

ABSTRACT
Non-coding transcription across the antisense strands of genes is an abundant, pervasive process in eukaryotes from yeast to humans, however its biological function remains elusive. Here, we provide commentary on a recent study of ours, which demonstrates a genome-wide role for antisense transcription: establishing a unique, dynamic chromatin architecture over genes. Antisense transcription increases the level of nucleosome occupancy and histone acetylation at the promoter and body of genes, without necessarily modulating the level of protein-coding sense transcription. It is also associated with high levels of histone turnover. By allowing genes to sample a wider range of chromatin configurations, antisense transcription could serve to make genes more sensitive to changing signals, priming them for responses to developmental programs or stressful cellular environments. Given the abundance of antisense transcription and the breadth of these chromatin changes, we propose that antisense transcription represents a fundamental, canonical feature of eukaryotic genes.
KEYWORDS: antisense transcription, chromatin dynamics, gene regulation, histone acetylation, nucleosomes, promoters
It is well established that the transcription of genomes is not limited to the transcription of genes alone. Transcription is a universally pervasive and interleaved process,1 with transcription events initiating not just from gene promoters but from those regions between genes and within genes as well, and from regulatory sequences such as enhancers.2,3 What is more, in recent years it has become apparent that transcription events often initiate at the 3′ ends of genes, in a manner that appears carefully orchestrated by the transcription initiation machinery, revealing an inherent symmetry to gene structure (Fig. 1A).4,5 This leads to transcription along the antisense strand of the gene, giving rise to non-coding, antisense transcripts, the function of which is poorly understood.
FIGURE 1.
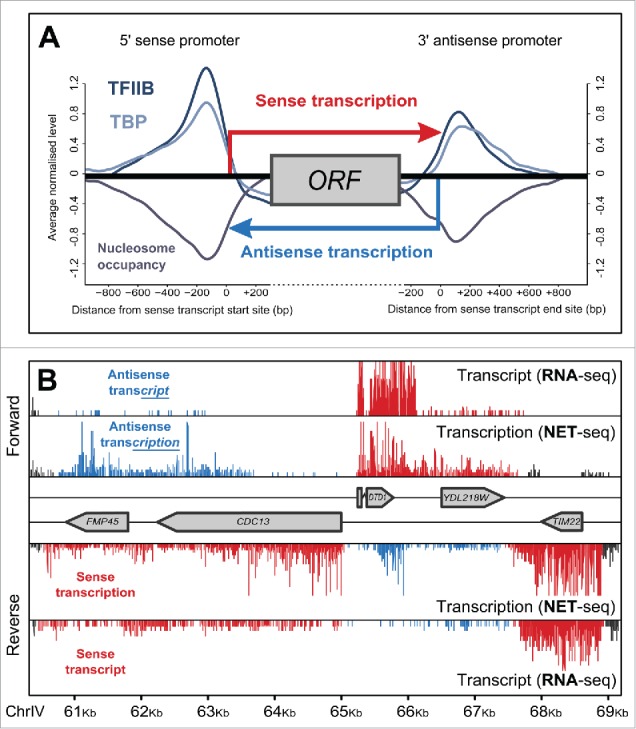
(A) The symmetrical nature of genes. Eukaryotic genes typically display transcription across both the sense and antisense strand (red and blue arrows respectively), driven from 2 convergent, opposed promoters. Shown are the average levels of TBP and TFIIB, 2 factors that play a critical role in transcription initiation, as well as nucleosome occupancy, relative to the start site and end site of the sense transcript.4 This reveals the presence of 2 promoter architectures at both ends of the gene, one directing sense transcription and the other antisense transcription, referred to here as the sense and antisense promoters respectively. (B) Transcription occurs extensively on both the sense and antisense strands of genes across the yeast genome. Shown is the level of transcript arising from a given region of S. cerevisiae chromosome IV, measured by RNA-seq, and the level of transcription measured by NET-seq. Reads have been colored as reflecting either sense (red) or antisense (blue) transcription/transcript based on the orientation of their nearest gene. Though antisense transcription is abundant this is not reflected in the level of antisense transcript, which tend to be extensively degraded.
