

Abstract
By combining ancestral sequence reconstruction and in vitro evolution, Smock et al. identified single motifs that assemble into a functional five-bladed β-propeller, and a likely route for conversion into the more complex, extant single chain fusion. Interestingly, although sequence diversification destabilized five-motif fusions, it also destabilized aggregation-prone intermediates, increasing the level of functional protein in vivo.
Keywords: protein fold, sequence evolution, beta-propeller, repeat protein structure
The protein universe is filled with structures comprising small repeating structural motifs, including the TIM barrel, β-propeller, parallel β-helix and ankyrin repeat [1]. The individual motifs that comprise a repeat fold are not stable in isolation; rather, interactions between motifs stabilize the native structure [2], raising questions as to how these folds arose during evolution. The consensus hypothesis--that a short, functional gene segment was duplicated and (in some cases) rearranged to form the extant folds we know today--has proven surprisingly difficult to recapitulate in the laboratory. A new study by the Tawfik group [3] exploits an interdisciplinary combination of ancestral inference, in vitro evolution, biophysics and bioinformatics to illuminate one plausible evolutionary path to a five-bladed β-propeller fold.
Of all repeat protein folds, β-propellers are particularly remarkable for their structural plasticity. Each propeller blade consists of a four-β-strand motif totaling ~50 aa, but this motif can be used to build a propeller that has 4, 5, 6, 7 or even 8 blades (Figure 1). Moreover, the blade motif is remarkably tolerant to circular permutation, strand-swapping between blades [4] and large insertions (including entire additional propellers [5]) between its strands, which enables β-propeller proteins to fulfill an unusually broad range of binding and catalytic functions but raises additional questions as to how these folds evolved.
Figure 1.
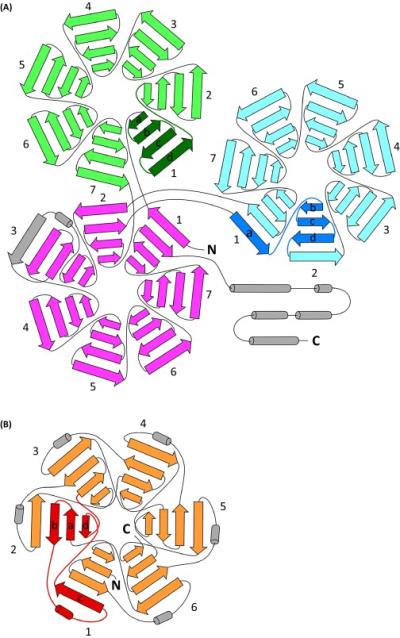
Examples of structural diversity within extant β-propeller structures. Each propeller blade consists of four β-strands that can be connected in diverse ways. (A) DDB1, a subunit of a ubiquitin ligase, is composed of three seven-bladed propellers with different β-strand topologies [5], including the simplest ‘intact’ blade topology (green) and a ‘Velcro’ frame (blue). Both the green and blue propellers are inserted between β-strands of the first two blades of the remaining propeller (magenta), which follows the Velcro frame overall but includes additional topological complexity in the third blade. (B) The six-bladed propeller of the DNA gyrase A C-terminal domain [4] (orange). For propellers with consistent topological repeats, a single repeat is shown in a darker color to highlight the topology, with β-strands labeled a-b-c-d from N- to C-terminus.
As the basis for their studies, Smock et al. used tachylectin-2, a five-bladed propeller from horseshoe crab that binds N-acetylglucosamine (GlcNAc) on cell surface glycoproteins. A key objective was the identification of a single-motif peptide capable of assembling into a functional pentameric propeller. The dual requirements for (i) assembly from identical monomers and (ii) function were a formidable challenge. While others had designed functional repeat proteins from larger (2+ motifs) components [6] or achieved assembly from identical monomer peptides without preserving function [7], the combination remained elusive. Tawfik's previous attempt, which focused on tachylectin-2's fourth repeat, failed to assemble from individual peptide motifs [8].
Using tachylectin-2's repeats as a starting point, along with a distantly related protein from a sea anemone, the authors constructed a library of 6000 likely ancestral motif sequences, expressed these in Escherichia coli, and screened E. coli cell lysates for GlcNAc binding to identify functional members of the library. It is noteworthy that the readout from this assay (total binding capacity) concatenates many individual parameters that could confer a selective advantage, including gene expression level, foldability (native state stability and the ability to avoid competing aggregation and degradation pathways) and binding affinity. This screen identified a clone that assembled into a stable pentameric propeller fold and exhibited low but measurable levels of GlcNAc binding activity. Interestingly, the functional single motif required another significant departure from wild type tachylectin-2 and many other extant propellers: an ‘intact’ propeller blade frame, rather than N-terminal strand-swapping (‘Velcro’ frame) (Figure 1). Moreover, although tandem fusions of the best library candidates greatly improved GlcNAc binding, converting these identical repeat proteins to the Velcro frame yielded only modest changes. Together, these results support a model where duplication of short individual repeats to form a five-repeat monomer occurred first, prior to circular permutation.
