

Abstract
Chemotherapy-induced cognitive impairment, known also as “chemobrain”, is a medical complication of cancer treatment that is characterized by a general decline in cognition affecting visual and verbal memory, attention, complex problem solving skills, and motor function. It is estimated that one-third of patients who undergo chemotherapy treatment will experience cognitive impairment. Alterations in the release and uptake of dopamine and serotonin, central nervous system neurotransmitters that play important roles in cognition, could potentially contribute to impaired intellectual performance in those impacted by chemobrain. To investigate how chemotherapy treatment affects these systems, fast-scan cyclic voltammetry (FSCV) at carbon-fiber microelectrodes was used to measure dopamine and serotonin release and uptake in coronal brain slices containing the striatum and dorsal raphe nucleus, respectively. Measurements were taken from rats treated weekly with selected doses of carboplatin and from control rats treated with saline. Modeling the stimulated dopamine release plots revealed an impairment of dopamine release per stimulus pulse (80% of saline control at 5 mg/kg and 58% at 20 mg/kg) after 4 weeks of carboplatin treatment. Moreover, Vmax, the maximum uptake rate of dopamine, was also decreased (55% of saline control at 5 mg/kg and 57% at 20 mg/kg). Nevertheless, overall dopamine content, measured in striatal brain lysates by high performance liquid chromatography, and reserve pool dopamine, measured by FSCV after pharmacological manipulation, did not significantly change, suggesting that chemotherapy treatment selectively impairs the dopamine release and uptake processes. Similarly, serotonin release upon electrical stimulation was impaired (45% of saline control at 20 mg/kg). Measurements of spatial learning discrimination were taken throughout the treatment period and carboplatin was found to alter cognition. These studies support the need for additional neurochemical and behavioral analyses to identify the underlying mechanisms of chemotherapy-induced cognitive disorders.
Keywords: Carboplatin, chemobrain, chemotherapy, dopamine, serotonin, fast-scan cyclic voltammetry, reserve pool
Adjuvant chemotherapy has long been a key treatment strategy for either eradicating or slowing the proliferation of cancer cells. Unfortunately, chemotherapeutic treatment approaches, while effective in killing cancer cells, are also toxic to other cell populations in the body. This toxicity has been addressed by redesigning chemotherapeutic compounds to maximize effective treatment and minimize the harmful impact. Nevertheless, chemotherapy still leads to multiple side effects that decrease patients’ quality of life not only during treatment, but also long after the treatment has concluded. Cells in the central nervous system (CNS) are particularly vulnerable to the effects of chemotherapy. This vulnerability applies to both dividing and nondividing cell populations after both localized and peripheral treatment with chemotherapy.1−7
Chemotherapy-induced cognitive impairment, also known as “chemobrain”, is a syndrome caused by the toxicity of chemotherapy agents to the CNS.8 Improvements in treatment and early detection capabilities have improved the survivability of many cancers, especially breast cancer. With this improved survival rate, the identification of postchemotherapy cognitive impairment has increased dramatically. About 18% of breast cancer patients that receive standard chemotherapy treatment9 and 30% of all cancer patients that receive high level chemotherapy treatment experience chemobrain.10 Another report suggests that up to one-third of females who have undergone chemotherapy treatment for breast cancer report symptoms of chemobrain.11 These symptoms include loss of verbal and visual memory, a decrease in mental flexibility, attention deficits, and a loss of motor function.12,13
Although the underlying causes of chemobrain are still unknown, a variety of mechanisms have been proposed. One possibility is that the expression of genes that heighten the probability of getting cancer may also increase the likelihood for vulnerability to the negative side effects of chemotherapy treatment.14 These genetic factors include blood-brain barrier dysfunction, impaired DNA repair mechanisms, and dysregulation of the immune system.14−18 It has also been proposed that chemotherapy-induced DNA damage can lead to an increase in cytokine production that promotes a chronic state of inflammation leading to cognitive impairments.19−23 Chemobrain may also be caused, in part, by the disruption of blood flow throughout the vasculature of the brain. This phenomenon could result in ischemia, which may directly affect cognition due to oxidative stress.24 Moreover, it has been suggested that the inherent toxicity of many chemotherapeutic agents may impair neurotransmitter signaling.14 Both dividing and nondividing progenitor cells and oligodendrocytes in the brain have also been shown to be highly sensitive to some chemotherapeutics, even more so than the cancer itself.25
Dopamine (DA) and serotonin (5-HT) are CNS neurotransmitters involved in a variety of neurological functions. DA plays roles in reward,26 cognition,27 and locomotor control.27 Alterations in DA system function have been observed in response to chemically induced oxidative insult,28 genetic modifications that model oxidative stress,29 and neurodegenerative disease.28,30−35 Serotonin (5-HT) has been implicated in mood and cognition, as well as other neurological functions.36−38 Similar to the case of DA, functional alterations of the 5-HT system have been observed both in models of oxidative stress39 and neurodegenerative disease.39,40 Therefore, we hypothesized that treatment with chemotherapeutic agents would impair DA and 5-HT release. The identification of such impairments would have important consequences for the cognitive processes that involve DA and 5-HT signaling, as well as imply that signaling by other neurotransmitter systems also may be affected.
Carboplatin (cis-diammine(1,1-cyclobutanedicarboxylato)platinum(II)), a chemotherapeutic agent commonly employed in the treatment of various cancers, including those of the head, neck, breast, ovaries, bladder, and colon,40−42 has become a common alternative to cisplatin, which had caused severe side effects during and after treatment.43 Despite its reduced toxicity, carboplatin has also been implicated in chemobrain.8,44,45 To assess the effects of carboplatin treatment on the function of dopaminergic and serotonergic terminals, male Wistar rats were injected intravenously with carboplatin once a week for 4 weeks. Measurements of electrically evoked DA release and uptake, measured with FSCV in coronal brain slices, revealed that release was impaired even though reserve pool DA and the total content of DA were unchanged. Furthermore, Vmax was diminished, indicating that the uptake process is negatively impacted by exposure to carboplatin. Similar to DA, stimulated 5-HT release was also impaired. Moreover, spatial learning measurements in operant conditioning chambers revealed that carboplatin treatment altered cognitive function. These findings provide insight into how a commonly used chemotherapeutic agent impacts subsecond neurotransmitter release and uptake dynamics, and suggest its involvement in chemobrain.
Results and Discussion
Striatal Dopamine Release
Impairments in the release of DA, 5-HT, glutamate, GABA, and other neurotransmitters could be detrimental to many neurological functions, including cognition, mood, and the control of movement. Clinical studies have suggested that DA may play a role in chemobrain.46,47 Additionally, preclinical research implies the involvement of DA impairment in chemobrain.14,47−51 It has been well-established that dopaminergic function in the basal ganglia is important in cognition.27,52−57 Therefore, we investigated the release and uptake properties of DA, which are readily measured electrochemically with FSCV.
