


Abstract
Identifying coding genes is an essential step in genome annotation. Here, we utilize existing whole genome alignments to detect conserved coding exons and then map gene annotations from one genome to many aligned genomes. We show that genome alignments contain thousands of spurious frameshifts and splice site mutations in exons that are truly conserved. To overcome these limitations, we have developed CESAR (Coding Exon-Structure Aware Realigner) that realigns coding exons, while considering reading frame and splice sites of each exon. CESAR effectively avoids spurious frameshifts in conserved genes and detects 91% of shifted splice sites. This results in the identification of thousands of additional conserved exons and 99% of the exons that lack inactivating mutations match real exons. Finally, to demonstrate the potential of using CESAR for comparative gene annotation, we applied it to 188 788 exons of 19 865 human genes to annotate human genes in 99 other vertebrates. These comparative gene annotations are available as a resource (http://bds.mpi-cbg.de/hillerlab/CESAR/). CESAR (https://github.com/hillerlab/CESAR/) can readily be applied to other alignments to accurately annotate coding genes in many other vertebrate and invertebrate genomes.
INTRODUCTION
Due to advances in DNA sequencing technology, the number of sequenced genomes has increased substantially in the last years, exemplified by the recent sequencing of 48 bird genomes (1). Ongoing projects such as Genome-10K (2) and i5K (3) aim at sequencing many more vertebrates and arthropod species. To use these genomes for biomedical and evolutionary research, it is essential to comprehensively annotate which genomic regions are functional and what their molecular function is (4–7). An essential step in genome annotation is the identification of coding genes.
To annotate coding genes in the genome of a newly sequenced species, a number of different approaches can be used (8,9). On the experimental side, genes can be detected by sequencing full length or parts of mRNAs and mapping the sequenced transcripts or reads back to the genome. However, only those genes that have a sufficiently high expression in the sampled tissues and cell types can be identified. To complement this approach, ab initio methods predict genes based on statistical sequence patterns using only the given genome (10–12). While both of these approaches can find exons and genes that are unique to this species, most coding genes are well conserved across related species. This is exploited in homology-based approaches.
One group of homology-based approaches aligns cDNA or protein sequences of known genes to related genomes using a spliced alignment. These cross-species spliced alignment approaches attempt to find the location of exons often considering the reading frame of the encoded protein and the splice sites (13–15). The detected protein homologies are an essential part in gene annotation methods and pipelines (16–20). However, applying homology-based approaches or even an entire gene annotation pipeline requires a targeted effort for every genome of interest.
The second group of homology-based approaches searches alignments of related genomes for signatures of selection to preserve a coding sequence (21–25). These comparative gene and exon finding approaches make use of sequence conservation and the fact that synonymous changes and reading frame-preserving insertions/deletions (indels) are more frequent in conserved coding exons. These methods generally use multiple genome alignments where many ‘query’ (also called informant) species are aligned to a single ‘reference’ species, which is typically a model organism. These query genomes provide the comparative sequence information to annotate exons and genes in the reference. Applied to alignments of mammalian or Drosophilid genomes, such approaches detected many previously unknown coding exons and genes missed by transcriptional profiling in the reference species human or Drosophila melanogaster (26,27). It is important to note that with very few exceptions (28–30) these comparative gene and exon finders use the given multiple alignment to only annotate genes in the selected reference genome. The reason is that in order to annotate exons across species, the conservation of all exons must be assessed for every species. Indeed, even if a genomic region evolves under selection to preserve a coding sequence in several species, this does not imply that this region is a functional exon in every aligned species.
Despite this, genome alignments are very attractive for mapping gene annotations from a well-annotated genome to all aligned species for the following reasons. First, several genome alignments already exist where numerous related species are aligned to a well-annotated reference genome. Examples include 23 Drosophilids aligned to D. melanogaster (31), 47 birds aligned to the chicken genome (1), or 99 vertebrates aligned to the human genome (32). However, many species in these alignments lack a high-quality gene annotation. Second, genome alignment makes use of synteny (33–35), which helps to distinguish orthologs from paralogs and pseudogenes that are in a different syntenic context. This is an advantage over spliced alignment approaches that map each known gene individually without considering synteny. Third, coding exons are often conserved (36–38), even in distant species (39) and thus typically align to many species.
Using pairwise genome alignments to map genes from a well-annotated reference genome to related species by projecting the coordinates of aligned exons was first developed at the University of California, Santa Cruz in the TransMap approach (40–42). As argued above, exons that are conserved in the related species should lack exon-inactivating mutations that destroy splice sites, shift the reading frame or create in-frame stop codons. In principle, determining exon conservation by analyzing the aligned sequence is simple. In practice, however, determining exon conservation is difficult, since this requires that alignments of truly conserved exons lack any exon-inactivating mutations. This requirement is not true for genome alignments. As we show in the following, genome alignments have two limitations that hinder their direct application to assess coding exon conservation.
The first limitation is that genome alignments, by design, align entire genomes and are unaware of reading frame and splice sites in case the aligned region corresponds to a coding exon. Instead, genome alignments aim at optimizing sequence similarity by using scoring schemes that reward matches between identical bases and penalize substitutions and insertions/deletions (indels) (43). Consequently, the resulting exon alignments can have exon-inactivating mutations even if the exon is conserved. In these cases, there is often an alternative alignment that lacks exon-inactivating mutations. For example, the alignment of the human and cow genome shows a frameshifting 4 bp insertion in DKC1, despite the fact that this exon is an annotated RefSeq gene in cow (Figure 1A). An alternative alignment with the same sequence identity exists where this deletion is located in the intron, showing that this exon is indeed conserved. Similarly, the human-mouse alignment of MISP shows two frameshifting indels, just separated by six bases (Figure 1B). These frameshifts are likely spurious as an alternative alignment without these indels and with similar sequence identity exists. Thus, due to alignment ambiguities and aiming at optimizing sequence similarity, standard alignment is not optimal for assessing if coding exons are conserved across species.
Figure 1.

Limitations of genome alignments for assessing exon conservation. (A) The genome alignment shows a 4 bp frameshifting insertion (red font). This is an alignment ambiguity, as an equivalent alignment exists where this insertion is in the intron (‘ideal alignment’). Upper case letters are exonic bases, lower case letters are intronic bases. (B) The genome alignment shows two close frameshifts that compensate each other. These two frameshifts are likely spurious and did not happen in evolution as an alternative alignment with 12 versus 13 identical bases (grey background) exist that lacks these indels. (C) Two examples where the acceptor (left) or donor (right) splice site is mutated. In both cases, the exon is conserved but its splice site has shifted by 9 bp into the intron. The ideal alignment would align the original and the shifted splice site. By aligning non-orthologous but ‘functionally equivalent’ splice site bases, the ideal alignment correctly identifies the exon boundaries in the other species. (D) In contrast to (B), two real compensating frameshifts change the reading frame for 15 codons. The alignment with both frameshifts has a much higher number of identical bases (grey background) than the alignment without both frameshifts, which strongly suggests that these compensating frameshifts did occur in evolution.
