

Summary
De novo ASXL1 mutations are found in patients with Bohring-Opitz syndrome, a disease with severe developmental defects and early childhood mortality. The underlying pathologic mechanisms remain largely unknown. Using Asxl1-targeted murine models, we found that Asxl1 global loss as well as conditional deletion in osteoblasts and their progenitors led to significant bone loss and a markedly decreased number of bone marrow stromal cells (BMSCs) compared with wild-type littermates. Asxl1−/− BMSCs displayed impaired self-renewal and skewed differentiation, away from osteoblasts and favoring adipocytes. RNA-sequencing analysis revealed altered expression of genes involved in cell proliferation, skeletal development, and morphogenesis. Furthermore, gene set enrichment analysis showed decreased expression of stem cell self-renewal gene signature, suggesting a role of Asxl1 in regulating the stemness of BMSCs. Importantly, re-introduction of Asxl1 normalized NANOG and OCT4 expression and restored the self-renewal capacity of Asxl1−/− BMSCs. Our study unveils a pivotal role of ASXL1 in the maintenance of BMSC functions and skeletal development.
Keywords: Bohring-Opitz syndrome, ASXL1 mutation, bone marrow stromal cell, self-renewal and differentiation, skeletal development
Graphical Abstract
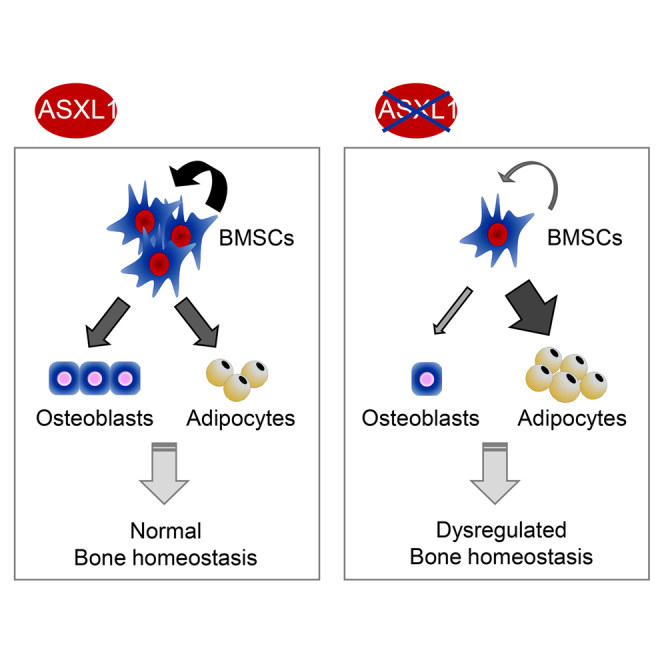
Highlights
-
•
Asxl1 loss impairs BMSC self-renewal and cell fate
-
•
Asxl1 loss leads to dramatic bone loss
-
•
Asxl1 loss alters the expression of genes critical for cell fates of BMSCs
-
•
Re-introducing Asxl1 restores self-renewal and lineage commitment in Asxl1−/− BMSCs
In this article, Yang, Wang, and colleagues show that loss of Asxl1 led to multiple skeletal developmental defects, closely reminiscent of Bohring-Opitz syndrome. They identified that the skeletal defects were associated with an impaired self-renewal and skewed lineage commitment of bone marrow stromal cells (BMSCs). These findings indicate a pivotal role of ASXL1 in the maintenance of BMSC functions and skeletal development.
Introduction
Bone marrow stromal cells (BMSCs) are multi-potent progenitor cells with self-renewal capabilities and multi-lineage differentiation potentials, including osteogenesis and adipogenesis (Bianco et al., 2008, Teitelbaum, 2010, Uccelli et al., 2008, Ye et al., 2012). The relation between osteogenesis and adipogenesis in BMSCs is critical for normal bone homeostasis. Skewed cell fate of BMSCs can lead to developmental defects. For example, inhibition of adipogenesis may enhance bone growth and repair (Kawai and Rosen, 2010, McCauley, 2010). More recently, Mendez-Ferrer and others reported that the final cell-fate decision of BMSCs relies on an orchestrated activation of lineage-specific genes and repression of genes governing cell stemness or commitment to other lineages (Mendez-Ferrer et al., 2010, Takada et al., 2009, Wei et al., 2011).
Bohring-Opitz syndrome (BOS) is a heterogeneous genetic condition characterized by severe developmental delay, characteristic craniofacial appearance, fixed contractures of the upper limbs, abnormal posture, feeding difficulties, severe intellectual disability, fetal microsomia, and failure to thrive (Bohring et al., 2006, Hastings et al., 2011, Oberklaid and Danks, 1975). Most patients die in early childhood due to developmental deficits, unexplained bradycardia, obstructive apnea, or pulmonary infections (Hastings et al., 2011). In 2011, Hoischen et al. (2011) identified de novo nonsense mutations of the additional sex combs-like 1 gene (ASXL1) in patients with BOS. Somatic ASXL1 alterations have also been reported in elderly patients with myeloid malignancies, including myelodysplastic syndrome, chronic myelomonocytic leukemia, and acute myeloid leukemia (Carbuccia et al., 2009, Gelsi-Boyer et al., 2009).
ASXL1 belongs to the enhancer of trithorax group (TrxG) and polycomb group (PcG) (ETP), and genetically interacts with CBX2 in mice (Fisher et al., 2010). PcG and trxG proteins are key regulators for the expression of numerous developmental genes by silencing or activating gene expression, respectively. The ETP genes encode proteins required for both maintenance of activation and silencing, as shown by simultaneous anterior and posterior transformations caused by failure to activate or repress Hox genes. In an Asxl1 mutant mouse model, Fisher et al. (2010) reported an alteration of the axial skeleton in newborn pups involving anterior and posterior transformations. However, the cellular and molecular mechanisms by which ASXL1 mutation causes BOS remain unclear.