The striatum was divided into four quadrants in the coronal plane, and electrically stimulated DA release and uptake were quantified in the quadrants. The dorsal striatum is associated with motivation and reward as well as motor function58,59 whereas the ventral striatum has shown an association with reinforcement learning.60 Measurements were taken in the four quadrants to account for potential regional differences in release, which are known to have a dorsal to ventral gradient in multiple rodent species and nonhuman primates.61
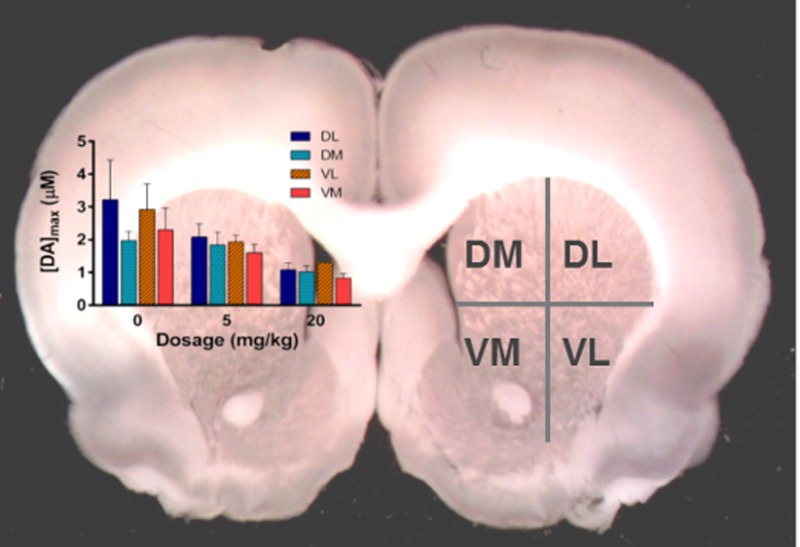
Electrically stimulated DA release in the dorsal lateral (DL), dorsal medial (DM), ventral medial (VM), and ventral lateral (VL) quadrants of the striatum was measured with FSCV in coronal brain slices (Figure 1A). Four measurements were taken at random locations within each quadrant and averaged. Representative data for 0, 5, and 20 mg/kg carboplatin treatment groups are shown in Figure 1B. The representative cyclic voltammograms indicate that the analyte measured was DA. Each color plot consists of a series of unfolded, stacked cyclic voltammograms. The current response is color-coded (z-axis). The absence of faradaic currents, other than those due to DA oxidation and reduction, suggest that no additional electroactive species are released in response to electrical stimulation in either the brain slices from carboplatin-naïve animals or from 20 mg/kg carboplatin-treated animals.
Figure 1.

Dopamine release comparison between saline-treated and carboplatin-treated rats. (A) Image of a coronal brain slice with labeled quadrants of the striatum. (B) Representative color plots and stimulated DA release plots, sampled along the horizontal dashed lines on the color plots, are shown. Cyclic voltammograms, sampled along the vertical dashed lines on the color plots, are positioned directly above the stimulated release plots and confirm the presence of DA.
The presence of DA was confirmed by the cyclic voltammograms sampled at the point of peak release on each plot. There was no significant effect of region on stimulated DA release (Figure 2A). Given the lack of regional effects, DA levels were averaged across striatal quadrants, revealing a statistically significant carboplatin treatment effect on DA release in the striatum (detailed statistical information can be found in the Supporting Information): 72 ± 10% of vehicle control at 5 mg/kg and 41 ± 7% % at 20 mg/kg (Figure 2B). The attenuation of DA release was, therefore, found to be generalized throughout the entire striatum. Since DA is involved in motor function and cognition,25 it is possible that the symptoms of chemobrain, such as short-term memory loss and fatigue, could be influenced by a generalized depression of the release of dopamine into the striatal synapses for chemotherapy treated patients.27,62,63 However, the suppression of other neuronal populations, including GABAergic neurons and glutamatergic neurons, cannot be ruled out.
Figure 2.

Dopamine release measurement at multiple doses. (A) [DA]max at each dose shown in each of the four regions of the striatum. (B) [DA]max averaged across all brain regions at each dose (***p < 0.001, 20 mg/kg versus 0 mg/kg, n = 5–9 rats per group).
Determination of [DA]p and Vmax
To quantify DA uptake in carboplatin-treated rats, the stimulated release plots were modeled. This modeling process allowed the determination of Vmax, the maximum rate of DA uptake, and [DA]p, DA release per electrical stimulus pulse. The determination of [DA]p corrects for electrode performance and DA uptake by the dopamine transporter that occurs concomitant with stimulated release. The curve-fitting process is described in Methods, and a representative fit of a stimulated release plot is shown in Figure 3A. A statistically significant attenuation in [DA]p was found across all regions of the striatum after 4 weeks of carboplatin treatment: 67 ± 10% of saline control at 5 mg/kg and 44 ± 8% at 20 mg/kg (Figure 3B and C). Similar to the uncorrected release data in which [DA]max was determined, no significant effect was found between the four regions of the striatum probed. Additionally, a substantial effect of carboplatin dose on Vmax (Figure 3D and E) was found: 55 ± 9% of saline control at 5 mg/kg and 57 ± 8% at 20 mg/kg. Similar to the [DA]p data, there was no statistical difference in Vmax between the regions of the striatum sampled. To illustrate the overall effect of carboplatin treatment on striatal DA release within the striatum, graphical representations of [DA]p and Vmax are shown in Figure 3C and E, respectively, with these values averaged across the four quadrants.
Figure 3.

Dopamine release and uptake are attenuated by carboplatin treatment. (A) Representative example of a fitting curve used for modeling. The arrow indicates the point of stimulation. (B) [DA]p measured from each dosing group in each region. No regional differences in the striatum were found, but generalized release was sharply attenuated. (C) [DA]p averaged across all brain regions at each dose. (D) Vmax measured from each dosing group in each region. No significant difference in Vmax was found between regions. (E) Vmax averaged across all brain regions at each dose (*p < 0.5, ***p < 0.001, n = 5 rats per group).
These data show that DA per pulse, as well as DA uptake, is diminished in carboplatin-treated rats. These findings suggest that the decrease in stimulated DA release that occurred in response to electrical stimulation did not arise from enhanced DA clearance, given the competitive relationship between DA release and uptake, by the dopamine transport system. According to Michaelis–Menten kinetics, Vmax is directly proportional to the number of dopamine transporter protein molecules.64 Therefore, it is possible that the total number of functioning dopaminergic terminals decreases with carboplatin treatment—a finding that is not surprising given the inherent toxicity of carboplatin. It is important, however, to obtain measures of how much DA is present within the terminals since this parameter would be a measure of how much DA is available for release.
Striatal Dopamine Content
Striatal tissue samples were analyzed by high performance liquid chromatography (HPLC) with electrochemical detection to measure the total content of DA and 3,4-dihydroxyphenylacetic acid (DOPAC) (Table 1). We found no significant dose effect of carboplatin treatment on DA content (one-way ANOVA, p > 0.05; 0, 5, and 20 mg/kg, n = 5), DOPAC content (one-way ANOVA, p > 0.05; 0, 5, and 20 mg/kg, n = 5 rats per group), or DA/DOPAC (one-way ANOVA, p > 0.05; 0, 5, and 20 mg/kg, n = 5 rats per group) in striatal tissue samples. Several conclusions can be drawn from these data. First, since DOPAC levels, as well as the DA/DOPAC ratio, did not change with carboplatin treatment, it is unlikely that the metabolism of DA to DOPAC is altered by exposure to this chemotherapeutic agent. Second, these results suggest that overall DA content is not affected by chronic treatment with carboplatin. Thus, DA is present in proper amounts in carboplatin treated rats, but it is not released efficiently.
