

Abstract
Virus entry into host cells relies on interactions between viral and host structures including lipids, carbohydrates and proteins. Particularly, protein–protein interactions between viral surface proteins and host proteins as well as secondary host protein–protein interactions play a pivotal role in coordinating virus binding and uptake. These interactions are dynamic and frequently involve multiprotein complexes. In the past decade mass spectrometry based proteomics methods have reached sensitivities and high throughput compatibilities of genomics methods and now allow the reliable quantitation of proteins in complex samples from limited material. As proteomics provides essential information on the biologically active entity namely the protein, including its posttranslational modifications and its interactions with other proteins, it is an indispensable method in the virologist's toolbox. Here we review protein interactions during virus entry and compare classical biochemical methods to study entry with novel technically advanced quantitative proteomics techniques. We highlight the value of quantitative proteomics in mapping functional virus entry networks, discuss the benefits and limitations and illustrate how the methodology will help resolve unsettled questions in virus entry research in the future.
Keywords: Virus entry, Protein–protein interactions, Quantitative proteomics, Protein interaction networks
1. Protein–protein interactions during virus entry
Viruses need to enter permissive host cells in order to propagate and spread. The penetration of cells requires an initial interaction of viral and host surface structures. Typical viral attachment proteins (VAP) are transmembrane glycoproteins for enveloped viruses or capsid proteins for naked viruses. Alternatively, some viruses incorporate phosphatidylserine into their envelope and use this lipid for host cell interaction (Amara and Mercer, 2015, Jemielity et al., 2013, Kondratowicz et al., 2011, Mercer and Helenius, 2008, Moller-Tank et al., 2013, Shimojima et al., 2012). For the majority of viruses, however, proteins bind to cellular receptors, i.e., proteins or carbohydrates, and the expression of a particular receptor is often determining tissue and species tropism of a virus. In this review we focus on the role of protein–protein interactions (PPI) during viral penetration as protein–lipid and protein–carbohydrate interactions in cell entry of viruses have been discussed elsewhere (Amara and Mercer, 2015, Lozach et al., 2007, Stencel-Baerenwald et al., 2014).
Historically, the first virus receptor found was a glycan. In 1981 Helenius and coworkers described sialic acid as the influenza A virus receptor (Matlin et al., 1981). In the following years it became clear that most virus receptors are proteins or protein-linked entities, such as glycosaminoglycans. As a consequence, protease treatment rendered susceptible host cells non-susceptible for many viruses. With the discovery of CR2 as Epstein Barr virus (EBV) and CD4 as human immunodeficiency virus type 1 (HIV-1) receptor the hunt for virus receptors began (Fingeroth et al., 1984, Maddon et al., 1986). Since then technological developments like antibody based affinity purification (AP), mass spectrometry (MS) of proteins, DNA mediated transformation and molecular cloning led to the discovery of dozens of receptors for human pathogenic viruses (Fig. 1 ). Nevertheless, for many viruses the cognate receptor(s) remain enigmatic and a “one fits all” method for their discovery has not yet been described.
Fig. 1.
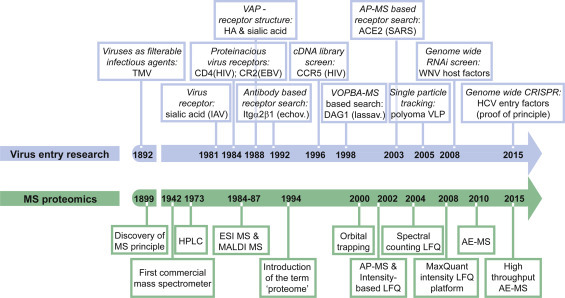
Timeline of the milestones in virus entry research and pioneering MS technology development.
Virus entry factors, which interact with VAPs directly, can be differentiated into attachment factors and entry receptors. Attachment factors facilitate entry by concentrating the virus on the cell surface. Virus receptors not only mediate cell attachment but the PPI of VAP and receptor also guides subsequent entry steps, e.g. internalization (Fig. 2 and Box 1 ). Sometimes the VAP- receptor interaction unleashes the fusogenic property of viral glycoproteins by inducing conformational changes in the VAP ectodomains. A well-studied example is the priming of the hepatitis C virus (HCV) glycoprotein E2 by the CD81 receptor (Sharma et al., 2011). For viruses, which fuse in intracellular vesicular compartments, a drop in pH typically acts as primer for fusion. In some cases host proteases interact with and cleave VAPs rendering them fusogenic. Proteolytic processing of VAPs either occurs during VAP biosynthesis or during virus entry. The latter applies to ebola virus, which requires processing of its envelope glycoprotein by cathepsins in endosomes (Chandran et al., 2005). Moreover, interactions of VAPs with secondary receptors, occurring after membrane surfing of viruses towards cellular junctions may trigger internalization (Coyne and Bergelson, 2006). After endocytosis, secondary PPIs with endosomal receptors can in addition to the pH drop render viral glycoproteins fusogenic (Carette et al., 2011, Cote et al., 2011). Subsequently viruses replicating in the nucleus are transported along the cytoskeleton towards the nuclear pore, where the genome is released into the nucleus. Transport relies again on PPI of virus structural proteins and microtubular motor proteins. All viruses additionally need to undergo uncoating, i.e., the viral nucleocapsid needs to disassemble to release the genome into the cell. Uncoating is poorly understood for most viruses, but as detailed below, also requires interaction of viral capsids with host proteins. Fig. 2 highlights the most important steps in virus entry and critical PPI for each step.
Fig. 2.

Schematic representation of virus entry pathways. PPIs between viral and cellular factors orchestrate viral attachment, trafficking, fusion and uncoating. Classical virus infection pathways are illustrated for a generic enveloped virus. Note that naked virions infect cells in a similar manner with viral capsid proteins mediating critical PPIs with cellular proteins (not shown). PPIs facilitating virus attachment, surfing, fusion and transport are depicted. In some cases proteolytic processing mediated by host factors triggers viral membrane fusion. Moreover, secondary PPIs can lead to assembly of multiprotein receptor platforms that allow internalization and ultimately membrane fusion.
Box 1. Definitions and abbreviations in virus entry.
Susceptible cell: cell that expresses virus receptors and entry factors and thus allows entry of a virus.
Permissive cell: cell that expresses all host factors required for replication, assembly and release of a virus and thus can generate infectious progeny after infection.
Virus entry: process of delivery of the viral genome to the viral replication compartment in the cells. Depending on the replication site and the virus entry route, virus entry includes cell attachment, endocytosis, membrane penetration, trafficking in the cytoplasm, nuclear import, release of viral structural proteins like tegument and genome uncoating.
Virus attachment factor: cell surface protein interacting with a virus. Attachment factors facilitate entry by concentrating virions at the cell surface and can promote subsequent interactions with entry receptors.
Virus entry receptor: cellular protein, lipid or glycan that binds the virus particle and mediates virus uptake and/or triggers critical conformational changes crucial for infection. Viral receptors are often localized to the plasma membrane, but can also reside in endosomal compartments.
Virus entry factor: operationally defined as any cellular factor that in some way participates in the virus entry process, but not necessarily interacts with the virus.
Viral attachment protein (VAP): viral surface protein that binds to the virus receptor. VAPs of enveloped viruses are transmembrane glycoproteins, while VAPs of naked viruses are viral capsid proteins.
Capsid (also: Core): protein shell of a virus arranged around the viral genome or complexed to it; build by repeat units of single proteins or protein complexes. The capsid proteins are encoded by the viral genome.
Nucleocapsid: complex of viral genome and capsid, which may for enveloped viruses be surrounded by a lipid bilayer.
Envelope (also: Peplos): host cell derived lipid bilayer surrounding the nucleocapsid of enveloped viruses. The envelope typically displays viral genome encoded transmembrane proteins. These envelope proteins are VAPs and in some cases enzymes (neuraminidases) that cleave cellular carbohydrates to facilitate virus shedding or ion channel proteins that prime viral uncoating. Recently, it was shown that some classical naked viruses, like hepatitis A virus, could be enveloped as well.
Virion surfing: directional movement of virions within the cell plasma membrane, i.e. on the cell surface or cell projections like filipodia, towards the viral entry sites
Virus fusion: membrane mixing of the lipid bilayer of enveloped viruses with a host cell lipid bilayer, i.e. either the plasma membrane or the endosomal membrane, resulting in the release of viral nucleocapsids into the cytoplasm.
Uncoating: disassembly of the viral capsid to release the viral genome into the cytosol or nucleus of a host cell. Large DNA viruses additionally require tegument disassembly prior to disassembly of the capsid.
In brief, most processes during virus entry are coordinated by PPIs. In fact, eighty percent of the approximately 20,000 protein coding genes of a cell are estimated to perform their function through interactions with other proteins. Roughly 10,000 distinct interaction types, i.e., structurally similar interactions, and 130,000–650,000 binary protein interactions can occur in a cell (Aloy and Russell, 2004, Stumpf et al., 2008, Venkatesan et al., 2009). Thus, knowledge on protein complexes is essential for the understanding of cellular mechanisms and for the targeted interference with a protein's function. In some instances this knowledge has been successfully translated into therapeutic approaches for treatment of viral infections. The best known examples are the HIV-1 CCR5 coreceptor antagonist maraviroc and the HIV-1 gp41 fusion inhibitors (Dorr et al., 2005, Wild et al., 1993). Importantly, many interactions are mediated by short 10 amino acid long unstructured peptide motifs, are transient, and have relatively low affinity. Classical high stringency tandem AP protocols thus often missed peptide motif based protein interactions. Novel shotgun proteomics of one step affinity enriched samples provides now the opportunity to unravel a multitude of previously missed interactions. Typical host peptide motifs, which coordinate PPIs and occur in several hundred human proteins, are the functional unit of src-homology 2 domain (SH2-domain), SH3-domain, the post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1)-domain (PDZ-domain) and the so called WW-domains which carry two tryptophan residues as signature (Tompa et al., 2014). Notably, viruses and other pathogens often make use of these and other peptide motifs to hijack cellular pathways during their life cycle including virus entry (Davey et al., 2011, Hagai et al., 2014).