We have recently reported that Asxl1 null mice are smaller in size and exhibit anophthalmia (Wang et al., 2014). In this study, we aimed to unveil the cellular and molecular mechanisms underlying the pathogenesis of ASXL1 loss-mediated skeletal defects. Our study demonstrated that nullizygous loss of Asxl1 led to multiple skeletal developmental defects, including runting, markedly reduced bone mineral density (BMD), microcephaly, and hypoplastic supraorbital ridges, closely reminiscent of BOS. We further identified that the defective skeletal development was associated with an impaired self-renewal and skewed lineage commitment of BMSCs, away from osteoblast and favoring adipocyte differentiation. Moreover, RNA-sequencing (RNA-seq) analysis demonstrated that Asxl1 loss altered the expression of genes that are critical for stem cell self-renewal. Importantly, re-introduction of Asxl1 into Asxl1−/− BMSCs restored BMSC self-renewal and lineage commitment. These data indicate a pivotal role of ASXL1 in the maintenance of BMSC functions and skeletal development.
Results
Loss of Asxl1 Impairs BMSC Self-Renewal and Differentiation Capacity
Self-renewal and multi-lineage differentiation are the two key characteristics of BMSCs. We first assessed whether Asxl1 loss alters BMSC cellular functions. qPCR was performed to ensure that Asxl1 was successfully deleted. While Asxl1 mRNA was detected in wild-type (WT) BMSCs, no Asxl1 mRNA was detected in Asxl1−/− BMSCs, indicating successful deletion of Asxl1 (Figure S1A). Asxl1 null mice were generated by replacing part of the Asxl1 exon 1 sequence with nlacZ/nGFP (inserted 6 bp upstream of the start codon) as previously reported (Wang et al., 2014). The targeted allele results in transcription of nGFP mRNA instead of Asxl1 (the endogenous ATG was disrupted). We examined GFP expression in the BMSCs from Asxl1+/− mice by flow-cytometric analysis to determine whether Asxl1 is expressed in BMSCs. The CD45−CD105+CD44+CD73+CD146+CD90+ cell population, defined as BMSCs (Barry and Murphy, 2013, Joyce and Pollard, 2009), were GFP positive (Figure S1B), suggesting expression of ASXL1 in BMSCs.
Having confirmed ASXL1 expression in WT BMSCs, we next examined the effect of Asxl1 deletion on BMSC expansion. [3H]Thymidine incorporation assays revealed that Asxl1 ablation diminished BMSC proliferation (Figure 1A). Mendez-Ferrer et al. (2010) reported that Nestin+ BMSCs form non-adherent mesenspheres that can be serially re-plated in culture due to their self-renewal capability. We next performed clonal mesensphere cultures to determine whether loss of Asxl1 altered BMSC self-renewal capacity. The clonal mesensphere formation potential of Asxl1−/− BMSCs was significantly reduced compared with WT BMSCs when equal numbers of BMSCs were plated (Figure 1B). WT mesenspheres were 414 ± 28.5 μm in diameter and the secondary spheres were 133 ± 20 μm in diameter (Figures 1C and 1D). In contrast, Asxl1−/− mesenspheres were significantly smaller (187.5 ± 24 μm in diameter) and exhibited a decreased capacity to form secondary spheres (64 ± 4 μm in diameter) (Figures 1C and 1D). Flow cytometric analysis revealed that the cells of the mesensphere colonies were Nestin+, CD51+, CD105+, and CD146+ (Figure S1C), confirming that these cells were indeed BMSCs.
Figure 1.

Asxl1 Is Required for BMSC Self-Renewal and Differentiation
(A) Proliferation assays show impaired growth of Asxl1−/− BMSCs compared with WT (n = 4 mice per genotype from at least three independent experiments).
(B) The frequency of non-adherent mesenspheres from Asxl1−/− BMSCs is significantly decreased compared with WT BMSCs. An equal number of BMSCs were plated (n = 5 mice per genotype from three independent experiments).
(C) Self-renewal capacity of BMSCs was assayed by clonal non-adherent sphere formation. Asxl1−/− BMSCs (n = 10 mice) exhibit a significantly decreased sphere diameter compared with WT BMSCs (n = 8 mice). Scale bar represents 100 μm.
(D) The formation of secondary spheres from Asxl1−/− BMSCs is also significantly reduced compared with WT BMSCs (n = 5 mice per genotype).
(E) ALP staining shows significantly decreased osteoblast differentiation by Asxl1−/− BMSCs (n = 11 mice) as compared with WT BMSCs (n = 9 mice).
(F) Oil red O staining shows significantly increased adipocyte differentiation by Asxl1−/− BMSCs compared with WT BMSCs (n = 11 mice per genotype). Scale bar represents 50 μm.
Data are presented as mean ± SEM. ∗∗p < 0.005, ∗∗∗p < 0.001. See also Figure S1.
BMSCs have multi-lineage differentiation potential, including the ability to form osteoblasts and adipocytes (Pittenger et al., 1999). To evaluate the effect of Asxl1 deletion on BMSC differentiation, we performed osteoblast and adipocyte differentiation assays. Liquid culture of BMSCs with different culture conditions verified that Asxl1 deletion diminished osteoblast differentiation, as evidenced by minimal alkaline phosphatase-positive (ALP+) expression and increased oil red O-positive (+) cells in Asxl1−/− cultures compared with WT cultures (Figures 1E and 1F, respectively). Chondrocyte differentiation assays showed that Asxl1 deletion did not affect chondrocyte differentiation from BMSCs (data not shown). Collectively, these data indicate that loss of Asxl1 impairs BMSC self-renewal and skews BMSC lineage commitment away from osteoblasts but in favor of adipocytes.