Table 1. Total DA and DOPAC content and DA/DOPAC ratio from homogenized striatal lysates. The number of rats analyzed (n) is indicated in parentheses.
| DA (μg DA/mg protein) | DOPAC (μg DOPAC/mg protein) | DA/DOPAC | |
|---|---|---|---|
| 0 mg/kg (n = 5) | 1.1 ± 0.3 | 0.31 ± 0.09 | 4.1 ± 0.5 |
| 5 mg/kg (n = 5) | 1.1 ± 0.3 | 0.30 ± 0.09 | 3.6 ± 0.6 |
| 20 mg/kg (n = 5) | 0.88 ± 0.3 | 0.25 ± 0.08 | 2.9 ± 0.7 |
Dopamine Reserve Pool Content
The generalized three-pool model of neurotransmitter-containing vesicles within neurons consists of a readily releasable pool, a recycling pool, and a reserve pool.65−67 The readily releasable pool represents only 1–2% of vesicles within a neuron.67 In the context of voltammetric recordings, DA from the readily releasable pool in striatal slices is released and detected by FSCV upon application of a single electrical stimulus pulse. After depleting the readily releasable pool of vesicles, these vesicles are replenished by the recycling pool, which accounts for 5–20% of the total number of vesicles within neurons.68−70 The reserve pool constitutes 80–90% of the total vesicles within neurons and is thought to be mobilized in response to increased levels of synaptic activity.65−67
Experimentally, reserve pool DA can be mobilized by intense electrical stimulation29,31,70−72 or pharmacological manipulation.31,72 Here, we isolated the reserve pool of DA in striatal brain slices from saline-treated (see raw data in Figure 4A) and carboplatin-treated (see raw data in Figure 4B) rats by blocking DA synthesis with α-methyl-p-tyrosine methyl ester (αMPT) while applying single-pulse electrical stimulations every 5 min.31 The compound αMPT inhibits DA production by blocking tyrosine hydroxylase, the rate-limiting enzyme in DA synthesis.73 When αMPT was present in the perfusion buffer, the DA signal decreased in response to repeated stimulation, leaving only the reserve pool of DA within the terminals. The signal dissipation from 100% to 10% was not significantly different between slices from carboplatin-treated rats and vehicle-treated rats (carboplatin, 120.0 ± 2.3 min, n = 5 rats; vehicle, 140.8 ± 19.1 min, n = 6 rats; p = 0.50, unpaired t test), suggesting that releasable DA vesicles are present in similar amounts across the two groups.
Figure 4.

Quantitation of reserve pool DA. Representative raw data traces of reserve pool content analysis in brain slices from (A) saline-treated and (B) carboplatin-treated (20 mg/kg) rats. For a typical measurement, slices were continuously exposed to αMPT, a tyrosine hydroxylase inhibitor, to block DA synthesis. Electrically evoked DA release was measured every 5 min until the signal diminished (occurring at 150 min in the data shown). Traces are shown here every 30 min for clarity. The addition of AMPH then caused the efflux of reserve pool DA. The cyclic voltammogram (upper right of each trace) confirms the presence of DA at the peak of the AMPH-induced efflux. (C) Bar graph representing the average of the [DA]AMPH measurements (p > 0.05, n = 5 rats per group).
Subsequent application of AMPH, in the continued presence of αMPT, resulted in the spontaneous, quantifiable efflux of reserve pool of DA. The concentration of DA measured as a result of αMPTand AMPH treatment was 2.6 ± 0.5 μM in brain slices prepared from control rats and 2.5 ± 0.6 μM in slices from 20 mg/kg carboplatin-treated rats (Figure 4C). There was no significant difference in this DA efflux (unpaired t test, p > 0.05, n = 6 control rats and 5 rats treated with 20 mg/kg carboplatin), suggesting that the mechanism leading to stimulated DA attenuation in the striatum is not based on storage impairment.
Additionally, the physical striatal area was not grossly altered by carboplatin treatment (Figure 1, Supporting Information), suggesting that the striatum and DA terminals have not undergone significant atrophy.
Given these DA content results, we conclude that evoked release is not diminished due to a decrease in overall tissue content or reserve pool. Thus, DA is present in the terminals; however, it is likely that the ability of neurons to release it is impaired.
Dorsal Raphe Serotonin Release
To examine further the mechanism underlying chemobrain, we also measured the stimulated release of 5-HT within the dorsal raphe nucleus. Serotonin plays a vital role in cognition, including memory and learning processes74,75 and synaptic plasticity.76 Furthermore, the interplay between 5-HT and other neurotransmitter systems, including DA, has been shown.77,78 Due to the specific involvement in cognition and interplay with the DA system, we chose to study electrically stimulated 5-HT release in the dorsal raphe nucleus by FSCV. Five measurements were taken within the dorsal raphe from each brain slice and averaged. Representative data from the 0 and 20 mg/kg carboplatin treatment groups are shown in Figure 5A. The representative cyclic voltammograms indicate that the analyte measured is 5-HT. As shown in Figure 5B, the peak concentration of serotonin released following electrical stimulation was significantly diminished in 20 mg/kg carboplatin-treated rats compared to saline-treated rats (45 ± 9% of saline control, n = 5 carboplatin-treated and 5 vehicle-treated rats, p < 0.05, unpaired t test).
Figure 5.

Serotonin release impairment in carboplatin-treated rats. (A) Representative plots of stimulated 5-HT release. Cyclic voltammograms above the stimulated release plots confirm the presence of 5-HT. (B) Bar graph showing attenuation of peak 5-HT release in 20 mg/kg carboplatin-treated rats (45% compared to saline-treated controls) (**p < 0.01, n = 5 rats per group).
Our results suggest that impaired 5-HT release may contribute to the cognitive deficits experienced by patients who have undergone chemotherapy treatment. Taken together with DA release impairments, these results indicate that chemotherapy may induce a general effect upon the mechanism of neurotransmitter release. Thus, it is likely that the release of other neurotransmitters, such as glutamate and GABA, is impaired; consequently, the impact of these deficits on cognitive function should also be considered.
Spatial Learning Measurements
To test for carboplatin related deficits in spatial learning, rats were evaluated on a spatial learning paradigm in which five response options (i.e., nose-pokes detected by infrared beam) were presented in a horizontal row. When the rats poked their noses to a target preselected by the investigator, a food reward was delivered. This paradigm consistently results in Gaussian response distributions centered on the target location, with steeper Gaussian functions indicating more robust spatial learning.79Figure 6 shows the spread of responses to nontarget nose pokes (A), responses at the nontarget positions, 2 and 4 (B), overall rates of nose pokes (C), and rats’ weights during the testing phase (D). As can be seen, the responses to nontarget nose poke locations (expressed as a proportion of the rate of nose pokes on the target location) fit well into a Gaussian function for rats treated with saline (r2 = 0.99) and 20 mg/kg carboplatin (r2 = 0.96). The parameters for these Gaussian functions significantly differed between groups (F[2,64] = 4.339, p < 0.05). Moreover, t test of responding at individual targets found that the proportion of target responses on nose poke location 2 (t[60] = 2.0651, p < 0.05) and 4 (t[60] = 2.41287, p < 0.05) differed significantly between carboplatin- and vehicle-treated rats (Figure 6B). No significant differences were obtained for overall rate of nose pokes (Figure 6C; p = 0.65) or rats’ weights during the testing phase (Figure 6D; p = 0.29).
Figure 6.

Spatial learning measurements obtained using operant conditioning chambers. The spread of responses (A) is expressed as a proportion of the rates of nose pokes at each location and is fit to a Gaussian function for both the vehicle-treated (r2 = 0.99) and carboplatin-treated (r2 = 0.96) rats (F [2,64] = 4.339, p < 0.05, n = 7, vehicle versus carboplatin). The responses at the nontarget positions, 2 and 4 (B), overall rates of nose pokes (C), and rats’ weights during the testing phase (D) are also shown (*p < 0.05, n = 7 rats per group).
Consistent with the neurochemical data, a treatment course of carboplatin resulted in altered cognitive function. Here, carboplatin resulted in small but significant alterations in spatial learning/memory that resulted in increased responding to nontarget nose poke locations on a spatial learning paradigm. The degree of cognitive impairment observed following carboplatin treatment is consistent with chemotherapy patients’ symptoms, which often present as subtle impairments in parameters such as learning, concentration, reasoning, and executive function.8 An important aspect of these cognitive measurements is that the overall rates of responding did not significantly differ between carboplatin- and vehicle-treated groups (p > 0.05). Additionally, rats did not lose weight during or after treatment, suggesting that food consumption and general feeling of well-being was not impacted. Taken together, these results strongly suggest that the decreased responding of carboplatin-treated rats on locations 2 and 4 was not due merely to a detriment of overall health, but rather to the more specific effect of cognitive impairment.
