

Abstract
Advanced stages of neuroblastoma, the most common extracranial malignant solid tumor of the central nervous system in infants and children, are refractive to therapy. Ectopic expression of melanoma differentiation associated gene-7/Interleukin-24 (mda-7/IL-24) promotes broad-spectrum antitumor activity in vitro, in vivo in pre-clinical animal models and in a Phase I clinical trial in patients with advanced cancers, without harming normal cells. mda-7/IL-24 exerts cancer-specific toxicity (apoptosis or toxic autophagy) by promoting ER stress and modulating multiple signal transduction pathways regulating cancer cell growth, invasion, metastasis, survival and angiogenesis. To enhance cancer-selective expression and targeted anti-cancer activity of mda-7/IL-24 we created a tropism-modified Cancer Terminator Virus (Ad.5/3-CTV), which selectively replicates in cancer cells producing robust expression of mda-7/IL-24. We now show that Ad.5/3-CTV induces profound neuroblastoma anti-proliferative activity and apoptosis in a caspase 3/9-independent manner both in vitro and in vivo in a tumor xenograft model. Ad.5/3-CTV promotes these effects through a unique pathway involving apoptosis inducing factor (AIF) translocation into the nucleus. Inhibiting AIF rescued neuroblastoma cells from Ad.5/3-CTV-induced cell death, whereas pan-caspase inhibition failed to promote survival. Ad.5/3-CTV infection of neuroblastoma cells increased ATM phosphorylation instigating nuclear translocation and increased γ–H2AX, triggering nuclear translocation and intensified expression of AIF. These results were validated further using two ATM small molecule inhibitors that attenuated PARP cleavage by inhibiting γ–H2AX, which in turn inhibited AIF changes in Ad.5/3-CTV-infected neuroblastoma cells. Taken together, we elucidate a novel pathway for mda-7/IL-24-induced caspase-independent apoptosis in neuroblastoma cells mediated through modulation of AIF, ATM and γ–H2AX.
Keywords: Neuroblastoma, MDA-7/IL-24, CTV, AIF, ATM
Introduction
Neuroblastoma is the most frequent extracranial solid tumor in children under five years of age, affecting 1 in 7000 children. It is speculated that these tumors develop as a result of rapid proliferation of neuroblasts during fetal growth (1). Neuroblastoma arises along the sympathetic nervous system and adrenal medulla (2) from neural crest cells of sympathetic origin. They represent heterogeneous masses, both clinically and biologically. When occurring in infants, neuroblastoma may relapse unpredictably in a majority of cases, while in older patients these tumors frequently persist as benign ganglioneuromas (2). At the time of diagnosis, approximately half of the patients are assumed to be high-risk due to distant metastasis. Neuroblastoma can be classified into four stages. In stage I and II, disease is confined to the primary lesion. Stage III and IV are characterized by disease outside the primary lesion. Once neuroblastoma attains advanced stage (III or IV), it expands persistently even when subjected to rigorous multimodal therapies (1, 3). Presentation of tumors at advanced stages combined with the absence of surgical options culminates in very poor patient prognosis. Although some improvements in the overall cure rate of neuroblastomas have been realized using intensive multimodality therapies these therapies promote significant short- and long-term toxicities (1, 3, 4). Only about 2% of neuroblastoma patients with stage III or IV remain disease free with relapse occurring shortly after completing chemotherapy, indicating a negligible effect of these agents long-term (5). Therapy failures are believed to be a consequence of development of resistance, underscoring the need for less toxic and more efficient therapeutic strategies (6). Accordingly, a comprehensive knowledge of mechanisms governing proliferation, differentiation, and cell death may expand our understanding of the molecular pathogenesis of neuroblastoma, which may result in novel biologically-based therapies diminishing toxicity and maximizing efficacy.
mda-7/IL-24 is a unique member of the IL-10-related cytokine gene family (7) displaying wide spectrum anti-tumor activity in diverse cancers without harming normal cells or tissues (8, 9). mda-7/IL-24 was initially cloned using subtraction hybridization combined with induction of cancer cell terminal differentiation (10). Forced expression of mda-7/IL-24 in cancer cells promotes direct cancer toxicity through induction of apoptosis or toxic autophagy (11) and indirect antitumor effects through inhibition of angiogenesis (8, 12), promoting antitumor immune responses (8), sensitization of cancer cells to radiation- and chemotherapy-induced killing (13), and by promoting potent ‘antitumor bystander activity’ through autocrine/paracrine secretion (14). mda-7/IL-24 displays nearly universal antitumor properties in vitro and in vivo in almost every cancer context (15, 16), which led to successful entry into clinical trials (17, 18). These properties of mda-7/IL-24 make it a potential candidate gene for the treatment of neuroblastoma, where adenovirus administration in a single human neuroblastoma cell line (SH-SY5Y) inhibited both in vitro and in vivo xenograft growth (19). To enhance the utility of mda-7/IL-24 for gene therapy of cancer, we are employing a conditionally replication-competent Ad carrying mda-7/IL-24 (20). In this Cancer Terminator Virus (CTV) (21) adenoviral replication is controlled by the promoter of a cancer-selective rodent gene, progression elevated gene-3 (PEG-3) (22). To enhance even further the utility of the CTV we have engineered the adenovirus to more effectively infect cancer cells, creating tropism modified chimeric CTVs (23).
Adenoviruses (Ads) use CAR (Coxsackie-Adenovirus Receptors) to infect normal and cancer cells, however cancer cells express varying levels of CAR on their cell surface. To improve the low efficiency of Ad infection of tumor cells, “tropism modification” approaches have been developed (23). One such vector Ad.5/3 displayed equal efficacy when compared with wild type Ad.5, thereby providing an expanded range of utility for Ad.5/3, in both low and high CAR expressing cells (23, 24). For that reason, we used a modified Ad.5/3-CTV (Ad.5/3-PEG-E1A-mda-7) to evaluate therapeutic applications in human neuroblastoma cells.
We presently describe a previously unrecognized pathway involved in mda-7/IL-24-mediated induction of caspase-independent apoptosis induction in neuroblastoma cells. This pathway involves modulation of AIF expression and translocation into the nucleus of neuroblastoma cells that is mediated through induction of ATM followed by phosphorylation and nuclear translocation of γ–H2AX into the nucleus. These findings provide new insights into the mechanism of action of a near ubiquitous cancer-suppressing gene supporting its’ applications for potential therapy of neuroblastoma.