While transcriptomics can reveal long-term alterations of the cellular state, virus entry usually occurs within minutes and typically relies on rapid changes of protein conformation, localization, interactions and post-translational modifications (PTM). Thus studying the proteome state, preferentially in a time-resolved manner, is ideally suited for understanding the mechanisms of virus entry.
Proteomics has already led to the characterization of global cellular alterations induced by virus infection. For instance, virus induced changes in the protein composition of subcellular HCV replication compartments revealed several host replication and assembly factors (Huang et al., 2007, Mannova et al., 2006, Paul et al., 2013, Yi et al., 2006, Yi et al., 2011). Interaction proteomics at a large scale was moreover performed for all 18 HIV-1 proteins outside of the context of HIV-1 infection and revealed important virus manipulations of cellular pathways (Jager et al., 2012). Another global interaction proteomics study revealed host protein interactions of viral immune modulating proteins uncovering viral immune evasion strategies (Pichlmair et al., 2012). Although in principle possible, global interaction proteomics during virus attachment and entry has not yet been extensively pursued. As virus entry is the process in the viral life cycle, which can most easily be synchronized by inhibitors or temperature shifts, we believe that proteomic analyses can close many gaps of knowledge in virus entry research. Below we discuss successful applications of proteomics to understand virus uptake and highlight future perspectives.
The various entry routes of viruses have been extensively reviewed (Grove and Marsh, 2011, Mercer et al., 2010, Amara and Mercer, 2015). Similarly, we refer the reader elsewhere for methods to define protein interactions other than MS based proteomics, i.e., native PAGE, yeast two hybrid, protein microarrays, fluorescence resonance energy transfer, surface plasmon resonance, structure-based approaches and phage display (Ngounou Wetie et al., 2014). Here, we focus on the PPIs and PTMs that mediate virus uptake and on past and present MS proteomics based techniques to study virus invasion.
2. Classical biochemical approaches for discovery of virus receptors
To gain access to a susceptible host cell the vast majority of viruses interacts with proteinacious receptors on the cell surface. Consequently, biochemical protein-based approaches successfully discovered virus receptors in the past (Table 1 ). Typically, these approaches build on virus overlay protein blot assays (VOPBA). VOPBA relies on probing of cell lysates or cell membrane extracts, separated by gel electrophoresis and transferred onto nitrocellulose membranes, with virus particles (Boyle et al., 1987). Virus bound proteins are in this case either detected by radiolabelling of the virus or by specific antibodies against the virus glycoproteins. In both cases MS fingerprinting of a corresponding polyacrylamide gel band identifies the putative receptor. A prime example for VOPBA approaches is the identification of alpha-dystroglycan as receptor for lymphocytic choriomenigitis virus (Cao et al., 1998). The VOPBA qualitative proteomics approach has the advantage that the protein is left intact until the last step of identification and thus information on the molecular weight and posttranslational modifications like glycosylation is available. However, it suffers from low sensitivity, as retrieval of proteins or peptides from polyacrylamide gels is typically low. Moreover interactions could be lost under denaturing conditions.
Table 1.
Examples of virus receptors identified by protein-based approaches.
| Virus | Baltimore class | Family | Receptors identified by protein-based approaches | Receptor identification strategy | Reference |
|---|---|---|---|---|---|
| Varizella zoster virus (VZV) | 1 | Herpesviridae | Insulin-degrading enzyme | AP, MS | Li et al. (2006) |
| Karposi sarcoma herpesvirus (KSHV) | 1 | Herpesviridae | Ephrin A2 | AP, MS | Hahn et al. (2012) |
| Hepatitis C virus | 4 | Flaviviridae | Scavenger receptor class B type I | AP, biochemical assays | Scarselli et al. (2002) |
| Japanese encephalitis virus (JEV) | 4 | Flaviviridae | Heat shock protein 70 | VOPBA, MS | Das et al. (2009) |
| Severe acute respiratory syndrome virus (SARS) | 4 | Coronaviridae | Angiotensin-converting enzyme 2 | AP, MS | Li et al. (2003) |
| Mapucho, Guanarito, Junin and Sabia virus | 5 | Arenaviridae | Transferrin receptor 1 | AP, MS | Radoshitzky et al. (2007) |
| Henipahvirus | 5 | Paramyxoviridae | Nephrin B2 | AP, MS | Negrete et al. (2005) |
| Lymphocytic chorio-meningitis virus | 5 | Arenaviridae | alpha-dystroglycan | VOPBA, MS | Cao et al. (1998) |
| Hepatitis B virus, hepatitis D virus (HBV, HDV) | 7 | Hepadnaviridae | sodium taurocholate cotransporting polypeptide | VOPBA, MS | Yan et al. (2012) |
An alternative to VOPBA-based receptor identification is the affinity purification (AP) of virus glycoprotein – receptor complexes. This method overcomes the caveat of protein denaturation and analyzes interactions in their native state. For instance, Ig-fusion proteins of truncated soluble viral glycoproteins were successfully used to co-purify the New World arenavirus receptor transferrin receptor 1 (Radoshitzky et al., 2007). Other examples for qualitative interaction proteomics are the identification of receptors for Nipah virus (ephrin B2), KSHV (ephrin A2) and SARS (angiotensin-converting enzyme 2) (Hahn et al., 2012, Li et al., 2003, Negrete et al., 2005). Here, glycoprotein−Fc fusion proteins served as baits for AP of the receptor post cell lysis. In a third approach, Yan et al. used biotinylated receptor binding peptides derived from the hepatitis B virus (HBV) glycoprotein, equipped them with photoactive amino acids and tandem affinity captured the HBV and hepatitis delta virus (HDV) receptor sodium taurocholate cotransporting polypeptide (Yan et al., 2012). Generally, biotinylation of surface proteins prior to AP or amine-reactive crosslinking are strategies to increase specificity of the AP and help avoid detection of biologically irrelevant interactions after cell lysis. A refinement of both methods is the use of biotinylated ligands equipped with hydrazine groups for crosslinking of cell surface proteins glycans. To this end, a trifunctional reagent for ligand derivatization, termed TRICEPS, has been developed by Frei et al. (2012). Critical negative controls further include, non-susceptible cell lines, which do not bind the virus, and glycoprotein mutants or peptide variants, which are deficient for receptor binding. In principle, MS fingerprinting of the purified proteins is the sole method to unambiguously identify the prey after AP. It should however be noted that analysis of the prey with respect to its sensitivity to glycosidases, proteases, heparinases and lipid raft altering agents can lead to an educated guess, which upon further validation might identify a virus receptor. A prominent example is the identification of scavenger receptor class B type I for HCV (Scarselli et al., 2002). Similar to VOPBA approaches, receptor identification after AP relied on qualitative proteomics in the past, i.e. gel purification of the co- precipitated proteins, followed by MS fingerprinting of a defined gel band. Such interaction proteomics approaches typically require a truncated and epitope tagged virus glycoprotein for efficient affinity enrichment (AE) of cellular receptors. High affinity antibodies against the viral glycoprotein may supersede epitope tagging of the bait.
Both VOPBA-based and AP-based interaction proteomics are well suited for the identification of primary receptor interactions. However, interactions with late receptors, which require a conformational change of the glycoprotein and are more difficult to synchronize, cannot be captured easily with these techniques. Also virus-triggered secondary PPIs are not resolved by these classical methods. The development of shotgun proteomics techniques described below thus presents a major breakthrough, which holds the promise of detecting higher order, reduced affinity and even transient virus receptor interactions.
3. Quantitative interaction proteomics (interaction systems virology)
In the past decade, the development of quantitative shotgun proteomics techniques revolutionized the protein interactions field. In contrast to previous qualitative approaches, shotgun techniques avoid extensive purification from gels and instead analyze a complex mixture of proteins, thereby tremendously increasing sensitivity and throughput. Technological development of high performance liquid chromatography (LC) and ultra-high resolution ion trap MS together reduced measurement time, increased peptide sensitivity and measurement reproducibility and thus made shotgun techniques possible. Moreover, mass accuracy of new generation ion trap mass spectrometers increased up to 0.001 Da, guaranteeing high confidence peptide identification and analysis of posttranslational modifications (PTMs). LC-MS thus emerged as prime in depth proteomics method leaving behind alternative techniques like two-dimensional gel electrophoresis, low-resolution MALDI and protein arrays (Mann and Kelleher, 2008). Shortly, in shotgun interaction proteomics affinity enriched protein mixtures are as a whole digested to peptides and analyzed by LC–MS. To separate the specific binding partners from the excess of background proteins, specific and control purifications are directly compared (Vermeulen et al., 2008). This is achieved either by stable isotope labeling of amino acids in culture (SILAC) or by the more recent computational based label-free quantification methods (LFQ) described in the subsequent paragraph. Most recent techniques use one-step AE protocols, which preserve weak and transient interactions (see below for details). Fig. 1 highlights the milestones in the development of MS- based proteomics techniques and Box 2 describes common proteomics terms. While quantitative shotgun proteomics has been used in a variety of fields including signal transduction, cell adhesion and even viral immune modulation, we are just starting to appreciate its power for the study of the viral replication cycle and in particular virus entry (Kruger et al., 2008, Pichlmair et al., 2012, Weekes et al., 2014, Zanivan et al., 2013).
Box 2. Definitions and abbreviations in mass spectrometry based proteomics.
Proteome: entity of proteins expressed at a given cellular state or in a given tissue. The term was coined in analogy to ‘genome’ in 1996.
MS: mass spectrometry.
ESI: electrospray ionization; ionization technique based on spraying of a sample solution into a strong electric field.
MALDI: matrix-assisted laser desorption/ionization; ionization technique based on mixing of samples with a matrix, which absorbs ultraviolet light and converts it to heat energy leading to sample vaporization.