Conclusions
To our knowledge, this is the first published study that investigates dopamine and serotonin release alterations occurring as a result of chemotherapeutic drug administration. We found an impairment of striatal DA release and uptake in response to treatment with carboplatin that were not accompanied by significant regional differences in this attenuation. Thus, DA release impairment was generalized across the entire striatum. Because Vmax was diminished, it is unlikely that the decrease in stimulated DA measured was a result of enhanced clearance. DA reserve pool content and overall content of DA were unaffected by treatment. Therefore, we conclude that DA is present in the striatum, but it is not released as efficiently. We also showed that 5-HT release was impaired in carboplatin-treated rats, suggesting that multiple neurotransmitter systems are altered as a result of chemotherapy treatment. Moreover, our spatial learning paradigm result indicate that carboplatin treatment results in cognitive impairment of these rats.
The particular mechanisms underlying neurotransmitter release impairments following carboplatin treatment have not yet been identified. However, it is possible that multiple factors contribute, including influences by other neurotransmitters and neuromodulators that have become dysregulated due to carboplatin treatment, damage to proteins that mediate exocytosis, and morphological degradation of neuronal terminals. In future studies it will be important to address these issues to identify specific cellular mechanisms that contribute to release impairment. Additionally, it will be critical to design behavioral and neurochemical experiments so that we can determine the relevance of neurotransmitter release alterations in specific brain regions, such as the striatum and dorsal raphe nucleus, to cognitive ability.
In summary, our spatial learning paradigm results indicate a decrease in cognitive performance; thus, it is possible that diminished neurotransmitter release capability negatively impacts cognitive ability. Collectively, these findings (1) suggest neurotransmitter release impairment as a possible mechanism of cognitive dysfunction in patients treated with chemotherapeutic agents and (2) support the need for more refined behavioral and neurochemical analyses to elucidate how release is impaired and which neurotransmitter systems are affected.
Methods
Animals
All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the University of Kansas Institutional Animal Care and Use Committee. Male Wistar rats (Charles River Laboratories, Inc., Wilmington, MA) were housed two or three per cage in the University of Kansas Animal Care Unit. Food and water was available ad libitum. Rats were maintained on a 12 h light/dark cycle with lights on at 6:00 AM and lights out at 6:00 PM. A temperature of 70 ± 2 °C and humidity level of 50 ± 20% were maintained. Rats were approximately 9 weeks old upon arrival. For neurochemical measurements, rats were 10 weeks old at time of first treatment with carboplatin or saline and 14 weeks old at the time of analysis with FSCV. For behavioral measurements, rats were about 12 weeks old at time of carboplatin treatment. A total of 43 rats were used for neurochemical measurements and 14 rats were used for spatial learning measurements to allow sufficient statistical power for each analysis. All behavioral sessions were conducted during the light phase on the light-dark cycle.
Drugs
Carboplatin (lot number C2538), d-amphetamine sulfate (lot number 065 K1894), and α-methyl-dl-tyrosine methyl ester hydrochloride (lot number 037 K1402) were purchased from Sigma-Aldrich (St. Louis, MO). For reserve pool measurements, AMPH was dissolved in phosphate buffered saline (0.9% NaCl, 2.5 mg/mL) prior to use. For all 5-HT and reserve pool DA experiments, pharmaceutical grade carboplatin (lot number 61703-339-50) was purchased from Hospira (Lake Forest, IL).
Electrode Fabrication
Carbon-fiber cylindrical microelectrodes were fabricated as previously described.28,80 Briefly, individual 7 μm carbon fibers, purchased from Goodfellow Cambridge Ltd. (Huntingdon, England), were loaded into glass capillaries (4 in, 1.2 mm OD; A-M Systems, Inc. Carlsborg, WA) and pulled using a heated coil puller (Narishige International USA, East Meadow, NY). Carbon-fiber tips were then cut with a scalpel 25 μm from the end of the glass seal. Electrodes were sealed by dipping into a well-mixed epoxy mixture of 0.24 g of EPI-CURE 3234 Curing Agent (lot FCXC4114/0886GG) and 2.00 g of EPON Resin 815C (lot HADN0003/1307GG). Excess resin was removed by dipping several times in toluene, and electrodes were then baked for 1 h at 100 °C. The electrodes were backfilled with 0.5 M potassium acetate to establish electrical connections between the carbon-fibers and the inserted silver wires. For 5-HT detection, electrodes were coated with Nafion (Nafion perfluorinated ion-exchange resin, 5 wt % solution in a mixture of lower aliphatic alcohols and water, Sigma-Aldrich, St. Louis, MO) via electrodeposition using an adaptation of previously described methods.81,82 Electrodes were dipped in Nafion, and a potential of 1.0 V was applied for 30 s. Nafion-coated electrodes were then cured at 70 °C for 10 min and stored for no longer than 1 week prior to use.
Chemotherapy Treatment
Male Wistar rats received one injection (iv, tail vein) of carboplatin once a week for four consecutive weeks. All carboplatin solutions were made up in saline (2.5 and 15 mg/mL) prior to injection. There were three experimental groups that included treatments with 0.9% biological saline, 5.0 mg/kg carboplatin, and 20 mg/kg carboplatin. Both dosage and treatment regimen were chosen to mimic clinical dosing regimens and to allow the drug effects to stabilize. Although the dose of carboplatin varies dramatically in a clinical setting, a 20 mg/kg dose of carboplatin, which corresponds to a clinical dose of 120 mg/m2,83 falls within the middle of a typical dose range in humans.84
To examine further the effect of carboplatin treatment on DA reserve pool efflux and stimulated 5-HT release, Male Wistar rats received one injection (iv, tail vein) of carboplatin once a week for four consecutive weeks. For this round of treatment, there were two experimental groups consisting of treatment with 0.9% biological saline or a 20 mg/kg dose of pharmaceutical grade carboplatin (10 mg/mL, Hospira, Inc.).
Brain Slices
Brain slices were harvested as previously described.32 Briefly, rats were deeply anesthetized by isoflurane inhalation and decapitated. The brain was then immediately removed and placed into ice-cold artificial cerebral spinal fluid (aCSF). The aCSF solution contained the following concentrations: 2.5 mM KCl, 126 mM NaCl, 1.2 mM NaH2PO4, 25 mM NaHCO3, 2.4 mM CaCl2, 1.2 mM MgCl2, 20 mM HEPES, 11 mM d-glucose. The pH was adjusted to 7.4. To ensure the tissue received ample oxygen, the aCSF was continuously bubbled with 95% O2/5% CO2 throughout the experiment. After chilling for 1 min, the cerebellum was removed and the brain was bisected longitudinally using a sterile razor blade. The striatum was dissected from the sample and was stored at −80 °C and saved for HPLC analysis. The other hemisphere of the brain was then glued to a plate against a cube of agar for support. Several 300 μm coronal brain slices were then obtained using a vibratome (Leica Microsystems, Bannockburn, IL). In a typical recording session, a single striatal brain slice was transferred to a perfusion chamber where oxygenated aCSF, maintained at 34 °C using a thermostated perfusion chamber and in-line heater, flowed over the slice at 2 mL/min. For all 5-HT release measurements, brain slices containing the dorsal raphe were obtained and transferred to a perfusion chamber using the method described. Slices were equilibrated for at least 1 h before collecting measurements.
Striatal Area Measurements
After tissue harvesting, coronal slices containing the striatum was transferred to a Petri dish containing aCSF. A glass slide was immediately placed on top of the slice to prevent the tissue from curling. The slice was then placed under a stereomicroscope (Nikon SMZ745, Japan). Brain slices were imaged by an eyepiece camera (AmScope MU300, Irvine, CA) mounted on the stereomicroscope. The striatum was selected and outlined as show in Supplemental Figure 1 using Photoshop CS6 (Adobe, San Jose, CA). The area of the striatum in pixels was determined and converted to cm2 via calibration (See Supporting Information Figure 1).