The second limitation in using genome alignments to assess coding exon conservation comes from their objective which is to align orthologous bases (33). This is useful for inferring evolutionary history, but is not optimal for assessing conservation of coding exons, where splice sites can shift during evolution. Such splice site shifts likely happen if a new (maybe alternative) splice site arises and subsequently the ancestral splice site is mutated. In these cases, the genome alignment will align the splice site of an exon in the reference to the orthologous bases in the related species; however these bases do not function as the splice site anymore. This would appear like a splice site mutation in the genome alignment and results in incorrectly annotated exon boundaries, as shown for two examples in Figure 1C and Supplementary Figure S1. However, the splice site in the reference and the shifted site in the related species are ‘functionally equivalent’. To identify the correct exon boundaries in the related species, it would be beneficial to align these functionally equivalent splice sites (Figure 1C).
To improve the use of genome alignments to assess which coding exons of a well-annotated reference genome are conserved in which related genomes, one could realign the exonic sequence considering reading frame and splice sites, and aligning functionally equivalent bases if necessary. For the examples in Figure 1A–C, this ‘ideal alignment’ will show an intact reading frame and splice sites, and would provide the correct exon boundary coordinates in the related genome. However, it is important to note that real frameshifting indels do occur in evolution. Such frameshifts can have two consequences. First, two frameshifts can compensate each other if the downstream frameshift restores the original reading frame, thus preserving the exon (Figure 1D). Second, and more important, frameshifts can result in gene inactivation, exemplified by a frameshifting deletion that inactivates the masticatory muscle gene MYH16 in human (44) or frameshifts that inactivate the vomeronasal organ gene TRPC2 in human and other primates (45). Therefore, to avoid mistakenly classifying inactivated exons as conserved, the realignment should not enforce intact reading frames and splice sites by all means.
Here, we show that the examples in Figure 1 are not exceptions and that genome alignments contain thousands of spurious frameshift and splice site mutations in exons of conserved genes. To utilize genome alignments to map exon annotations from a reference to many aligned genomes, we have developed CESAR (Coding Exon-Structure Aware Realigner) that realigns coding exons with a Hidden-Markov-Model (HMM) and directly incorporates the reading frame and splice site annotation of each exon. Extensive tests on simulated and real data show that CESAR is able to align shifted splice sites and to distinguish real from spurious frameshifts. We show that CESAR detects thousands of additional exons without inactivating mutations and that such exons match real exons with very high accuracy. Finally, we demonstrate the potential of this approach on the UCSC alignment of 100 vertebrate genomes (32). By realigning 188 788 human coding exons of 19 865 genes with CESAR, we map on average 88% and 71% of the human genes to 61 other mammals and to 38 non-mammalian vertebrates, respectively. These comparative gene annotations in 99 non-human vertebrates are an important resource that is available at http://bds.mpi-cbg.de/hillerlab/CESAR/.
MATERIALS AND METHODS
Structure of CESAR
An HMM is a powerful probabilistic method that can represent the intron–exon structure and the reading frame of the coding sequence. To realign the query sequences to a given exonic sequence of the reference genome, we built an exon-specific HMM whose states represent the codons in the given reference sequence and used the Viterbi algorithm to find the most probable alignment. Similar to HMMs for gene finding (10), our general HMM structure has states to emit the up- and downstream intron, states to emit the splice sites, and states to emit the exonic sequence (Figure 2). Splice sites comprise 22 acceptor and 6 donor states that model the nucleotide distribution of the last 22 and first 6 intron positions, respectively. Conceptually similar to profile HMMs (46), the exon body comprises clusters of states that emit and insert full or partial codons, thus modeling frameshifting insertions and deletions (Figure 2). In contrast to profile HMMs, the HMM is built only from the single exon sequence of the reference genome. Codon deletions are modeled by transitions that skip codon-emitting states. While intronic and splice site states are generic, the number of codon emitting clusters is determined by the given exonic sequence.
Figure 2.

Schematic representation of CESAR. The Hidden-Markov-Model consists of states that emit the up- and downstream intronic bases, the splice sites and the exon body in between. The exon body consists of states that match entire codons with emission probabilities reflecting the similarity to the codon in the reference exon (47), states that emit partial 1 or 2 bp codons that represent frameshifting deletions, states that insert any of the 61 non-stop codons, and nucleotide insertion states that insert in-frame stop codons or insert frameshifts. Codon deletions are modeled by transitions that skip between 1 and 10 codon units (blue transitions; only 1 to 3 codon deletions are illustrated here for clarity). The non-emitting (silent) black-circle states allow deleting more than 10 successive codons, similar to delete states in a profile HMM (46). All transitions representing exon-inactivating mutations (splice site mutations or frameshifting indels) are shown in red, transitions to codon insertion states are green and transitions that loop in insert states are black. The grey transitions are not free parameters but are fixed by the constraint that the sum of all out-going transition probabilities of a state must be 1.
We intended to build a model that is minimal in the number of states and transitions, yet sufficient to capture all different types of evolutionary events. For example, we did not model a 5 bp deletion by a single direct transition, however the HMM is able to report such a 5 bp deletion by a single codon deletion followed by a 1 bp partial codon emission (which models a 2 bp deletion). Also, we did not explicitly model insertions inside a codon, which would substantially increase the number of states and transitions. Instead, our model allows insertions only between codons, which is sufficient to find frame-preserving and frameshifting indels. Finally, partial codons split by intron boundaries are emitted as single nucleotides, which is important to find correct splice sites and the correct reading frame, but we scored them as nucleotides and not as codons, as done in GeneWise (13).
Parameterization
Our model consists of the following five free different transition probabilities: (i) codon insertion probability (green transitions in Figure 2), (ii) subsequent codon insertion probability (black transition), (ii) subsequent nucleotide insertion probability (black transition), (iv) total codon deletion probability (blue transitions) and (v) probability of exon-inactivating mutations (red transitions), which is the probability of disrupting a splice site, introducing a frameshifting indel or emitting a stop codon in a full codon emission state. The total codon deletion probability is the sum of all probabilities that delete between 1 and 10 codons at once. The 10 individual probabilities were empirically derived from the ratio of 1 to 10 codon indels observed in RefSeq exons between human and rhesus (Supplementary Table S1).
We determined which of these five transition probabilities achieved the highest accuracy on a simulated data set containing exons with and without real frameshifts and exons with splice site shifts (described below). In order to realign exonic sequences from a whole genome alignment that includes species at various distances, we determined the 5 transition probabilities separately for 10 data sets that represent evolutionary distances from 0.1 to 1.0 substitutions per neutral site in step sizes of 0.1. However, in all 10 simulated data sets, the following probabilities were consistently the best-performing parameters: 0.01, 0.8, 0.25, 0.025 and 0.001 for codon insertion, subsequent codon insertion, subsequent nucleotide insertion, total codon deletion and exon-disrupting probability, respectively. Therefore, we used these transition probabilities for all tests.