While it is now clear that antisense transcription occurs to some extent at most genes, what is less clear is why. To begin to elucidate a possible role of antisense transcription, it is important to establish just how abundant a process it is. Attempts to estimate genome-wide levels of antisense transcription have been confounded by the fact that the majority of antisense transcripts are extensively degraded by exonucleases.6,7 Recently, however, a technique has been developed that allows us to assess strand-specific, nascent-transcription on a genome-wide scale. Native Elongating Transcript Sequencing (NET-seq, Fig. 1B) is a modification of RNA-seq in which RNA polymerase II (RNAPII) is immunoprecipitated, and the nascent RNA in its active site purified and sequenced.2 Using NET-seq measurements in S. cerevisiae, we previously demonstrated that the level of antisense transcription across a gene is, on average, one-tenth of the level of protein-coding sense transcription,4 increased to one-fifth when considering those genes with a previously defined antisense transcript (approximately a third of genes).7,8 Considering that coding transcription plays a fundamental role in protein biosynthesis, while antisense transcription does not, this is a staggering ratio, posing the immediate question: if so much energy is being invested in producing transcripts that are themselves being extensively degraded, then why is this transcription happening in the first place?
ANTISENSE TRANSCRIPTION IS ASSOCIATED WITH A UNIQUE CHROMATIN ARCHITECTURE
Since most antisense transcripts are extensively degraded in spite of their prolific synthesis, one might reasonably hypothesize that it is the act of antisense transcription, rather than the transcripts themselves, that exert a meaningful biological function. In our recent publication,9 we asked how genes in S. cerevisiae differ depending on the amount of antisense transcription they experience, to begin to tease apart what this function might be. Using a combination of genome-wide data, computational analyses and experimental, single-gene studies, we identified a broad array of relationships between antisense transcription and other fundamental gene features. These relationships are specific to antisense transcription – they do not apply to protein-coding sense transcription – and revolve around changes in the chromatin structure of the gene body and the canonical gene promoter (henceforth called the sense promoter – i.e. the promoter directing sense transcription). The changes are summarized in Figure 2. Antisense transcription is associated with high nucleosome occupancy at the sense promoter, coincident with increases in histone turnover and levels of the chromatin remodeling enzymes/complexes Isw1, Isw2 and INO80. It is also associated with high levels of histone acetylation (H3K4ac, H3K9ac, H3K14ac, H3K56ac and H4ac) over the sense promoter and gene body, and low levels of H3K36me3, H3K79me3 and H2BK123 ubiquitination over the gene body. To put it simply, antisense transcription is associated with an increase in dynamic, remodeled, and highly acetylated chromatin across the sense promoter and gene body.
FIGURE 2.
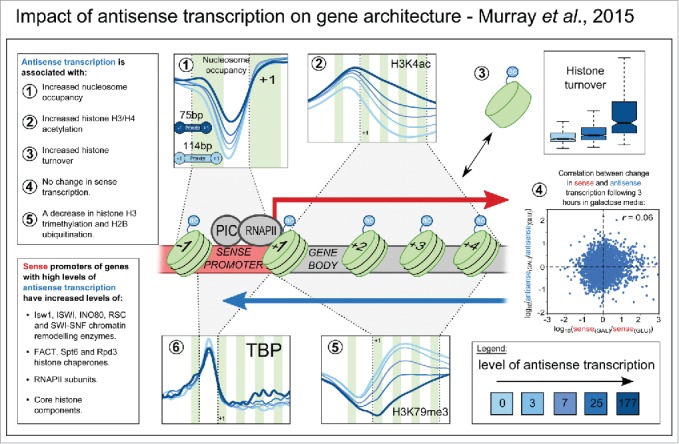
A summary of the findings presented in Murray et al.9 Shown are the average levels of nucleosome occupancy (1), H3K4 acetylation (2), histone turnover (3), H3K79me3 (5) and TBP (6) in 5 different gene classes selected on the basis of their level of antisense transcription. The median level of antisense transcription in these groups relative to one another is shown in the bottom right inset. Also shown in (4) is a scatter plot of the change in sense transcription versus the change in antisense transcription in 5,183 S. cerevisiae genes following a change in growth conditions (a shift from glucose- to galactose-containing medium). Green vertical strips represent approximate positions of nucleosomes. Arrows show the direction of sense and antisense transcription (red and blue respectively). PIC = pre-initiation complex. RNAPII = RNA polymerase II. TBP = TATA-box binding protein.