Materials and methods
Cells and reagents
Human neuroblastoma cancer cell lines SK-N-AS and SK-N-SH were obtained from ATCC (Manassas, VA) and NB1691 cells were obtained from Dr. Alan Houghton of St. Jude Children’s Research Hospital (Memphis, TN). NB1691 cell line was authenticated using the “CellCheck” service provided by the Research Animal Diagnostic Laboratory and compared with initial STR profile generated by the collaborator (IDEXX BioResearch). The cumulative culture length of the cells (SK-N-SH and SK-N-AS) was less than 6 months after recovery. Early passage cells were used for all experiments. All the cell lines were frequently tested for mycoplasma contamination using a mycoplasma detection kit from Sigma. SK-N-AS cells were cultured in DMEM with non-essential amino acids, SK-N-SH cells were cultured with RPMI 1640 and NB1691 cells were cultured with DMEM (Invitrogen, Carlsbad, CA) supplemented with 10 % fetal bovine serum (FBS), 50-units/mL penicillin, and 50 μg/mL streptomycin (Life Technologies Inc., Frederick, MD). Cells were incubated in a humidified 5 % CO2 atmosphere at 37 °C. The antibodies specific for AIF (#4642), ATM (#2873), pATM (ser 1981, #13050), BCL-2 (#2876), BAX (2772), PARP (#9542), Caspase 3 (#9662), Caspase 8 (#9746), H2AX (#7631) and γ-H2AX (ser 139, #9718) (Cell signaling Technology, Boston, MA), MDA-7/IL-24 (#K101, GenHunter, Nashville, TN), HRP-conjugated secondary antibodies (Dako, Carpinteria, CA), β-Actin (#NB600-501, Novus Biologicals, Inc., Littleton, CO), were used in this study. The other materials used in this study were Transcriptor First Strand cDNA Synthesis Kit, In Situ Cell Death Detection Kit, Fluorescein (#11684795910, Roche Applied Science, Indianapolis, IN), MTT cell growth assay kit (#CT02, Millipore Corporation, Billerica, MA). KU60019 (#SML1416) and KU-55933 (#SML-1109) (Sigma, St Louis, MO).
Transfection with plasmids
All transfection experiments were performed with FuGene HD transfection reagent according to the manufacturer’s protocol (Roche, Indianapolis, IN). Briefly, plasmid/siRNA was mixed with FuGene HD reagent (1:3 ratio) in 500 μL of serum free medium and left for 30 min to allow for complex formation. The complex was then added to the 100-mm plates, which had 2.5 mL of serum-free medium (2 μg plasmid per mL of medium). After 6 hours of transfection, complete medium was added, and cells were cultured for another 24 h (1).
Cell proliferation assay (MTT assay)
Cell growth rate was determined using a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as a measurement of mitochondrial metabolic activity as described earlier (8). Cells were treated with mock, Ad.5/3-Null, Ad.5/3-E1A or the indicated doses of Ad.5/3-CTV and incubated at 37 °C. After 0–96 h, MTT reagent was added, and cells were incubated for 4 h at 37 °C. After removing the medium, formazan crystals were dissolved in DMSO, absorbance at 550 nm was read using a microplate spectrophotometer and the results were expressed graphically.
Terminal deoxy nucleotidyl transferase-mediated nick labeling (TUNEL) assay
Induction of apoptosis in Neuroblastoma cancer cells as well as in xenograft tumor tissue sections of mice treated with mock, Ad.5/3-Null, Ad.5/3-E1A or Ad.5/3-CTV was evaluated using TUNEL enzyme reagent following the manufacturer’s instructions and as described previously (25). Briefly, 5 × 103 Neuroblastoma cancer cells were cultured and treated with mock, Ad.5/3-Null, Ad.5/3-E1A or Ad.5/3-CTV for 72 hours, fixed in 4 % paraformaldehyde in PBS for 1 hour at room temperature (RT), and permeabilized with 0.1 % Triton-X 100 in 0.1 % sodium citrate in PBS for 2 min (for cells) or 10 min (for tissue sections) on ice. The samples were incubated in TUNEL reaction mixture in a humidified atmosphere at 37 °C for 1 hour in the dark. Images were captured with an Olympus research fluorescence microscope attached to a CCD camera and cells were counted. The positive-staining apoptotic cells were counted from 5 microscopic fields per tumor tissue from 3 animals per treatment.
Western blotting
Western blotting analysis was performed as described previously (25). Briefly, Mock, Ad.5/3-Null, Ad.5/3-E1A or Ad.5/3-CTV-treated neuroblastoma cancer cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 1 mM sodium orthovanadate, 0.5 mM PMSF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin. Equal amounts of total protein fractions of lysates were resolved by SDS-PAGE and transferred to PVDF membranes. The blot was blocked with 5% non-fat dry milk/5% BSA and probed overnight with primary antibodies followed by HRP-conjugated secondary antibodies. An ECL system was used to detect chemiluminescent signals. All blots were re-probed with β-Actin antibody to confirm equal loading.
In vivo studies
To directly evaluate the effect of Mock, Ad.5/3-Null, Ad.5/3-E1A or Ad.5/3-CTV on tumor growth in vivo, we subcutaneously implanted 5 X 106 NB1691 cells on both flanks of 4- to 6-week-old athymic nude mice. The tumors on the left flank were challenged with 8 intra-tumoral injections of Mock, Ad.5/3-Null, Ad.5/3-E1A or Ad.5/3-CTV for three weeks (3 injections for 2 weeks and 2 injections in the last week) after 7 days post tumor cell implantation when the tumors reached palpable sizes. Tumor growth was monitored in mice by measuring tumor size with calipers on each flank every alternate day until completion of the experiment. Each treatment group had two sets of animals. One set was sacrificed 2 days after the final dose of treatment (1 mouse from each group) and another set (N=5) was followed until the control tumor group reached a point where it needed to be sacrificed according to our IACUC protocol. After completion of the experiment the tumors were fixed and the sections were used for immunohistochemical analysis.
Statistical analysis
All data are presented as mean ± standard deviation (S.D.) of at least three independent experiments, each performed at least in triplicate. One-way analysis of variance (ANOVA) combined with the Tukey post hoc test of means was used for multiple comparisons. Statistical differences are presented at probability levels of p < 0.05, p < 0.01 and p < 0.001.
