfer therapy. Our results also suggest that effector cells of naïve origin are relatively resistant to immunosuppressive effects of TGF-β with a trend in lower Foxp1 expression. High-dose IL-2 priming of naïve T cells successfully expands tumor-specific effector cells that exert favorable cytotoxic function, and serves as a potential means for overcoming some of the current barriers in immunotherapy.
Results
Successful expansion of human naïve, memory, and TIL CD8+ T cells in vitro
CD8+ T cell subsets could be defined by the surface markers CD62L, CD45RO, CCR7, and CD45RA to naïve (CCR7+CD45RA+ or CD62L+CD45RO−) and memory (CD45RA− or CD45RO+) cells. Tumor-infiltrating CD8+ T cells were CD44+, mostly with loss of homing receptors CD62L and CCR7 expression, and composed of two main sub-populations CD45RA− and CD45RA+, suggesting a relatively differentiated status. After MACS isolation and stimulation by CD3/CD28 Dynabeads on anti-CD2 coated plates, successful expansion was demonstrated in all three groups by either CFSE peak division of NTeff and MTeff subsets, or enlarged populations of CD44+CD8+ TILeff cells (Fig. 1A). CFSE staining of TILeff proliferation was observed but peak division was indistinct due to the small number of initial TILs (data not shown). Activation kinetics differed for cell subsets where exponential expansion and accumulation was observed starting at day 3 for NTeff and later at day 5 for MTeff and TILeff cells, and thus further analyses of all effector cells were performed on day 5 post-stimulation.
Figure 1. Characteristics of human naïve, memory, and TIL effector CD8+ T cells.

(A) Isolation of CD8+ T cell subsets and proliferation assays. MACS isolation and FACS Aria analyses displayed in vitro expansion of naïve (CCR7+CD45RA+), memory (CD45RA−), and TIL (CD44+) subpopulations, 5 days post TCR stimulation. CFSE assay showed positive proliferation results after 3 to 5 days of culture. (B) Activation markers of effector cell subsets. Effector naïve and memory CD8+ cells (NTeff and MTeff, respectively) are characterized by CD62L-CD25+CD44+OX40+ expression. Re-stimulated CD8+ TIL (TILeff) populations also showed similar phenotypes. (C) OX40+ cell percentage of effector cell subsets. NTeff showed significantly higher values compared to MTeff and TILeff (*P < 0.05 vs TILeff, **P < 0.005 vs MTeff). (D) Telomere length comparison showed the longest telomeres in NTeff and shortest in TILeff cells (*P < 0.05). RTLs of effector cells were measured against CCRF-CEM control cells (1:1 ratio, total 5.0 × 105 cells) with a DAKO Telomere PNA Kit. Data are representative of three to four independent experiments and are presented as mean ± SD.
Phenotypic characteristics of effector cell populations were assessed by surface expression of activation markers CD62L, CD25, CD44, and OX40. Effector cells from all progenitors (naïve, memory, and TILs) exhibited an effector phenotype with significant up-regulation of activation markers in comparison to their progenitors, and were considered activated as CD62L−CD25+CD44+OX40+ populations (Fig. 1B). Despite variability in the percentage of effector cells produced among donors, a significantly higher percentage of OX40 expressing cells was observed for NTeff in comparison to MTeff (p < 0.05) or TILeff (p < 0.005) cells (Fig. 1C). To further compare the proliferative potential among cell populations, relative telomere length (RTL), which correlate with replicative capacity, of NTeff, MTeff, and TILeff cells were investigated against CCRF-CEM control cell line. Telomere length was greatest in NTeff, shorter in MTeff, and shortest in TILeff cell subsets (Fig. 1D). This result was consistent with previously published reports of longer telomeres in human naive T cells, leading to increased proliferation of NTeff cells in comparison to MTeff cells after in vitro stimulation9.
Exhaustion phenotypes differ among in vitro generated human and murine effector cells NTeff, MTeff, and TILeff
Inhibitory receptors on T cell surfaces such as PD-1, CTLA-4, and KLRG-1, have been shown to facilitate T cell exhaustion by interaction with ligands on antigen presenting cells or tumor cells3,4,5. We therefore compared the expression of these inhibitory receptors on the three effector cell subtypes. All effector cells showed significant increase of exhaustion phenotypes during proliferation, but NTeff cells showed significantly less expression of PD-1 and CTLA4 compared to MTeff and TILeff subsets (Fig. 2A). Check point inhibitors showed varying levels of expression dependent on the time elapsed from activation with peak expression on days 4–5 for PD-1, days 5–7 for CTLA-4, and days 4–7 for KLRG-1 (data not shown). To investigate functional relevance of exhaustion phenotypes, we then evaluated the secretory function of cytotoxic cytokines such as granzyme B, perforin, and IFN- ɤ from different human effectors. During days 3 to 5 post-stimulation, the secretion of granzyme B and perforin gradually increased in all effector cells derived from naïve, memory, and TIL populations (Fig. 2C). Among fully activated day 5 effector cells, NTeff possessed the highest expression level of perforin and granzyme B, and similar levels of IFN-ɤ (data not shown) compared to MTeff and TILeff cells. In the murine model, effectors derived from CD8+ naïve and memory phenotype (MP) T cells, and TILs collected from OVAp-expressing EL4-EG7 tumors were successfully proliferated with CD44+OX40+ phenotypes. Expression levels of inhibitory receptors PD-1, KLRG1, and LAG3 in murine CD8+ effectors showed similar patterns to those of human cells (Fig. 2B).