Bottom up proteomics: identification of proteins based on MS peptide fingerprints after digestion of a protein into peptides using a sequence-specific enzyme such as trypsin.
Top down proteomics: MS analysis of entire proteins.
Shotgun proteomics: bottom-up proteomics technique to identify proteins in a complex mixture. Proteins are digested to peptides, peptides separated by liquid chromatography and then identified by tandem MS.
Quantitative proteomics: shotgun proteomics with the additional dimension of measuring the quantity of a protein in a complex sample. Relative quantities in two or more samples are determined either by SILAC or LFQ (see below). Absolute quantification can be achieved using spike in standards or a proteomics ruler (see paragraph on protein complex stoichiometry).
Interaction proteomics: unbiased identification of protein–protein interactions by affinity enrichment followed by MS.
SILAC: stable isotope labeling by amino acids in cell culture; first quantitative method for the comparison of cellular protein amounts in two to three different cellular states. SILAC is based on the isotope labeling of cellular proteins by feeding cells with heavy isotope amino acids (e.g. N-15 and C-13 labeled arginine and lysine).
Label-free quantification (LFQ): quantitative proteomics technique based on either signal intensity measurement or spectral counting followed by computational matching of peptides from multiple samples and signal normalization.
PPI: protein-protein interactions.
PTM: posttranslational modification. Common protein modifications include among others phosphorylation, lipidation, glycosylation, methylation, acetylation, ubiquitinylation and SUMOylation.
AP-MS: affinity purification of proteins using antibodies against endogenous epitopes or epitope tags followed by mass spectrometric identification or quantification of the bound proteins.
AE-MS: affinity enrichment of proteins using fast single step methods, which preserve transient and low affinity interactions, followed by mass spectrometric identification or quantification of bound proteins.
3.1. SILAC versus label-free methods
MS based methods are commonly used to identify and quantify proteins, to profile protein-interactions or whole proteomes. Therefore, proteins are isolated and proteolytically digested, the resulting peptides are separated via LC, and then analyzed by tandem mass spectrometry (MS/MS) (bottom-up approach or shotgun proteomics) (Vermeulen et al., 2008). Alternatively a top-down approach can be used, where the protein is fragmented during the mass spectrometric analysis (Boeri Erba, 2014). For quantitative proteomics, most often bottom-up high resolution MS techniques are used in combination with either labeling or label-free methods:
A powerful and the most commonly used label-based method for the quantification of proteins is SILAC (Ong et al., 2002). In short, an isotope label is metabolically incorporated into peptides and proteins of a cell. Typically, essential amino acids with heavy isotopes (e.g., 15N, 13C) are fed to cells over several passages until nearly all unlabeled amino acids are replaced. As the physicochemical properties of light and heavy amino acids are nearly identical, cells labeled with the isotopic analogs behave similar or even identical to unlabeled cells. Based on the isotope label SILAC allows the direct comparison of two cell populations (heavy and light) from two distinct experimental conditions; for instance, from cells with surface-bound virus or cells without bound virus (Gerold et al., 2015). Prior to proteomic analysis, i.e., whole cell lysate analysis or AP of protein complexes, the differently labeled samples are mixed. This is advantageous as all samples are processed equally excluding sample handling biases. Proteins are then digested, peptides reduced and alkylated, separated by LC and analyzed by MS. Peptides derived from heavy or light cells are subsequently differentiated by their characteristic mass offset. Relative abundances of proteins in each sample are calculated based on the heavy and light signal intensities in the same spectrum. Typically, SILAC is used for relative quantification of protein levels under two different cellular conditions. Of note, medium labeled amino acids are additionally available and allow expanding a SILAC experiment to three cellular conditions (Hilger and Mann, 2012). The high accuracy of SILAC typically requires analysis of only two experimental replicates. In addition, two replicates of a label swap experimental condition allow assessing label-specific effects. Thus, a typical dual SILAC experiment starts with eight experimental samples mixed into four SILAC pairs. Those are duplicates of the forward label pair (light-condition 1; heavy-condition 2) and duplicates of the reverse label pair (light-condition 2; heavy-condition 1). Fig. 3 A depicts the SILAC workflow.
Fig. 3.
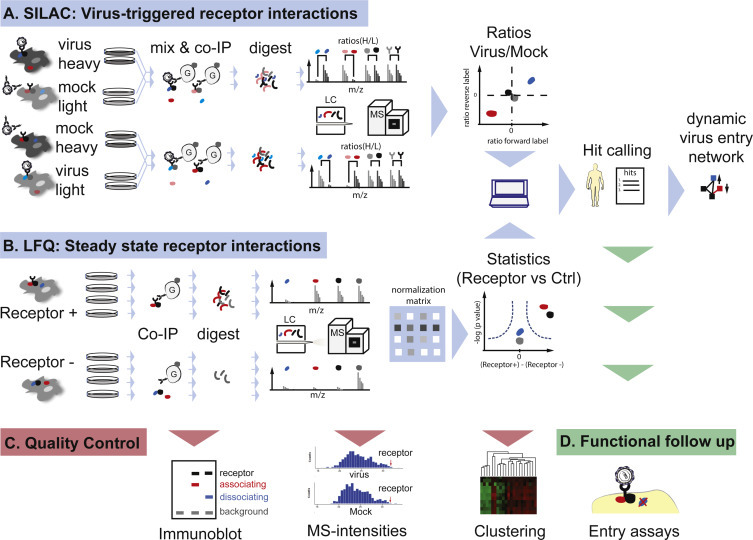
High resolution quantitative MS-based interaction proteomics using stable isotope labeling (A) or label-free (B) technologies. Consecutive quality control steps (C) and follow up analysis (D) are indicated.
SILAC followed by LC-MS/MS can answer various questions in virology. For example, it can be used to compare the proteome of uninfected to that of virus infected cells (e.g., Zhang et al., 2013). Also cells at different time points post infection can be analyzed to understand how viruses alter host cell protein expression (Weekes et al., 2014). For instance, Berard et al. performed a temporal quantification of cells infected with herpes simplex virus type 1 (HSV1) and by this showed how HSV1 modulates the metabolic function of the host cell (Berard et al., 2015). Beside this, SILAC followed by LC-MS/MS can also be used to identify cellular targets of viral enzymes, as it has been done for the identification of the HCV protease NS3-4A targets: by comparing whole proteomes from hepatoma cells with and without the NS3-4A protease, Morikawa et al. discovered that ‘probable glutathione peroxidase 8’ is cleaved by the viral protease (Morikawa et al., 2014). SILAC is also suitable to analyze how viruses alter the proteome of distinct cellular compartments. For instance, Xie et al. defined the modification of lipid raft composition in HBV infection (Xie et al., 2012).
In combination with AP of a protein of interest, SILAC followed by LC–MS/MS can also discover protein-interactions and dynamic changes in interaction complexes (reviewed e.g., in (Gingras et al., 2007, Trinkle-Mulcahy, 2012)). High-throughput AP-MS has led to important findings regarding virus entry, for instance the identification of receptors or receptor-complexes. A successful example is the AP-MS analysis of the HCV receptor CD81, which revealed HRas as a CD81-binding partner and signaling molecule needed for HCV entry (Zona et al., 2013).
A clear benefit of SILAC is its high reproducibility, robustness and relatively easy execution. SILAC is applicable to most cell culture cells and sample conditions. Even SILAC mice have been developed in the past for ex vivo proteomics analyses (Zanivan et al., 2012). Caveats of SILAC are the limited sample number of maximum three and the additional labeling step requiring culturing of cells for two to three weeks prior to analysis. Thus some primary human cells, which rapidly dedifferentiate ex vivo, are not suited for SILAC. Similarly, clinical samples cannot be labeled and are thus not amenable to SILAC analysis.
Increasingly popular alternatives to SILAC are LFQ methods. These can determine the relative or absolute abundance of a protein in complex samples. The first semi-quantitative label-free method developed, which has since then been continuously refined, is based on the measurement of MS/MS peptide intensities (Bondarenko et al., 2002). To this end, peak areas of all identified peptides from one protein are summed up and normalized to one or several proteins with constant concentration or even to all identified proteins in the sample. Bondarenko et al. showed that there is a linear correlation of the abundance of a protein and its total reconstructed peak area. An alternative semi-quantitative method used is spectral sampling (Liu et al., 2004). It was shown that highly abundant proteins are sampled more frequently by LC–MS/MS and that the spectral copy number obtained increases linearly with the concentration of a protein. Accordingly, the number of MS/MS spectra gained for one protein can be counted and compared between different samples measured using the same experimental conditions. Several computational approaches to predict specific protein interactions in label-free datasets have been developed, among them Significance Analysis of INTeractome (SAINT) and Mass spectrometry interaction STatistics (MiST) (Choi et al., 2011, Jager et al., 2012). These tools compute interaction probabilities based on spectral counts of all interactions that a prey-bait pair is involved in or based on abundance, reproducibility and specificity of a prey-bait pair, respectively. Recent developments in MS technology and analysis algorithms now allow highly sensitive, efficient and robust intensity-based label-free protein quantification (reviewed e.g., in (Nahnsen et al., 2013)). For this, samples are processed in parallel and not mixed, as in SILAC. Thus, an unlimited number of samples can be compared. Normalization is achieved post LC–MS/MS analysis by comparing all peptide signals from all MS runs and assuming that most protein signals remain unaltered in the different sample conditions. Thus, no external standards are needed (Cox et al., 2014). However, acquisition of accurate datasets requires the analysis of at least three, typically four, biological replicates (Fig. 3B).