Results
CTV expressing mda-7/IL-24 inhibits neuroblastoma cell proliferation in vitro
To examine mda-7/IL-24 overexpression on neuroblastoma tumor growth in vitro and in vivo a genetic approach was used with a tropism modified Cancer Terminator Virus (Ad.5/3-CTV) (23, 26), i.e., a conditionally replicating adenovirus that ectopically expresses mda-7/IL-24. Adenovirus entry is cell surface receptor dependent so we determined receptor status of CAR (for Ad.5), desmoglein and CD46 (for Ad.3) of the three neuroblastoma cell lines, SK-N-AS, NB1691 and SK-N-SH, used in this study. CAR expression was variable with similar lower levels in SK-N-AS and NB1691 cells and higher levels in SK-N-SH cells (Supplemental Fig. 1). CD46 was expressed at a comparable level in all three-cell lines and Desmoglein levels followed a similar pattern as CAR, with highest expression in SK-N-SH cells. When transgene expression was monitored (MDA-7/IL-24 and E1A) following Ad.5/3-CTV infection, all three neuroblastoma cell lines expressed the expected transgenes as compared to mock and Ad.5/3-Null infected cells (Fig. 1A). A dose-dependent increase in MDA-7/IL-24 protein levels was seen in Ad.5/3-CTV infected cells compared to controls. To confirm that the increased levels of MDA-7/IL-24 protein represented an up-regulation of mda-7/IL-24 mRNA transcription, we assessed mRNA transcript levels in the Ad.5/3-CTV-treated cells. Ad.5/3-CTV treated neuroblastoma cells showed a strong increase in mda-7/IL-24 mRNA levels when compared to mock, Ad.5/3-null, or Ad.5/3-E1A treated cells (data not shown). Next, we assessed the effects of Ad.5/3-CTV on neuroblastoma cell growth using MTT assays. Although Ad.5/3-E1A inhibited cell proliferation to some extent through its oncolytic activity, forced expression of mda-7/IL-24 using Ad.5/3-CTV resulted in the greatest dose-dependent decrease in cell proliferation compared to other treatments (Fig. 1B).
Figure 1. Ad.5/3-CTV inhibits neuroblastoma cell growth.

(A) Neuroblastoma cells were infected with either Ad.5/3-Null (25 pfu), Ad.5/3-E1A (25 pfu) or Ad.5/3-CTV (12.5 or 25 pfu) for 72 hours and cell lysates were evaluated by western blotting for E1A and MDA-7/IL-24 protein using specific antibodies. (B) Neuroblastoma cells were plated in 96-well plates in quadruplicate and infected with virus as indicated in 1A for the indicated times. Cell growth was measured using MTT assay and shown as relative proliferation rate compared with control cells. *, p<0.01 versus control.
Ad.5/3-CTV induces cell death in neuroblastoma cells
Ectopic expression of mda-7/IL-24 induces apoptosis in a wide-array of cancer types (16). Overexpression of mda-7/IL-24 in the neuroblastoma cell lines increased TUNEL positive cells compared to that of cells treated with mock, Ad.5/3-Null or Ad.5/3-E1A treatment (Fig. 2A). Further confirmation of apoptosis-induction was shown by FACS analysis where the sub-G1 population (DNA content) was enhanced in Ad.5/3-CTV-infected cells. DNA frequency distribution histograms in which the sub-G1 region corresponded to apoptotic cells indicated that Ad.5/3-CTV increased the number of apoptotic cells to 40–50 % in SK-N-AS, 35–50% in SK-N-SH and 40–50% in NB1691 cells, as compared with 5% in Ad.5/3-Null-infected controls or ~20% in Ad.5/3-E1A infected cells (Fig. 2B). PARP cleavage was also evident by Western blotting analysis predominantly in Ad.5/3-CTV-infected neuroblastoma cells (Fig. 2C). We also analyzed the expression of mda-7/IL-24-downstream molecules and signals involved in cell death following infection of neuroblastoma cells with Ad.5/3-CTV. In many cancers, mda-7/IL-24 is known to enhance pro-apoptotic genes and decrease anti-apoptotic genes in the Bcl-2 family of proteins (27). A similar expression profile was found in neuroblastoma cells following Ad.5/3-CTV infection, i.e., enhancement of BAX and P-JNK (pro-apoptotic proteins) and decreased expression of BCL-2 and BCL-xL (anti-apoptotic proteins) (Supplemental Fig. 2). Of note, these changes were only evident following Ad.5/3-CTV infection and not with Ad.5/3-null or Ad.5/3-E1A infection of neuroblastoma cells.
Figure 2. Ad.5/3-CTV induces apoptosis in neuroblastoma cells.
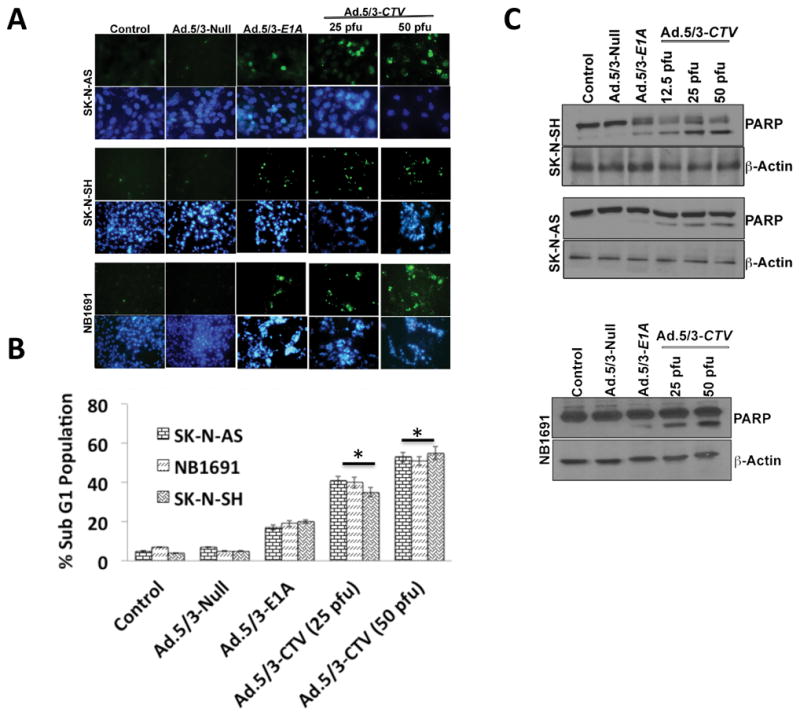
(A) Neuroblastoma cells were cultured in 8-well chamber slide and treated with 25 pfu of Ad.5/3-Null or Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 72 hours. Cells were fixed and TUNEL assays were performed. Data presented as TUNEL positive cells in a defined microscopic field as compared with un-treated control cells. (B) Neuroblastoma cells were infected as described in 2A for 72 hours and were collected and subjected to FACS analysis with propidium iodide staining for DNA content and data presented in a graphical manner from three independent experiments. Columns: mean of triplicate experiments. Bars: S.D., *, p<0.001 versus control. (C) Neuroblastoma cells were treated as described in 2A for 72 hours. Cells were collected and western blot analysis was performed for PARP using specific antibody and β-Actin served as loading control.
mda-7/IL-24 induces caspase-independent apoptosis in vitro in neuroblastoma cells
Although mda-7/IL-24 has well established tumor-suppressor and apoptosis-promoting properties in a broad spectrum of human cancer cells, the molecular mechanism by which mda-7/IL-24 induces apoptosis is quite diverge and involves different pathways depending on the tumor type (27). Caspase-dependent apoptosis is the common modality of programmed cell death (28), so we determined whether mda-7/IL-24-induced cell death in neuroblastoma was caspase dependent. Treatment of neuroblastoma cells with Ad.5/3-CTV did not induce caspase-3 or caspase-9 activation when analyzed by Western blotting or using a luminescence-based assay (Figs. 3A and B). To determine a potential involvement of the extrinsic apoptosis pathway we monitored caspase 8 expression following Ad.5/3-CTV infection and found no activation in these cell lines (Supplemental Fig. 3). In an additional confirmatory study, neuroblastoma cells were treated with z-vad fmk, a pan-caspase inhibitor, prior to Ad.5/3-CTV infection and cultured for an additional 48 hours. The cells were then analyzed via western blotting for PARP cleavage, which demonstrated similar PARP cleavage with or without treatment with the pan-caspase inhibitor prior to Ad.5/3-CTV infection (Fig. 3C). This result further supports the caspase-independent cell death mechanism following ectopic expression of MDA-7/IL-24 in neuroblastoma cells.