Figure 2. Exhaustion phenotypes of effector CD8+ T cell subpopulations in human and mice.

Effector cells (OX40+) derived from naïve cells (NTeff) expressed lower levels of exhaustion markers PD1, CTLA4, KLRG1, and LAG3 compared to effectors from memory cells or TILs (MTeff or TILeff) (A) in human (*P < 0.05 for PD-1, **P < 0.005 for CTLA4) and (B) in mice at 5 days post-stimulation with anti-CD3/CD28 and anti-CD2. MFI levels are the average result of three experiments and are presented as mean ± SD. (C) In vitro functional analysis of human effector cells by cytokine production. An equal number of progenitors (2 × 105 cells per subtype) activated to OX40+ effectors were characterized by flow cytometry for direct comparison among subtypes. NTeff cells demonstrated a greater increase in perforin+ granzyme B+ populations compared to MTeff and TILeff subsets at two time points (day 3 and 5 post-stimulation). Data are representative of three independent experiments.
Expression kinetics of transcription factors T-bet and Eomes differ among CD8+ T cell effector populations
For further characterization of the CD8+ T cell subpopulations, the expression levels of two T-box transcription factors, T-bet and Eomes, were examined using intracellular staining for FACS analysis and confirmed with Western blot. Although naïve CD8+ T cells expressed lower levels of T-bet and Eomes in comparison to memory cells before stimulation, activated effectors NTeff, MTeff, and TILeff showed relatively similar MFI levels of T-bet and Eomes, demonstrating the capacity of naïve or memory cells to induce similar effector populations (Fig. 3A). However, considering the lower MFI for pre-stimulated naïve cells, this cell population displays a greater increase in T-bet and Eomes expression levels post-stimulation compared to memory T cells.
Figure 3. Expression of T-box transcriptional factors and characteristics of human effector CD8+ T cells under TGF-β conditioning.

(A) Intracellular staining of T-bet and Eomes expressed on TCR stimulated effectors NTeff, MTeff, and TILeff. MFI levels are shown for day 0 (initial peak) and day 5 (right side peak) post-stimulation. For TILs, day 0 peaks are lost due to insufficient cell amount pre-expansion. (B) Western blot analysis of naïve and memory T cells, and TILs on days 0, 3, 5 and 8 post TCR activation. Kinetics of T-bet and Eomes expression in differentiating CD8+ T cells suggest rapid and sustained stimulation for NTeff populations. Effector groups from day 5 post-stimulation are treated with TGF-β (10 ng/mL) for 48 hours and checked for Foxp1 expression (day 7 post-stimulation) by (C) intracellular FACS staining and (D) Western blot. Increase in Foxp1 MFI levels pre and post TGF-β conditioning was minimal for naïve effector cells and NTeff showed the least Foxp1 protein expression, although not statistically significant. MFI levels and protein density ratios are the average result of three experiments and shown as mean ± SD. (E) Annexin V assay by flow cytometry. Quadrant plots show the percent distribution of early and late apoptotic cells (first quadrant) with or without 48-hour exposure to TGF-β. Naïve cell derived effectors (NTeff) showed minimal increase in apoptotic cell populations relative to MTeff and TILeff subgroups. All experiments were repeated three times and are presented by one representative figure.
To clarify and confirm the expression profile of these T-box transcription factors in the effector subpopulations, Western blot analysis was performed using whole cell lysates of naïve and memory T cells, and TILs on days 0, 3, 5 and 8 post TCR activation. Our experiments revealed clear differences in the kinetics of T-bet and Eomes expression during CD8+ T cell activation (Fig. 3B). Effectors derived from naïve cells increased in T-bet expression appreciably starting on day 3, reaching maximum on day 8, whereas memory cell derived effectors gained the highest expression of T-bet earlier on day 3 and were quickly down-regulated on days 5 through 8. TILeff cells demonstrated poor expression of T-bet throughout stimulation time. While naïve and memory cells showed increasing Eomes expression with stimulation, TILs exhibited decreasing intensity during day 5 through 8 post-activation. Such kinetics suggest that NTeff cells express higher levels of both T-bet and Eomes, and expression is sustained for longer periods during activation, in comparison to other effector groups MTeff and TILeff. Collectively, naïve T cell derived effectors appear to differentiate relatively rapidly, proliferate actively, and have potential to be converted into long-lived memory cells.