Asxl1 Deletion Reduces BMSC Frequency and Alters BMSC Fates in Mice
To examine whether Asxl1 deletion alters the BMSC frequency in vivo, we next performed flow cytometric analysis on bone marrow cells stained with a cocktail of antibodies, including CD45, CD44, CD105, CD73, CD146, and CD90 (Barry and Murphy, 2013, Joyce and Pollard, 2009). A significant reduction in the CD45−CD44+CD105+CD73+CD146+CD90+ BMSC population was observed in the bone marrow of Asxl1−/− mice compared with WT littermates (Figure 2A). Consistently, colony-forming-unit fibroblast (CFU-F) assay, a well-established method to determine BMSC frequency in vivo, revealed a significantly reduced frequency of CFU-F in Asxl1−/− bone marrow compared with WT controls (Figures 2B and 2C). To determine whether the decreased CFU-F frequency was due solely to the decreased frequency of BMSCs in Asxl1−/− bone marrow, we performed CFU-F assays with an equal number of BMSCs. Significantly fewer CFU-F were formed by Asxl1−/− BMSCs compared with WT BMSCs (Figure S1D). These data indicate that Asxl1 deletion decreases the BMSC pool and their CFU-F forming ability in the bone marrow in vivo.
Figure 2.

Loss of Asxl1 Impairs BMSC Self-Renewal and Differentiation in Mice
(A) Quantitation of percentage of CD45−CD105+CD44+CD73+CD146+CD90+ cell populations (BMSCs) in the bone marrow of WT (n = 9 mice) and Asxl1−/− (n = 10 mice, 3 weeks old) by flow cytometry.
(B and C) The frequencies of CFU-F per femur (B) and per 1 × 106 BMMNCs (C) from WT and Asxl1−/− mice are shown (n = 6 mice per genotype).
(D) The frequencies of CFU-osteoblast (ALP+) per femur are shown (top panel). Representative ALP staining of WT and Asxl1−/− osteoblastic colonies cultured in osteogenic culture medium from BMMNCs. WT (n = 8 mice) and Asxl1−/− (n = 7 mice).
(E) The ratio of oil red O+ adipocytes to total colonies demonstrates enhanced adipocyte differentiation in Asxl1−/− BMSCs compared with WT BMSCs (n = 3 mice per genotype). Scale bar represents 200 μm.
(F) Pie chart illustrates that loss of Asxl1 impairs BMSC self-renewal and skews BMSC lineage commitment away from osteoblasts, favoring adipocytes. Data represent the means of four independent experiments.
Data are presented as mean ± SEM. ∗∗∗p < 0.001. See also Figure S1.
To further evaluate the effect of Asxl1 loss on BMSC differentiation in vivo, we subsequently performed CFU-osteoblast and CFU-adipocyte differentiation assays using primary bone marrow mononuclear cells (BMMNCs) from WT and Asxl1−/− mice. Compared with WT controls, Asxl1−/− BMMNCs gave rise to a significantly reduced number of ALP+ osteoblastic colonies (Figure 2D). In contrast, the total number of oil red O+ colonies and the ratio of oil red O+ colonies to CFU-F were dramatically increased in Asxl1−/− cultures (Figure 2E). Collectively, these data indicate that loss of Asxl1 impairs BMSC self-renewal and skews BMSC lineage commitment in vivo (Figure 2F).
Genetic Deletion of Asxl1 Leads to Multiple Skeletal Deficits Reminiscent of Human Bohring-Opitz Syndrome
Asxl1-knockout mice are smaller in size, and some mice have anophthalmia (Wang et al., 2014). Using alizarin red S/Alcian blue 8GX staining, we showed that loss of Asxl1 resulted in ∼30% reduction in body size compared with littermate controls (Figure S2A). A kinetic quantitative measurement of body size and body weight revealed significant delays in growth of Asxl1−/− mice over time compared with WT littermates (Figures S2B and S2C). Asxl1−/− mice also exhibited a significant reduction in skull size compared with their littermate controls, reminiscent of the microcephaly in BOS patients (Figure S2D). Peripheral dual-energy X-ray absorptiometry (pDEXA) revealed significantly reduced BMD in the distal femurs of Asxl1−/− mice compared with their littermate controls (Figure S2E). Microcomputed tomography (μCT) demonstrated a marked reduction of bone volume fraction in Asxl1−/− mice compared with their littermate controls (Figure S2F). Furthermore, hypoplastic supraorbital ridges, a characteristic of BOS, were observed in Asxl1−/− mice (Figure S2G).
Labeling bones with fluorochrome markers provides a method to study the dynamic changes of bone formation (Warden et al., 2005, Wu et al., 2011). We next examined bone remodeling in WT and Asxl1−/− mice with a fluorescent labeling assay. An ∼45% reduction in the bone formation rate per bone surface (BFR/BS) was observed in Asxl1−/− mice compared with WT control mice (Figures S2H and S2I). These data demonstrate that loss of Asxl1 in mice impairs skeletal development.
Conditional Deletion of Asxl1 in Osteoblasts Recapitulates the Developmental Defects Observed in Asxl1−/− Mice
To further delineate the cell-autonomous role of ASXL1 in mediating BMSC cellular functions, we crossed Asxl1 floxed allele mice (Asxl1fl/fl) (Abdel-Wahab et al., 2013) with transgenic mice harboring Osterix-Cre (Osx-Cre, specifically expressed in the pre-osteoblastic and osteoblastic lineages) to generate OsxCre;Asxl1fl/fl mice (OsxCre;Asxl1Δ/Δ) (Figure S3A). Similar to Asxl1−/− mice, X-ray images of OsxCre;Asxl1Δ/Δ mice at 3 weeks of age demonstrated dwarfism compared with Asxl1fl/fl control mice (Figure 3A). Serial measurement of body weight revealed a consistently lower body weight at varying ages of OsxCre;Asxl1Δ/Δ mice compared with age-matched littermate control mice (Figure 3B). pDEXA scans demonstrated reduced whole-body BMD and distal femoral BMD in OsxCre;Asxl1Δ/Δ mice compared with their littermate controls (Figures S3B and 3C). μCT further revealed diminished trabecular bone volume and trabecular number along with increased trabecular spacing in OsxCre;Asxl1Δ/Δ mice compared with WT controls (Figures 3D, 3E, and S3C). In addition, the cortical bone thickness of the mid-shaft femur was significantly reduced compared with control mice (Figure S3D). A greater than 50% reduction in the mineral apposition rate and a 43% reduction in the BFR/BS were observed in OsxCre;Asxl1Δ/Δ mice compared with control mice (Figures 3F and 3G). These data reinforce the cell-autonomous role of Asxl1 deletion in defective skeletal development.