MS combined with LFQ algorithms is suitable for virological research and has been used e.g., to show that subsets of dendritic cells (DCs) differ in their ability to sense single stranded RNA (ssRNA) viruses (Luber et al., 2010). Briefly, Luber at al. compared the whole cell proteome of DC subsets by high-resolution MS combined with LFQ. They found, that DC subsets differently express proteins involved in pattern recognition pathways and that this correlates with their ability to sense ssRNA viruses, such as Sendai virus and influenza A virus. Label- free methods also allow the analysis of protein complexes. Importantly, extensive purification of complexes prior to LC–MS/MS is no longer necessary for LFQ. Instead one AE step is sufficient for precise complex profiling, when controls are designed appropriately (Keilhauer et al., 2015). False positive interaction partners, which are proteins unspecifically binding to the affinity resin, are computationally eliminated by comparing datasets to those of a control AE sample from cells without bait. Fig. 3B outlines the here described LFQ workflow.
When compared to label-based methods, LFQ techniques have several advantages: nearly all kinds of samples can be measured, including clinical material and primary cells; sample number is unlimited; no special sample processing steps are needed; slight differences in sample preparation and measurement between the different samples are tolerated.
Both, labeling and label-free quantification methods identify and quantify proteins with high confidence and lead to highly accurate results. Each step of the proteomics workflow can be quality controlled, e.g., by immunoblotting, computational analysis of peptide intensity distributions, and clustering analysis of biological replicate datasets (Fig. 3C). To study virus entry both SILAC and LFQ are valuable tools. LFQ is ideally suited to study steady state virus receptor interactions. To this end, virus receptor-complexes are purified from cells expressing the receptor and cells lacking the receptor. Comparison of both datasets then allows to define receptor interacting proteins in a given cell line. SILAC on the other hand can sensitively distinguish low degree quantitative changes, as they occur when virus receptor complexes change upon virus binding. Briefly, heavy labeled cells are incubated with virus and light cells are left untreated (forward label); and vice versa (reverse label). After sample mixing, AP of receptor complexes and LC–MS analysis, changes in interaction strength of each protein in the receptor complex can be determined. Importantly, the untreated control should ideally contain all extracellular components as the virus sample, i.e. the ideal control is incubation with virus, which had been neutralized by blocking antibodies, or incubation with a “mock” virus preparation if such antibodies are not available. Using SILAC, we recently identified transient, virus-triggered interactions of the HCV receptor CD81. Among the 26 dynamic CD81 interactors we identified serum response factor binding protein 1 as a host factor aiding HCV entry (Gerold et al., 2015). Of note, similar studies will soon be possible with advanced LFQ methods. Fig. 3 illustrates how quantitative proteomics can identify steady state and dynamic virus-triggered receptor interactions. In summary, SILAC and LFQ are powerful tools for analyzing virus-host interactions and global proteomic changes upon virus infection. Due to the recent improvements in high resolution MS and label-free analysis tools, the highly accurate, cost effective and broadly applicable LFQ methods will soon become the method of choice for profiling proteins, their interactions and whole proteomes.
3.2. Steady state virus receptor interactions
Steady state interactions of cellular proteins, which preexist before virus attachment, can be crucial for virus infection. This has been shown in the past mainly by biochemical assays. For instance, the interaction of moesin and CD46, which is needed for measles virus entry, was demonstrated by AP followed by immunoblot analysis (Schneider-Schaulies et al., 1995). Since MS-based proteomics became the method of choice to analyze protein interactions, several studies used mostly non-quantitative proteomic techniques to characterize interactions of virus receptors. For example Chakraborty et al. (2012) analyzed immuoprecipitates of the Kaposís sarcoma- associated herpesvirus (KSHV) receptor and lipid raft protein integrin α3β1 via SDS-PAGE and MS of selected gel bands. They identified the association of integrin α3β1 with several proteins, including EphA2. The identified associations were enriched in lipid rafts of KSHV infected human dermal microvascular endothelial cells, compared to uninfected cells. With virological assays Chakraborty et al. then showed that this enrichment is important for effective KSHV infection, especially for macropinocytosis and trafficking of the virus.
Also quantitative MS-based techniques in combination with traditional virological assays can be used to decipher the role of steady state protein interactions in different steps of the viral life cycle. An excellent example is the identification of a PPI network consisting of the HCV receptors CD81 and Claudin1 and the membrane proteins HRas, Rap2B and ITGB1, which are required for efficient entry of the virus into hepatocytes (Zona et al., 2013). The authors identified the HCV receptor network by SILAC of human hepatoma cells with and without CD81 receptor expression. The role of the identified proteins in the HCV entry process was then studied by virological assays in conjunction with RNA silencing and small molecule inhibitors.
Using a similar workflow, host restriction factors can likewise be discovered. One example is the association of EWI-2 and α-actinin-4 in T-cells. Gordon-Alonso et al. (2012) identified the complex by analyzing EWI-2 co-precipitated proteins via non-quantitative MS, and later on showed that the complex negatively regulates HIV 1 entry. Similarly, the cleavage product of EWI-2, EWI-2wint, has been shown to interact with the HCV receptor CD81 and by this inhibit HCV entry (Rocha-Perugini et al., 2008). As EWI-2wint is not expressed in the target cells of HCV, which are hepatic cells, this interaction could contribute to the viral cell tropism.
In summary, the analysis of steady state virus receptor interactions can reveal protein complexes critical for virus uptake and in some cases help understand host and tissue tropism.
3.3. Virus-triggered receptor interactions
Upon binding of viruses to their receptors, additional secondary PPIs are triggered, most of which lead to virus entry. These interactions have at least five distinct functions depending on the virus entry strategy. First, binding of the multivalent virus particle to the cell surface can induce receptor clustering, as has been shown for phleboviruses and influenza A viruses (Eierhoff et al., 2010, Lozach et al., 2011). While it is unclear whether influenza A virus triggered clustering of sialylated receptor tyrosine kinases like EGFR and c-Met is required for endocytic virus uptake, phlebovirus induced clustering of DC-SIGN seems to trigger signaling needed for uptake. Other viruses, which have been suggested to induce receptor clustering, include coxsackievirus B and Lassa virus (Coyne and Bergelson, 2006, Moraz et al., 2013). HIV-1 entry also relies on clustering of its CD4 and CXCR4 receptors on the plasma membrane. Specifically, the HIV gp120 glycoprotein signals for recruitment of actin adaptor proteins filamin and debrin, which regulate actin rich cap formation at the virus entry site (Gordon-Alonso et al., 2013, Jimenez-Baranda et al., 2007). Notably, actin depolymerization is induced at later stages of the HIV entry process, highlighting that actin remodeling during virus entry is highly dynamic and tightly regulated. While proteomes of detergent resistant membrane microdomains have been determined in naïve and virus replicating cells resulting in the identification of more than 200 proteins (Foster et al., 2003, Xie et al., 2012), few quantitative proteomics dataset are currently available for receptor platforms upon virus binding to the cell surface. For the HCV receptor CD81, we recently described the virus triggered receptor interactome and identified cytoskeleton regulators, cell junction proteins and a clathrin adaptor protein (Gerold et al., 2015).
Second, viruses can induce signaling leading to actin cortex disassembly. This is particularly important for viruses, which directly penetrate the cell at the plasma membrane and need to overcome the physical barrier of the actin cortex. Viruses entering by endocytosis might similarly require local actin cortex disassembly at the site of the incoming vesicle. Although the concept that the cortex can prevent or delay transit of viruses has already been suggested by Marsh and colleagues in 1997 (Marsh and Bron, 1997), only few studies address virus induced actin cortex remodeling. A well-studied example is HIV, which upon Env binding to the CXCR4 coreceptor induces Galphai signaling and subsequent cofilin activation leading to actin depolymerization thereby facilitating nuclear translocation of HIV particles (Yoder et al., 2008). Of note, not only actin cortex but also actin stress fiber disruption can aid virus entry. For instance cytomegaloviruses binding to EGFR and integrin alphaVbeta3, induces cofilin dependent actin stress fiber disruption thereby facilitating nuclear translocation of incoming virions (Wang et al., 2005).
Third, viruses, which use multiple receptors for entry, can induce their lateral translocation along the plasma membrane towards the site of endocytic uptake. A prominent example of such surfing is coxsackievirus B, which after binding to the apical decay-accelerating factor (DAF) on epithelial cells induces Abl kinase signaling and subsequent Rac-dependent actin reorganization. This leads to the lateral translocation of the coxsackievirus B—DAF complex to tight junctions, where the secondary receptor coxsackievirus-adenovirus receptor (CAR) is localized. After CAR engagement coxsackievirus B is endocytosed. This endocytosis is also triggered by PPIs as initial DAF binding additionally induces Fyn kinase, which phosphorylates caveolin thereby facilitating uptake (Coyne and Bergelson, 2006). HCV seems to use a similar multistep strategy to enter after initial binding to the basolateral side of hepatocytes, where the receptors SR-BI and CD81 reside. Through a yet insufficiently described mechanism HCV also triggers lateral translocation of the CD81–virus complex to tight junctions, where the two additional entry factors CLDN1 and OCLN reside and endocytosis occurs (Brazzoli et al., 2008). Receptor tyrosine kinases like EGFR are thought to provide the trigger for lateral membrane translocation through HRas signaling (Zona et al., 2013). Moreover, surfing of HCV and retroviruses on filopodia has been observed and is governed by PPI with the actin cell cortex (Coller et al., 2009, Lehmann et al., 2005).
Fourth, viruses, which enter through endocytic routes, can induce endocytic uptake mechanisms including clathrin-mediated, caveolin-dependent, macropinocytic or alternative uptake routes like the clathrin-independent carriers in parvovirus entry (Nonnenmacher and Weber, 2011). On the one hand, some viruses like human rhinovirus 2 and phleboviruses hijack endogenous constitutive receptor recycling pathways. After endocytosis phleboviruses, which hijack DC-SIGN for cell entry into DCs, however dissociate from the receptor in the early endosome thereby avoiding antigen processing and presentation (Lozach et al., 2011). On the other hand, viruses can actively trigger their endocytosis like influenza A virus. After interaction of influenza virions with sialylated membrane receptors, the clathrin adaptor Epsin-1 is recruited in a ubiquitin-dependent manner towards the receptor complex and induces clathrin-coated pits (Chen and Zhuang, 2008). PI3K activation was independently reported to be required for influenza uptake, thus more than one signaling pathway might coordinate uptake of a virus. This is in line with reports, showing that influenza virus can utilize more than one endocytosis pathway to gain access to host cells (Rust et al., 2004, de Vries et al., 2011). Larger viruses like poxviruses enter by macropinocytosis and actively induce this uptake pathway through Rac-1 and p21-activated kinase 1 (Pak-1) dependent actin remodeling (Mercer and Helenius, 2008). Formation of plasma membrane protrusions is however not specific to poxviruses, but occurs also during entry of herpes viruses, papillomaviruses and flaviviruses (Clement et al., 2006, Coller et al., 2009, Smith et al., 2008).