Foxp1 expression and apoptosis studies under TGF-β conditioning show favorable results for CD8+ NTeff cells
To determine the immunosuppressive resistance potential of CD8+ NTeff cells in the tumor microenvironment, we examined expression of Foxp1 among effector cells derived from naïve, memory, and TIL progenitors in the presence of TGF-β. After 5 days of proliferation, effector cells were exposed to TGF-β (10 ng/mL) for 48 hours and Foxp1 expression (day 7 post-stimulation) was evaluated by intracellular FACS staining. MTeff and TILeff populations showed increased Foxp1 expression with TGF-β conditioning, whereas NTeff cells maintained their lower expression levels in three independent experiments (Fig. 3C). To confirm the increased expression of Foxp1 in CD8+ effector cells in the tumor microenvironment, Western blot analysis was performed for effector T cell subsets under TGF-β exposure using non-stimulated naïve cells as the control. As expected, NTeff populations expressed the lowed amount of Foxp1 protein expression relative to other effector groups, consistent with intracellular staining results (Fig. 3D). After conditioning with TGF-β, flow cytometry detection of apoptosis with Annexin V displayed limited induction of apoptotic cell populations for NTeff cells relative to MTeff and TILeff subgroups (Fig. 3E). Taken together, these results suggest that CD8+ effector subgroups differ in Foxp1 expression and apoptotic response to TGF-β exposure, and that NTeff cells maintain resistance to inhibitory factors induced from the tumor microenvironment.
Cytotoxic activity against targeted tumor cells is superior for effector cells of naïve T cell origin in both human and mouse models
To investigate the cytotoxic function of tumor-specific human CTLs, antigen (U266 MM cell line) loaded autologous mature dendritic cells (mDCs) were co-cultured for 7 days with IL-2 primed naïve and memory CD8+ T cells and the generated CTLs were assessed via IFN-ɤ ELISPOT assay and LDH Cytotoxicity Detection Kit. A total of five cell groups were harvested for cytotoxic analyses including CD8+ (Naïve D14, Naïve D7, Naïve D0, and Memory) and CD3+ CTLs. Briefly, Naïve D14 and Naïve D7 cells refer to CD8+ naïve cells co-cultured with high-dose IL-2 (1 μg/mL) for 14 and 7 days respectively, whereas Naïve D0 and Memory cells are co-cultured with conventional dose IL-2 (10 ng/mL) and are of naïve and memory T cell origin, respectively (refer to Methods). IFN-ɤ ELISPOT was measured in the presence and absence of MHC class I specific monoclonal antibody (W6/32, 20 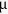 g/mL) with CTL alone as the control. High dose IL-2 primed Naïve D14 effector cells showed the highest secretion of IFN-ɤ against target U266 cells when co-cultured for one day at a 10:1 ratio of effector (E) to target (T) cells (Fig. 4A.). Although CD3+ cells also secreted higher levels of IFN-ɤ relative to Naïve D7, Naïve D0, and Memory CD8+ cells, they also displayed non-specific secretion as observed in the control CTL alone group. Direct cytotoxicity testing against U266 target cells using the LDH cytotoxic assay revealed enhanced CTL activity for Naïve D14 cells that was significantly greater than other effector CTLs when E:T ratios were at least 10:1 (Fig. 4B.). In the murine model, in vivo CTL activity against inoculated OVAp-expressing EL4-EG7 lymphoma cells in B6 mice was examined for NTeff, MTeff, and TILeff CD8+ effector subsets obtained from OT-1 Thy1.1 mice. Tumor growth was negative for at least 35 days after tumor inoculation in mice injected with NTeff cells, whereas MTeff, and TILeff injected mice showed positive tumor growth within the same time frame (Fig. 4C). Tumor growth became positive in one NTeff injected mouse at 42 days post inoculation (data not shown).
g/mL) with CTL alone as the control. High dose IL-2 primed Naïve D14 effector cells showed the highest secretion of IFN-ɤ against target U266 cells when co-cultured for one day at a 10:1 ratio of effector (E) to target (T) cells (Fig. 4A.). Although CD3+ cells also secreted higher levels of IFN-ɤ relative to Naïve D7, Naïve D0, and Memory CD8+ cells, they also displayed non-specific secretion as observed in the control CTL alone group. Direct cytotoxicity testing against U266 target cells using the LDH cytotoxic assay revealed enhanced CTL activity for Naïve D14 cells that was significantly greater than other effector CTLs when E:T ratios were at least 10:1 (Fig. 4B.). In the murine model, in vivo CTL activity against inoculated OVAp-expressing EL4-EG7 lymphoma cells in B6 mice was examined for NTeff, MTeff, and TILeff CD8+ effector subsets obtained from OT-1 Thy1.1 mice. Tumor growth was negative for at least 35 days after tumor inoculation in mice injected with NTeff cells, whereas MTeff, and TILeff injected mice showed positive tumor growth within the same time frame (Fig. 4C). Tumor growth became positive in one NTeff injected mouse at 42 days post inoculation (data not shown).
Figure 4. Tumor-specific CTL function of in vitro human and in vivo murine CD8+ T cells.