Figure 3.

Loss of Asxl1 in Pre-osteoblasts and Their Progenies Leads to BOS-like Phenotypes in Mice
(A) High-resolution X-ray images demonstrate runting of OsxCre;Asxl1Δ/Δ mice compared with Asxl1fl/fl littermates at 3 weeks of life.
(B) Serial body weight measurement of OsxCre;Asxl1Δ/Δ mice and Asxl1fl/fl littermates (n = 6 mice per genotype). Mice were weighed weekly on the same date and time.
(C) Distal femur BMD of 3-week-old Asxl1fl/fl (n = 4 mice) and OsxCre;Asxl1Δ/Δ (n = 6 mice).
(D and E) μCT analysis shows a significantly reduced bone volume (D) and trabecular bone (E) in OsxCre;Asxl1Δ/Δ (n = 6 mice) compared with Asxl1fl/fl controls (n = 4 mice). Scale bar represents 200 μm.
(F and G) Dynamic bone histomorphometry was performed using alizarin labeling. The mineral apposition rate (MAR, F) and bone formation rate (BFR/BS, G) are significantly decreased in OsxCre;Asxl1Δ/Δ (n = 6 mice) versus controls (n = 4 mice). Scale bar represents 20 μm.
(H) Quantitation of percentage of PDGFRα+CD45−TER119−CD31− BMSCs in the bone marrow of OsxCre;Asxl1Δ/Δ (n = 5 mice) and littermate controls (n = 4 mice, 5 weeks old).
(I) The frequencies of CFU-F per femur from Asxl1fl/fl (n = 5 mice) and OsxCre;Asxl1Δ/Δ (n = 7 mice) are shown.
(J) The frequencies of CFU osteoblasts per femur are shown (top panel). Representative ALP staining of osteoblastic colonies cultured in osteogenic culture medium from OsxCre;Asxl1Δ/Δ (n = 5 mice) and Asxl1fl/fl (n = 3 mice) BMMNCs.
(K) Ratio of ALP+ osteoblasts to total colonies demonstrates impaired osteoblast differentiation in OsxCre;Asxl1Δ/Δ (n = 5 mice) compared with littermate controls (n = 3 mice).
Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001. See also Figures S2 and S3.
To determine whether the BMSC frequency is affected in OsxCre;Asxl1Δ/Δ mice, we examined the percentage of PDGFRα+CD45−TER119−CD31− BMSCs by flow cytometry (Worthley et al., 2015, Zhou et al., 2014). A significant reduction in BMSCs was observed in the bone marrow of OsxCre;Asxl1Δ/Δ mice compared with littermate controls (Figures 3H and S3E). PDGFRα+CD45−TER119−CD31− BMSCs from OsxCre;Asxl1Δ/Δ and controls contained a similar proportion of Sca1+ and CD51+ cell populations (Figure S3E). Consistently, a significant reduction in frequency of CFU-F was observed in the bone marrow of OsxCre;Asxl1Δ/Δ mice compared with littermate control mice (Figure 3I). When plating an equal number of cells from CFU-F, we found a significant reduction of plating efficiency in OsxCre;Asxl1Δ/Δ BMSCs compared with control BMSCs (Figure S3F), suggesting an impaired self-renewal capacity in OsxCre;Asxl1Δ/Δ mice. CFU-osteoblast differentiation assays further confirmed that BMMNCs from OsxCre;Asxl1Δ/Δ mice gave rise to a significantly reduced number of ALP+ osteoblastic colonies than those of control mice (Figure 3J). The ratio of ALP+ osteoblast colonies to total colonies was significantly decreased in OsxCre;Asxl1Δ/Δ mice compared with control mice, further confirming the impaired osteoblast differentiation in OsxCre;Asxl1Δ/Δ mice (Figure 3K). Collectively, these data further validate our finding that loss of Asxl1 decreases the BMSC pool and impairs osteoblast differentiation.
Asxl1 Loss Dysregulates Transcriptional Programs to Induce BMSC Lineage Commitment
To determine whether loss of Asxl1 alters gene expression profiles in BMSCs, we next performed RNA-seq on WT and Asxl1−/− BMSCs. Compared with WT BMSCs, Asxl1−/− BMSCs exhibited a distinct gene expression signature with a total of 338 dysregulated genes. Among those, 182 genes were upregulated and 156 genes downregulated (Figure 4A). Gene ontology (GO) analysis and enrichment mapping (Merico et al., 2010, Pinto et al., 2010) demonstrated that the downregulated genes in Asxl1−/− BMSCs were enriched in skeletal development and morphogenesis (Figure 4B). Furthermore, gene set enrichment analysis (GSEA) (Subramanian et al., 2005) indicated that loss of Asxl1 significantly decreased expression of stem cell signature genes in BMSCs (Figure 4C). By contrast, upregulated genes were enriched in cell development/differentiation and tissue morphogenesis (Figure S4).
Figure 4.

Asxl1 Deletion Alters the Expression of Genes Critical for BMSCs Self-Renewal and Differentiation
(A) The heatmap shows clustering of differentially expressed genes between WT and Asxl1−/− BMSCs (p < 0.05, FDR < 0.25). Light purple and light blue indicate 182 up- and 156 downregulated genes, respectively.
(B) The enrichment map helps visualize the network of KEGG pathways and GO terms enriched with downregulated genes in Asxl1−/− BMSCs. Nodes and ovals indicate enriched functional gene sets (p < 0.01 and FDR < 0.25 in DAVID) and functional clusters, respectively. Edges indicate overlap between the enriched gene sets; thickness represents significance. Only edges with a Fisher's exact test nominal p value smaller than 10−4 were visualized. Color intensity is proportional to enrichment significance.
(C) The GSEA plot shows decreased gene expression of the embryonic stem cell signature (Wong et al., 2008) in Asxl1−/− BMSCs compared with WT BMSCs. The normalized enrichment score (NES), p value, and FDR are shown.