Electrochemical Measurements Using FSCV
Procedures for measuring DA and 5-HT release and uptake with background-subtracted FSCV have been described in detail previously.31,32,82,85 Briefly, a precalibrated cylinder carbon-fiber microelectrode was inserted 100 μm into the brain slice using micromanipulators and a stereoscope. The electrode was positioned between two biphasic stimulating electrodes (A-M Systems Inc., Carlsborg, WA) in the striatum. For DA detection, triangular waveform starting at −0.4 V, scanning up to +1.0 V, and back to −0.4 V, was applied to the carbon-fiber microelectrode at a scan rate of 300 V/s and an update rate of 10 Hz. For 5-HT detection, a waveform of +0.2 V, up to +1.0 V, down to −0.1 V and back up to +0.2 V was applied at a scan rate of 800 V/s and an update rate of 10 Hz. An Ag/AgCl reference electrode was used. DA release was evoked by applying a single, biphasic current pulse (current of 350 μA, 4 ms total duration) to the stimulating electrode. The peak current after stimulation was used for all release measurements. The current measured from DA oxidation was plotted versus potential and the successive voltammograms were plotted versus time. To account the for the natural heterogeneity of DA release in the striatum, measurements were taken in four different regions of the striatum four times, therefore obtaining 16 measurements at unique positions for each slice. Two or three slices were analyzed per rat.
To monitor reserve pool dopamine efflux, single pulse measurements were taken every 5 min while treating the brain slice with αMPT. These measurements were taken until stimulated DA release was completely diminished. At this point, the brain slice was perfused with 20 μM AMPH and 50 μM αMPT. DA release was then continuously measured for 25 min without electrical stimulation.
Modeling Stimulated Release Plots
Dopamine release and uptake kinetics were analyzed using modeling software written by R.M. Wightman (University of North Carolina, Chapel Hill, NC). This modeling software was used to measure dopamine per pulse ([DA]p), which is dopamine release per electrical stimulus corrected for electrode performance and uptake, and Vmax, which is the maximum rate of dopamine uptake. The time of stimulation is loaded into the software; however, the actual rise in the DA oxidation current typically does not occur immediately after stimulation. This delay, which may be on the order of 100 ms, is influenced by several factors, including the time it takes for DA to diffuse to the electroactive surface and adsorption of DA to the electrode surface. The mechanism of DA reuptake has been well-defined previously.30 Briefly, DA uptake obeys Michaelis–Menten kinetics86−88 under the following reaction paradigm:
where extracellular dopamine, (DA)o, is converted to intracellular dopamine, (DA)i. T represents the dopamine transporter. The curves were fitted to the equation that describes the rate of DA uptake:
where [DA] represents the extracellular concentration of DA in the brain, [DA]p is the change in [DA] at the electrode surface in response to each electrical stimulus pulse, and Vmax and KM are constants in the Michaelis–Menten equation that describe how the transporter functions.64 A KM value of 0.2 μM was used during the modeling operation. The parameters Vmax and [DA]p were then adjusted to fit the traces, as shown in Figure 3A.
Striatal Dopamine Content
Striatal tissue samples were stored at −80 °C until use. Samples were homogenized in 0.1 M perchloric acid and centrifuged at 7200g for 10 min. The same day dopamine tissue content was determined using HPLC coupled with electrochemical detection. The supernatant was manually injected onto a Phenomenex Luna 2.5u C18(2)-HST column (100 × 3.00 mm) for separation followed by detection using an ESA 5014B microdialysis cell (E1 = −150 mV; E2 = +230 mV). A guard cell (ESA 5020) was used in-line before the injection loop and was set at +350 mV. The mobile phase was delivered at a flow rate of 0.38 mL/min by a Shimadzu LC-20AD HPLC pump (Shimadzu Scientific Instruments, Columbia, MD). The mobile phase composition was: 75.2 mM sodium phosphate (monobasic, monohydrate), 1.39 mM 1-octanesulfonic acid (sodium salt, anhydrous), 0.125 mM ethylene diamine tetraacetic acid, 0.0025% triethylamine, and 10% acetonitrile; pH 3.0 adjusted with 85% phosphoric acid. Dopamine peak areas were integrated and quantified against known standards using LC Solutions Shimadzu Software. Dopamine levels were normalized to total protein levels as determined (in duplicate) by the commercially available BCA protein assay kit from Thermo Scientific (Thermo Fisher Scientific Inc., Waltham, MA). Final values were reported as μg dopamine/mg protein.
Spatial Learning Measurements
Deficits in learning and memory are commonly reported after patients undergo chemotherapy.89−91 Moreover, neuropsychological testing has found impairments in visuo-spatial skills up to one year after treatment.92 As a result, behavioral testing paradigms designed to identify these common clinical concerns in laboratory rodents must provide a robust and face valid test of any proposed neurobiological mechanism thought to cause chemobrain. Unfortunately, the behavioral paradigms primarily applied to chemobrain poorly address the learning processes disrupted in chemobrain. Many of the paradigms test simple associative learning,7−9 which falls short of the complexity entailed in the cognitive deficits seen in chemobrain.
Within an operant learning framework, if an organism is reinforced for responding on a particular target within a horizontally arranged array, patterns of responding are Gaussian in form and centered on the target response.79 These generalization gradients are consistently observed, with spatial learning failures manifest as wider and less focused gradients.93 The present experiment examined these generalization gradients in rats treated with carboplatin versus saline vehicle.
Sessions occurred in operant conditioning chambers (33.0 cm long, 24.1 cm wide, 29.2 cm high; Med Associates, Inc., St. Albans, VT). The rear wall was curved with five (2.54 cm × 2.54 cm) nose poke receptacles fitted with stimulus lights and infrared sensors 2.54 cm apart arranged horizontally 2 cm from the floor. Centered on the front wall, 2 cm above the floor grid was a pellet receptacle (3 cm × 4 cm) into which a pellet dispenser could dispense grain based pellets (45 mg; Bio-Serv, Frenchtown, NJ). A 28 V DC houselight centered on the back wall (26.7 cm from the floor) provided general illumination. Chambers were housed in sound attenuating cubicles with fans to mask extraneous noise. All experimental events were programed and recorded using MED-PC IV software controlled by a PC.
Sessions occurred 6 days a week at approximately the same time each day and ended either after the rats earned 100 reinforcers or 1 h had passed. At the beginning of each session, the house light and the stimulus lights in each of the five nose poke receptacles was turned on. In each session, one of the five nose pokes was made active. Specifically, triggering the infrared sensor in that receptacle resulted in a brief tone (0.1 s), the extinguishing of all nose poke cue lights for 5 s, the lighting of the pellet receptacle, and the delivery of one 45 mg pellet. Responding on the other four nose poke receptacles was recorded but did not result in any differential consequences. The target nose poke remained constant for each session, with the order of conditions varying quasi-randomly.
Statistics
Statistical analyses were conducted using GraphPad Prism software (GraphPad Software Inc., San Diego, CA). For statistical analyses, N = the number of rats. Data are reported as means ± SEMs.
Acknowledgments
The authors thank Prof. Susan Lunte (University of Kansas) and Prof. Craig Lunte (University of Kansas) for intellectual contributions regarding the conduct of this research.
Glossary
Abbreviations
- FSCV
fast-scan cyclic voltammetry
- DA
dopamine
- [DA]p
dopamine per pulse
- Vmax
maximal uptake rate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.5b00029.
Image showing the striatal area as unaffected in carboplatin-treated rats; one-way and two-way ANOVA data (PDF)
Author Contributions
All authors contributed to the writing and editing of the manuscript. The experiments were conceived of by M.A.J. and D.P.J. Total dopamine content was determined by T.M. and B.N. Dopamine and serotonin release analyses were carried out by S.V.K., R.A.L., R.C.G., J.L.D., G.L.O., M.D.N., and M.A.J. The behavioral experiments were carried out by M.J.S., S.V.K., R.C.G., and D.P.J..