Lastly, several viruses need to interact with host enzymes to trigger their uptake. A prominent example is ebola virus, which requires proteolytic processing of its envelope glycoprotein GP1 by cathepsin B and L to render it fusion competent (Chandran et al., 2005). Similarly coronavirus relies on cathepsin cleavage of its spike protein to permit fusion (Simmons et al., 2005). In addition to lysosomal enzymes, cell surface proteases can prime VAPs for fusion, like shown for some influenza A virus strains and SARS coronavirus (Bertram et al., 2010, Bertram et al., 2011). Host protein disulfide isomerases similarly regulate entry by catalyzing disulfide shuffling in viral envelope proteins, as shown for dengue virus, HIV-1 and Newcastle disease virus (Jain et al., 2008, Ryser et al., 1994, Stantchev et al., 2012, Vega-Almeida et al., 2013). Of note, high resolution proteomics can not only reveal transient interactions of VAP with enzymes, but also has the potential to identify proteolytic cleavage sites and redox modifications in VAPs.
It is conceivable that virus induced protein interactions during entry not only serve to promote the virus uptake pathway, but can also help cloak viruses and lead to immune evasion. For instance, the HIV capsid recruits two cofactors, cleavage and polyadenylation specificity factor subunit 6 (CPSF6) and cyclophilins (Nup358 and CypA), thereby preventing antiviral IFN responses (Rasaiyaah et al., 2013). More complex viruses can further express viral gene products on the infected cell surface, which then bind receptors on bystander cells and modulate immunity. For instance, HCMV protein pUL11 binds to CD45 on T cells thereby reducing T cell proliferation (Gabaev et al., 2011). While we here focus on the role of virus - host PPI in entry, it should be noted that quantitative virus entry proteomics at the same time has the potential to reveal immunomodulatory mechanisms. Similarly, PPIs of viral tegument and capsid proteins with host proteins occurring after membrane fusion are amenable to quantitative proteomics. We refer the reader elsewhere for the detailed description of post-fusion PPIs coordinating nuclear trafficking (Dohner et al., 2005, Yarbrough et al., 2014).
Comprehensive proteomics analyses of virus induced PPIs leading to actin remodeling and endocytic uptake of virions are lacking to date. However, several studies underline the value of proteomics to analyze stimulus specific interactions in cellular signaling pathways. For instance, the ligand-induced changes of the two Wnt signaling pathway members adenomatous polyposis coli protein (APC) and axin-1 identified 28 and 18 dynamic interaction partners, respectively (Hilger and Mann, 2012). For other virus life cycle steps, like e.g., CMV particle assembly, interaction proteomics uncovered important mechanistic insights (Moorman et al., 2010). Further, interaction proteomics of viral immunomodulatory proteins revealed virus induced alterations in human protein interaction networks and underlying viral perturbation strategies (Pichlmair et al., 2012). Similar studies hold the promise of uncovering common and specific viral strategies to alter and exploit host cell functions during cell entry.
3.4. PTMs
Post-translational modifications (PTMs) play two major roles during virus entry into a host cell. First, virus receptor PTMs can be critical for the localization to specific membrane microdomains. For instance, palmitoylation of the HCV receptor CD81 and its associated proteins EWI-2 and EWI-2wint regulate their interaction with each other and the localization to cholesterol rich membrane domains thereby influencing HCV entry (Montpellier et al., 2011, Zhu et al., 2012). Second, viruses rely on cell signaling for their uptake and such cellular signals are often transmitted via PTMs like reversible protein phosphorylation. A widespread example common to viruses of diverse families including herpesviruses, papillomaviruses and flaviviruses is the triggering of receptor tyrosine kinase signaling (Hahn et al., 2012, Lupberger et al., 2011, Surviladze et al., 2013, Zheng et al., 2014). In most cases signaling through growth factor receptors regulates actin dynamics required for virus trafficking into the cell. Some respiratory viruses however use EGFR signaling to suppress antiviral signaling in the airway epithelium (Ueki et al., 2013). Other phosphorylation signals that coordinate virus entry are the Src kinase pathway in HCMV penetration (Nogalski et al., 2013) and the Lassa virus induced phosphorylation of its receptor dystroglycan (Moraz et al., 2013). Certain kinase activities can be a hallmark of a specific uptake pathway, as for instance PAK-1 for macropinocytic virus entry (Amstutz et al., 2008). In particular for large DNA viruses not only host proteins are phosphorylated early during virus infection, but also viral gene products. For instance, herpes simplex virus type 1 (HSV-1) protein pUL46 is heavily phosphorylated six hours post infection and interacts with several viral and host kinases as determined by quantitative interaction proteomics (Lin et al., 2013).
Apart from protein phosphorylation, other PTMs like ubiquitination are thought to regulate virus uptake, but are poorly characterized. Ubiquitination of viral proteins seems to induce viral uncoating in at least two instances. Influenza A virus M1 protein is ubiquitinated by the E3 ligase Itch and this is a prerequisite for endosomal escape (Su et al., 2013). Vaccinia virus on the other hand uses ubiquitinated incoming viral core proteins to trigger proteasomal degradation of its protein coat (Mercer et al., 2012). All aforementioned studies, except for the HSV-1 study, demonstrate PTMs of receptors, downstream signaling molecules or viral proteins by immunoblotting, thus requiring prior knowledge of the signaling pathways.
In contrast, MS-based identification and quantification of PTMs upon virus binding to a host cell is completely unbiased and thus broadly applicable. Another advantage over previous immunoblot based methods is the global and quantitative measurement of PTMs by MS in conjunction with the ability to pinpoint the modified amino acid. Clearly, high read depth is required for PTM proteomics as the search space for the peptides explodes when taking into account all possible peptide PTMs and all of their combinations. Still successful large-scale measurements of protein phosphorylation (Ficarro et al., 2002, Olsen et al., 2006), N-glycosylation (Kaji et al., 2007), lysine methylation (Ong et al., 2004) or acetylation (Kim et al., 2006), ubiquitination (Peng et al., 2003) and sumoylation (Andersen et al., 2009, Impens et al., 2014) have been achieved. To overcome the caveat of low abundance of modified peptides, enrichment methods for instance using anti-phosphotyrosine antibodies are available. Importantly, when measuring PTMs in interaction proteomics samples, which are of much lower complexity than whole cell lysates, PTM identification is facilitated.
For stimuli other than viruses, PTM proteomics has yielded comprehensive datasets. For instance, phosphoproteomics of epidermal growth factor (EGF) stimulated HeLa cells unraveled 6600 phosphorylation sites on 2244 proteins, with 14% of the interactions showing an at least 2-fold modulation by EGF. The majority of proteins further showed multiple PTM sites with different kinetics highlighting how one protein can integrate numerous signals (Olsen et al., 2006). This and other studies underline the complexity of receptor signaling. How extensively virus-receptor interactions alter the phosphoproteome of a host cell, was demonstrated in a first global description for HIV-1 entry (Wojcechowskyj et al., 2013). A total of 239 phosphorylation sites in 175 proteins changed just one minute after HIV-1 binding and the study disclosed several previously unknown HIV-1 entry factors. It is conceived that most viruses will trigger PTM-dependent signaling during their entry process and this is either critical for cell penetration or for modulating cellular immunity. Recent developments in PTM proteomics now allow the global mapping of virus induced changes in host proteome PTMs and can thereby disclose signaling pathways active during cell entry.
3.5. Protein complex stoichiometry
The stoichiometric composition of receptor complexes is critical for membrane microdomain localization, signal coordination and ligand binding. Interaction proteomics can provide information on the relative abundances of a protein in a complex. Typically prey protein abundance in the pull down is corrected for the prey abundance in a control pull down and subsequently normalized to the bait protein. This approach was used to estimate the stoichiometry of the YAP and TAZ protein complexes in the human Hippo pathway (Kohli et al., 2014).
Beyond complex stoichiometry measurements, shotgun proteomics now allows to estimate which fraction of a protein is sequestered into a protein complex. Mann and colleagues recently developed a method to estimate protein copy numbers in label-free proteomes from whole cells. This “proteomic ruler” technique determines the absolute abundance of a protein in a cell, based on the fact that the total amount of histones, which is defined by the cell ploidity and genome size, can be used for normalization of any detected protein in a proteomic dataset of sufficient depth. The minimum depth of a whole cell proteome to guarantee reliable histone based scaling is 12,000 peptides. This scaling method is similarly accurate as previous spiked-in protein epitope signature tags (PrESTs) based methods, which required cell counting and cell labeling (Wisniewski et al., 2014). Thus, whole cell proteomic datasets can provide information on total protein copy numbers in a given cell. Based on this knowledge the receptor complex stoichiometry can be estimated by comparing relative abundances of co-purified proteins in an AE proteomics dataset. Successful receptor complex stoichiometry calculation has recently been demonstrated for subcellular fractionation proteomics (Borner et al., 2014). Thus, shotgun proteomics goes beyond pure identification of virus entry receptor complexes, but is also suitable to estimate complex stoichiometry and degree of sequestration of a protein into a complex.