(A) In vitro comparison of tumor-specific CTL function of IL-2 primed human CD8+ T cells against U266 MM cells by IFN-ɤ ELISPOT assay. CTL alone represents the control group and U266 refers to cytotoxic activity of effectors against U266 target cells (E:T ratio of 10:1) in the absence of MHC class I restriction, expressed as the number of IFN-ɤ spots observed. Naïve T cells activated under high-dose IL-2 for 14 days (Naïve D14) displayed highest activity against U266 target cells. (B) LDH cytotoxicity assay of IL-2 primed human CD8+ T cells. Specific cytotoxicity lysis percentages against U266 MM target cells were highest for Naïve D14 effectors compared to other groups when E:T ratios were at least 10:1 (*p < 0.05). (C) In vivo CTL activity in the murine model. Following injection of NTeff, MTeff, and TILeff CD8+ T cells, OVAp-expressing EL4-EG7 cells (8 × 105 cells/200 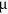 L) were inoculated subcutaneously in B6 recipient mice in a 1:2 ratio of target tumor to effector cells. All B6 mice injected with TILeff had positive mass (1 cm3) formation after 28 days, whereas tumor growth was suppressed in NTeff harboring B6 mice for longer than 35 days. All data are representative of three independent experiments and shown as mean ± SD.
L) were inoculated subcutaneously in B6 recipient mice in a 1:2 ratio of target tumor to effector cells. All B6 mice injected with TILeff had positive mass (1 cm3) formation after 28 days, whereas tumor growth was suppressed in NTeff harboring B6 mice for longer than 35 days. All data are representative of three independent experiments and shown as mean ± SD.
Discussion
Current adoptive immunotherapy involves mainly the use of CD3-collected T cells or autologous genetically engineered TILs, with increasing prospect for memory CD8+ cells18,19. Autologous CD8+ subpopulations hold the advantage of avoiding contamination of immunosuppressive regulatory T cells and decrease in terminal differentiation with repeated clonal proliferation in cancer patients. In order to demonstrate the superior background of tumor-specific CTLs derived from naïve CD8+ populations we focused on clarifying the different exhaustion characteristics, including expression check point inhibitors and transcription factors, and divergence of cytotoxic function among different effector groups NTeff, MTeff, and TILeff. We found that naïve CD8+ effectors have relatively favorable resistance patterns to immunosuppression and strong CTL potency against tumor targets. This may be explained by the “fresh” nature of these cell groups that display lower levels of inhibitory immune checkpoint receptors such as PD-1, CTLA-4, and KLRG1, leading to a greater activation and replicative potential20. Our results were consistent with reports by Hinrichs et al.21 indicating effectors derived from naïve CD8+ T cells may be the preferable candidate for T-cell adoptive immunotherapy based on evaluation of KLRG1, CD57, CD27 expression and telomere length. Such markers represent tendency of MTeff or TILeff cells to quickly enter into a T cell “exhaustion” phase with elevated levels of inhibitory receptors following long-term exposure to tumor antigen and ultimately acquire a non-functional state4,22,23.
Another point of consideration is the expression of two T-box transcriptional factors, T bet and Eomes, as well as their effect on CTL differentiation. Among various transcription factors that function in pairs to regulate effector and memory CD8+ T cell development, short- and long-term effects of T-bet and Eomes have been relatively well identified in the murine model, but are still limited to the context of infection and disease in humans16,24,25,26,27. Previous studies have emphasized on the role of high T-bet expression, which is associated with long-term resilience, low expression of inhibitory receptors, and protection from CD8+ T-cell exhaustion27,28, as well as Eomes induced inhibition of cell death leading to generation or survival of memory cells17,29. In our experiments, data showed that NTeff cells gradually increase and maintain high expression levels of both T-bet and Eomes throughout activation, whereas other effectors quickly down-regulated these factors. It has been suggested that high expression of T-bet correlates with a granzyme B+perforin+ phenotype where high perforin secretion was only detected in T-bethiEomeshi/lo cells15. Stable expression of Eomes levels in NTeff and MTeff subsets show their ability to become memory-precursor effector cells (MPECs), whereas TILeff cells enter a terminal differentiation phase via down regulation of Eomes. Collectively, our data indicates that NTeff populations co-express sustained, high levels of T-beta and Eomes, and a granzyme B+perforin+ phenotype representative of full cytotoxic function.
To clarify the relationship between patterns of Foxp1 expression and TGF-β, we activated T cells by in vitro TCR stimulation and evaluated Foxp1 levels of these effectors before and after TGF-β exposure with intention to mimic the immunosuppressive TME. As shown in a recent article7, overexpression of Foxp1 is thought to be mediated by TGF-β signaling through Smad protein interaction, which in turn mediates c-Myc and c-Jun transcriptional repression, and is expected to be upregulated after homing to the TME. However, in this study, only whole mouse CD8+ cells were investigated and did not address subsets of human CD8+ T cells. As inferred from our results, CD8+ NTeff are less influenced by tumor-derived inhibitory factor TGF-β, i.e., effectors derived from naïve cells are less affected by tumor suppression mechanisms driven by Foxp1 in response to TGF-β. Importantly, 48 hours post treatment of TGF-β, a smaller percentage of NTeff cells entered apoptosis compared to MTeff and TILeff subsets, which further offers evidence for the superior features of naïve cell derived effectors in resistance to the TGF-β induced apoptosis effect. These findings provide indication for the use of TGF-β inhibitors29, NTeff engineered to express a dominant-negative mutant of the TGF-β receptor30, and Foxp1-deficient NTeff, with objectives to enhance the immunosuppressive resistance of lymphocytes in the TME in various clinical applications.