In addition to these associations, we were further able to demonstrate that antisense transcription can in fact cause these chromatin changes. Using an experimental system in which we varied the level of antisense transcription over the sense promoter of the GAL1 gene, we demonstrated that decreasing the level of antisense transcription led to a decrease in the level of histone H3 and histone H3 acetylation, and an increase in H3K36me3. Also, by combining NET-seq data and ChIP-seq data obtained in a variety of mutants known to have altered genome-wide patterns of antisense transcription (set1Δ, set2Δ, eaf3Δ and rco1Δ)2 we found that changes in antisense transcription resulting from these mutations were coincident with changes in nucleosome occupancy, histone acetylation and H3K36me3.
Taken together, our results demonstrate that antisense transcription can itself establish a dynamic, acetylated chromatin architecture. We present a unique function of antisense transcription not associated with other forms of gene regulation. Given the abundant, pervasive nature of antisense transcription, we therefore propose that antisense transcription should be considered a fundamental, canonical feature of genes – that a typical gene has a 3′, regulatable antisense promoter that can drive antisense transcription and therefore establish a dynamic chromatin environment.
ANTISENSE TRANSCRIPTION IS NOT A UNIVERSAL REPRESSOR OF SENSE TRANSCRIPTION
A persisting idea in the discourse surrounding antisense transcription is that antisense transcripts and/or transcription itself should have a negative regulatory effect on sense transcription. This is in part due to a small number of early, single-gene studies suggesting antisense transcription might be repressive.10,11 It is perhaps also due to the pleasing notion that an antisense-transcribing polymerase complex might collide with and disrupt either the sense-transcribing polymerase, or else the pre-initiation complex on the sense promoter. However, Hongay et al.12 found that antisense transcription did not occur at the same time as sense transcription within the same cells, while Castelnuovo et al.13 and Nguyen et al.14 found that the PHO84 or HMS2 sense and antisense transcripts were very rarely present simultaneously within the same cell. Furthermore, mathematical models of transcriptional interference by Sneppen et al.15 have shown that in order for transcription in one direction to impact transcription in the opposite direction, very high levels of transcription are required, possibly above what is typically observed.
In Murray et al.9 we demonstrate that antisense transcription is not correlated with sense transcription genome-wide, either positively or negatively. This lack of correlation remained when considering only subsets of genes with different levels of antisense transcription. Furthermore, an increase in antisense transcription was not associated with changes in the levels of TBP or the general transcription factor TFIIB at the sense promoter, suggesting that the levels of key components of the pre-initiation complex are not being modulated by antisense-transcription. Perhaps the most striking demonstration of this lack of correlation was shown using NET-seq data obtained in a number of different mutant strains of S. cerevisiae,2 all of which showed changes in their patterns of antisense transcription to varying extents. Using this data, we found that there was no correlation (or inverse correlation) between changes in antisense transcription and changes in sense transcription – i.e., the extent of the change in antisense transcription at a given gene in the mutant strain relative to wild-type could not be used to predict how the sense transcription levels would change.