4. Modeling of virus entry networks
The era of systems biology and proteomics puts the virologist into the unique position of drawing a comprehensive picture of all molecular interactions during virus infection of a host cell. Large scale genome and transcriptome-based methods for the characterization of virus-host interactions, including mRNA microarrays, RNA interference and cDNA library screens, led to the discovery of a multitude of host factors. We currently lack information on the physical interconnection of these host factors and thus it is difficult to identify critical players in virus entry pathways. Open access databases on PPIs like the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) integrate public databases like IntAct and Reactome and can facilitate the mapping of host factors into known networks (Croft et al., 2014, Orchard et al., 2014, Szklarczyk et al., 2015). However, many virus host factors do not map to previously described networks. Also, virus induced changes of cellular PPIs are not deposited in current databases. Some repositories, however, curate host-pathogen interactions, e.g., VirHostNet and the HIV-centric GPSprot site (Fahey et al., 2011, Guirimand et al., 2015). Importantly, viruses do not play according to the rule, i.e., they may induce PPI that do not normally exist in non-infected cells. For instance, they can change the subcellular localization of a host factor and thereby connect pathways, which in an uninfected cell operate independently. Thus, existing databases on host PPI may have only limited predictive value for infected cells, in particular at steps of the life cycle that heavily depend on the cell's machinery like virus entry. Again this calls for repositories that either specialize in or integrate PPI from infected cells.
The above described technological advances now enable rapid acquisition of large sets of protein interaction data from one-step AEs. Thus, proteomics is now high throughput compatible and amenable to systems biology studies (Hosp et al., 2015) providing the unprecedented chance to globally define protein–protein networks during virus infection. Critical for high confidence network generation and data deposition is the analysis of a large number of biological replicates, experimental conditions and controls including immunoprecipitates from bait deficient cells. Furthermore, several laboratories performed control pull-downs under various experimental conditions and established a “contaminant repository for AP” (CRAPome) (Mellacheruvu et al., 2013). However, background binders can critically differ between experimental setups and thus a universal CRAPome is problematic, making internal controls more reliable (Keilhauer et al., 2015). Clearly, newly acquired datasets on virus infection interactomes should be integrated into established host protein–protein networks. Currently, the above mentioned VirHostNet is probably the most comprehensive database for virus-host interactions, which directly links interaction partners to UniProt (Guirimand et al., 2015). As many pathogens share mechanisms for exploiting host cells, a central database for host-pathogen interactions, multiplexing datasets for viruses, bacteria, fungi and parasites would allow the rapid identification of critical nodes and putative broad spectrum drug targets. Clearly, publically available databases should reliably catalogue virus-host interactions and make these easily accessible to the virologist community through user-friendly web interfaces.
Ultimately, genomics and proteomics techniques provide essential complementary datasets, which can be integrated to reveal a most complete picture of molecular events during virus infection. Functional genomics screens can weigh the importance of a host factor in an interaction network and reveal redundant pathways. Annotation clusters can highlight the enrichment of certain cellular processes, molecular functions or protein domains (Mercer et al., 2012). Novel open access bioinformatics solutions building on available resources like STRING and DAVID can help harmonizing available datasets. Scripting and visualization environments like Cytoscape, R and Bioconductor, which were previously developed for transcriptomics and genomics data integration, are also suitable for pathway mapping of proteomics data (Cline et al., 2007, Gentleman et al., 2004). Proteomics-specific and publically available programs like Perseus provide an additional tool for data analysis and integration (Cox and Mann, 2012). Meta-analysis of omics datasets together with sharing of data in a timely manner will be critical to successfully model the complexity of virus-host interactions in the future (Law et al., 2013).
Such networks will then allow the identification of essential nodes, bottlenecks and hubs for selection of optimal drug targets. Each node in a PPI network represents a protein, while the edges indicate interactions. Bottlenecks are nodes in a network, which critically connect subnetworks and thus restrict information flow, like e.g. central kinases in a signaling pathway. Hubs instead are highly interlinked and therefore central points of control, but typically only control one branch of a signaling pathway. While bottlenecks are less attractive drug targets as their inhibition would shut down complete endogenous pathways, hub inhibition can lead to an efficient block of infection (Box 3 ). Furthermore, drug targeting of pathogen relevant PPIs using small molecule drugs or peptidomimetics seems an attractive strategy to combat infection (de Chassey et al., 2012a, Law et al., 2013). In particular, targeting of interfaces between viral and host proteins, holds the promise of minimal side effects. But even host PPIs, which are triggered or exploited during virus infection, represent attractive targets. In contrast to direct acting antivirals, which often lead to rapid emergence of viral resistance mutations, host proteins and thus their interactions represent a high evolutionary barrier (Gerold and Pietschmann, 2014, von Hahn et al., 2011). In particular, host proteases and kinases are attractive drug targets and both protein classes can be identified by advanced interaction proteomics and PTM proteomics, respectively. Taken together, increasing knowledge on interactomes during virus infection and the compilation of interactome datasets can spur the development of novel antiviral therapeutics. The experimental validation of central network hubs, also as potential drug targets, is discussed below.
Box 3. Network term definitions.
Node: component of a network, e.g., a gene or a protein.
Hub: node, which is highly connected and therefore a point of control of a specific subnetwork.
Bottleneck: node, which connects subnetworks and thus critically restricts information flow.
Edge: connection of two nodes in a network, e.g., representing a physical interaction of two proteins.
5. Hypothesis generation and experimental validation
Protein interaction network analysis allows in the next step to hypothesize, which biological pathway is critical for virus uptake and whether certain proteins are central hubs in the network. The hypothesis can then be tested by disrupting the function of one of the interaction partners and studying the resulting phenotype (Fig. 3D). To this end, blocking antibodies have been used, but the method is limited to proteins with accessible extracellular domains. Alternatively, small molecule inhibitors, if available, can block the protein of interest. Due to their microreversibility and rapid mode of action, such molecular probes have in the past proven indispensable to dissect the kinetics of virus-host cell interplay.
The most commonly used strategy to address a protein's function was in the past the disruption or reduction of its expression by RNA interference (RNAi) (Elbashir et al., 2001, Fire et al., 1998). Here, short RNA molecules sequence-specifically target complementary mRNAs and induce their degradation. RNAi has been used to address the role of protein interactions identified by MS in virus entry (Zona et al., 2013). However, RNAi approaches can have off-target effects (reviewed e.g., in (Mohr et al., 2014)) and the slow kinetics of action of two to three days can lead to compensatory adaptation of cells. This might be the reason why large-scale RNAi screens performed poorly in studies of virus entry, which operates at a minute timescale (de Chassey et al., 2012b).
Since 2012, targeted genome editing became possible making use of a bacterial adaptive antiviral immune mechanism. Clustered regularly interspaced short palindromic repeats (CRISPR) in combination with the RNA guided DNA endonuclease CRISPR associated protein 9 (Cas9) is able to induce specific DNA double strand breaks (Gasiunas et al., 2012, Jinek et al., 2012). Therefore, the system can be used to generate site-specific modifications in genomes, e.g. to knock-out specific proteins (reviewed e.g. in (Doudna and Charpentier, 2014)). The CRISPR-Cas9 technology has already been applied to analyze the function of different proteins in the viral entry process. For example CRISPR-Cas9 knock-out of PDZD8 verified its role in HIV-1 and murine leukemia virus infections (Zhang and Sodroski, 2015). In a different approach, a genome wide lentiviral CRISPR-Cas9 library was developed to screen for cellular factors essential for entry and cell-to-cell transmission of HCV (Ren et al., 2015). In this screen, the known HCV entry factors CD81, claudin-1 and occludin were identified. While knock-out strategies are limited to host factors with non-essential endogenous function and similar to RNAi compensatory effects can occur, a genetically clean knock-out system holds the promise of unambiguously defining essential virus entry factors. Future studies will show whether knockout screens have higher reproducibility than RNAi screens and are suitable for virus entry research.
Both techniques, RNAi and CRISPR-Cas9, display specific advantages and disadvantages, as reviewed in (Boettcher and McManus, 2015). Therefore, the appropriate technique, including small molecule inhibitors, dominant negative variants and blocking antibodies, has to be chosen carefully depending on the experimental setup and the target genes.
After hypothesis testing, drug targets can be defined and searches for compounds interfering with critical network nodes initiated. Alternatively, novel baits can be selected and fueled into a new round of AE-MS, thereby widening the interaction network. Such iterative processes will ultimately complete the picture of PPI during virus invasion (Fig. 4 ).
Fig. 4.

Interaction proteomics based systems virology workflow.
6. Benefits and shortcomings of proteomics approaches
Proteomics is the only method to directly characterize the biologically active entity of most biological processes, the proteins. In an AE setup, it provides information on primary protein interactions and higher order complexes depending on the stringency of purification. It can further yield the order of binding as well as interaction interfaces, when crosslinkers are used (Chen et al., 2010, Leitner et al., 2010). As detailed above, interaction proteomics can reveal receptor complex stoichiometry as well as PTM of the analyzed proteins. With increasing instrument sensitivity not only stable protein interactions, but also transient and virus-induced interactions are accessible. Protein networks between two cellular states, e.g., with and without receptor bound virus, can be compared in a quantitative manner. In the past, detection of subtle differences in protein abundance required isotope labeling, but label-free methods are nowadays sensitive enough for most applications. A clear advantage over RNA interference or image-based screens is that the host cell is in its endogenous state without the requirement for labeling of proteins or change of their expression level. Recent developments moreover make proteomics high throughput screen compatible (Hosp et al., 2015), so that virus entry processes could be studied in a large panel of cell lines as well as in a time resolved manner.
As every method, proteomics has its own shortcomings. Certain proteins are detected as common contaminants and these false positives have to be eliminated using proper controls and by comparison to contaminant databases. When searching for interactions of endogenous proteins, antibodies of high specificity are required. In the future, CRISPR knockin methods might overcome this caveat and allow expression of affinity tagged proteins from their natural locus. Alternatively, controlled expression systems, although not under endogenous gene control, can help express tagged proteins at endogenous levels. Clearly, interaction proteomics just highlights one biological function of a given protein, i.e., its interaction network. Thus the method should be considered as complimentary tool in the virologist's toolbox. To address whether an interaction is critical for virus infection, follow up analysis using established (RNAi, inhibitors, dominant negative mutants) or newly developed techniques (CRISPR) is needed. Nonetheless, we believe that interaction proteomics will prove essential to model molecular networks during virus infection, assign critical nodes, generate and test hypotheses on the molecular function of the proteins in the network and thereby reveal essential host factors, which can be targeted by antiviral drugs.