The emission probabilities of codon emitting states were taken from the codon substitution matrix (47), thus the emission of a specific ‘full codon emission’ state reflects the similarity to the respective codon in the given reference exon. The probability of emitting a stop codon (exon-inactivating probability) was added to emission vectors of codon match states. If a codon in the given reference sequence has one or more ambiguous bases, we used an emission vector that averages the emission vectors of all possibilities for this codon (for GNC, we will average GAC, GTC, GGC and GCC). Codons that have one or more ambiguous (N) bases in the query are emitted with a probability that is the average emission probability of all possibilities for this codon. Emission probabilities of nucleotides at the different splice sites were empirically determined from human RefSeq exons. Since splice sites of U12 introns can have dinucleotides other than GT/GC and AG (48), we captured the special U12 donor and acceptor nucleotide distribution in a separate profile. We used a uniform distribution for the probabilities of inserting bases in the nucleotide insertion state. To get codon insertion probabilities, we calculated for each codon the probability that any codon mutates into this codon based on the codon substitution matrix (47). These 61 probabilities are given in Supplementary Table S2. For first coding exons that do not have an acceptor site but a start codon, we replaced the acceptor states with states that emit the start codon. Likewise, for last coding exons that do not have a donor site, we replaced the donor states with states that emit a stop codon.
Simulated data sets to determine CESAR transition probabilities
It would be ideal to have exon alignments where the true alignment is known. Since the true alignment is never known with certainty for real exons, we used simulated data for parameter estimation, similar to (49). Since the simulated data sets from (49) lack alignments with splice site shifts and real frameshifts, we created simulated data sets using Evolver (http://www.drive5.com/evolver/), a method previously used to compare genome alignment methods (50). Evolver simulates whole genome evolution including substitutions, insertions and deletions, and genomic rearrangements. Important for our purpose is that (i) Evolver has an explicit model of protein evolution and maintains the gene structure for genes under selection, and (ii) Evolver outputs the true alignment. We used the mouse chromosome 19 containing 645 genes with 6105 coding exons and evolved it for different evolutionary distances ranging from 0.1 to 1.0 substitutions per neutral site in a step size of 0.1. In this way, we produced 10 genome alignments, each one corresponding to a particular evolutionary distance. Then, we extracted the alignments of coding exons, which are considered to be the true alignment in the following experiments. To test if CESAR is sensitive with respect to the length of the sequence flanking the exon, we extracted 50 bp, 100 bp and 500 bp upstream and downstream flanks of the exon from the evolved genome. Our tests showed that the length of the flanks has very little influence on the alignment. Therefore, we conservatively used the larger 500 bp sequence context in all subsequent tests.
We created different data sets in order to evaluate alignment accuracy for the different technical and biological scenarios described in Figure 1. First, we created an ‘intact exon’ data set of 1000 exons where no splice site shifts and no frameshifting indels occurred. Evolver maintains the gene structure of a gene that evolves under selection, implying that the splice sites and the reading frame is preserved but nucleotide substitutions and frame-preserving indels will occur proportional to the evolutionary distance. Our intact exons data set consists of these exon alignments.
Second, we created a so called ‘no-frameshift’ data set, motivated by our observation that standard sequence alignments contain numerous pairs of frameshifting indels that are separated by only a few bp and that compensate each other without having an in-frame stop codon in between (Figure 1B). To this end, we first extracted all compensating frameshift events from the human-mouse pairwise alignment for the UCSC knownGene set (51) and plotted the distance between such pairs of events (Figure 3). The distance histogram shows that 85% have a distance of 12 bp or less, which are most likely alignment ambiguities and not two real frameshifts. To create the no-frameshift data set, we randomly sampled 500 intact exon alignments, inserted a 1 bp frameshifting insertion at a random position in the evolved exon sequence, and deleted 1 bp to create the second compensating frameshift 6 to 12 bp up- or downstream. We ensured that the manipulated exonic sequence could be translated into a protein. Since these two close compensating frameshifts result in at most four different amino acids in an otherwise intact exon, an exon structure aware aligner should not introduce any frameshift. The true alignment is therefore the alignment that does not have any frameshift.
Figure 3.

Close pairs of compensatory frameshifts are abundant in genome alignments. The distance between two compensating frameshifts is plotted as a histogram for the human-mouse alignment. Compensating frameshifts are pairs of frameshifts where the second frameshift returns to the original reading frame and the sequence between the two frameshifts is translatable (no in-frame stop codon in the new reading frame).
Third, we created the counterpart data set with two real compensating frameshifts, motivated by the 371 human-mouse cases with a distance of 30 bp or larger (Figure 3) that are likely real evolutionary events. Such real compensatory events are also characterized by a higher similarity at the nucleotide level compared to the protein sequence in the region between the two frameshifts (Figure 1D). This ‘two frameshift’ data set differs from the no-frameshift data set only in the distance of 30 to 45 bp between the two frameshifts that we introduced in 500 randomly chosen exons. Since both frameshifts are considered to be real, the true alignment should show two frameshifts.
Fourth, we created a ‘one-frameshift’ data set where real frameshifts destroy the exon's reading frame (44,45). To this end, we introduced a single bp insertion in the middle 80% of 500 randomly chosen intact exons. The true alignment for such cases contains exactly one frameshift.
Fifth, we created a ‘splice site shift’ data set, motivated by the examples in Figure 1C. If the splice site of an exon in the reference is aligned to a shifted splice site in the query, there will be an insertion or deletion close to this splice site, depending on whether the splice site shifted into the intron or into the exon. Therefore, to simulate splice site shifts, we inserted or deleted a multiple of 3 bp from one boundary of 500 randomly chosen intact exons. When mimicking a splice site shift into the intron, which makes an exon longer, we ensured that the insert does not contain a stop codon. The lengths of the introduced insertions or deletions were sampled from the distribution of splice site shift distances of 360 real exons where a splice site shift happened in the mouse, rat, cow or dog genome (according to the RefSeq and Ensembl annotation) (Supplementary Figure S2). All these splice site shifts are also supported by ESTs/mRNAs. To ensure that these splice site shifts are genuine and not artifacts arising from sequencing errors, we assessed if the cow, rat and dog splice sites are in genomic regions with high sequencing quality scores (no quality scores are available for mouse). We used liftOver to map the exon coordinates plus a 20 bp flanking sequence to an assembly (bosTau7 to bosTau4, rn5 to rn4 and canFam3 to canFam2) where sequencing quality scores are available. Indeed, all cow, rat and dog splice site shifts are located in regions with high quality scores above 20 (probability of calling a base correctly is 0.99).
Data sets to test spliced alignment methods
To investigate if existing spliced alignment methods (Spaln, Exonerate, GeneWise and Pairagon (13–15,52)) that map proteins or the entire coding sequences to the genome can be used to realign coding exons, we first created an independent data set by running Evolver again with a different seed to obtain 10 new genome alignments for the 10 different evolutionary distances (0.1 to 1.0 substitutions per neutral site). Since spliced aligners are not designed to align individual exons but entire cDNAs, we created cDNAs comprising all protein-coding exons of a gene, and aligned them to the genomic locus that spans the entire cDNA. To compare spliced aligners and CESAR, we focused our evaluation on a single, randomly chosen internal exon of each gene. In these tests, the only difference between spliced aligners and CESAR is that CESAR aligned the single exon only to the genomic locus of this exon with a 500 bp flank. Then, we evaluated the accuracy of the alignment of this exon under consideration. For Spaln and Exonerate, we tested both aligning the coding and the protein query sequence to the genome (coding2genome and protein2genome mode). GeneWise always requires a protein sequence as input. The parameters used to run the different tools are listed in Supplementary Table S3. To test frameshifts and splice site shifts, we manipulated a single internal exon per gene. All data sets, including the cDNA, the surrounding genomic sequence and the true alignments are available at http://bds.mpi-cbg.de/hillerlab/CESAR/.