Clearly, the relationship between sense and antisense transcription is more nuanced and complex than a simple repression model. Of course, at some genes an increase in antisense transcription following one of these 4 mutations is coincident with a decrease in sense transcription, while at other genes the 2 either both increase or both decrease. We propose that, while the universal role of antisense transcription is to set up a dynamic chromatin environment at the sense promoter, the effects of this new environment might be context dependent, and so gene-specific, as shown in Figure 3A i) and ii).
FIGURE 3.
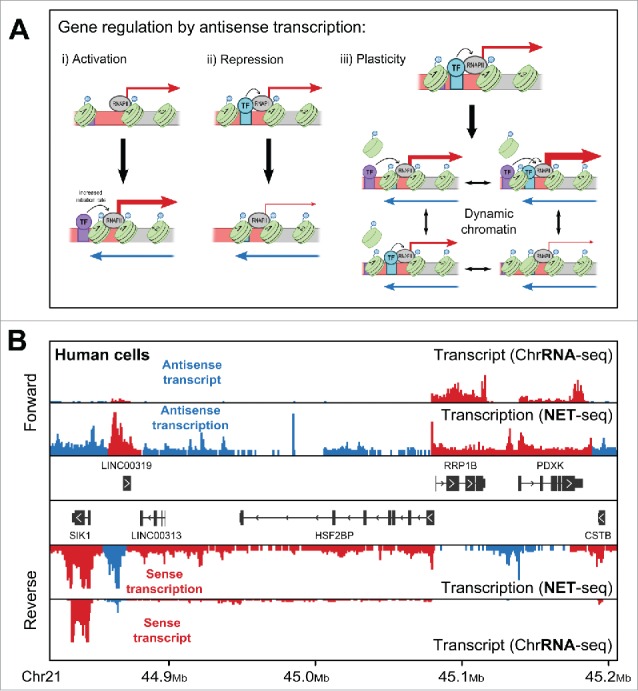
(A) Three, mutually inclusive models for how antisense transcription might regulate genes. i) A gene with a single, nucleosome-occluded transcription factor (TF) binding site (purple box) in its promoter shows a moderate level of sense transcription. In the presence of antisense transcription (blue arrow), chromatin remodeling leads to the movement of this nucleosome, resulting in increased sense transcription (indicated by the larger red arrow). In ii) the situation is reversed, with antisense transcription leading to occlusion of a different binding site (turquoise box), causing reduced sense transcription. iii) Here, antisense transcription sets up a dynamic chromatin at the promoter allowing switching between different chromatin states, each with a distinct sense transcription output. In a population of cells this would result in noisier gene expression. (B) A view of transcription of both strands (forward and reverse) of a region of chromosome 21 in human HeLa cells, analogous to Figure 1B. Shown are the levels of transcript, measured using ChrRNA-seq (a modification of RNA-seq that aims to measure both processed and unprocessed transcript levels), and transcription, using NET-seq.32 Reads have been colored as reflecting either sense (red) or antisense (blue) transcription/transcript based on the orientation of their nearest gene.
THE MECHANISM OF ANTISENSE TRANSCRIPTION-MEDIATED REGULATION
It is evident that antisense transcription plays a genome-wide role in chromatin modulation. What remains unclear is how antisense transcription might affect such changes. Our work demonstrates that antisense transcription has associations with chromatin that are distinct from sense-transcription/chromatin associations. For example, histone turnover is much more strongly associated with antisense transcription than it is with sense transcription, while H3K36me3 is positively correlated with sense transcription in the gene body but negatively correlated with antisense transcription. This raises the intriguing possibility that antisense transcription and sense transcription might in some way be fundamentally distinct processes. But how might they differ?