This research was funded by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health Award Number P20GM103638-02 (to M.A.J.), a National Institutes of Health Exploratory Developmental Research Grant Award Number R21NS077485-02 (to M.A.J.), an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health Grant Award Number P20GM103418 (to R.A.L.), and an Institutional Research Grant from the American Cancer Society Grant Award Number QK86026N (to D.P.J.). The authors also thank the University of Kansas and the R. N. Adams Institute for Bioanalytical Chemistry for funding and support.
The authors declare no competing financial interest.
Supplementary Material
References
- Shapiro W. R.; Young D. F. (1983) Neurological complications of antineoplastic therapy. Acta Neurol. Scand. Suppl. 100, 125–132. [PubMed] [Google Scholar]
- Keime-Guibert F.; Napolitano M.; Delattre J.-Y. (1998) Neurological complications of radiotherapy and chemotherapy. J. Neurol. 245, 695–708. 10.1007/s004150050271. [DOI] [PubMed] [Google Scholar]
- Verstappen C. C.; Heimans J. J.; Hoekman K.; Postma T. J. (2003) Neurotoxic complications of chemotherapy in patients with cancer. Drugs 63, 1549–1563. 10.2165/00003495-200363150-00003. [DOI] [PubMed] [Google Scholar]
- Minisini A.; Atalay G.; Bottomley A.; Puglisi F.; Piccart M.; Biganzoli L. (2004) What is the effect of systemic anticancer treatment on cognitive function?. Lancet Oncol. 5, 273–282. 10.1016/S1470-2045(04)01465-2. [DOI] [PubMed] [Google Scholar]
- Poppelreuter M.; Weis J.; Külz A.; Tucha O.; Lange K.; Bartsch H. (2004) Cognitive dysfunction and subjective complaints of cancer patients. Eur. J. Cancer 40, 43–49. 10.1016/j.ejca.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wefel J. S.; Kayl A. E.; Meyers C. A. (2004) Neuropsychological dysfunction associated with cancer and cancer therapies: a conceptual review of an emerging target. Br. J. Cancer 90, 1691–1696. 10.1038/sj.bjc.6601772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J.; Han R.; Yang Y.; Mayer-Pröschel M.; Noble M. (2006) of Biology. J. Biol. 5, 22. 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyriou A. A.; Assimakopoulos K.; Iconomou G.; Giannakopoulou F.; Kalofonos H. P. (2011) Either called “chemobrain” or “chemofog,” the long-term chemotherapy-induced cognitive decline in cancer survivors is real. J. Pain Symptom Manage. 41, 126–139. 10.1016/j.jpainsymman.2010.04.021. [DOI] [PubMed] [Google Scholar]
- Meyers C. A.; Abbruzzese J. L. (1992) Cognitive functioning in cancer patients effect of previous treatment. Neurology 42, 434–434. 10.1212/WNL.42.2.434. [DOI] [PubMed] [Google Scholar]
- Schagen S. B.; van Dam F. S.; Muller M. J.; Boogerd W.; Lindeboom J.; Bruning P. F. (1999) Cognitive deficits after postoperative adjuvant chemotherapy for breast carcinoma. Cancer 85, 640–650. . [DOI] [PubMed] [Google Scholar]
- Reid-Arndt S. A. (2009) Breast cancer and “chemobrain”: the consequences of cognitive difficulties following chemotherapy and the potential for recovery. Mo. Med. 106, 127–131. [PubMed] [Google Scholar]
- Nelson C. J.; Nandy N.; Roth A. J. (2007) Chemotherapy and cognitive deficits: mechanisms, findings, and potential interventions. PAX 5, 273–280. 10.1017/S1478951507000442. [DOI] [PubMed] [Google Scholar]
- Boykoff N.; Moieni M.; Subramanian S. K. (2009) Confronting chemobrain: an in-depth look at survivors’ reports of impact on work, social networks, and health care response. J. Cancer Surviv. 3, 223–232. 10.1007/s11764-009-0098-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahles T. A.; Saykin A. J. (2007) Candidate mechanisms for chemotherapy-induced cognitive changes. Nat. Rev. Cancer 7, 192–201. 10.1038/nrc2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J.; Han R.; Yang Y.; Mayer-Pröfischel M.; Noble M. (2006) CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol. 5, 22. 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroziak K.; Robak T. (2004) Pharmacogenomics of MDR1/ABCB1 gene: the influence on risk and clinical outcome of haematological malignancies. Hematology 9, 91–105. 10.1080/10245330310001638974. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer S.; Burk O.; Von Richter O.; Arnold H. P.; Brockmöller J.; Johne A.; Cascorbi I.; Gerloff T.; Roots I.; Eichelbaum M.; et al. (2000) Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. U. S. A. 97, 3473–3478. 10.1073/pnas.97.7.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Zglinicki T. (2002) Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. 10.1016/S0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Meyers C. A.; Albitar M.; Estey E. (2005) Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 104, 788–793. 10.1002/cncr.21234. [DOI] [PubMed] [Google Scholar]
- Kelley K. W.; Bluthé R.-M.; Dantzer R.; Zhou J.-H.; Shen W.-H.; Johnson R. W.; Broussard S. R. (2003) Cytokine-induced sickness behavior. Brain, Behav., Immun. 17, 112–118. 10.1016/S0889-1591(02)00077-6. [DOI] [PubMed] [Google Scholar]
- Scheibel R. S.; Valentine A. D.; O’Brien S.; Meyers C. A. (2004) Cognitive dysfunction and depression during treatment with interferon-alpha and chemotherapy. J. Neuropsychiatry Clin. Neurosci. 16, 185–191. 10.1176/jnp.16.2.185. [DOI] [PubMed] [Google Scholar]
- de Visser K. E.; Eichten A.; Coussens L. M. (2006) Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37. 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Joshi G.; Aluise C. D.; Cole M. P.; Sultana R.; Pierce W. M.; Vore M.; St Clair D. K.; Butterfield D. A. (2010) Alterations in brain antioxidant enzymes and redox proteomic identification of oxidized brain proteins induced by the anti-cancer drug adriamycin: implications for oxidative stress-mediated chemobrain. Neuroscience 166, 796–807. 10.1016/j.neuroscience.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramassamy C.; Averill D.; Beffert U.; Bastianetto S.; Theroux L.; Lussier-Cacan S.; Cohn J. S.; Christen Y.; Davignon J.; Quirion R.; et al. (1999) Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein E genotype. Free Radical Biol. Med. 27, 544–553. 10.1016/S0891-5849(99)00102-1. [DOI] [PubMed] [Google Scholar]
- Dietrich J.; Han R.; Yang Y.; Mayer-Proschel M.; Noble M. (2006) CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol. 5, 22. 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. (2002) Getting Formal with Dopamine and Reward. Neuron 36, 241–263. 10.1016/S0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Bäckman L.; Nyberg L.; Lindenberger U.; Li S.-C.; Farde L. (2006) The correlative triad among aging, dopamine, and cognition: Current status and future prospects. Neurosci. Biobehav. Rev. 30, 791–807. 10.1016/j.neubiorev.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Kraft J. C.; Osterhaus G. L.; Ortiz A. N.; Garris P. A.; Johnson M. A. (2009) In vivo dopamine release and uptake impairments in rats treated with 3-nitropropionic acid. Neuroscience 161, 940–949. 10.1016/j.neuroscience.2009.03.083. [DOI] [PubMed] [Google Scholar]