Figure 3. Ad.5/3-CTV induces caspase-independent cell death in neuroblastoma cells.

(A) Neuroblastoma cells were infected with 25 pfu of Ad.5/3-Null or Ad.5/3-E1A or with the indicated dose of Ad.5/3-CTV for 72 hours. Cells were collected and western blot analysis was performed for caspase-3 and caspase-9 using specific antibodies and β-Actin served as loading control. Staurosporine served as a positive control for caspase 3 activation. (B) Neuroblastoma cells were treated as described in 3A for 72 hours, collected and caspase-3 activation assays were performed according to the manufacturer’s protocol. Staurosporine served as a positive control. Results represent three independent experiments displayed in a graphical manner. Columns: mean of triplicate experiments; bars, S.D. (C) Neuroblastoma cells were pre-treated with 20 μM Z-VAD-FMK and were infected as described in 3A for 72 hours. Cells were collected and western blotting analysis was performed for PARP using specific antibody and β-Actin served as loading control. Results are representative of three independent experiments.
mda-7/IL-24-mediated apoptosis in neuroblastoma involves AIF activation and nuclear translocation
The tumor suppressor p53 plays an important role in suppressing tumorigenesis by inducing genomic stability, cell cycle arrest or apoptosis (29). Apoptosis inducing factor (AIF) is a mitochondrial protein, which, when translocated to the nucleus, results in apoptosis, mainly in a caspase-independent context through the induction of chromatin condensation and DNA fragmentation (30). p53 is an established regulator of AIF in caspase-independent cell death and AIF can contribute to p53-mediated cell death (31). p53 levels were modestly increased in a dose-dependent manner following Ad.5/3-CTV infection as compared with the mock or Ad.5/3-Null-infected cells (Supplemental Fig. 4). AIF levels were increased in a dose-dependent manner in Ad.5/3-CTV-infected neuroblastoma cells (Fig. 4A) suggesting the potential involvement of AIF in caspase-independent cell death.
Figure 4. Ad.5/3-CTV promotes AIF-mediated cell death in neuroblastoma cells.

(A) Neuroblastoma cells were infected with 25 pfu of Ad.5/3-Null or Ad.5/3-E1A or with the indicated dose of Ad.5/3-CTV for 72 hours. Cells were collected and western blotting analysis was performed for AIF using specific antibodies and β-Actin served as loading control. Results are representative of three independent experiments. (B) Neuroblastoma cells were cultured in 8-well chamber slide and treated as described in 4A for 72 hours. These cells were then subjected to immunofluorescence analysis of AIF using anti-AIF antibody and Alexa Flour-594 secondary antibody (red fluorescence). Nuclei were stained with DAPI (blue fluorescence). Fluorescent cells were visualized and photographed from 10 different fields and representative images are shown. (C) Subcellular distribution of AIF was determined using western blot analysis. Neuroblastoma cells were infected with 25 pfu of Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 72 hours. The cytoplasmic and nuclear fractions were isolated and examined by western blotting for AIF using specific antibodies. HDAC3 served as loading control for nuclear extract and β-Actin served as loading control for cytoplasmic extracts. (D) Neuroblastoma cells were pre-treated with AIF inhibitor and infected with 25 pfu of Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 48 hours. Cells were collected and western blotting analysis was performed for AIF and PARP using specific antibodies and β-Actin served as loading control. Results are representative of three independent experiments.
Previous studies suggest that BCL-2 proteins facilitate the insertion of BAK or BAX into the mitochondrial membrane to form functional oligomers, which result in depolarization of the inner mitochondrial membrane and subsequent AIF nuclear translocation, which promotes caspase-independent apoptosis (28). To determine if mda-7/IL-24 exerts similar effects on AIF nuclear translocation, immunostaining and cell fractionation methods were used. AIF displayed a granular pattern in the mitochondria of untreated controls, whereas after treatment with Ad.5/3-CTV AIF was detected in the nucleus (Fig. 4B). We confirmed this result using cellular fractionation, which clearly showed AIF accumulation in nuclear lysates and concomitant decreased levels in the cytoplasmic fraction following infection with Ad.5/3-CTV (Fig. 4C).
Ad.5/3-CTV-induced, AIF-mediated cell death in neuroblastoma cells was confirmed using an AIF inhibitor, N-Phenylmaleimide. Treatment with the AIF inhibitor at a concentration of 50 μM/L for 1 hour prior to treatment with Ad.5/3-CTV for 48 hours reduced cell death as monitored by FACS analysis (data not shown). This phenomenon was further confirmed by western blotting analysis for PARP cleavage. Treatment with an AIF-inhibitor prior to Ad.5/3-CTV infection resulted in decreased PARP cleavage (Fig. 4D). Collectively, these results indicate that Ad.5/3-CTV induces caspase-independent AIF-mediated apoptosis in neuroblastoma cells.
ATM-γ–H2AX axis mediates AIF-induced cell death in neuroblastoma cells
The mechanism that regulates AIF induction and nuclear translocation leading to caspase-independent apoptotic functions is not well understood. Previous studies suggest that γ-H2AX plays a pivotal role in AIF-mediated necroptosis in MEFs (32). To test this hypothesis, we checked the effects of Ad.5/3-CTV on expression and activation of H2AX in SK-N-AS and NB1691 neuroblastoma cells. Ad.5/3-CTV infection increased the levels of H2AX as well as phosphorylation of H2AX (γ-H2AX) when compared to control, Ad.5/3-Null, or Ad.5/3-E1A infected cells (Fig. 5A and Supplemental Fig. 5). To further decipher the molecular mechanism of H2AX phosphorylation, we evaluated the levels and activation of ATM in Ad.5/3-CTV treated SK-N-AS, SK-N-SH and NB1691 neuroblastoma cells which showed increased ATM levels as well as phosphorylation of ATM when compared to control, Ad.5/3-Null or Ad.5/3-E1A treated cells (Fig. 5B and Supplemental Fig. 6). In total, these results indicate that AIF-mediated caspase-independent apoptosis requires ATM-induced histone H2AX phosphorylation in Ad.5/3-CTV treated neuroblastoma cells.