While it has been demonstrated that antisense promoters recruit the same components of the RNAPII pre-initiation complex machinery as sense promoters,5 there are other ways in which the polymerase complex could differ, which could explain how antisense transcription affects the observed changes in chromatin, and also why antisense transcripts are so often extensively degraded.6,7 One possibility is that the subunit composition of the elongating polymerase could itself differ. For example, RNAPII contains a subunit, Rpb4, which is only found in approximately 20% of cells under optimal conditions, but is found at higher percentages under conditions of stress,16 demonstrating that RNAPII subunit composition can be regulated to functional ends. RNAPII subunit composition has been shown to be altered by the HMG-protein Nhp6a and the mediator component Med3p,17 while the RING finger ubiquitin ligase Asr1 can disengage the Rpb4/Rpb7 heterodimer from the polymerase complex via ubiquitination.18 RNA processing factors have also been found to modulate chromatin. The cap binding complex, for example, has been shown to support Set2-dependent H3K36me3.19 Differential processing of antisense transcripts might also result in the polymerase complex having unique chromatin-regulating activities – for example, the transcript might be capped but not associated with cap binding proteins.
How might an antisense-transcribing RNAPII recruit chromatin remodelers? Although such enzymes are generally thought to be recruited by direct interaction with transcriptional activators, or by binding to specific histone modifications, there is evidence that they might be recruited directly by RNAPII. The FACT chromatin remodeling complex has been shown to travel with RNAPII through the bodies of genes,20 while INO80, in cooperation with Spt6, has been shown to associate with the ORFs of stress-response genes in a manner that is dependent upon elongating RNAPII, but independent of histone modifications.21 It is entirely plausible then that an antisense-processing transcription complex might introduce chromatin remodeling enzymes and histone chaperones to the sense promoters and gene body, and in doing so bring about the observed changes in chromatin.
One finding from our recent work that may shed further light is the fact that, while sense transcription is strongly correlated with phosphorylation on Ser2 of the RNAPII C-terminal domain (CTD), antisense transcription is not. Modification of the CTD is known to play important roles at all stages of the transcription process,22 including roles in the recruitment of histone modifying enzymes, chromatin remodeling enzymes and transcription elongation factors. This change in CTD modifications could be what marks the antisense-transcribing RNAPII complex as distinct from the sense-transcribing complex, resulting in differential recruitment of chromatin modulators and so explaining their unique associations. Moreover, these differences could explain differential processing and fate of antisense transcripts.
ROLES FOR DYNAMIC CHROMATIN
Our work demonstrates a clear genome-wide role for antisense transcription in the establishment of a unique, dynamic and acetylated chromatin architecture across the sense promoter and gene body. What remains uncertain, however, is what the role of such an architecture might be, and why and when specific genes might have need to turn on antisense transcription and so adopt this architecture.
One likely consequence of having dynamic chromatin is an increase in gene expression variation. There are numerous different sorts of gene expression variation, such as plasticity, which is the variation in the expression of a given gene across different environments, and noise, which is the variation in expression of a gene across a clonal population of cells. If one imagines that a gene has a finite number of chromatin configurations that it can adopt, each with a distinct potential expression output, then having a dynamic chromatin might allow a gene to be constantly switching between these configurations, sampling different chromatin states. This would result in a greater variety of expression values across a population, leading to increased noise, and might also allow access for a greater variety of potential transcription factor binding events in response to changing environments, thus increasing plasticity, as shown in Figure 3Aiii). Indeed, using available genome-wide measurements of noise obtained by Newman et al.23 and plasticity by Tirosh and Barkai,24 we have found that the level of antisense transcription across a gene is well correlated with both features (unpublished data). In support of this, Xu et al.25 also showed that genes with defined overlapping antisense transcripts showed a higher variation of gene expression across 5 different environmental conditions compared to those genes that did not. Another possibility is that antisense transcription might enforce the stochastic, ‘burstier’ mode of sense transcription that is characterized by infrequent, rapid bouts of transcription initiation, which can be observed as bright dots using RNA-FISH, and which is known to be inherently noisy.26 Noisy gene expression is thought to be a fundamental component of cell differentiation during organismal development,27 and of the stress response in yeast,28 while Thattai and Oudenaarden29 developed mathematical models to show that heterogeneous populations could achieve higher growth rates than homogenous populations under rapidly changing environmental conditions. Antisense transcription might therefore represent a means by which genes can adopt a more stochastic mode of expression to the benefit of the organism.