7. Research perspective
Genetic screens identified numerous virus receptors, but their actual binding to the respective virus has often not been formerly proven. Quantitative interaction proteomics approaches hold the promise of closing this gap of knowledge. Due to technological and computational developments in the past decade proteomics is now on par with genomics and allows high throughout comprehensive analyses of complex samples. Recently, a 96-well compatible and streamlined interaction proteomics protocol has been developed (Hosp et al., 2015). Given the fact that for many viruses the target cells are either not known or are of multiple host and tissue origin, the parallel analysis of many biological samples will in the future allow proteomics screening approaches without prior knowledge of receptor expression or susceptibility.
A second consequence of the vast increase in MS sensitivity is that patient material and material from animal experiments, which is typically limited, can be analyzed in depth. Transcriptomics data only accounts for a subset of pathogen induced changes in a host as it lacks information on PTMs and PPI. Proteomics of ex vivo samples thus holds the promise of providing a more complete picture of the virus host interplay in an infected organism. It will be interesting to demonstrate to what extend the changes in host and virus proteomes observed thus far in cell culture models, reflect the in vivo situation.
Notably, proteomics can not only elucidate protein networks during virus infection, but also serve to identify diagnostic and prognostic markers of virus infection and disease progression (Mancone et al., 2013, Rhea et al., 2010). It also allows the identification of targets of chemical compound libraries by interaction proteomics (Ong et al., 2009). MS-based proteomics is thus valuable for virologists with a molecular biology, cell biology, and clinical focus. We believe that increasing availability of proteomics hardware, protocols and analysis software is currently spurring a shift from the largely genomics dominated field of virus infection research towards proteomic characterization of cellular perturbations by viruses.
Acknowledgements
We thank Stefan Kunz for critical reading of the manuscript. This work was funded by a fellowship from the German Research Foundation (DFG, GE 2145/3-1) to G.G., who was further supported by the Ina-Pichlmayr Program of Hannover Medical School. Work in T.P’s. laboratory is supported by grants from the European Research Council (ERC-2011-StG_281473-VIRAFRONT) by the Deutsche Forschungsgemeinschaft (CRC 900, project A6), by the Bundesministerium für Bildung und Forschung (GINAICO; 16GW0105; and DZI-F), by the Helmholtz Alberta Initiative for Infectious Disease Research (HAI- IDR), and by the Helmholtz Association (SO-024). J.B. is supported by the Hannover Biomedical Research School (HBRS) and the Centre for Infection Biology (ZIB).
Contributor Information
Gisa Gerold, Email: gisa.gerold@twincore.de.
Thomas Pietschmann, Email: thomas.pietschmann@twincore.de.
References
- Aloy P., Russell R.B. Ten thousand interactions for the molecular biologist. Nat. Biotechnol. 2004;22:1317–1321. doi: 10.1038/nbt1018. [DOI] [PubMed] [Google Scholar]
- Amara A., Mercer J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015;13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstutz B., Gastaldelli M., Kalin S., Imelli N., Boucke K., Wandeler E., Mercer J., Hemmi S., Greber U.F. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 2008;27:956–969. doi: 10.1038/emboj.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J.S., Matic I., Vertegaal A.C. Identification of SUMO target proteins by quantitative proteomics. Methods Mol. Biol. 2009;497:19–31. doi: 10.1007/978-1-59745-566-4_2. [DOI] [PubMed] [Google Scholar]
- Berard A.R., Coombs K.M., Severini A. Quantification of the host response proteome after herpes simplex virus type 1 infection. J. Proteome Res. 2015;14:2121–2142. doi: 10.1021/pr5012284. [DOI] [PubMed] [Google Scholar]
- Bertram S., Glowacka I., Blazejewska P., Soilleux E., Allen P., Danisch S., Steffen I., Choi S.Y., Park Y., Schneider H. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 2010;84:10016–10025. doi: 10.1128/JVI.00239-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram S., Glowacka I., Muller M.A., Lavender H., Gnirss K., Nehlmeier I., Niemeyer D., He Y., Simmons G., Drosten C. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011;85:13363–13372. doi: 10.1128/JVI.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeri Erba E. Investigating macromolecular complexes using top-down mass spectrometry. Proteomics. 2014;14:1259–1270. doi: 10.1002/pmic.201300333. [DOI] [PubMed] [Google Scholar]
- Boettcher M., McManus M.T. Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol. Cell. 2015;58:575–585. doi: 10.1016/j.molcel.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko P.V., Chelius D., Shaler T.A. Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography–tandem mass spectrometry. Anal. Chem. 2002;74:4741–4749. doi: 10.1021/ac0256991. [DOI] [PubMed] [Google Scholar]
- Borner G.H., Hein M.Y., Hirst J., Edgar J.R., Mann M., Robinson M.S. Fractionation profiling: a fast and versatile approach for mapping vesicle proteomes and protein–protein interactions. Mol. Biol. Cell. 2014;25:3178–3194. doi: 10.1091/mbc.E14-07-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle J.F., Weismiller D.G., Holmes K.V. Genetic resistance to mouse hepatitis virus correlates with absence of virus-binding activity on target tissues. J. Virol. 1987;61:185–189. doi: 10.1128/jvi.61.1.185-189.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzoli M., Bianchi A., Filippini S., Weiner A., Zhu Q., Pizza M., Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W., Henry M.D., Borrow P., Yamada H., Elder J.H., Ravkov E.V., Nichol S.T., Compans R.W., Campbell K.P., Oldstone M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and lassa fever virus. Science. 1998;282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Veettil M.V., Bottero V., Chandran B. Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E1163–E1172. doi: 10.1073/pnas.1119592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Zhuang X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. U. S. A. 2008;105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.A., Jawhari A., Fischer L., Buchen C., Tahir S., Kamenski T., Rasmussen M., Lariviere L., Bukowski-Wills J.C., Nilges M. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Larsen B., Lin Z.Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement C., Tiwari V., Scanlan P.M., Valyi-Nagy T., Yue B.Y., Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B. Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller K.E., Berger K.L., Heaton N.S., Cooper J.D., Yoon R., Randall G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009;5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote M., Misasi J., Ren T., Bruchez A., Lee K., Filone C.M., Hensley L., Li Q., Ory D., Chandran K. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high- throughput data. BMC Bioinform. 2012;13(Suppl. 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne C.B., Bergelson J.M. Virus- induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42:D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Laxminarayana S.V., Chandra N., Ravi V., Desai A. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology. 2009;385:47–57. doi: 10.1016/j.virol.2008.10.025. [DOI] [PubMed] [Google Scholar]
- Davey N.E., Trave G., Gibson T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011;36:159–169. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- de Chassey B., Meyniel-Schicklin L., Aublin-Gex A., Andre P., Lotteau V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr. Opin. Virol. 2012;2:606–613. doi: 10.1016/j.coviro.2012.09.001. [DOI] [PubMed] [Google Scholar]
- de Chassey B., Meyniel-Schicklin L., Aublin-Gex A., Andre P., Lotteau V. Genetic screens for the control of influenza virus replication: from meta-analysis to drug discovery. Mol. BioSyst. 2012;8:1297–1303. doi: 10.1039/c2mb05416g. [DOI] [PubMed] [Google Scholar]
- de Vries E., Tscherne D.M., Wienholts M.J., Cobos-Jimenez V., Scholte F., Garcia-Sastre A., Rottier P.J., de Haan C.A. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011;7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohner K., Nagel C.H., Sodeik B. Viral stop- and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol. 2005;13:320–327. doi: 10.1016/j.tim.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Dorr P., Westby M., Dobbs S., Griffin P., Irvine B., Macartney M., Mori J., Rickett G., Smith-Burchnell C., Napier C. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small- molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Eierhoff T., Hrincius E.R., Rescher U., Ludwig S., Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010;6:e1001099. doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fahey M.E., Bennett M.J., Mahon C., Jager S., Pache L., Kumar D., Shapiro A., Rao K., Chanda S.K., Craik C.S. GPS-Prot: a web-based visualization platform for integrating host- pathogen interaction data. BMC Bioinform. 2011;12:298. doi: 10.1186/1471-2105-12-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro S.B., McCleland M.L., Stukenberg P.T., Burke D.J., Ross M.M., Shabanowitz J., Hunt D.F., White F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- Fingeroth J.D., Weis J.J., Tedder T.F., Strominger J.L., Biro P.A., Fearon D.T. Epstein- Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. U. S. A. 1984;81:4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Foster L.J., De Hoog C.L., Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei A.P., Jeon O.Y., Kilcher S., Moest H., Henning L.M., Jost C., Pluckthun A., Mercer J., Aebersold R., Carreira E.M. Direct identification of ligand-receptor interactions on living cells and tissues. Nat. Biotechnol. 2012;30:997–1001. doi: 10.1038/nbt.2354. [DOI] [PubMed] [Google Scholar]
- Gabaev I., Steinbruck L., Pokoyski C., Pich A., Stanton R.J., Schwinzer R., Schulz T.F., Jacobs R., Messerle M., Kay-Fedorov P.C. The human cytomegalovirus UL11 protein interacts with the receptor tyrosine phosphatase CD45, resulting in functional paralysis of T cells. PLoS Pathog. 2011;7:e1002432. doi: 10.1371/journal.ppat.1002432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerold G., Meissner F., Bruening J., Welsch K., Perin P.M., Baumert T.F., Vondran F.W., Kaderali L., Marcotrigiano J., Khan A.G. Quantitative proteomics identifies serum response factor binding protein 1 as a host factor for hepatitis C virus entry. Cell Rep. 2015;12:864–878. doi: 10.1016/j.celrep.2015.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerold G., Pietschmann T. The HCV life cycle: in vitro tissue culture systems and therapeutic targets. Dig. Dis. 2014;32:525–537. doi: 10.1159/000360830. [DOI] [PubMed] [Google Scholar]