- Ortiz A. N.; Oien D. B.; Moskovitz J.; Johnson M. A. (2011) Quantification of reserve pool dopamine in methionine sulfoxide reductase A null mice. Neuroscience 177, 223–229. 10.1016/j.neuroscience.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. A.; Rajan V.; Miller C. E.; Wightman R. M. (2006) Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 97, 737–746. 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Ortiz A. N.; Kurth B. J.; Osterhaus G. L.; Johnson M. A. (2010) Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J. Neurochem. 112, 755–761. 10.1111/j.1471-4159.2009.06501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz A. N.; Osterhaus G. L.; Lauderdale K.; Mahoney L.; Fowler S. C.; von Hörsten S.; Riess O.; Johnson M. A. (2012) Motor function and dopamine release measurements in transgenic Huntington’s disease model rats. Brain Res. 1450, 148–156. 10.1016/j.brainres.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore G. T.; Jackson-Lewis V.; Vukosavic S.; Mandir A. S.; Vila M.; McAuliffe W. G.; Dawson V. L.; Dawson T. M.; Przedborski S. (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 5, 1403–1409. 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Masliah E. (2000) Dopaminergic loss and inclusion body formation in -synuclein mice: Implications for neurodegenerative disorders. Science 287, 1265–1269. 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Morgan D. G.; May P. C.; Finch C. E. (1987) Dopamine and serotonin systems in human and rodent brain: effects of age and neurodegenerative disease. J. Am. Geriatr. Soc. 35, 334–345. 10.1111/j.1532-5415.1987.tb04641.x. [DOI] [PubMed] [Google Scholar]
- Coppen A. J.; Doogan D. P. (1988) Serotonin and its place in the pathogenesis of depression. J. Clin. Psychiatry 49, 4–11. [PubMed] [Google Scholar]
- Coppen A. (1967) The biochemistry of affective disorders. Br. J. Psychiatry 113, 1237–1264. 10.1192/bjp.113.504.1237. [DOI] [PubMed] [Google Scholar]
- Stockmeier C. A.; Shapiro L. A.; Dilley G. E.; Kolli T. N.; Friedman L.; Rajkowska G. (1998) Increase in serotonin-1A autoreceptors in the midbrain of suicide victims with major depression—postmortem evidence for decreased serotonin activity. J. Neurosci. 18, 7394–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T.; Wagner G. C.; Nakashima T. (2003) Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J. Pharmacol. Sci. 92, 178–195. 10.1254/jphs.92.178. [DOI] [PubMed] [Google Scholar]
- Eisenberger M.; Hornedo J.; Silva H.; Donehower R.; Spaulding M.; Van Echo D. (1986) Carboplatin (NSC-241-240): an active platinum analog for the treatment of squamous-cell carcinoma of the head and neck. J. Clin. Oncol. 4, 1506–1509. [DOI] [PubMed] [Google Scholar]
- Kavanagh J. J.; Nicaise C. (1989) Carboplatin in refractory epithelial ovarian cancer. Semin. Oncol. 16, 45–48. [PubMed] [Google Scholar]
- Wagstaff A. J.; Ward A.; Benfield P.; Heel R. C. (1989) Carboplatin. Drugs 37, 162–190. 10.2165/00003495-198937020-00005. [DOI] [PubMed] [Google Scholar]
- Smith I. E.; Harland S. J.; Robinson B. A.; Evans B. D.; Goodhart L. C.; Calvert A. H.; Yarnold J.; Glees J. P.; Baker J.; Ford H. T. (1985) Carboplatin: a very active new cisplatin analog in the treatment of small cell lung cancer. Cancer Treat. Rep. 69, 43–46. [PubMed] [Google Scholar]
- Schagen S. B.; Muller M. J.; Boogerd W.; Mellenbergh G. J.; van Dam F. S. A. M. (2006) Change in cognitive function after chemotherapy: a prospective longitudinal study in breast cancer patients. JNCI J. Natl. Cancer Inst. 98, 1742–1745. 10.1093/jnci/djj470. [DOI] [PubMed] [Google Scholar]
- Craig C. D.; Monk B. J.; Farley J. H.; Chase D. M. (2014) Cognitive impairment in gynecologic cancers: A systematic review of current approaches to diagnosis and treatment. Supportive Care Cancer 22, 279–287. 10.1007/s00520-013-2029-7. [DOI] [PubMed] [Google Scholar]
- Staat K.; Segatore M. (2005) The phenomenon of chemo brain. Clin. J. Oncol. Nurs. 9, 713–721. 10.1188/05.CJON.713-721. [DOI] [PubMed] [Google Scholar]
- Merriman J. D.; Von Ah D.; Miaskowski C.; Aouizerat B. E. (2013) proposed mechanisms for cancer- and treatment-related cognitive changes. Semin. Oncol. Nurs. 29, 260–269. 10.1016/j.soncn.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefel J. S.; Kesler S. R.; Noll K. R.; Schagen S. B. (2015) Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. Ca-Cancer J. Clin. 65, 123–138. 10.3322/caac.21258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigers R.; Fardell J. E. (2011) Neurobiological basis of chemotherapy-induced cognitive impairment: A review of rodent research. Neurosci. Biobehav. Rev. 35, 729–741. 10.1016/j.neubiorev.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Madhyastha S.; Somayaji S. N.; Rao M. S.; Nalini K.; Bairy K. L. (2002) Hippocampal brain amines in methotrexate-induced learning and memory deficit. Can. J. Physiol. Pharmacol. 80, 1076–1084. 10.1139/y02-135. [DOI] [PubMed] [Google Scholar]
- Evenden J. (2013) Cognitive impairments and cancer chemotherapy: Translational research at a crossroads. Life Sci. 93, 589–595. 10.1016/j.lfs.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Gerfen C. R. (1992) The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci. 15, 285–320. 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Ferguson S. M.; Eskenazi D.; Ishikawa M.; Wanat M. J.; Phillips P. E. M.; Dong Y.; Roth B. L.; Neumaier J. F. (2011) Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat. Neurosci. 14, 22–24. 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M.; de Carvalho F. D.; Chohan M. O.; Shegda M.; Chuhma N.; Rayport S.; Ahmari S. E.; Moore H.; Kellendonk C. (2014) Dopamine D2 Receptors Regulate the Anatomical and Functional Balance of Basal Ganglia Circuitry. Neuron 81, 153–164. 10.1016/j.neuron.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T.; Kimura K.; Wada N.; Funabiki K.; Nakanishi S. (2010) distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66, 896–907. 10.1016/j.neuron.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Hikosaka O.; Takikawa Y.; Kawagoe R. (2000) Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol. Rev. 80, 953–978. [DOI] [PubMed] [Google Scholar]
- Kravitz A. V.; Tye L. D.; Kreitzer A. C. (2012) Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci. 15, 816–818. 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow N. D.; Wang G.-J.; Fowler J. S.; Gatley S. J.; Logan J.; Ding Y.-S.; Hitzemann R.; Pappas N. (1998) Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am. J. Psychiatry 155, 1325–1331. 10.1176/ajp.155.10.1325. [DOI] [PubMed] [Google Scholar]
- Palmiter R. D. (2008) Dopamine signaling in the dorsal striatum is essential for motivated behaviors. Ann. N. Y. Acad. Sci. 1129, 35–46. 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jocham G.; Klein T. A.; Ullsperger M. (2011) Dopamine-mediated reinforcement learning signals in the striatum and ventromedial prefrontal cortex underlie value-based choices. J. Neurosci. 31, 1606–1613. 10.1523/JNEUROSCI.3904-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari E. S.; Huggins K. N.; Mathews T. A.; Jones S. R. (2012) Conserved dorsal–ventral gradient of dopamine release and uptake rate in mice, rats and rhesus macaques. Neurochem. Int. 61, 986–991. 10.1016/j.neuint.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bãckman L.; Ginovart N.; Dixon R. A.; Wahlin T.-B. R.; Wahlin A.; Halldin C.; Farde L. (2000) Age-related cognitive deficits mediated by changes in the striatal dopamine system. Am. J. Psychiatry 157, 635–637. 10.1176/ajp.157.4.635. [DOI] [PubMed] [Google Scholar]