IS ANTISENSE TRANSCRIPTION PREVALENT IN MAMMALIAN SYSTEMS?
There is convincing evidence that the sort of antisense transcription described here is not a unique feature of yeast, but is also present in mammals and other organisms such as flies. A study in human cells by Conley & Jordan30 identified many thousands of antisense promoters using CAGE analysis, which characterizes the 5′ ends of transcripts, and found that such promoters were generally present toward the 3′ ends of genes. Antisense transcription from these promoters was abundant, though not as high as from sense promoters, in agreement with our findings in yeast,4 varied between different cell types, and showed evidence of activatory histone marks at their promoters that correlated with the level of RNAPII occupancy. Perhaps most importantly, they found that the activity of sense promoters did not correlate with the levels of activity of their associated antisense promoter. Mammalian antisense transcription has also been shown to extend into the sense promoter, as in yeast. Recent studies by Mayer et al.31 and Nojima et al.32 employing nascent RNA-seq or NET-seq in human cells, found evidence of extensive antisense transcription into the sense promoters of genes. Strikingly, when comparing NET-seq profiles in HeLa cells with those obtained by RNA-seq, one sees a very similar picture to that in yeast – i.e. the level of nascent transcription on both strands of genes is much less biased toward the coding strand than one would predict based on transcript level (Fig. 3B), with a lot of antisense transcription across the sense promoter of coding genes. Antisense transcription was found to associated with decreased DNase I sensitivity over the sense promoter in mammals,31 indicative of increased nucleosome occupancy, agreeing with our findings in yeast.9
There is evidence that antisense transcription in mammals might also be involved in mediating the response of a sense promoter to changing environmental cues. Xu et al.25 found that genes with evidence of antisense transcription displayed greater variation in sense transcription across 5 different human cell lines – i.e., that these genes were more transcriptionally plastic. Furthermore, Sigova et al.33 identified several thousand noncoding divergent transcripts in murine embryonic stem cells that underwent changes in transcription level during differentiation, while Tong et al.34 have found evidence that antisense transcripts may play a role in pluripotency maintenance. Thus, as in yeast, antisense transcription may play a crucial role in increasing chromatin dynamics and so priming genes toward changing cellular environments.
CONCLUDING REMARKS
The recent study described here demonstrates a fundamental role for the antisense transcription of genes in establishing dynamic chromatin across a gene.9 Based on this, and the abundant nature of antisense transcription genome-wide,4 we propose that antisense transcription should be considered a canonical feature of genes, as opposed to an idiosyncrasy particular to just a subset of genes. Future work should seek to understand the function of the dynamic chromatin established by antisense transcription. For example, the relationship between antisense transcription and a wide array of histone acetylation marks is particularly exciting. Every acetylation mark we investigated was strongly correlated with antisense transcription, while H3K9, 14, 18, 23 and H4 acetylation all showed an increase in response to increased antisense transcription. It is tempting to suggest that this increase in acetylation is due to increased histone deposition – soluble histones tend to be hyperacetylated, thus incorporation of new histones would replace the pre-existing modifications with acetylation instead.35 However, a recent study suggested that histone turnover does not alter acetylation levels, or vice-versa, despite their strong correlations.36 Understanding the relationship between antisense transcription, acetylation and histone turnover may go some way toward explaining the role of these 3 fundamental processes in gene regulation. Clearly, antisense transcription represents an additional, pervasive mechanism of regulation, present in both yeast and mammals, which demands that we reassess our view of genes and gene regulation.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Françoise S Howe for her helpful comments and careful proof-reading.