Figure 5. Ad.5/3-CTV-induced AIF-mediated cell death requires ATM and γ–H2AX phosphorylation.

Neuroblastoma cells were infected with 25 pfu of Ad.5/3-Null or Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 72 hours. (A) Cells were collected and western blotting was performed for γ–H2AX and H2AX using specific antibodies and β-Actin served as loading control. (B) Western blotting was performed for determining pATM and ATM protein levels using specific antibodies and β-Actin served as loading control. (C) Neuroblastoma cells were untreated or treated overnight with KU-60019 (3 μM) and infected with 25 pfu Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 48 hours. Cells were collected and western blotting was performed for MDA-7/IL-24, pATM, γ–H2AX, AIF and PARP using specific antibodies and β-Actin served as loading control. Results are representative of three independent experiments. (D) Neuroblastoma cells were pre-treated with AIF inhibitor and infected with 25 pfu of Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 48 hours. Cells were collected and western blotting analysis was performed for pATM using specific antibodies and β-Actin served as loading control. Results are representative of three independent experiments. (E) Neuroblastoma cells were untreated or treated over night with KU-60019 (3 μM) and infected with 25 pfu Ad.5/3-Null or Ad.5/3-E1A or the indicated dose of Ad.5/3-CTV for 48 hours. Cells were fixed and TUNEL assays were performed. TUNEL positive cells were counted and data presented as TUNEL positive cells per microscopic field in a graphical manner, columns, average of TUNEL positive cells per 5 different microscopic fields; bars, S.D. *, p<0.001 versus control; @, p<0.01 versus Ad.5/3-CTV alone at corresponding doses.
To confirm further the involvement of ATM as an upstream regulator of H2AX- and AIF-mediated cell death in Ad.5/3-CTV treated neuroblastoma cells we used small molecule ATM inhibitors, KU-60019 and KU-55933, to block the ATM kinase. ATM phosphorylates numerous proteins at specific positions, including H2AX at S139 (γ-H2AX). Neuroblastoma cells were treated over night with either KU-60019 (3 μM) or KU-55933 (5 μM, Supplemental Fig. 7) prior to Ad.5/3-CTV infection. Treatment with KU60019 prior to Ad.5/3-CTV infection inhibited the phosphorylation of ATM thereby inhibiting γ-H2AX in SK-N-AS and NB1691 cells (Fig. 5C). This also resulted in decreased PARP cleavage reflecting decreased apoptosis, which was confirmed by TUNEL assay (Fig. 5D and Supplemental Fig. 7). Taken together, these results indicate that ATM acts as an upstream regulator of H2AX phosphorylation, which results in AIF-mediated cell death in neuroblastoma cells following treatment with Ad.5/3-CTV.
mda-7/IL-24 inhibits neuroblastoma tumor growth in vivo
To directly evaluate the effect of mda-7/IL-24 delivered by Ad.5/3-CTV on tumor growth in vivo, we implanted NB1691 cells subcutaneously on both sides of nude mice. The tumors on the left flank were challenged with intra-tumoral injections of Ad.5/3-Null, Ad.5/3-E1A, or Ad.5/3-CTV (mda-7/IL-24 transducing virus). Tumor growth was monitored in mice by measuring tumors every alternate day with Vernier calipers. There was a significant decrease in tumor volume in mice treated with Ad.5/3-CTV compared with mice treated with Ad.5/3-Null or Ad.5/3-E1A (Fig. 6A). Although Ad.5/3-E1A reduced tumor growth to some extent on the injected left side, it was evident that “bystander activity” in the non-injected right tumor was only observed in animals in which the left tumors were treated with Ad.5/3-CTV (Fig. 6). These results confirm previously published data that MDA-7/IL-24 exhibits potent anti-tumor “bystander activity”. To determine whether MDA-7/IL-24 caused AIF-mediated apoptosis in vivo, tumor sections were immunoassayed for MDA-7/IL-24, pATM, γ–H2AX and AIF. Apoptotic content was determined by TUNEL analysis. Consistent with our in vitro observations, tumor sections from Ad.5/3-CTV-treated mice showed increased staining for MDA-7/IL-24, pATM, γ–H2AX and AIF (Fig. 6C). Furthermore, the apoptotic index of tumor cells quantified by the number of TUNEL staining positive cells increased with Ad.5/3-CTV treatment (Fig. 6B).
Figure 6. Intratumoral injections of Ad.5/3-CTV induce AIF-mediated cell death and inhibit human neuroblastoma xenograft tumor growth.

NB1691 human neuroblastoma cells were implanted subcutaneously in both flanks of nude mice and left-sided tumors were treated with 8 intratumoral injections including mock (solvent), Ad.5/3-E1A or Ad.5/3-CTV as described in Materials and Methods. A total of 6 animals were studied in each group. Once the control animals’ tumors reached maximum allowable limit, tumors were collected fixed in formalin and embedded in paraffin. (A) Tumor volumes from the left and right flank were quantified and the results are presented in a graphical manner. Line represents average of all the tumor volumes of the group at the indicated time points: Bars, S.D. *, p<0.05 versus control; **, p<0.001 versus control. (B) Formalin fixed paraffin embedded tissue sections were stained for H&E and TUNEL as per standard protocol; representative images of the indicated treatment groups are shown. (C) Immunohistochemical analysis of MDA-7/IL-24, AIF, γ–H2AX and pATM from tumor sections as described in Materials and Methods. Representative sections shown.
Discussion
Neuroblastoma is a heterogeneous clinical entity, ranging from subgroups that have a very favorable prognosis with a high probability of spontaneous regression to those that display a very poor prognosis despite aggressive therapies (1, 33). Considering this conundrum and the high incidence of recurrence in advanced stages of neuroblastoma (stage III and IV) (4, 6), defining appropriate strategies for treating this cancer particularly in advanced stages is a priority. Presently, we evaluated a broad-spectrum anti-tumor protein MDA-7/IL-24 (11, 12, 16) delivered by a tropism-modified chimeric cancer terminator virus (Ad.5/3-CTV) (23, 26) in neuroblastoma cells. Ad.5/3-CTV induced decreased neuroblastoma cell growth and increased apoptosis in vitro and decreased tumor growth in vivo, supporting potential applications for the therapy of this cancer. Mechanistic studies uncovered a new pathway by which mda-7/IL-24 can promote apoptosis in cancer cells, i.e., through induction and translocation of AIF into the nucleus.
mda-7/IL-24 shows potent antitumor activity that is mediated through multiple pathways in diverse cancers (7, 16). Mechanisms of mda-7/IL-24 toxicity include ER stress and tumoral cell apoptosis by suppression of anti-apoptotic Bcl-2 family members (27, 34), which was also evident in this study. Previously, MDA-7/IL-24 treatment has been shown to increase ROS production in many cancer types (18, 35). It is well known that ROS generation is closely associated with early stages of apoptosis and mitochondrial dysfunction (36). Earlier reports suggest that ROS generation, together with Cyt C release from mitochondria, promote cell death (37, 38). This results in increased permeability of the outer mitochondrial membrane, decreasing transmembrane potential, and activation of AIF (32) eventually inducing caspase-independent apoptosis (39). Ad.5/3-CTV-delivered mda-7/IL-24 to neuroblastoma cells increased the levels of AIF. Our results show, in agreement with previously published data that AIF is translocated into the nucleus inducing caspase-independent cell death in mda-7/IL-24 overexpressed neuroblastoma cells. This was further confirmed using AIF and pan-caspase inhibitors. Inhibition of AIF by small molecule inhibitor attenuated PARP cleavage, inhibiting cell death in neuroblastoma cells upon treatment with Ad.5/3-CTV. In addition, applying a pan-caspase inhibitor did not alter mda-7/IL-24-induced PARP cleavage; further validating caspase-independent cell death induced by this cytokine in neuroblastoma cells.