(D) qPCR analysis shows that expression levels of the pluripotent marker genes Nanog, Pou5f1, and Sox2 are significantly lower in Asxl1−/− BMSCs than in WT BMSCs (n = 6 mice per genotype).
(E) Western blot analysis shows that the protein levels of NANOG, OCT4, and SOX2 are decreased in Asxl1−/− BMSCs compared with WT BMSCs.
(F) qPCR analysis shows decreased expression levels of the osteoblast marker genes (Runx2, Bglap, Sp7, and Alpl) in Asxl1−/− BMSCs versus WT BMSCs (n = 10 mice per genotype).
(G) qPCR analysis shows significantly increased expression of the adipocyte marker genes (Pparγ, Cepbα, Fabp4, and Lpl) in Asxl1−/− BMSCs compared with WT BMSCs (n = 4 mice per genotype).
Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001. See also Figure S4.
To further validate the altered expression of key genes required for BMSC self-renewal and differentiation, we performed qPCR and western blotting with WT and Asxl1−/− BMSCs. qPCR revealed that the mRNA expression of genes critical for stem cell pluripotency, including Nanog, Pou5f1, and Sox2, was significantly reduced in Asxl1 null BMSCs compared with WT BMSCs (Figure 4D), correlating with the impaired self-renewal of Asxl1 null BMSCs. Consistently, western blot analysis revealed markedly reduced NANOG, OCT4, and SOX2 protein levels in Asxl1 null BMSCs compared with WT cells (Figure 4E). Furthermore, mRNA expression of Runx2, Sp7, Bglap, and Alpl, genes critical for osteogenic differentiation, was also reduced in Asxl1 null BMSCs compared with WT BMSCs (Figure 4F). By contrast, mRNA expression of Pparγ, Cebpα, Fabp4, and Lpl, genes controlling adipogenic differentiation, was dramatically increased in Asxl1-deficient BMSCs compared with WT BMSCs (Figure 4G). Altogether, these data suggest that ASXL1 regulates BMSC fates through controlling the expression of genes critical for BMSC self-renewal and lineage commitment.
Re-expression of Asxl1 Rescues Self-Renewal and Osteoblast/Adipocyte Differentiation in Asxl1−/− BMSCs
To determine whether impaired self-renewal of Asxl1−/− BMSCs was a direct consequence of the Asxl1 loss, we transduced WT Asxl1 (with a FLAG tag for easy detection) into Asxl1−/− BMSCs using a lentiviral system. Asxl1−/− BMSCs transduced with empty vector were used as a control. The expression of ASXL1 was confirmed by western blot analysis (Figure 5A) and qPCR (Figure S5). Re-expression of WT Asxl1 in Asxl1−/− BMSCs significantly increased mesensphere size (Figure 5B) and restored levels of NANOG and OCT4 (Figure 5C), suggesting a restoration of self-renewal capacity of Asxl1 null BMSCs.
Figure 5.

Re-expression of ASXL1 Rescues the Aberrant Cellular Phenotypes of Asxl1−/− BMSCs
(A) Western blot analysis shows the expression of FLAG-ASXL1 protein in Asxl1−/− BMSCs transduced with empty vector control or ASXL1 expression vector (FLAG-Asxl1).
(B) Mesensphere assay shows the self-renewal capacity of WT and Asxl1−/− BMSCs expressing either empty vector or FLAG-Asxl1 as indicated. Top bars represent the mean mesensphere diameter (μm) from three independent experiments. Representative mesensphere photomicrographs are labeled accordingly. Scale bar represents 200 μm.
(C) Western blot analysis for NANOG and OCT4 in WT and Asxl1−/− BMSCs expressing either empty vector or FLAG-Asxl1.
(D) ALP staining of osteoblast cultures of WT BMSCs and Asxl1−/− BMSCs expressing empty vector or FLAG-Asxl1 as indicated. Top bars represent the relative ALP+ activity from four independent experiments. Bottom panel shows representative photomicrographs of ALP staining cultures. Scale bar represents 200 μm.
(E) Oil red O staining of adipocyte cultures of WT or Asxl1−/− BMSCs expressing either empty vector or FLAG-Asxl1 as indicated. Top bars represent the number of oil red O+ cells in cultures of WT or Asxl1−/− BMSC expressing either empty vector or FLAG-Asxl1 from four independent experiments. Bottom panel shows representative photomicrographs of oil red O+ staining cells. Scale bar represents 50 μm.
(F) qPCR shows the expression of osteoblast differentiation genes (Runx2, Sp7, and Alpl) in WT and Asxl1−/− BMSCs transduced with FLAG-Asxl1 or empty vector in six independent experiments.
(G) qPCR shows the expression of adipocyte differentiation genes in WT and Asxl1−/− BMSCs with or without transduction of FLAG-Asxl1 from four independent experiments.
Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001. See also Figure S5.
To determine whether re-expression of Asxl1 could rescue the aberrant lineage commitment of Asxl1−/− BMSCs, we repeated the osteoblast and adipocyte differentiation assays. Re-expression of Asxl1 in Asxl1−/− BMSCs increased osteoblast differentiation (Figure 5D) while inhibiting adipocyte differentiation (Figure 5E) compared with empty vector controls. qPCR confirmed that the expression levels of genes controlling osteoblast and adipocyte differentiation, including Runx2, Sp7, Pparγ, and Cebpα, were restored (Figures 5F and 5G).
Discussion
De novo nonsense mutations of ASXL1 account for approximately 75% of BOS cases (Hoischen et al., 2011, Magini et al., 2012, Russell et al., 2015). This disease is fatal, and the underlying cellular and molecular mechanisms remain unknown. Here, we report that global loss of Asxl1 or conditional deletion in osteoblasts and their progenitors leads to multiple developmental defects, including dwarfism, hypoplastic supraorbital ridges, microcephaly, low bone mass, and growth retardation. In addition, Asxl1 loss impairs self-renewal capacity and skews differentiation away from osteoblasts while in favor of adipocytes. Furthermore, deletion of Asxl1 dysregulates the expression of genes controlling BMSC fates. Our study, therefore, demonstrates a pivotal role of ASXL1 in bone homeostasis.