The essential role of high-dose IL-2 in cancer has now been widely discussed, especially because IL-2 is suspected to optimize all stages of CD8+ T cell response, including primary expansion, contraction, memory generation, and secondary expansion31. In addition, high-dose IL-2 may strongly induce the proliferation as well as stimulation of naïve CD8+ T cells both in vitro and in vivo32. In our research, high-dose IL-2 (1 μg/mL) stimulated naïve CD8+ T cells, which were then co-cultured with autologous DCs, could successfully generate strong CTLs within a short duration. By evaluation of ELISPOT and LDH assay, Naïve D14 and Naïve D7 both generated CTLs with a high number of IFN-ɤ spots and better (Naïve D14) or similar (Naïve D7) cytotoxic effects compared to CD3+ CTLs. As mentioned, high concentrations of IL-2 (0.1–1 μg/mL) are necessary for stimulation of CD8+ naïve T cells through the antigen-independent manner13. Although our antigen-loaded DCs could induce antigen-specific CTLs from CD8+ naive T cells, higher than conventional dose IL-2 was required not only for proliferation of the limited number of antigen-specific naïve CD8+ T cells but also for generation of potent CTLs. During cytokine stimulation, high-dose IL-2 plays an important role in both pre-activation (during 7 days co-culture with naïve T cells) and co-activation (during 7 days co-culture with IL-2 primed T cells and DCs). In the co-activation period, high-dose IL-2 could potentially be used with other ɤc cytokines (IL-7, IL-15) for greater generation of CTLs. However, the optimal exposure times of high-dose IL-2 to tumor-specific naïve CD8+ T cells remains to be determined.
Taken together, our results provide a relatively complete framework for collecting effector cells derived from naïve origin and present a practical method to effectively generate strongly functional NTeff populations with the use of high-dose IL-2 (1 μg/mL). Nowadays, human T lymphocytes, which can be genetically engineered prior to adoptive transfer, can express virtually any target gene, by application of techniques involving genes encoding T-cell receptors (TCRs) or chimeric antigen receptors (CARs) to provide the desired T-cell specificity11. CAR-redirected T lymphocytes (CAR-T cells) are mostly limited to memory and effector T cells33,34,35 due to potent cytotoxic function. However, the appropriate substrate of effector T cells for clinical use is still controversial and the development of antigen escape variants may represent the limitation of monoclonal specificity. To obtain an adequate amount of effector cells with exceeding cytotoxic function, we suggest that naïve cells should be considered as a candidate for CAR-T therapy.
In summary, primary effector cells derived from naïve CD8+ T cells showed the highest potential for adoptive cell transfer therapy, with lower expression of inhibitory surface markers, higher secretion of cytotoxic cytokines, and improved resistance to TGF-β induced suppression in the TME, in comparison to secondary effectors derived from memory or TIL CD8+ cells. High-dose IL-2 priming amplifies the number of tumor-specific naïve CD8+ T cells without clonal exhaustion and results in the generation of potent tumor-reactive CTLs with capacity to overcome tumor-derived immune suppression.
Methods
Ethical approval of studies and informed consent
All experimental protocols were approved by the institutional ethical committee at ChonnamNational University Hwasun Hospital (CNUHH2014-146) and methods were carried out in accordance with the approved guidelines. Human peripheral blood and tumor masses were donated from healthy donors or lung cancer patients after obtaining written informed consent from all subjects. Animal experiments were performed with ethical approval from the Chonnam National University Animal Research Committee (HCRL15001-2).
Isolation of human CD8+ T cell subsets and tumor-infiltrating lymphocytes (TILs)
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors by density gradient centrifugation with Lymphoprep (AXIS-SHIED Rodelokka, Oslo, Norway). CD3+ and CD8+ lymphocytes were isolated by a magnetic-activated cell sorter (MACS) using CD3 Microbeads and a CD8 T cell isolation kit (MiltenyiBiotec), respectively. The cells were then labeled with anti-CD8, anti-CCR7, and anti-CD45RA (Ebiosciences) and sorted into naïve and memory CD8+ T cell subsets on a BD fluorescence activated cell sorting (FACS) Aria sorter (BD Biosciences). For extraction of TILs, tissue from lung tumor samples were minced and digested with collagenase (2.5 mg/ml collagenase I) at 37 °C for one hour. Cell suspension was then twice filtered through 100-μm and 40-μm cell strainers (BD Biosciences) to obtain single cells. TILs were isolated from single cells utilizing density gradient centrifugation with Lymphoprep and further purified using CD8 MicroBeads (MiltenyiBiotec).
Generation of human CD8+ effector subgroups and proliferation assay
Isolated naïve, memory, and TIL subpopulations were mixed in a 1:1 ratio with Human T-Activator CD3/CD28 Dynabeads (Gibco, Life Technologies) and cultured on CD2-coated plates (CD2-Biotin functional grade, MiltenyiBiotec). Carboxyl fluorescein succinimidyl ester (CFSE) studies were performed by labeling cells with 2.5 μM CFSE (Life Technologies) before stimulation. Three and 5 days post-stimulation, CFSE division of cells was determined by flow cytometry and analyzed using FlowJo Version 10.0.7 software. After 5 days of stimulation, activation of effector cells was determined by FACS analysis based on CD62L, CD25, CD44, and OX40 expression.