Cells respond to DNA damage by phosphorylating a variant of the H2A protein family, H2AX (40). H2AX assists chromatin to facilitate DNA repair by providing binding sites for downstream repair factors (41). AIF is a flavoprotein that is in the mitochondrial inter-membrane space and performs a major role in mediating caspase-independent cell death (42, 43). Upon receiving a cell death stimuli, AIF is cleaved within the mitochondria by calpains and cathepsins (44), released into the cytosol possibly through a mitochondrial permeability transition pore, and translocated into the nucleus where it induces chromatin condensation and DNA fragmentation through complex formation with H2AX and cyclophilin A (45). A variety of apoptotic stimuli have been documented to induce AIF mitochondria-to-nucleus translocation including DNA damaging agents, hypoxia/ischemia, oxidative stress and excitotoxins (such as glutamate) (46). However, the signaling pathways that cause AIF nuclear translocation have not been fully elucidated. In this regard, we studied the effects of Ad.5/3-CTV on key apoptotic proteins, AIF and poly (ADP-ribose) polymerase-1 (PARP1), which constitutes a relatively novel, yet, crucial pathway of caspase-independent apoptosis in MDA-7/IL-24 treatment.
H2AX, a member of the histone H2A family is characterized by a phosphorylable SQE motif in its C-terminal tail (32, 47). It is also established that DNA fragmentation induces phosphorylation of H2AX histone at serine 139 (48). Even though H2AX is mainly associated with DNA-damage repair and DNA packaging, it is also a key regulator of programmed cell death (32, 49). To decipher the mechanism by which MDA-7/IL-24 leads to AIF nuclear translocation we assessed the levels of γ-H2AX in Ad.5/3-CTV treated cells and found increased activation levels of H2AX. It is known that ATM is a primary kinase involved in the phosphorylation of H2AX and also that ATM is one of the earliest kinases to be activated in the cellular response to double-strand breaks. We found activation of ATM, H2AX and AIF translocation following Ad.5/3-CTV treatment and these cellular modifications were confirmed by pharmacological inhibition of ATM and AIF. Data obtained through our studies indicate that ATM activation is important in triggering H2AX phosphorylation and AIF activation leading to caspase-independent cell death in Ad.5/3-CTV treated neuroblastoma cells. In contrast to neuroblastoma cells, infection of human breast cancer (MDA-MB-231 and ZR-751) and melanoma (C8161 and SK-Mel) cells with Ad.5/3-CTV did not enhance AIF expression (Supplemental Fig. 8). Precisely how ATM is activated in MDA-7/IL-24-mediated apoptosis in neuroblastoma cells is a key question requiring further experimentation. One recent study by Baritaud et al. revealed the significance of ATM and DNA-PK in regulating γ-H2AX in AIF-mediated caspase-independent necroptosis (32). In particular, they showed that ATM inhibition prevents the H2AX phosphorylation observed after MNNG addition and, subsequently, blocked AIF-mediated cell death. This is in agreement with our observations, which show that ATM small molecule inhibitors attenuated Ad.5/3-CTV-induced PARP cleavage and H2AX phosphorylation, and inhibited AIF changes in neuroblastoma cells (Fig. 5C). Conversely, an AIF small molecule inhibitor reduced Ad.5/3-CTV-induced ATM phosphorylation and cell death (Fig. 4D and 5D) in neuroblastoma cells. Taken together, our results suggest that ATM and AIF are functionally related in a positive feedback loop in which they regulate each other in neuroblastoma cells following Ad.5/3-CTV infection (Fig 7). Another recent study reported that treatment with pro-oxidant resulted in caspase-independent, AIF-dependent apoptosis in ATM-null primary CLL tumors (50). These results from others and us suggest that ATM and AIF can work independently or together to induce caspase-independent cell death.
Figure 7. ATM and γ–H2AX phosphorylation induces AIF-mediated cell death in Ad.5/3-CTV-treated neuroblastoma cells.
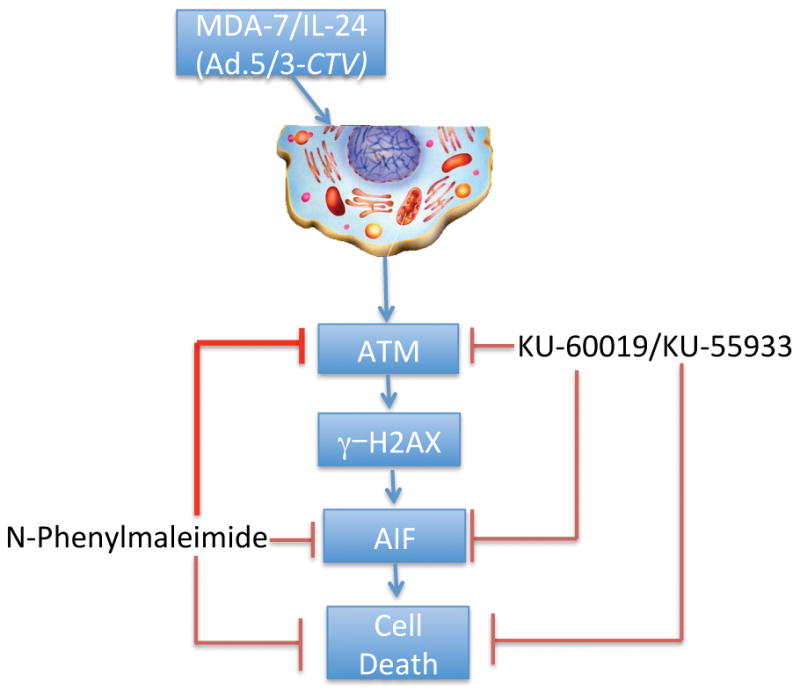
Schematic representation of Ad.5/3-CTV-induced cell death in neuroblastoma cells.