- Brozoski T. J.; Brown R. M.; Rosvold H.; Goldman P. S. (1979) Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 205, 929–932. 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- Mathews C., van Holde K., and Ahern K. G. (1996) Biochemistry, Pearson, London. [Google Scholar]
- Neves G.; Lagnado L. (1999) The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J. Physiol. 515, 181–202. 10.1111/j.1469-7793.1999.181ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker R. S.; Regehr W. G. (2002) Short term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- Rizzoli S. O.; Betz W. J. (2005) Synaptic vesicle pools. Nat. Rev. Neurosci. 6, 57–69. 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Harata N.; Ryan T. A.; Smith S. J.; Buchanan J.; Tsien R. W. (2001) Visualizing recycling synaptic vesicles in hippocampal neurons by FM 1–43 photoconversion. Proc. Natl. Acad. Sci. U. S. A. 98, 12748–12753. 10.1073/pnas.171442798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H.; Kidokoro Y. (2003) Two synaptic vesicle pools, vesicle recruitment and replenishment of pools at the Drosophila neuromuscular junction. J. Neurocytol. 32, 551–565. 10.1023/B:NEUR.0000020610.13554.3c. [DOI] [PubMed] [Google Scholar]
- Richards D. A.; Guatimosim C.; Rizzoli S. O.; Betz W. J. (2003) Synaptic vesicle pools at the frog neuromuscular junction. Neuron 39, 529–541. 10.1016/S0896-6273(03)00405-7. [DOI] [PubMed] [Google Scholar]
- Heien M.; Wightman R. M. (2006) Phasic dopamine signaling during behavior, reward, and disease states. CNS Neurol. Disord.: Drug Targets 5, 99–108. 10.2174/187152706784111605. [DOI] [PubMed] [Google Scholar]
- Venton B. J. (2006) Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J. Neurosci. 26, 3206–3209. 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector S.; Sjoerdsma A.; Udenfriend S. (1965) Blockade of endogenous norepinephrine synthesis by α-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. J. Pharmacol. Exp. Ther. 147, 86–95. [PubMed] [Google Scholar]
- Seyedabadi M.; Fakhfouri G.; Ramezani V.; Mehr S. E.; Rahimian R. (2014) The role of serotonin in memory: interactions with neurotransmitters and downstream signaling. Exp. Brain Res. 232, 723–738. 10.1007/s00221-013-3818-4. [DOI] [PubMed] [Google Scholar]
- Nic Dhonnchadha B. Á.; Cunningham K. A. (2008) Serotonergic mechanisms in addiction-related memories. Behav. Brain Res. 195, 39–53. 10.1016/j.bbr.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch K.-P.; Waider J. (2012) Serotonin in the Modulation of Neural Plasticity and Networks: Implications for Neurodevelopmental Disorders. Neuron 76, 175–191. 10.1016/j.neuron.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Hall F. S.; Sora I.; Hen R.; Uhl G. R. (2014) Serotonin/Dopamine Interactions in a Hyperactive Mouse: Reduced Serotonin Receptor 1B Activity Reverses Effects of Dopamine Transporter Knockout. PLoS One 9, e115009. 10.1371/journal.pone.0115009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell L. L.; Cunningham K. A. (2015) Serotonin 5-HT2 Receptor Interactions with Dopamine Function: Implications for Therapeutics in Cocaine Use Disorder. Pharmacol. Rev. 67, 176–197. 10.1124/pr.114.009514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmolowicz D. P.; Hudnall J. L.; Darden A. C.; Lemley S. M.; Sofis M. J. (2015) On the time course of rapid and repeatable response induction. Behav. Processes 120, 116–119. 10.1016/j.beproc.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Kawagoe K. T.; Jankowski J. A.; Wightman R. M. (1991) Etched carbon-fiber electrodes as amperometric detectors of catecholamine secretion from isolated biological cells. Anal. Chem. 63, 1589–1594. 10.1021/ac00015a017. [DOI] [PubMed] [Google Scholar]
- Rice M. E.; Nicholson C. (1989) Measurement of nanomolar dopamine diffusion using low-noise perfluorinated ionomer-coated carbon fiber microelectrodes and high-speed cyclic voltammetry. Anal. Chem. 61, 1805–1810. 10.1021/ac00192a005. [DOI] [PubMed] [Google Scholar]
- Hashemi P.; Dankoski E.; Petrovic J.; Keithley R. B.; Wightman R. M. (2009) Voltammetric detection of 5-hydroxytryptamine release in the rat brain. Anal. Chem. 81, 9462–9471. 10.1021/ac9018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S.; Nihal M.; Ahmad N. (2007) Dose translation from animal to human studies revisited. FASEB J. 22, 659–661. 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Duffull S. B.; Robinson B. A. (1997) Clinical pharmacokinetics and dose optimization of carboplatin. Clin. Pharmacokinet. 33, 161–183. 10.2165/00003088-199733030-00002. [DOI] [PubMed] [Google Scholar]
- Jackson B. P.; Dietz S. M.; Wightman R. M. (1995) Fast-scan cyclic voltammetry of 5-hydroxytryptamine. Anal. Chem. 67, 1115–1120. 10.1021/ac00102a015. [DOI] [PubMed] [Google Scholar]
- Wightman R. M.; Amatorh C.; Engstrom R. C.; Hale P. D.; Kristensen E. W.; Kuhr W. G.; May L. J. (1988) Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience 25, 513–523. 10.1016/0306-4522(88)90255-2. [DOI] [PubMed] [Google Scholar]
- Jones S. R.; Garris P. A.; Kilts C. D.; Wightman R. M. (1995) Comparison of dopamine uptake in the basolateral amygdaloid nucleus, caudate-putamen, and nucleus accumbens of the rat. J. Neurochem. 64, 2581–2589. 10.1046/j.1471-4159.1995.64062581.x. [DOI] [PubMed] [Google Scholar]
- Kawagoe K. T.; Garris P. A.; Wiedemann D. J.; Wightman R. M. (1992) Regulation of transient dopamine concentration gradients in the microenvironment surrounding nerve terminals in the rat striatum. Neuroscience 51, 55–64. 10.1016/0306-4522(92)90470-M. [DOI] [PubMed] [Google Scholar]
- Argyriou A. A.; Assimakopoulos K.; Iconomou G.; Giannakopoulou F.; Kalofonos H. P. (2011) Either called “chemobrain” or “chemofog,” the long-term chemotherapy-induced cognitive decline in cancer survivors is real. J. Pain Symptom Manage. 41, 126–139. 10.1016/j.jpainsymman.2010.04.021. [DOI] [PubMed] [Google Scholar]
- Yang M.; Kim J.-S.; Song M.-S.; Kim S.-H.; Kang S. S.; Bae C.-S.; Kim J.-C.; Wang H.; Shin T.; Moon C. (2010) Cyclophosphamide impairs hippocampus-dependent learning and memory in adult mice: Possible involvement of hippocampal neurogenesis in chemotherapy-induced memory deficits. Neurobiol. Learn. Mem. 93, 487–494. 10.1016/j.nlm.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Weiss B. (2008) Chemobrain: A translational challenge for neurotoxicology. NeuroToxicology 29, 891–898. 10.1016/j.neuro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieneke M. H.; Dienst E. R. (1995) Neuropsychological assessment of cognitive functioning following chemotherapy for breast cancer. Psycho-Oncology 4, 61–66. 10.1002/pon.2960040108. [DOI] [Google Scholar]
- Blough D. S. (1969) Generalization gradient shape and summation in steady-state tests. J. Exp. Anal. Behav. 12, 91–104. 10.1901/jeab.1969.12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.