The balance between BMSC self-renewal and differentiation is critical for skeletal homeostasis (Ito and Suda, 2014, Sambasivan and Tajbakhsh, 2007). In this study, we found that Asxl1 null mice exhibited markedly reduced frequencies of BMSCs and CFU-F. Asxl1 loss in BMSCs reduced mesensphere formation and diminished re-plating capability, indicating impaired BMSC self-renewal. The frequency of CFU-F in Asxl1−/− bone marrow was even lower than the percentage of BMSCs as determined by flow-cytometric analysis. Thus, the dramatic reduction of CFU-F capacity of Asxl1−/− bone marrow is likely a combined effect of both reduced frequency and impaired self-renewal of BMSCs. In vitro differentiation assays also revealed markedly reduced osteoblast differentiation potential in Asxl1−/− BMSCs. In contrast, Asxl1−/− BMSCs exhibited an increased propensity for adipocyte differentiation, which is consistent with previous findings demonstrating that ASXL1 negatively regulates adipogenesis in a 3T3-L1 cell line (Park et al., 2011). These results implicate ASXL1 as a critical regulator of BMSC self-renewal and lineage commitment, providing a cellular mechanism underlying defective skeletal homeostasis associated with the Asxl1 deletion. ASXL1 mutations occur de novo in BOS patients, and it is therefore expected that multiple cell lineages are affected. To date, however, the specific cell lineages harboring ASXL1 mutations in BOS patients remain unknown, and further clinical studies are warranted.
The phenotype and fate of a given cell relies on the precise control of gene expression by complex transcriptional and epigenetic networks. Such regulation of gene expression is essential for proper differentiation, cellular function, development, and homeostasis (Feng et al., 2010, Reik, 2007). Histone modifications are complex, post-translational regulatory events that play pivotal roles in governing global gene expression programs. Although extensive studies have focused on the identification of intrinsic targets of transcription factors regulating cellular fate and functions of BMSCs, the epigenetic events that control BMSC identity and/or functions remain largely unknown. In vitro studies indicate that histone modifications (including methylation and acetylation) are a key epigenetic means for gene regulation that contribute to osteogenic lineage determination (Rui et al., 2015, Tan et al., 2009).
ASXL1 regulates epigenetic marks and transcription through interaction with polycomb complex proteins and various transcription activators and repressors (Boultwood et al., 2010, Cho et al., 2006, Scheuermann et al., 2010). We and others have previously reported that loss of Asxl1 reduces global H3K27me3 and H3K4me3 levels in myeloid progenitor cells (Abdel-Wahab et al., 2012, Abdel-Wahab et al., 2013, Wang et al., 2014). Further studies aiming to identify the chromatin and histone markers governing gene transcription for self-renewal and lineage-specific activation or repression in Asxl1 deleted BMSCs are ongoing.
Manipulation of epigenetic mechanisms holds great promise for the future treatment of metabolic bone diseases, cancer, and a multitude of other human disease states. A better understanding of the epigenetic regulation in these processes is required for the development of potential pharmacological agents with sufficient specificity to precisely modulate chromatin remodeling on a global scale. The Asxl1-deficient murine model provides a compelling system to achieve such insights in the context of the human disease BOS, for which the underling molecular basis was previously unknown.
Experimental Procedures
ASXL1 Murine Models
The generation of Asxl1:nlacZ/nGFP knockin and Asxl1fl/fl mice has been previously described (Abdel-Wahab et al., 2013, Wang et al., 2014). Osx-Cre transgenic mice were purchased from Jackson Laboratories. All mice were bred on a C57BL/6 genetic background. All protocols were approved by the Institutional Animal Care and Use Committee at University of Miami Miller School of Medicine.
BMSC Culture and Lineage Differentiation Assays
BMSCs were generated from each experimental group of mice as previously described (Wu et al., 2006). In brief, BMSCs were separated by low-density gradient centrifugation from 3- to 4-week-old mice and then resuspended and cultured in mouse MesenCult medium (MesenCult basal media plus 20% of MesenCult Supplemental; Stem Cell Technologies) at 37°C and 5% CO2. When the cultures reached 80%–90% confluence, cells were trypsinized and re-plated. BMSCs of identical passage number (between passages 3 and 5) were used for experiments.
For osteoblast differentiation, 5 × 104 BMSCs were cultured for 7 days in 6-well plates using osteogenic differentiation medium (MesenCult medium supplemented with 10−7 M dexamethasone, 50 μg/ml ascorbic acid, and 10 mM β-glycerophosphate). For induction of adipocyte differentiation in vitro, 1 × 105 BMSCs were plated in 6-well tissue-culture plates and cultured with adipogenic differentiation medium (MesenCult medium supplemented with 10−7 M dexamethasone, 450 μM isobutylmethylxanthine, 1 μg/ml insulin, and 200 μM indomethacin). For chondrocyte differentiation assays, 1 × 105 BMSCs were plated in 6-well plates and cultured with chondrogenic differentiation medium (osteogenic differentiation medium supplemented with 10 ng/ml transforming growth factor β3).
Mesensphere Assays
For clonal mesensphere formation, BMSCs were plated at clonal density (∼1,000 cells/cm2) in ultralow adherent 24-well plates (Corning) as previously described (Mendez-Ferrer et al., 2010, Pinho et al., 2013). The growth medium contained 15% chicken embryo extract (Fisher Scientific), 0.1 mM β-mercaptoethanol, 1% non-essential amino acids, 1% N2 and 2% B27 supplements (Gibco), basic fibroblast growth factor, insulin-like growth factor-1, epidermal growth factor, platelet-derived growth factor, and oncostatin M (Peprotech) (20 ng/ml) in DMEM/F12 (1:1)/human endothelial (Gibco) (1:2). The cultures were maintained at 37°C in a 5% CO2 water-jacketed incubator and left untouched for 1 week to prevent cell aggregation in low-density cultures. Half of the medium was changed weekly. The mesensphere colonies were detached with trypsin, and the cells were mechanically dispersed and re-plated back into ultralow adherent plates with culture medium. Secondary mesenspheres were counted after 7 days in culture.