Evaluating alignment accuracy
We noticed that alignment ambiguities make it difficult for both CESAR and other spliced aligners to locate the exact position of an indel. Such ambiguities arise if, for example, a GAG codon is deleted in a GAGGAG context or if there are substitutions close to the indel (Supplementary Figure S3A and 3B). Likewise, two 3 bp deletions that occurred in close proximity tend to be reported as a single 6 bp deletion (Supplementary Figure S3C). As such slight shifts in the indel position do not affect our ability to assess exon conservation, we defined ‘nearly identical’ alignments as alignments that are either identical to the true alignment or differ from the true alignments only in the position of an indel that we allow to be shifted by at most 6 bases up- or downstream. Note that such nearly identical alignments correctly identified both splice sites and show the right number of frameshifts.
Exon mutations of orthologous human, mouse, rat, cow and dog coding genes
We used Ensembl Biomart to extract 13 498 human genes that have 1:1 orthologs to mouse, rat, cow and dog. For each gene, we realigned all exons of all Ensembl transcripts with CESAR. Subsequently, we determined the transcript(s) with the minimal number of frameshifts and splice site mutations for each gene and took the longest of these transcripts in case of a tie. These transcripts contained a total of 149 331 coding exons (average of 11 exons per gene). For each of these exons, we determined the number of frameshifts and splice site mutations in the UCSC 100-way alignment (32) and after these exons had been realigned with CESAR.
Annotating human exons in 99 other vertebrates
We used the UCSC 100-way alignment where 99 vertebrates are aligned to the human hg19 genome assembly (32). We used the longest transcript of the UCSC knownGene annotation with 20 002 protein-coding genes. We excluded 137 genes, which have a frameshift in the human genome, either because of polymorphisms or programmed ribosomal frameshifting (such as the OAZ1 gene). The final set contained 19 865 genes having 188 788 exons.
For each exon and for each of the 99 species, we extracted the aligned exon sequence together with 50 bp genomic flanking to obtain the real or potentially shifted splice site sequences. Then, we used maf-join (53) to create a reference-guided alignment of all CESAR realigned exons. By design, CESAR aligns only coding exons, however, we add 20 unaligned bases flanking the exon to allow the inspection of splice sites. For each realigned exon, we checked if the exon is intact. An intact internal exon has an intact open reading frame and two consensus splice sites. Intact first (last) exons have a consensus donor (acceptor) splice site and an intact reading frame between the splice site and the annotated start (stop) codon. To annotate intact exons in the query genome, we identified the genomic coordinates by the query bases that align to the exon boundaries. This procedure was used to map genes annotated in human to 99 other vertebrates. The coordinates of the intact exons and annotated genes (genePred format) for the 99 species and the 100-way realignment (maf format, 7.9 GB) are available at http://bds.mpi-cbg.de/hillerlab/CESAR/ for download and visualization as a track hub in the UCSC genome browser.
RESULTS
CESAR has higher accuracy in aligning shifted splice sites and distinguishing real from spurious frameshifts than spliced alignment methods
The most critical part in using existing whole genome alignments to assess exon conservation and to map gene annotations across species is the accuracy of exon alignments. To improve this accuracy, we developed CESAR to realign coding exons, while considering both reading frame and splice sites of each exon as annotated in the reference species. Since aligning coding exon sequences is conceptually similar to spliced alignment, we first evaluated the accuracy of CESAR in comparison to other spliced aligners (see Materials and Methods). We tested the following widely used, state-of-the-art methods, all of which take the reading frame of the gene into account: GeneWise (13), Exonerate (14) and Spaln (15). While GeneWise requires a protein sequence as input, Exonerate and Spaln can align both coding nucleotide and its translated protein sequence to the genome. In addition, we tested Pairagon (52) that does not consider the reading frame but is one of the most accurate methods for aligning cDNAs across genomes. In contrast to CESAR, none of these spliced aligners found the ideal alignment for all four examples in Figure 1. In particular, the spliced aligners could not align the shifted splice sites in Figure 1C.
To systematically test these methods on a larger scale, we used five simulated data sets where the true alignment is known (see Materials and Methods). A detailed breakdown of the types of differences between the reported and true alignment is given in Supplementary Figure S4–S8. First, to test the suitability of our simulated data, we used intact exons with identical splice sites and without any frameshift. We found that all methods have high accuracy for close evolutionary distances (Figure 4A). The accuracy decreased for larger evolutionary distance where the sequence similarity between coding sequence and query genome is lower. CESAR's accuracy was slightly higher compared to Spaln and Pairagon. For larger evolutionary distances, the accuracy of Exonerate with coding sequence input mainly dropped because no alignment was found, while he accuracy of GeneWise dropped because incorrect splice sites were aligned; this behavior was found in the other tests as well.
Figure 4.