In conclusion, we demonstrate for the first time a new cell death pathway triggered by mda-7/IL-24 through ATM-mediated activation of H2AX and AIF resulting in caspase-independent apoptosis (Fig. 7) that appears unique to neuroblastoma cells, since this effect was not evident in human breast carcinoma or melanoma cells (Supplementary Fig. 8). Support for this stems from three lines of experimental evidence: (1) inhibition of AIF using AIF-inhibitors decreased MDA-7/IL-24-mediated apoptosis; (2) inhibition of caspases using a pan-caspase inhibitor did not block MDA-7/IL-24-induced cell death; and (3) inhibition of ATM altered the levels of AIF, resulting in inhibition of cell death in MDA-7/IL-24 overexpressing neuroblastoma cells. Accordingly, the use of Ad.5/3-CTV, which displays cancer-specific viral replication combined with robust production and secretion of MDA-7/IL-24, to selectively induce cytolysis in neuroblastoma cells may represent a potentially viable treatment option for this aggressive cancer.
Supplementary Material
Acknowledgments
Grant Support
The present study was supported in part by National Cancer Institute grant R01 CA097318 (PBF), the National Foundation for Cancer Research (PBF), a VCU Massey Cancer Center (MCC) development award (PBF) and NCI Cancer Center Support Grant to VCU Massey Cancer Center P30 CA016059 (SKD, DS, LE, PBF). D. Sarkar is a Harrison Scholar in the MCC and P.B. Fisher holds the Thelma Newmeyer Corman Chair in Cancer Research.
Footnotes
Conflict of Interest: There are no conflicts of interest for any of the authors on this paper.
Author’s Contributions: Conception and design: P. Bhoopathi, P.B. Fisher; Development of methodology: P. Bhoopathi, A.K. Pradhan; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P. Bhoopathi, N. Lee, X.-N. Shen; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): P. Bhoopathi, S.K. Das, D. Sarkar, L. Emdad, P.B. Fisher; Administrative, technical or material support (i.e., reporting or organizing data, constructing databases): S.K. Das, D. Sarkar, L. Emdad, X.-N. Shen, P.B. Fisher; Study Supervision: S.K. Das, D. Sarkar, L. Emdad, P.B. Fisher.
References
- 1.Bhoopathi P, Gorantla B, Sailaja GS, Gondi CS, Gujrati M, Klopfenstein JD, et al. SPARC overexpression inhibits cell proliferation in neuroblastoma and is partly mediated by tumor suppressor protein PTEN and AKT. PloS one. 2012;7(5):e36093. doi: 10.1371/journal.pone.0036093. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer research. 2002;62(22):6462–6. [PubMed] [Google Scholar]
- 3.Rosen EMCJ, Frantz CN, Kretschmar C, Levey R, Vawter G, Sallan SE. Improved survival in neuroblastoma using multimodality therapy. Radiother Oncol. 1984;2(3):189–200. doi: 10.1016/s0167-8140(84)80059-6. [DOI] [PubMed] [Google Scholar]
- 4.Saulnier Sholler GL, Bond JP, Bergendahl G, Dutta A, Dragon J, Neville K, et al. Feasibility of implementing molecular-guided therapy for the treatment of patients with relapsed or refractory neuroblastoma. Cancer medicine. 2015;4(6):871–86. doi: 10.1002/cam4.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahu U, Sidhar H, Ghate PS, Advirao GM, Raghavan SC, Giri RK. A Novel Anticancer Agent, 8-Methoxypyrimido[4′,5′:4,5]thieno(2,3-) Quinoline-4(3H)-One Induces Neuro 2a Neuroblastoma Cell Death through p53-Dependent, Caspase-Dependent and -Independent Apoptotic Pathways. PloS one. 2013;8(6):e66430. doi: 10.1371/journal.pone.0066430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masui K, Gini B, Wykosky J, Zanca C, Mischel PS, Furnari FB, et al. A tale of two approaches: complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis. 2013;34(4):725–38. doi: 10.1093/carcin/bgt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annual review of immunology. 2004;22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 8.Dash R, Bhoopathi P, Das SK, Sarkar S, Emdad L, Dasgupta S, et al. Novel mechanism of MDA-7/IL-24 cancer-specific apoptosis through SARI induction. Cancer research. 2014;74(2):563–74. doi: 10.1158/0008-5472.CAN-13-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer research. 2010;70(9):3667–76. doi: 10.1158/0008-5472.CAN-09-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang H, Lin JJ, Su ZZ, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995;11(12):2477–86. [PubMed] [Google Scholar]
- 11.Dash R, Bhutia SK, Azab B, Su ZZ, Quinn BA, Kegelmen TP, et al. mda-7/IL-24: a unique member of the IL-10 gene family promoting cancer-targeted toxicity. Cytokine & growth factor reviews. 2010;21(5):381–91. doi: 10.1016/j.cytogfr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menezes ME, Bhatia S, Bhoopathi P, Das SK, Emdad L, Dasgupta S, et al. MDA-7/IL-24: multifunctional cancer killing cytokine. Advances in experimental medicine and biology. 2014;818:127–53. doi: 10.1007/978-1-4471-6458-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emdad L, Sarkar D, Lebedeva IV, Su ZZ, Gupta P, Mahasreshti PJ, et al. Ionizing radiation enhances adenoviral vector expressing mda-7/IL-24-mediated apoptosis in human ovarian cancer. Journal of cellular physiology. 2006;208(2):298–306. doi: 10.1002/jcp.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar S, Quinn BA, Shen X, Dent P, Das SK, Emdad L, et al. Reversing translational suppression and induction of toxicity in pancreatic cancer cells using a chemoprevention gene therapy approach. Molecular pharmacology. 2015;87(2):286–95. doi: 10.1124/mol.114.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar D, Lebedeva IV, Gupta P, Emdad L, Sauane M, Dent P, et al. Melanoma differentiation associated gene-7 (mda-7)/IL-24: a ‘magic bullet’ for cancer therapy? Expert opinion on biological therapy. 2007;7(5):577–86. doi: 10.1517/14712598.7.5.577. [DOI] [PubMed] [Google Scholar]
- 16.Fisher PB. Is mda-7/IL-24 a “magic bullet” for cancer? Cancer research. 2005;65(22):10128–38. doi: 10.1158/0008-5472.CAN-05-3127. [DOI] [PubMed] [Google Scholar]
- 17.Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, et al. Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer biology & therapy. 2009;8(5):391–400. doi: 10.4161/cbt.8.5.7581. [DOI] [PubMed] [Google Scholar]
- 18.Lebedeva IV, Sauane M, Gopalkrishnan RV, Sarkar D, Su ZZ, Gupta P, et al. mda-7/IL-24: exploiting cancer’s Achilles’ heel. Molecular therapy : the journal of the American Society of Gene Therapy. 2005;11(1):4–18. doi: 10.1016/j.ymthe.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Zhuo B, Wang R, Yin Y, Zhang H, Ma T, Liu F, et al. Adenovirus arming human IL-24 inhibits neuroblastoma cell proliferation in vitro and xenograft tumor growth in vivo. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013;34(4):2419–26. doi: 10.1007/s13277-013-0792-1. [DOI] [PubMed] [Google Scholar]