Generation of Stably Asxl1-Transduced BMSCs
The full-length mouse Asxl1 cDNA with an N-terminal FLAG tag was cloned and then subcloned into the lentiviral vector. The empty vector was used as a control. All constructs were validated by direct sequencing prior to lentiviral generation. BMSCs were split approximately 16 hr prior to transduction and then transduced overnight with concentrated supernatants. Medium was replaced the next day, and transduced cells were cultured and expanded. EGFP+ cells were sorted using a FACS Vantage flow cytometer. Before further experiments, transduction and selection efficiency were verified by qPCR and western blot analysis.
Gene Expression Analysis by qPCR and RNA-Seq
Gene of interest mRNA levels were determined by real-time qPCR. Total RNA was extracted with TRIzol reagent (Ambion). qPCR was performed in triplicate using an ABI 7500 with SYBR Green PCR kits (Applied Biosystems). mRNA levels were normalized to housekeeping gene β-actin expression. All qPCR primers used are listed in Table S1.
RNA-Seq reads were aligned to the mouse genome reference sequence (GRCm38/mm10) using TopHat (Trapnell et al., 2012) with a tolerance of two mismatches. Cuffdiff (Trapnell et al., 2012) was used to detect the differentially expressed genes with a cutoff of p < 0.05 and false discovery rate (FDR) < 0.25. The identified differentially expressed genes were used for pathway enrichment analysis and functional annotation with the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources (Huang da et al., 2009). Heatmaps were generated in R using the gplot package. GSEA (Subramanian et al., 2005) was performed in MSigDB to generate the list of the most differentially expressed genes, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway signatures and stem cell signatures. Enriched gene sets were selected using a cutoff of p < 0.05 and FDR < 0.25 (Isserlin et al., 2014).
Western Blot Analysis
BMSC cell lysates were subjected to western blot analysis. Isolated proteins were fractionated using NuPAGE 4%–12% Bis-Tris Gels (Invitrogen) and electrotransferred to polyvinylidene fluoride membranes (Roche). Immunoblots were performed using specific antibodies (Table S2). After incubation with anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG (GE Healthcare) antibodies conjugated with horseradish peroxidase, signals were detected using ECL Chemiluminescence Substrate (Pierce).
Statistical Analysis
Differences between experimental groups were determined by Student's t test or ANOVA followed by Newman-Keuls multiple comparison tests as appropriate. p Values of less than 0.05 were considered significant.
Author Contributions
F.-C.Y., Q.-F.W., and M.X. supervised the study; P.Z., M.X., Q.-F.W., and F.-C.Y. designed the experiments; P.Z., C.X., S.D.R., Y.H., K.D., Z.L., F.H., C.Z., L.N., Y.Z., and S.C. performed the experiments; P.Z., C.X., S.D.R., Y.H., F.H., C.Z., K.S.M., T.A.G., O.A.W., M.X., Q.-F.W., and F.-C.Y. analyzed the data; P.Z., M.X., Q.-F.W., and F.-C.Y. wrote the manuscript. All authors reviewed, edited, and approved the manuscript.
Acknowledgments
This work was supported in part by grants from the NIH (CA172408 to F.-C.Y., M.X., CA185751 to F.-C.Y., M.X., and HL112294 to M.X.), the National Natural Science Foundation of China (grant 81270612 to C.X.), and the External Cooperation Program of BIC, Chinese Academy of Sciences (grant 153F11KYSB20150013 to Q.-F. W.). We thank the histological processing and analysis services provided by the Satellite Histological Core of Sylvester Comprehensive Cancer Center Core Facility.
Published: May 26, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.04.013.
Contributor Information
Qian-Fei Wang, Email: wangqf@big.ac.cn.
Feng-Chun Yang, Email: fxy37@med.miami.edu.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is NCBI Gene Expression Omnibus GEO GSE75787 and Genome Sequence Archive (GSA) in BIG Data Center (BIGD) PRJCA000236.
Supplemental Information
References
- Abdel-Wahab O., Adli M., LaFave L.M., Gao J., Hricik T., Shih A.H., Pandey S., Patel J.P., Chung Y.R., Koche R. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–193. doi: 10.1016/j.ccr.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab O., Gao J., Adli M., Dey A., Trimarchi T., Chung Y.R., Kuscu C., Hricik T., Ndiaye-Lobry D., Lafave L.M. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013;210:2641–2659. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry F., Murphy M. Mesenchymal stem cells in joint disease and repair. Nat. Rev. Rheumatol. 2013;9:584–594. doi: 10.1038/nrrheum.2013.109. [DOI] [PubMed] [Google Scholar]
- Bianco P., Robey P.G., Simmons P.J. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohring A., Oudesluijs G.G., Grange D.K., Zampino G., Thierry P. New cases of Bohring-Opitz syndrome, update, and critical review of the literature. Am. J. Med. Genet. A. 2006;140:1257–1263. doi: 10.1002/ajmg.a.31265. [DOI] [PubMed] [Google Scholar]
- Boultwood J., Perry J., Pellagatti A., Fernandez-Mercado M., Fernandez-Santamaria C., Calasanz M.J., Larrayoz M.J., Garcia-Delgado M., Giagounidis A., Malcovati L. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24:1062–1065. doi: 10.1038/leu.2010.20. [DOI] [PubMed] [Google Scholar]
- Carbuccia N., Murati A., Trouplin V., Brecqueville M., Adelaide J., Rey J., Vainchenker W., Bernard O.A., Chaffanet M., Vey N. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23:2183–2186. doi: 10.1038/leu.2009.141. [DOI] [PubMed] [Google Scholar]
- Cho Y.S., Kim E.J., Park U.H., Sin H.S., Um S.J. Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J. Biol. Chem. 2006;281:17588–17598. doi: 10.1074/jbc.M512616200. [DOI] [PubMed] [Google Scholar]