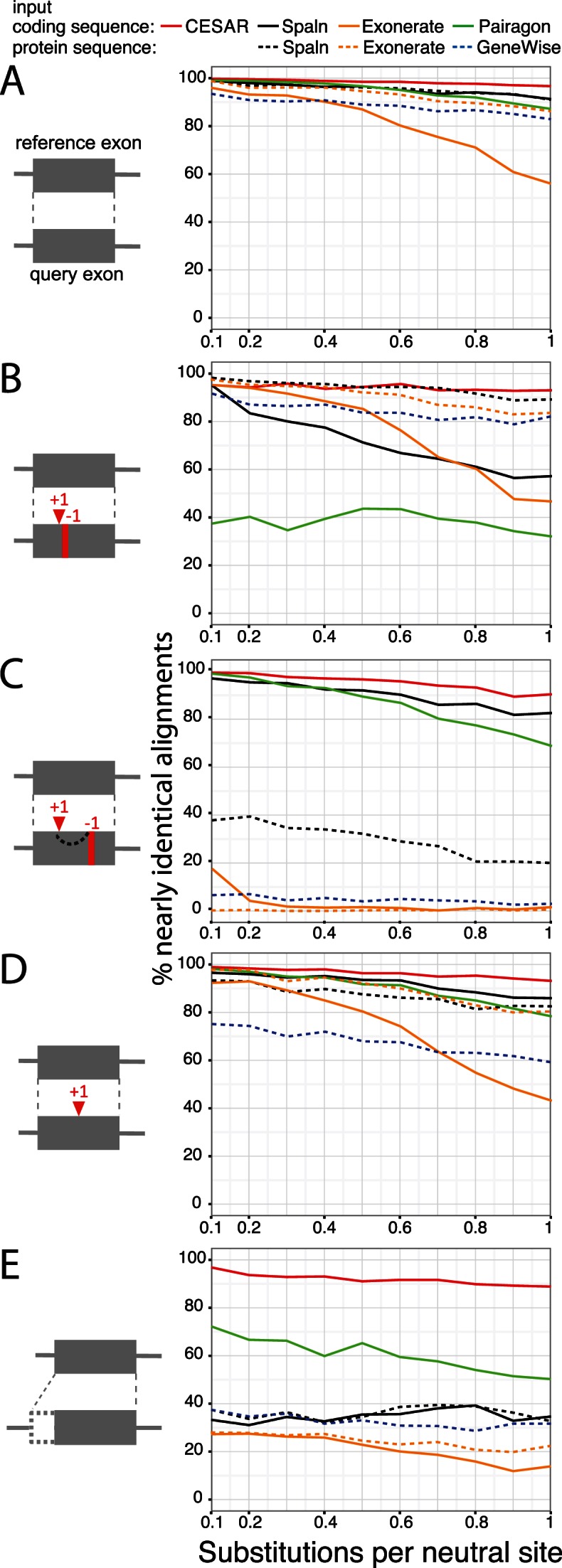
Comparative evaluation of CESAR's alignment accuracy. The accuracy of aligning five data sets of simulated exons with or without frameshifts and with shifted splice sites is shown. Nearly identical alignments are defined as being identical or differing from the true alignment only in a ≤6 bp shift in the position of indels. The five different data sets are: (A) intact exons without frameshifts and splice site shifts, (B) two spurious compensating frameshifts, (C) two real compensating frameshifts, (D) one real frameshift and (E) splice site shifts (see Materials and Methods).
Second, we tested the ability to avoid the numerous compensating frameshifts that are in close proximity and likely spurious (Figure 1B). As shown in Figure 4B, CESAR and Spaln (with protein sequence input) were the best performing methods with >90% accuracy, followed by GeneWise and Exonerate (protein input) with ≥80% accuracy. Spaln with coding sequence input and especially Pairagon, which by design is not aware of the reading frame, often produced incorrect alignments that contained these spurious frameshifts.
Third, we tested the ability to report frameshifts by analyzing a data set with two real compensating frameshifts separated by 30–45 bp and data set with a single real frameshift (Figure 4C and D). CESAR achieved >90% accuracy and slightly outperformed Spaln (coding sequence input) and Pairagon on both data sets. On the data set where two compensating frameshifts are expected, GeneWise, Spaln (protein input) and Exonerate frequently did not report any frameshifts. For the purpose of assessing exon conservation this is not a severe limitation as the exon is indeed conserved and is just translated in a different reading frame for 10–15 codons. However, on the data set with a single exon-inactivating frameshift, these methods also did not report this frameshift in several cases and often reported incorrect splice sites in an attempt to avoid the frameshift. This is a more severe error as one would incorrectly infer exon conservation from such an alignment.
Finally, we tested the ability to align the correct splice site if the splice site has shifted in the query genome. While CESAR achieved an accuracy of ≥89% (Figure 4E), all other methods had difficulties aligning the shifted splice site, which is most likely a consequence of aiming at aligning orthologous bases, which underlies the design of these methods. Pairagon was the second-best performing method with an accuracy of 50–72%.
In summary, in these tests CESAR was the only method with consistently high accuracy (≥89% for all evolutionary distances and all data sets) and was able to report correct alignments for most exons with shifted splice sites, even at large evolutionary distances. These tests also showed that the spliced aligners have different strengths and weaknesses. While Pairagon and Spaln with coding sequence input were accurate in reporting frameshifts when they occurred, they also reported two close compensating frameshifts when they are more likely explained by a few codon changes. The latter cases were handled well by GeneWise, Exonerate and Spaln with protein input, however, they failed to report many frameshifts that indeed occurred.
CESAR drastically reduces the number of spurious frameshifts in genome alignments
Next, we tested CESAR's ability to avoid spurious frameshifts on real data by applying it to 149 331 exons of 13 498 human coding genes that have a 1:1 orthology relationship between human and mouse, rat, cow and dog. Since these orthologous genes are also annotated in the other four species, most frameshifting indels and disrupted splice sites are likely spurious and we expected CESAR alignments to lack these exon-inactivating mutations. For mouse, we found that genome alignments contained 9046 frameshifts in 2.7% (3976) of the 149 331 coding exons. Realigning the exon sequence drastically reduced this number to 614 frameshifts in 0.3% (445) of the mouse coding exons (Figure 5A, Supplementary Table S4). A similar reduction was observed for rat, cow and dog, where CESAR reduced the number of exons containing frameshifts from ∼3% to 0.4–0.6% and the total number of frameshifts by ∼90% (Figure 5A, Supplementary Table S4). This shows that CESAR drastically reduces the number of spurious frameshifts in conserved genes.
Figure 5.

CESAR drastically reduces the number of exon inactivating mutations. (A) Number of frameshift mutations and (B) number of splice site mutations in genes that have a 1:1 orthology relationship and are annotated in mouse, rat, cow and dog.
Next, we analyzed the 614 frameshifts that remained in mouse after realigning. Of these 614, 7.5% (47) were pairs of real compensating frameshifts (Supplementary Figure S9 shows two examples). Of the remaining 567 frameshifts, 30.5% (173) and 44% (249) were located in the first or last 20% of the coding sequence, respectively (Supplementary Figure S10). Similar observations were made for gene inactivating mutations that occur in the human population (54), indicating that these frameshifts are real and that genes are more tolerant to frameshifts close to the start or end of the coding sequence. Finally, of the remaining 145 frameshifts that were not located in the first or last 20% of the coding sequence, 68 (47%) occurred in 35 human exons that are indeed not conserved in mouse. For example, the human NEDD4, SCML2, SH2D4A, CCDC15 genes are clearly conserved in mouse, yet each gene has an exon that is not conserved (Supplementary Figures S11–S14). Furthermore, there is the possibility that some of the reported frameshifts are actually sequencing errors in the mouse mm10 assembly and not real mutations. For example, we found that the AUTS2 gene has a frameshifting 1 bp deletion and the IFI30 gene has a frameshifting 1 bp insertion in the mouse assembly, but mRNAs, ESTs and all aligning Sanger sequencing reads do not support these frameshifts (Supplementary Figures S15 and S16). Overall, this shows that CESAR is able to report real frameshifts and that exon realignment substantially improves the accuracy in assessing exon conservation across species.
CESAR correctly identified 91% of real splice site shifts and drastically reduces the number of spurious splice site mutations
To test if CESAR is not only able to correctly align shifted splice sites in simulated but also real data, we tested it on 360 real exons where a splice site shift occurred in the mouse, rat, cow or dog. We found that CESAR correctly aligns the annotated shifted splice site in 91% (326) of these 360 exons. We manually inspected the other 9% (34 cases) and found that ∼47% (16 of 34) have splice site shifts of more than 24 bp. This shows that while CESAR was able to correctly align the majority of real shifted splice sites where the splice site shift is nearby, it had difficulties with splice site shifts over larger distances.