REFERENCES
- 1.Mellor J, Woloszczuk R, Howe FS. The interleaved genome. Trends Genet 2016; 32:57-71; PMID:26613890; http://dx.doi.org/21248844 10.1016/j.tig.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 2.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 2011; 469:368-73; PMID:21248844; http://dx.doi.org/ 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci 2014; 39:170-82; PMID:24674738; http://dx.doi.org/ 10.1016/j.tibs.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray SC, Serra Barros A, Brown DA, Dudek P, Ayling J, Mellor J. A pre-initiation complex at the 3′-end of genes drives antisense transcription independent of divergent sense transcription. Nucleic Acids Res 2012; 40:2432-44; PMID:22123739; http://dx.doi.org/ 10.1093/nar/gkr1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 2012; 483:295-301; PMID:22258509; http://dx.doi.org/ 10.1038/nature10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dijk EL, Chen CL, d'Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Né P, Loeillet S, et al.. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 2011; 475:114-7; PMID:21697827; http://dx.doi.org/ 10.1038/nature10118 [DOI] [PubMed] [Google Scholar]
- 7.Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 2009; 457:1038-42; PMID:19169244; http://dx.doi.org/ 10.1038/nature07747 [DOI] [PubMed] [Google Scholar]
- 8.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature 2009; 457:1033-7; PMID:19169243; http://dx.doi.org/ 10.1038/nature07728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murray SC, Haenni S, Howe FS, Fischl H, Chocian K, Nair A, Mellor J. Sense and antisense transcription are associated with distinct chromatin architectures across genes. Nucleic Acids Res 2015; 43:7823-37; PMID:26130720; http://dx.doi.org/ 10.1093/nar/gkv666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell 2007; 131:706-17; PMID:18022365; http://dx.doi.org/ 10.1016/j.cell.2007.09.014 [DOI] [PubMed] [Google Scholar]
- 11.Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M. A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell 2008; 32:685-95; PMID:19061643; http://dx.doi.org/ 10.1016/j.molcel.2008.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hongay CF, Grisafi PL, Galitski T, Fink GR. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 2006; 127:735-45; PMID:17110333; http://dx.doi.org/ 10.1016/j.cell.2006.09.038 [DOI] [PubMed] [Google Scholar]
- 13.Castelnuovo M, Rahman S, Guffanti E, Infantino V, Stutz F, Zenklusen D. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat Struct Mol Biol 2013; 20:851-8; PMID:23770821; http://dx.doi.org/ 10.1038/nsmb.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen T, Fischl H, Howe FS, Woloszczuk R, Serra Barros A, Xu Z, Brown D, Murray SC, Haenni S, Halstead JM, et al.. Transcription mediated insulation and interference direct gene cluster expression switches. Elife 2014; 3:1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sneppen K, Dodd IB, Shearwin KE, Palmer AC, Schubert RA, Callen BP, Egan JB. A Mathematical Model for Transcriptional Interference by RNA Polymerase Traffic in Escherichia coli. J Mol Biol 2005; 346:399-409; PMID:15670592; http://dx.doi.org/ 10.1016/j.jmb.2004.11.075 [DOI] [PubMed] [Google Scholar]
- 16.Choder M, Young R A. A portion of RNA polymerase II molecules has a component essential for stress responses and stress survival. Mol Cell Biol 1993; 13:6984-91; PMID:8413288; http://dx.doi.org/ 10.1128/MCB.13.11.6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue X, Lehming N. Nhp6p and Med3p regulate gene expression by controlling the local subunit composition of RNA polymerase II. J Mol Biol 2008; 379:212-30; PMID:18448120; http://dx.doi.org/ 10.1016/j.jmb.2008.03.069 [DOI] [PubMed] [Google Scholar]