- 20.Dash R, Dmitriev I, Su ZZ, Bhutia SK, Azab B, Vozhilla N, et al. Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) improves therapeutic efficacy in low CAR prostate cancer cells. Cancer gene therapy. 2010;17(7):447–56. doi: 10.1038/cgt.2009.91. [DOI] [PubMed] [Google Scholar]
- 21.Sarkar D, Su ZZ, Vozhilla N, Park ES, Gupta P, Fisher PB. Dual cancer-specific targeting strategy cures primary and distant breast carcinomas in nude mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(39):14034–9. doi: 10.1073/pnas.0506837102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su ZZ, Sarkar D, Emdad L, Duigou GJ, Young CS, Ware J, et al. Targeting gene expression selectively in cancer cells by using the progression-elevated gene-3 promoter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1059–64. doi: 10.1073/pnas.0409141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azab BM, Dash R, Das SK, Bhutia SK, Sarkar S, Shen XN, et al. Enhanced prostate cancer gene transfer and therapy using a novel serotype chimera cancer terminator virus (Ad.5/3-CTV) Journal of cellular physiology. 2014;229(1):34–43. doi: 10.1002/jcp.24408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park MA, Hamed HA, Mitchell C, Cruickshanks N, Dash R, Allegood J, et al. A serotype 5/3 adenovirus expressing MDA-7/IL-24 infects renal carcinoma cells and promotes toxicity of agents that increase ROS and ceramide levels. Molecular pharmacology. 2011;79(3):368–80. doi: 10.1124/mol.110.069484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhoopathi P, Quinn BA, Gui Q, Shen XN, Grossman SR, Das SK, et al. Pancreatic cancer-specific cell death induced in vivo by cytoplasmic-delivered polyinosine-polycytidylic acid. Cancer research. 2014;74(21):6224–35. doi: 10.1158/0008-5472.CAN-14-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das SK, Sarkar S, Dash R, Dent P, Wang XY, Sarkar D, et al. Cancer terminator viruses and approaches for enhancing therapeutic outcomes. Advances in cancer research. 2012;115:1–38. doi: 10.1016/B978-0-12-398342-8.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dash R, Richards JE, Su ZZ, Bhutia SK, Azab B, Rahmani M, et al. Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10-related cytokine. Cancer research. 2010;70(12):5034–45. doi: 10.1158/0008-5472.CAN-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(9):3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 29.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nature reviews Molecular cell biology. 2015;16(7):393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 30.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer biology & therapy. 2005;4(2):139–63. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 31.Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. The Journal of cell biology. 2002;158(3):507–17. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baritaud M, Cabon L, Delavallee L, Galan-Malo P, Gilles ME, Brunelle-Navas MN, et al. AIF-mediated caspase-independent necroptosis requires ATM and DNA-PK-induced histone H2AX Ser139 phosphorylation. Cell death & disease. 2012;3:e390. doi: 10.1038/cddis.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitanaka C, Kato K, Ijiri R, Sakurada K, Tomiyama A, Noguchi K, et al. Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. Journal of the National Cancer Institute. 2002;94(5):358–68. doi: 10.1093/jnci/94.5.358. [DOI] [PubMed] [Google Scholar]
- 34.Su Z, Emdad L, Sauane M, Lebedeva IV, Sarkar D, Gupta P, et al. Unique aspects of mda-7/IL-24 antitumor bystander activity: establishing a role for secretion of MDA-7/IL-24 protein by normal cells. Oncogene. 2005;24(51):7552–66. doi: 10.1038/sj.onc.1208911. [DOI] [PubMed] [Google Scholar]
- 35.Lebedeva IV, Su ZZ, Sarkar D, Kitada S, Dent P, Waxman S, et al. Melanoma differentiation associated gene-7, mda-7/interleukin-24, induces apoptosis in prostate cancer cells by promoting mitochondrial dysfunction and inducing reactive oxygen species. Cancer research. 2003;63(23):8138–44. [PubMed] [Google Scholar]
- 36.Chen Q, Chai YC, Mazumder S, Jiang C, Macklis RM, Chisolm GM, et al. The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell death and differentiation. 2003;10(3):323–34. doi: 10.1038/sj.cdd.4401148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1259–63. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen K, Xie J, Wang H, Zhang H, Yu M, Lu F, et al. Cambogin Induces Caspase-Independent Apoptosis through the ROS/JNK Pathway and Epigenetic Regulation in Breast Cancer Cells. Molecular cancer therapeutics. 2015;14(7):1738–49. doi: 10.1158/1535-7163.MCT-14-1048. [DOI] [PubMed] [Google Scholar]
- 39.Cande C, Vahsen N, Garrido C, Kroemer G. Apoptosis-inducing factor (AIF): caspase-independent after all. Cell death and differentiation. 2004;11(6):591–5. doi: 10.1038/sj.cdd.4401400. [DOI] [PubMed] [Google Scholar]
- 40.Podhorecka M, Skladanowski A, Bozko P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. Journal of nucleic acids. 2010;2010 doi: 10.4061/2010/920161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic acids research. 2008;36(17):5678–94. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23(16):2785–96. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 43.Cabon L, Galan-Malo P, Bouharrour A, Delavallee L, Brunelle-Navas MN, Lorenzo HK, et al. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell death and differentiation. 2012;19(2):245–56. doi: 10.1038/cdd.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delettre C, Yuste VJ, Moubarak RS, Bras M, Robert N, Susin SA. Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. The Journal of biological chemistry. 2006;281(27):18507–18. doi: 10.1074/jbc.M601751200. [DOI] [PubMed] [Google Scholar]
- 45.Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. The EMBO journal. 2010;29(9):1585–99. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2000;14(5):729–39. [PubMed] [Google Scholar]
- 47.Li A, Yu Y, Lee SC, Ishibashi T, Lees-Miller SP, Ausio J. Phosphorylation of histone H2A. X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. The Journal of biological chemistry. 2010;285(23):17778–88. doi: 10.1074/jbc.M110.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wen W, Zhu F, Zhang J, Keum YS, Zykova T, Yao K, et al. MST1 promotes apoptosis through phosphorylation of histone H2AX. The Journal of biological chemistry. 2010;285(50):39108–16. doi: 10.1074/jbc.M110.151753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. The Journal of biological chemistry. 2000;275(13):9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 50.Agathanggelou A, Weston VJ, Perry T, Davies NJ, Skowronska A, Payne DT, et al. Targeting the Ataxia Telangiectasia Mutated-null phenotype in chronic lymphocytic leukemia with pro-oxidants. Haematologica. 2015;100(8):1076–85. doi: 10.3324/haematol.2014.115170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.