Next, we analyzed the number of splice site mutations in the 149 331 coding exons of the 13 498 human genes with a 1:1 orthology relationship between human and mouse, rat, cow and dog. For mouse, we found that genome alignments contained 2891 splice site mutations in 1.9% (2792) of the 149 331 coding exons. CESAR reduced this number to 180 splice site mutations in 0.11% (172) of the coding exons (Figure 5B, Supplementary Table S4). Consistently, realigning exons also reduced the number of rat, cow and dog exons with mutated splice sites from 1.7–2% to 0.17–0.19% and reduced the total number of splice site mutations by ∼88% (Figure 5B, Supplementary Table S4). This shows that CESAR drastically reduces the number of spurious splice site mutations in conserved genes.
We investigated the remaining 180 splice site mutations that remained in realigned mouse exons. We found that 44.5% (80 of 180) are intron deletions, where an entire intron is precisely removed, probably by recombination with a processed transcript of the same gene (55). The remaining mutations contain actual splice site shifts that CESAR did not detect because the shifted splice site deviates from the consensus (Supplementary Figure S17) or because the shift involves large distances, exemplified by a 429 bp acceptor shift in an exon of SPATC1, reducing the size of this 540 bp human exon to 111 bp in mouse (Supplementary Figure S18). The other remaining splice site mutations often happen in coding exons that are not conserved in mouse, and some of them have additionally frameshifting indels (Supplementary Figure S14). These results confirm that realigning exons with the objective of aligning functionally equivalent splice site bases improves identifying correct splice site positions in other species.
CESAR detects thousands of additional intact exons in the mouse genome
To test if CESAR's ability to avoid spurious frameshifts and to identify shifted splice sites results in improved power to detect conserved exons, we realigned all 176 858 coding exons that align between human (reference) and mouse (query) in the genome alignment. To be conservative, we classified exons as intact if the realigned mouse sequence has consensus splice sites and intact reading frame (no frameshift, no internal stop codon).
After realigning with CESAR, 172 720 of the 176 858 exons were classified as intact. In contrast, only 167 327 exons were classified as intact when we used the sequence in the genome alignment without applying CESAR. This shows that CESAR detects an additional 5393 intact exons that have spurious mutations in the genome alignment.
Exons that lack inactivating mutations after realignment match mouse exons with high accuracy
Next, we tested if CESAR realignments make it possible to accurately map human exon annotations to mouse by using the mouse RefSeq and Ensembl annotation. For all intact exons, we used the bases at the aligned exon boundaries to identify the mouse exon coordinates and annotate these human exons in the mouse genome. All exons classified as non-intact are not mapped to mouse.
We found that the vast majority of these intact exons (99.1%, 170 267 of 171 765) overlap a mouse exon that is annotated in mouse RefSeq and Ensembl. Furthermore, for 97.6% (166 232 of 170 267) of these exons, both the boundaries were correctly identified. This shows that intact exons after realigning with CESAR match real mouse exons with very high accuracy.
Next, we focused on the 4138 exons that we classified as non-intact. We found that 51.2% (2120 of 4138) indeed do not overlap any exon annotated in mouse, showing that not all exons with aligning sequence correspond to exons in the query genome. However, 48.7% (2018) of the non-intact exons overlap an exon annotated in mouse. 86.9% (1753 of 2018) of these exons were conservatively classified as non-intact because CESAR could only correctly identify one but not the other boundary. The majority of these exons are first or last coding exons of multi-exon genes or single-exon genes (57.6%, 1009 of 1753), which is due to the following two reasons. First, the position of the start or stop codon in the mouse is shifted by an average of 97 bp (exemplified in Supplementary Figures S19, S20) and CESAR did not align the start/stop codons over these large distances. Second, a real frameshift did occur in many last exons, which leads to a different C-terminal peptide in mouse (Supplementary Figures S21, S22). In these cases, CESAR correctly reported the frameshift and according to our strict definition these exons are non-intact. This corroborates our observations that frameshifts are enriched the N- and C-terminus of a protein (Supplementary Figure S10) and suggests that the protein termini are under less evolutionary constraint because extensions or truncations are less likely to affect function.
Using CESAR to annotate human exons in 99 other vertebrates
To demonstrate the potential of using CESAR to annotate exons and genes in many other species, we applied our approach to the largest vertebrate genome alignment available, which includes 99 vertebrates that are aligned to the human genome (32). We realigned all 188 758 human coding exons of 19 865 genes with CESAR. For exons flanked by a U12 intron, we used a U12 specific splice site profile (Supplementary Figure S23). Figure 6 shows a genome browser visualization of a realigned exon.
Figure 6.

UCSC Genome Browser screenshot showing human exons realigned with CESAR. Top: Several exons of SLC24A3. Bottom: Realignment of one exon, together with unaligned flanking sequence on either side of the exon. Only a subset of the 99 non-human vertebrates is shown. Dots refer to bases that are identical to the aligned human base.
To annotate genes in 99 non-human vertebrates, we grouped all intact exons from the same gene into a gene model. As shown in Figure 7 and Supplementary Table S5, the percentage of 188 788 exons that we annotate ranges from 97% (green monkey) to 31% (lamprey). The percentage of the 19 865 genes for which we can annotate at least one exon ranges from 96% (chimpanzee) to 48% (lamprey). On average, 87.5% of the human genes have at least one intact exon in other mammals and 70.9% in non-mammalian vertebrates. Thus, the number of annotated exons and genes clearly depends on the evolutionary distance to human, because a higher proportion of coding exons will not align at all over larger distances and because the 19 865 genes include genes that are restricted to certain lineages such as mammals. However, the quality of the genome assembly also affects the number of annotated exons and genes. For example, the percentage of intact exons is higher for the green monkey compared to the chimpanzee (97% versus 95.3%). While the chimpanzee is evolutionarily closer to human, the green monkey has a better contig assembly (N50 value: 49 versus 90 kb). Similarly, the lower percentage of intact exons found in the scarlet macaw and the Atlantic cod is likely due to their short contigs (N50: 4 kb and 2 kb).
Figure 7.

Summary of comparative gene annotation in 99 non-human vertebrates. The proportion of the 19 865 human genes (blue triangles) and 188 788 exons (red circles) that we annotate in 99 vertebrate genomes after realignment by CESAR. (A) 61 mammalian species. (B) 38 non-mammalian species. Placental mammals, birds and teleost fish are highlighted by a light yellow background.
The human genes that we annotate in the 99 other species can be visualized in a genome browser, as exemplified in Figure 8 for 8 vertebrate genomes. These annotation tracks display the human gene symbol, which facilitates querying the literature and databases about the functional annotation of this human ortholog. These comparative gene annotations obtained with CESAR are an important genome annotation resource, especially for the genomes that currently lack homology-based gene annotations.
Figure 8.

Genome browser screenshot of human genes annotated in other vertebrates. The UCSC genome browser screenshot of a 605 kb locus in the human genome (hg19, chr19:34 287 751–34 893 318) with four genes is shown at the top. Genome browser annotation tracks of human exons mapped by CESAR are shown below for 8 of the 99 genomes covering different clades. The phylogenetic tree of all 100 species is shown on the right.