- 18.Daulny A, Geng F, Muratani M, Geisinger JM, Salghetti SE, Tansey WP. Modulation of RNA polymerase II subunit composition by ubiquitylation. Proc Natl Acad Sci U S A 2008; 105:19649-54; PMID:19064926; http://dx.doi.org/ 10.1073/pnas.0809372105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hossain MA, Chung C, Pradhan SK, Johnson TL. The yeast cap binding complex modulates transcription factor recruitment and establishes proper histone H3K36 trimethylation during active transcription. Mol Cell Biol 2013; 33:785-99; PMID:23230273; http://dx.doi.org/ 10.1128/MCB.00947-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwabish MA, Struhl K. Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol Cell 2006; 22:415-22; PMID:16678113; http://dx.doi.org/ 10.1016/j.molcel.2006.03.014 [DOI] [PubMed] [Google Scholar]
- 21.Klopf E, Paskova L, Solé C, Mas G, Petryshyn A, Posas F, Wintersberger U, Ammerer G, Schüller C. Cooperation between the INO80 complex and histone chaperones determines adaptation of stress gene transcription in the yeast Saccharomyces cerevisiae. Mol Cell Biol 2009; 29:4994-5007; PMID:19620280; http://dx.doi.org/ 10.1128/MCB.01858-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsin J, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 2012; :2119-37; PMID:23028141; http://dx.doi.org/ 10.1101/gad.200303.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newman JRS, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 2006; 441:840-6; PMID:16699522; http://dx.doi.org/ 10.1038/nature04785 [DOI] [PubMed] [Google Scholar]
- 24.Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res 2008; 18:1084-91; PMID:18448704; http://dx.doi.org/ 10.1101/gr.076059.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z, Wei W, Gagneur J, Clauder-Munster S, Smolik M, Huber W, Steinmetz LM. Antisense expression increases gene expression variability and locus interdependency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zenklusen D, Larson DR, Singer RH. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 2008; 15:1263-71; PMID:19011635; http://dx.doi.org/ 10.1038/nsmb.1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arias AM, Hayward P. Filtering transcriptional noise during development: concepts and mechanisms. Nat Rev Genet 2006; 7:34-44; PMID:16369570; http://dx.doi.org/ 10.1038/nrg1750 [DOI] [PubMed] [Google Scholar]
- 28.Petrenko N, Chereji R V., McClean MN, Morozov A V., Broach JR. Noise and interlocking signaling pathways promote distinct transcription factor dynamics in response to different stresses. Mol Biol Cell 2013; 24:2045-57; PMID:23615444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thattai M, Van Oudenaarden A. Stochastic gene expression in fluctuating environments. Genetics 2004; 167:523-30; PMID:15166174; http://dx.doi.org/ 10.1534/genetics.167.1.523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conley AB, King Jordan I. Epigenetic regulation of human cis-natural antisense transcripts. Nucleic Acids Res 2012; 40:1438-45; PMID:22371288; http://dx.doi.org/ 10.1093/nar/gkr1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, Churchman LS. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 2015; 161:541-54; PMID:25910208; http://dx.doi.org/ 10.1016/j.cell.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 2015; 161:526-40; PMID:25910207; http://dx.doi.org/ 10.1016/j.cell.2015.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG. Divergent transcription of long noncoding RNA / mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci U S A 2012; 110:2876-81; http://dx.doi.org/ 10.1073/pnas.1221904110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao T, Wu Z, Wang S, Chen L. Expression and function of natural antisense transcripts in mouse embryonic stem cells. Sci China Life Sci 2014; 57:1183-90; PMID:25209725; http://dx.doi.org/ 10.1007/s11427-014-4717-z [DOI] [PubMed] [Google Scholar]
- 35.Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell 2010; 37:469-80; PMID:20188666; http://dx.doi.org/ 10.1016/j.molcel.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrari P, Strubin M. Uncoupling histone turnover from transcription-associated histone H3 modifications. Nucleic Acids Res 2015; 43:3972-85; PMID:25845593; http://dx.doi.org/ 10.1093/nar/gkv282 [DOI] [PMC free article] [PubMed] [Google Scholar]