Motif sequences in wild type repeat proteins, including β-propellers, can deviate significantly from the consensus sequence. Past work has shown that deviations from the consensus sequence can help avoid incorrect, off-pathway inter-motif interactions during folding, improving folding yield [9]. To test the effects of sequence diversification in their designed five-bladed β-propeller, Tawfik and co-workers introduced low levels of motif diversity into a tandem fusion of identical repeats, and found that reducing internal sequence identity by just 2% significantly improved total binding capacity. Did motif diversification arise before or after circular permutation to the Velcro topology? While the more diverse 5-motif intact blade proteins had higher binding capacity than those with identical repeats, circular permutation significantly improved binding capacity only for the much more diverse wild type tachylectin-2 sequence, suggesting circular permutation may have appeared after diversification.
What specific contributions did diversification and motif strand topology make to total binding capacity? The authors found that total binding capacity correlated most closely with parameters related to foldability (soluble expression and in vitro refolding efficiency). They also identified an unexpected correlation between native stability and insoluble expression. A closer examination of the unfolding mechanism suggested a cause: the most stable proteins (the identical tandem repeats) populated a stable intermediate state upon unfolding in chemical denaturant. Although sequence diversification destabilized the native structure, it also destabilized this intermediate, perhaps reducing aggregation and thereby increasing folding efficiency.
These results highlight many remaining important questions regarding the evolution of tachylectin-2, β-propellers, and complex proteins in general. What mechanism(s) enable circular permutation from the intact blade to the Velcro frame? Tawfik and coworkers show one plausible intermediate, a 6-motif fusion, is also functional. However, given the ability of the propeller fold to accommodate a wide number of blades, is this extra motif added to the functional structure, or left hanging in the breeze? More broadly, what sequence tweaks lead to a propeller adopting a different number of blades, versus strand-swapping and circular permutation? Past studies suggest these effects are difficult to predict de novo [6]. In tachylectin-2, the GlcNAc binding sites lie between the blades of the propeller; what effect does this functional requirement have on the selective pressure to maintain a 5-blade topology? Can insights be gleaned from ancestral reconstructions of other diverse, ring-shaped protein complexes [10]?
The Velcro blade frame of wild type tachylectin-2 is one of many strand-swaps in extant propeller structures. How and why does evolution land on a particular strand-swapped topology? For example, although the β-strand topology for most propellers starts at the center and progresses to the outer periphery, in the C-terminal domain of DNA gyrase A, a 6-bladed propeller, the repeat starts at the second strand and ends with the innermost, and the outermost β-strand of each motif is swapped with the neighboring blade [4] (Figure 1). Intriguingly, increasing the number of long-range contacts in another multimeric repeat fold increased its kinetic stability (native state lifetime) [11]; does this effect drive selection for strand-swapped propellers? For polypeptides that include more than one propeller domain, what selection forces lead to simple linear arrays [12] versus much more complex intra-propeller fusions [5] (Figure 1)? Finally, although the authors confine the implications of their results to symmetrical folds (i.e., ‘closed’ folds with the N- and C-terminal motifs in contact), similar conclusions may apply to the evolution of elongated repeat folds, including Leucine-rich, TPR and ankyrin repeats, and parallel β-helices.
Most broadly (and applicable to any laboratory evolution study), how do the selective pressures that we apply in the laboratory (expression in a heterologous host, binding tested by ELISA, etc.) compare to the selective pressures experienced by a protein during its evolution in the primordial organism? We cannot replay history and therefore may never know the answer, or the precise evolutionary route taken. Nevertheless, recapitulating potential trajectories for the evolution of a protein in the laboratory gives us new tools to understand the physical and genetic forces that lead to function, and enabled these folds to emerge.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braselmann E, et al. Folding the proteome. Trends Biochem. Sci. 2013;38:336–343. doi: 10.1016/j.tibs.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Street TO, et al. Predicting coupling limits from an experimentally determined energy landscape. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4907–4912. doi: 10.1073/pnas.0608756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smock RG, et al. De novo evolutionary emergence of a symmetrical protein is shaped by folding constraints. Cell. 2016;164:476–486. doi: 10.1016/j.cell.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corbett KD, et al. The C-terminal domain of DNA gyrase A adopts a DNA-bending β-pinwheel fold. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7293–7298. doi: 10.1073/pnas.0401595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li T, et al. Structure of DDB1 in complex with a paramyxovirus V protein: Viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–117. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 6.Voet AR, et al. Computational design of a self-assembling symmetrical β-propeller protein. Proc. Natl. Acad. Sci. U.S.A. 2014;111:15102–15107. doi: 10.1073/pnas.1412768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Blaber M. Experimental support for the evolution of symmetric protein architecture from a simple peptide motif. Proc. Natl. Acad. Sci. U.S.A. 2011;108:126–130. doi: 10.1073/pnas.1015032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadid I, Tawfik DS. Funtional β-propeller lectins by tandem duplications of repetitive units. Protein. Eng. Des. Sel. 2011;24:185–195. doi: 10.1093/protein/gzq053. [DOI] [PubMed] [Google Scholar]
- 9.Borgia MB, et al. Single-molecule fluorescence reveals sequence-specific misfolding in multi-domain proteins. Nature. 2011;474:662–665. doi: 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finnigan GC, et al. Evolution of increased complexity in a molecular machine. Nature. 2012;481:360–364. doi: 10.1038/nature10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broom A, et al. Designed protein reveals structural determinants of extreme kinetic stability. Proc. Natl. Acad. Sci. U.S.A. 2015;112:14605–14610. doi: 10.1073/pnas.1510748112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn VE, et al. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell. 2011;21:862–873. doi: 10.1016/j.devcel.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]