DISCUSSION
Comparative gene prediction will be indispensable to annotate coding genes in numerous genomes that have been sequenced and will be sequenced in future. Genome alignments are highly useful to assess exon conservation across species, as they make use of synteny (33–35), which helps to distinguish many orthologous genes from their paralogs or pseudogenes. However, as we showed here, genome alignments contain thousands of spurious frameshifts and splice site mutations in exons that are conserved across species and consequently should not exhibit such mutations. We developed the Hidden-Markov-Model based method CESAR that realigns the exon sequence considering the reading frame and splice site position of the exon. We demonstrated that CESAR effectively avoids spurious mutations while being able to report real mutations, both on simulated and real data.
While CESAR is inspired by gene prediction and spliced alignment approaches (10,12,13,15), the purpose of CESAR is different in that it is designed to realign existing exon alignments. In addition, CESAR differs from gene prediction and spliced alignment approaches in the following aspects. First, we intended to develop an approach that is simple yet sufficient to capture the different types of spurious and real inactivating mutations. Consequently, while other methods have more sophisticated HMM layouts or use two integrated pair HMMs like GeneWise (13), we used a single HMM with a simple layout. Likewise, we used a single parameter for all exon-inactivating mutations (splice site disruptions, frameshifts, stop codon mutations) that reflects the balance between preserving reading frame and splice sites whenever possible while reporting real frameshifts that did occur in evolution. Second, in contrast to profile HMMs that would require a multiple sequence alignment of intact exons as input, our approach only requires the exon sequences of a single reference genome. Third, as shown in Figure 1C, CESAR is able to align ‘functionally equivalent’ splice site bases, in cases where the bases that are orthologous to the splice site in the reference genome are mutated in the query.
Similar to the most common alternative donor or acceptor splice variants (56–58), we found that most splice site shifts involve a distance of three or six bases (Supplementary Figure S2), which would lengthen or shorten the exon by one or two amino acids. While some of these subtle protein changes can modulate function (59), many of these short-distance shifts are likely to have a minimal impact on protein function or no impact at all. Thus, such exons are indeed conserved in the query species, and it is reasonable to align the shifted splice sites, as CESAR does. On the other hand, rare splice site shifts over a large distance pose a challenge for CESAR. While it would still be desirable to accurately align such large-distance shifted splice site instead of reporting a splice site mutation, large-distance shifts substantially elongate or shorten the exon, exemplified by an extreme 429 bp acceptor shift in a mouse exon of SPATC1 (Supplementary Figure S18). Since large-distance shifts are more likely to impact protein function, the reported splice site mutation at least points to exons where conservation in the query species is less certain.
To demonstrate the feasibility and utility of using genome alignments for comparative gene annotation, we used CESAR to realign 188 758 human exons in 99 non-human vertebrates, several of which have no comparative gene annotation yet. In order to produce a reliable annotation in the query species, our main objective was achieving a high precision, meaning a very high percentage of the intact exons should match real exons in the query species. Therefore, we used a conservative approach that only annotates intact exons without any inactivating mutation. However, as shown by comparison with the mouse gene annotation, this comes at the expense of missing first or last exons, where either large shifts of the start/stop codon or frameshifts affecting the proteins’ C-terminus happened. Future work could specifically train the CESAR HMM parameters for first and last exons to help aligning the start/stop codon also over larger distances. Similarly, it is conceivable to relax the strict requirement of no exon-inactivating mutation for first/last exons.
Using genome alignments to map genes to evolutionary distant species is more difficult for the following two reasons. First, an increasing number of coding exons will not align at all at a larger evolutionary distance. Consequently, they will be missed in the query annotation. However, highly sensitive local alignment parameters have been successfully used to detect remote homologies between conserved non-coding regions (60) and such sensitive parameters can uncover thousands of additional exon alignments between distant species (unpublished data). Second, all homology-based approaches are limited to annotating ancestral genes that existed in the common ancestor of reference and query species. Therefore, evolutionarily younger genes that are specific to lineages that do not include the reference species cannot be annotated. To annotate genes in distant species, CESAR can easily be applied to other existing genome alignments where evolutionarily closer species are used as the reference genome. For example, chicken, medaka and zebrafish have high-quality gene catalogs that include numerous lineage-specific genes that do not exist in human. Thus, existing genome alignments, where these species are the reference genome (1,32,60), can readily be used in combination with CESAR to accurately annotate coding genes many other bird and fish genomes. Given that gene annotation is an essential step to use genomes for biomedical research, CESAR contributes to reduce the growing gap between genome sequencing and genome annotation.
AVAILABILITY
CESAR's source code is available at https://github.com/hillerlab/CESAR/.
Supplementary Material
Acknowledgments
We thank Jacob Schreiber for valuable suggestions of the HMM model design and help with YAHMM. Furthermore, we would like to thank members of the Hiller lab and Holger Brandl for helpful comments on the manuscript, Xavier Prudent and Peter Schwede for their help in running the Evolver package, and the Computer Service Facilities of the MPI-CBG and MPI-PKS for their support.
FUNDING
Max Planck Society, the German Federal Ministry of Education and Research [031A099]; German Research Foundation [HI 1423/3-1]. Funding for open access charge: Max Planck Society; German Research Foundation [HI 1423/3-1].
Conflict of interest statement. None declared.