- Feng S., Jacobsen S.E., Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C.L., Lee I., Bloyer S., Bozza S., Chevalier J., Dahl A., Bodner C., Helgason C.D., Hess J.L., Humphries R.K. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev. Biol. 2010;337:9–15. doi: 10.1016/j.ydbio.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer V., Trouplin V., Adelaide J., Bonansea J., Cervera N., Carbuccia N., Lagarde A., Prebet T., Nezri M., Sainty D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- Hastings R., Cobben J.M., Gillessen-Kaesbach G., Goodship J., Hove H., Kjaergaard S., Kemp H., Kingston H., Lunt P., Mansour S. Bohring-Opitz (Oberklaid-Danks) syndrome: clinical study, review of the literature, and discussion of possible pathogenesis. Eur. J. Hum. Genet. 2011;19:513–519. doi: 10.1038/ejhg.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoischen A., van Bon B.W., Rodriguez-Santiago B., Gilissen C., Vissers L.E., de Vries P., Janssen I., van Lier B., Hastings R., Smithson S.F. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 2011;43:729–731. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Isserlin R., Merico D., Voisin V., Bader G.D. Enrichment Map - a Cytoscape app to visualize and explore OMICs pathway enrichment results. F1000Res. 2014;3:141. doi: 10.12688/f1000research.4536.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M., Rosen C.J. PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010;6:629–636. doi: 10.1038/nrendo.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magini P., Della Monica M., Uzielli M.L., Mongelli P., Scarselli G., Gambineri E., Scarano G., Seri M. Two novel patients with Bohring-Opitz syndrome caused by de novo ASXL1 mutations. Am. J. Med. Genet. A. 2012;158A:917–921. doi: 10.1002/ajmg.a.35265. [DOI] [PubMed] [Google Scholar]
- McCauley L.K. c-Maf and you won't see fat. J. Clin. Invest. 2010;120:3440–3442. doi: 10.1172/JCI44786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S., Michurina T.V., Ferraro F., Mazloom A.R., Macarthur B.D., Lira S.A., Scadden D.T., Ma'ayan A., Enikolopov G.N., Frenette P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberklaid F., Danks D.M. The Opitz trigonocephaly syndrome. A case report. Am. J. Dis. Child. 1975;129:1348–1349. doi: 10.1001/archpedi.1975.02120480062016. [DOI] [PubMed] [Google Scholar]
- Park U.H., Yoon S.K., Park T., Kim E.J., Um S.J. Additional Sex Comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2011;286:1354–1363. doi: 10.1074/jbc.M110.177816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S., Lacombe J., Hanoun M., Mizoguchi T., Bruns I., Kunisaki Y., Frenette P.S. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- Rui Y., Xu L., Chen R., Zhang T., Lin S., Hou Y., Liu Y., Meng F., Liu Z., Ni M. Epigenetic memory gained by priming with osteogenic induction medium improves osteogenesis and other properties of mesenchymal stem cells. Sci. Rep. 2015;5:11056. doi: 10.1038/srep11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B., Johnston J.J., Biesecker L.G., Kramer N., Pickart A., Rhead W., Tan W.H., Brownstein C.A., Kate Clarkson L., Dobson A. Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am. J. Med. Genet. A. 2015;167:2122–2131. doi: 10.1002/ajmg.a.37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R., Tajbakhsh S. Skeletal muscle stem cell birth and properties. Semin. Cell Dev. Biol. 2007;18:870–882. doi: 10.1016/j.semcdb.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Scheuermann J.C., de Ayala Alonso A.G., Oktaba K., Ly-Hartig N., McGinty R.K., Fraterman S., Wilm M., Muir T.W., Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada I., Kouzmenko A.P., Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009;5:442–447. doi: 10.1038/nrrheum.2009.137. [DOI] [PubMed] [Google Scholar]
- Tan J., Lu J., Huang W., Dong Z., Kong C., Li L., Gao L., Guo J., Huang B. Genome-wide analysis of histone H3 lysine9 modifications in human mesenchymal stem cell osteogenic differentiation. PLoS One. 2009;4:e6792. doi: 10.1371/journal.pone.0006792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum S.L. Stem cells and osteoporosis therapy. Cell Stem Cell. 2010;7:553–554. doi: 10.1016/j.stem.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccelli A., Moretta L., Pistoia V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- Wang J., Li Z., He Y., Pan F., Chen S., Rhodes S., Nguyen L., Yuan J., Jiang L., Yang X. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood. 2014;123:541–553. doi: 10.1182/blood-2013-05-500272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden S.J., Robling A.G., Sanders M.S., Bliziotes M.M., Turner C.H. Inhibition of the serotonin (5-hydroxytryptamine) transporter reduces bone accrual during growth. Endocrinology. 2005;146:685–693. doi: 10.1210/en.2004-1259. [DOI] [PubMed] [Google Scholar]
- Wei Y., Chen Y.H., Li L.Y., Lang J., Yeh S.P., Shi B., Yang C.C., Yang J.Y., Lin C.Y., Lai C.C. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.J., Liu H., Ridky T.W., Cassarino D., Segal E., Chang H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthley D.L., Churchill M., Compton J.T., Tailor Y., Rao M., Si Y., Levin D., Schwartz M.G., Uygur A., Hayakawa Y. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Estwick S.A., Chen S., Yu M., Ming W., Nebesio T.D., Li Y., Yuan J., Kapur R., Ingram D. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum. Mol. Genet. 2006;15:2837–2845. doi: 10.1093/hmg/ddl208. [DOI] [PubMed] [Google Scholar]
- Wu X., Chen S., He Y., Rhodes S.D., Mohammad K.S., Li X., Yang X., Jiang L., Nalepa G., Snider P. The haploinsufficient hematopoietic microenvironment is critical to the pathological fracture repair in murine models of neurofibromatosis type 1. PLoS One. 2011;6:e24917. doi: 10.1371/journal.pone.0024917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Fan Z., Yu B., Chang J., Al Hezaimi K., Zhou X., Park N.H., Wang C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11:50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.