Telomere lengths
Relative telomere lengths (RTL) of effector cells were determined by comparing test cells to a control cell line (human T-cell lymphoblast-like cell line CCRF-CEM, Sigma-Aldrich) using a DAKO Telomere PNA Kit (DAKO). Test cells (NTeff, MTeff, and TILeff) and control cells (cell line 1301) were washed in PBS and mixed in a 1:1 ratio. A total of 5 × 105 cells were resuspended in 300 μl of hybridization solution containing 70% formamide either without a probe (unstained control) or with a fluorescein-conjugated telomere PNA probe. The cells were heated for 10 min at 82 °C for DNA denaturation and hybridization was performed overnight at room temperature. Subsequently, cells were washed and resuspended in 0.5 ml of DAKO DNA staining solution and incubated at 2–8 °C for 2 hours. Samples were then analyzed by flow cytometry using logarithmic scale FL1-H for probe fluorescence and linear scale FL3-H for DNA staining. RTLs of the sample cells were calculated per manufacturer’s instructions and reported as mean ± standard deviation (SD) for triplicate samples.
Surface and intracellular staining using FACS
For surface staining, cells were labeled with mAbs against various targets including CD8, CD62L, CCR7, CD45RA, CD45RO, CD25, CD44, OX40, PD-1, CTLA-4, and KLRG-1 (Ebiosciences), incubated at 4 °C for 15 minutes, then washed twice with PBS 1% FBS (FACS buffer), and finally fixed in PBS containing 1% paraformaldehyde (Fix buffer). For T-bet (4B10) and Eomes (Dan11mag), and Foxp1 intracellular staining, cells were first labeled with surface markers CD8, CD62L, OX40 (Ebiosciences) for T-bet and Eomes, or CD25 (BD Biosciences), CD8, CD27, and CD44 (Ebiosciences) for Foxp1 detection, respectively, and then fixed and permeabilized with Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent (Ebiosciences) in a 96-round-well plate. Intracellular labeling for T-bet and Eomes (Ebiosciences), and Foxp1 (LifeSpan Technologies) was performed according to manufacturer’s instruction. For evaluation of Perforin, Granzyme B, and IFN-ɤ cytokine secretion, effector cells were incubated with brefeldin A for 4 hours at 37 °C to disrupt Golgi-mediated transport and accumulate cytokines. Cells were then surface stained, permeabilized with BD Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit (BD Biosciences), and intracellularly stained as recommended. Flow cytometry data was analyzed using FlowJo Version 10.0.7 software.
Apoptosis under TGF-β conditioning
For evaluation of apoptosis, individual effector CD8+ T cell populations were exposed under TGF-β (10 ng/mL) 48 hrs and stained with Annexin V FITC (FITC Annexin V Apoptosis Detection Kit I). Briefly, 1 × 106 cells were washed twice with cold PBS and stained with 5 μL of Annexin-V-FITC in binding buffer for 15 minutes at room temperature. Apoptotic cells were evaluated by flow cytometry according to the manufacturer’s protocol (BD Pharmigen).
Western blot protocol
For Western analysis, whole-cell protein lysates were obtained from effector CD8+ T cells at 4 time points (0, 3, 5, and 8 days post-stimulation) with lysis buffer Proprep (INTRON Biotechnology) by suspending 106 cells in 10 μl buffer and incubating on ice for 30 minutes in the presence of Halt Protease & Phosphatase inhibitors cocktail (Thermo Scientific). Cell lysates were resolved by 10% Bis-Tris SDS PAGE Gel, transferred onto PVDF membrane (Merck), blocked with 5% skim milk in TBST buffer containing 0.1% Tween-20, and probed with the following mAbs: T-bet (4B10, Santa Cruz), Eomes (Y-20, Santa Cruz), Foxp1 (polyclonal, Abcam), and β-actin (C4, Santa Cruz). Quantification of detected protein was performed with an Intelligent Dark Box unit (LAS-3000; Fujifilm) and normalized for loading with the amount of β-actin (1:5000) detected in each lane.
Generation of human tumor-specific CTLs
Mature dendritic cells (mDCs) were pulsed with apoptotic body (irradiated U266 cell line) at a 2:1 ratio (2 × 105 DC: 105 U266) from day 6 to day 8 and then presented to CD8+ T cells at a 1:10 ratio (2 × 105 DC: 2 × 106 CD8+ T cell). Naïve and memory T cells were isolated from the same donor and cultured in different conditions. To visualize cytokine-activated proliferation and determine the optimal co-culturing start time with antigen-loaded mDCs post high-dose IL-2 activation, activated naïve CD8+ T cells were labeled with CFSE and analyzed at various time points (Supplementary Fig. S1). Initiation of peak expansion at 7 days post high-dose IL-2 lead to the decision to start co-culture of cells with mDCs after 7 days of high-dose IL-2 pretreatment. Naïve cells were activated with high-dose IL-2 (1 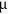 g/mL) in two conditions; 14 day exposure to high-dose IL-2 (Naïve D14; 7 days pretreatment and during 7 days of mDC co-culture) and 7 day exposure to high-dose IL-2 (Naïve D7; 7 days pretreatment only followed by 7 days of mDC co-culture without high-dose IL-2). For control, we cultured naïve and memory cells under equal conditions; 7 day mDC co-culture with IL-2 (10 ng/mL) and IL-7 (10 ng/mL), labeled Naïve D0 and Memory, respectively. After mDC co-culture, 4 groups of CD8+ CTLs (Naïve D14, Naïve D7, Naïve D0, and Memory) and CD3+ CTLs (cultured as previously described) were harvested for cytotoxic analysis.