REFERENCES
- 1.Jarvis E.D., Mirarab S., Aberer A.J., Li B., Houde P., Li C., Ho S.Y., Faircloth B.C., Nabholz B., Howard J.T., et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haussler D., O'Brien S., Ryder O., Barker F., Clamp M., Crawford A., Hanner R., Hanotte O., Johnson W., McGuire J., et al. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson G.E., Hackett K.J., Purcell-Miramontes M., Brown S.J., Evans J.D., Goldsmith M.R., Lawson D., Okamuro J., Robertson H.M., Schneider D.J. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- 4.modENCODE Consortium. Roy S., Ernst J., Kharchenko P.V., Kheradpour P., Negre N., Eaton M.L., Landolin J.M., Bristow C.A., Ma L., et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerstein M.B., Lu Z.J., Van Nostrand E.L., Cheng C., Arshinoff B.I., Liu T., Yip K.Y., Robilotto R., Rechtsteiner A., Ikegami K., et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brent M.R., Guigo R. Recent advances in gene structure prediction. Curr. Opin. Struct. Biol. 2004;14:264–272. doi: 10.1016/j.sbi.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Picardi E., Pesole G. Computational methods for ab initio and comparative gene finding. Methods Mol. Biol. 2010;609:269–284. doi: 10.1007/978-1-60327-241-4_16. [DOI] [PubMed] [Google Scholar]
- 10.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 11.Parra G., Blanco E., Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanke M., Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl. 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 13.Birney E., Clamp M., Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slater G.S., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotoh O. Direct mapping and alignment of protein sequences onto genomic sequence. Bioinformatics. 2008;24:2438–2444. doi: 10.1093/bioinformatics/btn460. [DOI] [PubMed] [Google Scholar]
- 16.Yeh R.F., Lim L.P., Burge C.B. Computational inference of homologous gene structures in the human genome. Genome Res. 2001;11:803–816. doi: 10.1101/gr.175701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curwen V., Eyras E., Andrews T.D., Clarke L., Mongin E., Searle S.M., Clamp M. The Ensembl automatic gene annotation system. Genome Res. 2004;14:942–950. doi: 10.1101/gr.1858004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantarel B.L., Korf I., Robb S.M., Parra G., Ross E., Moore B., Holt C., Sanchez Alvarado A., Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanke M., Schoffmann O., Morgenstern B., Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., White O., Buell C.R., Wortman J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siepel A., Haussler D. Proc. 8th Int'l Conf. on Research in Computational Molecular Biology. NY: ACM Press; 2004. pp. 177–186. [Google Scholar]
- 22.Gross S.S., Brent M.R. Using multiple alignments to improve gene prediction. J. Comput. Biol. 2006;13:379–393. doi: 10.1089/cmb.2006.13.379. [DOI] [PubMed] [Google Scholar]
- 23.Gross S.S., Do C.B., Sirota M., Batzoglou S. CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction. Genome Biol. 2007;8:R269. doi: 10.1186/gb-2007-8-12-r269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Washietl S., Findeiss S., Muller S.A., Kalkhof S., von Bergen M., Hofacker I.L., Stadler P.F., Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011;17:578–594. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin M.F., Jungreis I., Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E., et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark A., Lin M.F., Kheradpour P., Pedersen J.S., Parts L., Carlson J.W., Crosby M.A., Rasmussen M.D., Roy S., Deoras A.N., et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer I.M., Durbin R. Comparative ab initio prediction of gene structures using pair HMMs. Bioinformatics. 2002;18:1309–1318. doi: 10.1093/bioinformatics/18.10.1309. [DOI] [PubMed] [Google Scholar]
- 29.Alexandersson M., Cawley S., Pachter L. SLAM: cross-species gene finding and alignment with a generalized pair hidden Markov model. Genome Res. 2003;13:496–502. doi: 10.1101/gr.424203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.König S., Romoth L., Gerischer L., Stanke M. Simultaneous gene finding in multiple genomes. PeerJ PrePrints. 2015;3:e1594. doi: 10.1093/bioinformatics/btw494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speir M.L., Zweig A.S., Rosenbloom K.R., Raney B.J., Paten B., Nejad P., Lee B.T., Learned K., Karolchik D., Hinrichs A.S. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44:D717–D725. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dewey C.N. Whole-genome alignment. Methods Mol. Biol. 2012;855:237–257. doi: 10.1007/978-1-61779-582-4_8. [DOI] [PubMed] [Google Scholar]
- 34.Kent W.J., Baertsch R., Hinrichs A., Miller W., Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paten B., Herrero J., Beal K., Fitzgerald S., Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18:1814–1828. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke S.L., VanderMeer J.E., Wenger A.M., Schaar B.T., Ahituv N., Bejerano G. Human developmental enhancers conserved between deuterostomes and protostomes. PLoS Genet. 2012;8:e1002852. doi: 10.1371/journal.pgen.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J., Sanborn J.Z., Diekhans M., Lowe C.B., Pringle T.H., Haussler D. Comparative genomics search for losses of long-established genes on the human lineage. PLoS Comput. Biol. 2007;3:e247. doi: 10.1371/journal.pcbi.0030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanke M., Diekhans M., Baertsch R., Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 42.Kuhn R.M., Karolchik D., Zweig A.S., Wang T., Smith K.E., Rosenbloom K.R., Rhead B., Raney B.J., Pohl A., Pheasant M., et al. The UCSC Genome Browser database: update 2009. Nucleic Acids Res. 2009;37:D755–D761. doi: 10.1093/nar/gkn875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris R.S. Ph.D. Thesis. The Pennsylvania State University; 2007. Improved pairwise alignment of genomic DNA. [Google Scholar]
- 44.Stedman H.H., Kozyak B.W., Nelson A., Thesier D.M., Su L.T., Low D.W., Bridges C.R., Shrager J.B., Minugh-Purvis N., Mitchell M.A. Myosin gene mutation correlates with anatomical changes in the human lineage. Nature. 2004;428:415–418. doi: 10.1038/nature02358. [DOI] [PubMed] [Google Scholar]
- 45.Liman E.R., Innan H. Relaxed selective pressure on an essential component of pheromone transduction in primate evolution. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3328–3332. doi: 10.1073/pnas.0636123100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 47.Schneider A., Cannarozzi G.M., Gonnet G.H. Empirical codon substitution matrix. BMC Bioinformatics. 2005;6:134. doi: 10.1186/1471-2105-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alioto T.S. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res. 2007;35:D110–D115. doi: 10.1093/nar/gkl796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwata H., Gotoh O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 2012;40:e161. doi: 10.1093/nar/gks708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Earl D., Nguyen N., Hickey G., Harris R.S., Fitzgerald S., Beal K., Seledtsov I., Molodtsov V., Raney B.J., Clawson H., et al. Alignathon: a competitive assessment of whole-genome alignment methods. Genome Res. 2014;24:2077–2089. doi: 10.1101/gr.174920.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu F., Kent W.J., Clawson H., Kuhn R.M., Diekhans M., Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 52.Lu D.V., Brown R.H., Arumugam M., Brent M.R. Pairagon: a highly accurate, HMM-based cDNA-to-genome aligner. Bioinformatics. 2009;25:1587–1593. doi: 10.1093/bioinformatics/btp273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kielbasa S.M., Wan R., Sato K., Horton P., Frith M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–493. doi: 10.1101/gr.113985.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coulombe-Huntington J., Majewski J. Characterization of intron loss events in mammals. Genome Res. 2007;17:23–32. doi: 10.1101/gr.5703406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hiller M., Huse K., Szafranski K., Jahn N., Hampe J., Schreiber S., Backofen R., Platzer M. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat. Genet. 2004;36:1255–1257. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- 57.Hiller M., Huse K., Szafranski K., Rosenstiel P., Schreiber S., Backofen R., Platzer M. Phylogenetically widespread alternative splicing at unusual GYNGYN donors. Genome Biol. 2006;7:R65. doi: 10.1186/gb-2006-7-7-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bortfeldt R., Schindler S., Szafranski K., Schuster S., Holste D. Comparative analysis of sequence features involved in the recognition of tandem splice sites. BMC Genomics. 2008;9:202. doi: 10.1186/1471-2164-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hiller M., Platzer M. Widespread and subtle: alternative splicing at short-distance tandem sites. Trends Genet. 2008;24:246–255. doi: 10.1016/j.tig.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 60.Hiller M., Agarwal S., Notwell J.H., Parikh R., Guturu H., Wenger A.M., Bejerano G. Computational methods to detect conserved non-genic elements in phylogenetically isolated genomes: application to zebrafish. Nucleic Acids Res. 2013;41:e151. doi: 10.1093/nar/gkt557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.