g/mL) in two conditions; 14 day exposure to high-dose IL-2 (Naïve D14; 7 days pretreatment and during 7 days of mDC co-culture) and 7 day exposure to high-dose IL-2 (Naïve D7; 7 days pretreatment only followed by 7 days of mDC co-culture without high-dose IL-2). For control, we cultured naïve and memory cells under equal conditions; 7 day mDC co-culture with IL-2 (10 ng/mL) and IL-7 (10 ng/mL), labeled Naïve D0 and Memory, respectively. After mDC co-culture, 4 groups of CD8+ CTLs (Naïve D14, Naïve D7, Naïve D0, and Memory) and CD3+ CTLs (cultured as previously described) were harvested for cytotoxic analysis.
Cytotoxic functional evaluation of human tumor-specific CTLs
The U266 cell line was chosen as target cells for our cytotoxic assays based on its expression of E-Cadherin and PD-L1 (Supplementary Fig. S2), which are ligands of KLRG-1 and PD-1, respectively. ELISPOT assay (BD Biosience) was performed to quantify antigen-specific IFN-ɤ releasing effector T cells. 2 × 105 of the effector T cells were co-cultured with 2 × 104 U266 target cells in a 96-well nitrocellulose flat-bottom plate for 24 hours at 37 °C. IFN-ɤ ELISPOT was measured in the presence or absence of MHC class I specific monoclonal antibody (W6/32) with CTLs alone as the control, and spots were counted with ImmunoSpot Reader (Cellular Technology Ltd, Ohio). Data is presented as the mean number of spots ± SD of IFN- ɤ secreting cells per well of triplicate samples.
Evaluation of CD8+ and CD3+ CTL functional activity was performed using the Cytotoxicity Detection Kit LDH (Roche Applied Science, Basel, Switzerland) according to the manufacturer’s protocol36,37. Cytotoxicity of CTLs was calculated according to the following formula: % cell lysis = (experimental – effector spontaneous – low control) x 100/(high control – low control), where “experimental” corresponds to the experimental signal value, “effector spontaneous” to the spontaneous background signal value of the effector cells alone, “low control” to the spontaneous background signal value of target tumor cells alone, and “high control” to the maximum signal value of target cells in medium containing 1% Trixton X-100. Cytotoxicity assays were performed for varying effector cell (E) to target cell (T) ratios, and specific cytotoxicity lysis percentages are reported as the mean ± SD of triplicate samples.
T cell purification, in vitro stimulation, and in vivo CTL assay of the murine model
C57BL/6 (B6) mice were purchased from Orient Bio (Iksan, South Korea) and OT-1.Thy1.1 TCR transgenic mice on a B6 background were obtained from the Institute for Basic Science (IBS; Pohang, South Korea). All mice were used in experiments at 6 to 12 weeks of age according to protocol. Pooled lymph nodes from OT-1 transgenic mice were stained with fluorochrome-conjugated antibodies to CD4, CD8, CD25, CD44 and CD62L and sorted to obtain CD44−CD62L+ naïve or CD44+ memory-phenotype (MP) CD8+ T cells using FACS. CD8+ TILs were obtained from B6 mice that were inoculated subcutaneously with OVAp-expressing EL4-EG7 tumor cell lines. For in vitro stimulation, sorted naïve, MP, and TIL CD8+ T cells were cultured on plate-bound anti-CD3 and anti-CD28 with IL-2 (10 ng/mL) and IL-15 (10 ng/mL). Effector cell surface staining procedure for PD-1, KLRG-1, and LAG3 labeling was identical to human cell methods previously described. In vivo CTL functional assay was performed using OVAp-expressing EL4-EG7 tumor cell lines as target cells. EL4-EG7 tumor cells (0.8 × 105 cells/200 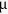 L) were inoculated subcutaneously in recipient B6 mice and 8 hours later, NTeff, MTeff, and TILeff were injected intravenously in a 2:1 ratio of effector to target cells. A tumor mass growth of greater than 1 cm3 was considered as positive tumor formation.
L) were inoculated subcutaneously in recipient B6 mice and 8 hours later, NTeff, MTeff, and TILeff were injected intravenously in a 2:1 ratio of effector to target cells. A tumor mass growth of greater than 1 cm3 was considered as positive tumor formation.
Statistical analysis
The two-tailed student’s t test was used to determine differences in the mean. P values of less than 0.05 were considered statistically significant.
Additional Information
How to cite this article: Nguyen, H. H. et al. Naïve CD8+ T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-β immunosuppression in the tumor microenvironment. Sci. Rep. 6, 28208; doi: 10.1038/srep28208 (2016).
Supplementary Material
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A1061513), by Chonnam National University Hwasun Hospital Institute for Biomedical Sciences (HCRI 14030-21), and by Chonnam National University Research Institute of Medical Sciences (2013-CURIMS-DR007).
Footnotes
Author Contributions D.-H.Y. and J.-H.C. designed the study; H.H.N., S.P. and H.H.C. performed the experiments; D.-H.Y., H.H.N. and T.K. analyzed the data; H.H.N. and T.K. prepared the manuscript; S.Y.S., J.-S.H., J.-S.A., H.-J.K., H.-O.K. and J.-J.L. critically reviewed the manuscript.
References
- Perret R. & Ronchese F. Memory T cells in cancer immunotherapy: which CD8+ T-cell population provides the best protection against tumours? Tissue Antigens 72, 187–194 (2008). [DOI] [PubMed] [Google Scholar]
- Wherry E. J. T cell exhaustion. Nature Immunology 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- Baistch L. et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation antigen-specificity and anatomical localization. Plos One 7, 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel C. et al. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current Opinion in Immunology 25, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caja F. & Vannucci L. TGFβ: A player on multiple fronts in the tumor microenvironment. J Immunotoxicol. 12, 300–7 (2015). [DOI] [PubMed] [Google Scholar]
- Flavell R. A., Sanjabi S., Wrzesinski S. H. & Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 10, 554–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen T. L. et al. Transforming growth factor β-mediated suppression of antitumor T cells requires foxp1 transcription factor expression. Immunity 41, 427–39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S. A. et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 310, 1676–1680 (1988). [DOI] [PubMed] [Google Scholar]
- Berger C. et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 118, 294–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff C. A. et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci. USA 102, 9571–9576 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian S. H. et al. Human effector CD8+ T cells derived from naïve rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 20, 808–814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch L., Fuertes-Marraco S. A., Legat A., Meyer C. & Speiser D. E. The three main stumbling blocks for anticancer T cells. Trends Immunol. 33, 364–72 (2012). [DOI] [PubMed] [Google Scholar]
- Cho J. H. et al. Unique features of naïve CD8+ T cell activation by IL-2. J. Immunology 191, 5559–5573 (2013). [DOI] [PubMed] [Google Scholar]
- Voisinne G. et al. T cells integrate local and global cues to discriminate between structurally similar antigens. Cell Rep. 26, 1208–19 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. et al. Endogenous antigen presentation impacts on T-box transcription factor expression and functional maturation of CD8+ T cells. Blood 120, 3237–3245 (2012). [DOI] [PubMed] [Google Scholar]
- Thaventhiran J. E., Fearon D. T. & Gattinoni L. Transcriptional regulation of effector and memory CD8+ T cell fates. Current Opinion in Immunology 24, 321–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A. et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 185, 4988–4992 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo N. P., Dudley M. E. & Rosenberg S. A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 12, 269–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch D. H., Fräßle S. P., Sommermeyer D., Buchholz V. R. & Riddell S. R. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 16, S1044–5323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E. J. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- Hinrichs C. S. et al. Adoptively transferred effector cells derived from naïve rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci. USA 106, 17469–17474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D. M. The blockade of immune checkpoints in cancer immunotherapy. Nature Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. S. & Ahmed R. Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol. 22, 223–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makedonas G. et al. Perforin and IL-2 upregulation define qualitative differences among highly functional virus-specific human CD8 T cells. PLoS Pathog. 6, e1000798 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertoghs K. M. et al. Molecular profiling of cytomegalovirus-induced human CD8+ T-cell differentiation. J Clin Invest. 120, 4077–4090 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersperger A. R. et al. Increased HIV-specific CD8 T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood 117, 3799–3808 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D. G., Bieganowska Bourcier K., Freeman G. J. &Hafler D. A. Examination of CD8+ T cell function in humans using MHC class I tetramers: similar cytotoxicity butvariable proliferation and cytokine production among different clonal CD8+ T cells specific to a single viral epitope. J. Immunol. 165, 6214–6220 (2000). [DOI] [PubMed] [Google Scholar]
- Intlekofer A. M. et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244 (2005). [DOI] [PubMed] [Google Scholar]
- Connolly E. C., Freimuth J. & Akhurst R. J. Complexities of TGF-β targeted cancer therapy. Int J Bio Sci. 8, 964–978(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L. & Kroemer G. Targeting foxp1 for reinstating anticancer immunosurveillance. Immunity previews, Immunity 41, 245–247 (2014). [DOI] [PubMed] [Google Scholar]
- Boyman O., Cho J. H. & Sprent J. The role of interleukin-2 in memory CD8 cell differentiation. Adv. Exp. Med. Biol. 684, 28–41 (2010). [DOI] [PubMed] [Google Scholar]
- Cho J. H. et al. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J. Exp. Med. 204, 1787–1801 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule M. A. et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14, 1264–1270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modifid T cells in lymphoma patients. J. Clin. Invest. 121, 1822–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M. et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri S., Mariani E., Meneghetti A., Cattini L.&Facchini A. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants.Clinical and Diagnostic Laboratory Immunology 8, 1131–1135 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. et al. A novel immunogenic CS1-specific peptide inducing antigen-specific cytotoxic T lymphocytes targeting multiple myeloma. British Journal of Haematology 157, 687–701 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.