- Gingras A.C., Gstaiger M., Raught B., Aebersold R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- Gordon-Alonso M., Rocha-Perugini V., Alvarez S., Ursa A., Izquierdo-Useros N., Martinez-Picado J., Munoz-Fernandez M.A., Sanchez-Madrid F. Actin-binding protein drebrin regulates HIV-1-triggered actin polymerization and viral infection. J. Biol. Chem. 2013;288:28382–28397. doi: 10.1074/jbc.M113.494906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Alonso M., Sala-Valdes M., Rocha-Perugini V., Perez-Hernandez D., Lopez-Martin S., Ursa A., Alvarez S., Kolesnikova T.V., Vazquez J., Sanchez-Madrid F. EWI-2 association with alpha-actinin regulates T cell immune synapses and HIV viral infection. J. Immunol. 2012;189:689–700. doi: 10.4049/jimmunol.1103708. [DOI] [PubMed] [Google Scholar]
- Grove J., Marsh M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011;195:1071–1082. doi: 10.1083/jcb.201108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirimand T., Delmotte S., Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015;43:D583–D587. doi: 10.1093/nar/gku1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagai T., Azia A., Babu M.M., Andino R. Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions. Cell Rep. 2014;7:1729–1739. doi: 10.1016/j.celrep.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A.S., Kaufmann J.K., Wies E., Naschberger E., Panteleev-Ivlev J., Schmidt K., Holzer A., Schmidt M., Chen J., Konig S. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat. Med. 2012;18:961–966. doi: 10.1038/nm.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger M., Mann M. Triple SILAC to determine stimulus specific interactions in the Wnt pathway. J. Proteome Res. 2012;11:982–994. doi: 10.1021/pr200740a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp F., Scheltema R.A., Eberl C., Kulak N.A., Keilhauer E.C., Mayr K., Mann M. A double-barrel LC–MS/MS system to quantify 96 interactomes per day. Mol. Cell. Proteomics. 2015 doi: 10.1074/mcp.O115.049460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Sun F., Owen D.M., Li W., Chen Y., Gale M. Jr., Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impens F., Radoshevich L., Cossart P., Ribet D. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12432–12437. doi: 10.1073/pnas.1413825111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S., Cimermancic P., Gulbahce N., Johnson J.R., McGovern K.E., Clarke S.C., Shales M., Mercenne G., Pache L., Li K. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., McGinnes L.W., Morrison T.G. Overexpression of thiol/disulfide isomerases enhances membrane fusion directed by the Newcastle disease virus fusion protein. J. Virol. 2008;82:12039–12048. doi: 10.1128/JVI.01406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielity S., Wang J.J., Chan Y.K., Ahmed A.A., Li W., Monahan S., Bu X., Farzan M., Freeman G.J., Umetsu D.T. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013;9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Baranda S., Gomez-Mouton C., Rojas A., Martinez-Prats L., Mira E., Ana Lacalle R., Valencia A., Dimitrov D.S., Viola A., Delgado R. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007;9:838–846. doi: 10.1038/ncb1610. [DOI] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji H., Kamiie J., Kawakami H., Kido K., Yamauchi Y., Shinkawa T., Taoka M., Takahashi N., Isobe T. Proteomics reveals N-linked glycoprotein diversity in Caenorhabditis elegans and suggests an atypical translocation mechanism for integral membrane proteins. Mol. Cell. Proteomics. 2007;6:2100–2109. doi: 10.1074/mcp.M600392-MCP200. [DOI] [PubMed] [Google Scholar]
- Keilhauer E.C., Hein M.Y., Mann M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS) Mol. Cell. Proteomics. 2015;14:120–135. doi: 10.1074/mcp.M114.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Kohli P., Bartram M.P., Habbig S., Pahmeyer C., Lamkemeyer T., Benzing T., Schermer B., Rinschen M.M. Label-free quantitative proteomic analysis of the YAP/TAZ interactome. Am. J. Physiol. Cell Physiol. 2014;306:C805–C818. doi: 10.1152/ajpcell.00339.2013. [DOI] [PubMed] [Google Scholar]
- Kondratowicz A.S., Lennemann N.J., Sinn P.L., Davey R.A., Hunt C.L., Moller-Tank S., Meyerholz D.K., Rennert P., Mullins R.F., Brindley M. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 2011;108:8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M., Kratchmarova I., Blagoev B., Tseng Y.H., Kahn C.R., Mann M. Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2451–2456. doi: 10.1073/pnas.0711713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law G.L., Korth M.J., Benecke A.G., Katze M.G. Systems virology: host-directed approaches to viral pathogenesis and drug targeting. Nat. Rev. Microbiol. 2013;11:455–466. doi: 10.1038/nrmicro3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M.J., Sherer N.M., Marks C.B., Pypaert M., Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005;170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Ali M.A., Cohen J.I. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell. 2006;127:305–316. doi: 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.E., Greco T.M., Dohner K., Sodeik B., Cristea I.M. A proteomic perspective of inbuilt viral protein regulation: pUL46 tegument protein is targeted for degradation by ICP0 during herpes simplex virus type 1 infection. Mol. Cell. Proteomics. 2013;12:3237–3252. doi: 10.1074/mcp.M113.030866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Sadygov R.G., Yates J.R. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- Lozach P.Y., Burleigh L., Staropoli I., Amara A. The C type lectins DC-SIGN and L-SIGN: receptors for viral glycoproteins. Methods Mol. Biol. 2007;379:51–68. doi: 10.1007/978-1-59745-393-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozach P.Y., Kuhbacher A., Meier R., Mancini R., Bitto D., Bouloy M., Helenius A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe. 2011;10:75–88. doi: 10.1016/j.chom.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Luber C.A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O'Keeffe M. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Lupberger J., Zeisel M.B., Xiao F., Thumann C., Fofana I., Zona L., Davis C., Mee C.J., Turek M., Gorke S. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddon P.J., Dalgleish A.G., McDougal J.S., Clapham P.R., Weiss R.A., Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- Mancone C., Ciccosanti F., Montaldo C., Perdomo A.B., Piacentini M., Alonzi T., Fimia G.M., Tripodi M. Applying proteomic technology to clinical virology. Clin. Microbiol. Infect. 2013;19:23–28. doi: 10.1111/1469-0691.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M., Kelleher N.L. Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18132–18138. doi: 10.1073/pnas.0800788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannova P., Fang R., Wang H., Deng B., McIntosh M.W., Hanash S.M., Beretta L. Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell. Proteomics. 2006;5:2319–2325. doi: 10.1074/mcp.M600121-MCP200. [DOI] [PubMed] [Google Scholar]
- Marsh M., Bron R. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997;110(Pt 1):95–103. doi: 10.1242/jcs.110.1.95. [DOI] [PubMed] [Google Scholar]
- Matlin K.S., Reggio H., Helenius A., Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J., Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- Mercer J., Schelhaas M., Helenius A. Virus entry by endocytosis. Ann. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- Mercer J., Snijder B., Sacher R., Burkard C., Bleck C.K., Stahlberg H., Pelkmans L., Helenius A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012;2:1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Mohr S.E., Smith J.A., Shamu C.E., Neumuller R.A., Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nat. Rev. Mol. Cell Biol. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller-Tank S., Kondratowicz A.S., Davey R.A., Rennert P.D., Maury W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 2013;87:8327–8341. doi: 10.1128/JVI.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpellier C., Tews B.A., Poitrimole J., Rocha-Perugini V., D'Arienzo V., Potel J., Zhang X.A., Rubinstein E., Dubuisson J., Cocquerel L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J. Biol. Chem. 2011;286:13954–13965. doi: 10.1074/jbc.M111.220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman N.J., Sharon-Friling R., Shenk T., Cristea I.M. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics. 2010;9:851–860. doi: 10.1074/mcp.M900485-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraz M.L., Pythoud C., Turk R., Rothenberger S., Pasquato A., Campbell K.P., Kunz S. Cell entry of Lassa virus induces tyrosine phosphorylation of dystroglycan. Cell. Microbiol. 2013;15:689–700. doi: 10.1111/cmi.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa K., Gouttenoire J., Hernandez C., Dao Thi V.L., Tran H.T., Lange C.M., Dill M.T., Heim M.H., Donze O., Penin F. Quantitative proteomics identifies the membrane-associated peroxidase GPx8 as a cellular substrate of the hepatitis C virus NS3-4A protease. Hepatology. 2014;59:423–433. doi: 10.1002/hep.26671. [DOI] [PubMed] [Google Scholar]
- Nahnsen S., Bielow C., Reinert K., Kohlbacher O. Tools for label-free peptide quantification. Mol. Cell. Proteomics. 2013;12:549–556. doi: 10.1074/mcp.R112.025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrete O.A., Levroney E.L., Aguilar H.C., Bertolotti-Ciarlet A., Nazarian R., Tajyar S., Lee B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature. 2005;436:401–405. doi: 10.1038/nature03838. [DOI] [PubMed] [Google Scholar]
- Ngounou Wetie A.G., Sokolowska I., Woods A.G., Roy U., Deinhardt K., Darie C.C. Protein-protein interactions: switch from classical methods to proteomics and bioinformatics-based approaches. Cell. Mol. Life Sci. 2014;71:205–228. doi: 10.1007/s00018-013-1333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogalski M.T., Chan G.C., Stevenson E.V., Collins-McMillen D.K., Yurochko A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013;9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonnenmacher M., Weber T. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe. 2011;10:563–576. doi: 10.1016/j.chom.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J.V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Ong S.E., Mittler G., Mann M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods. 2004;1:119–126. doi: 10.1038/nmeth715. [DOI] [PubMed] [Google Scholar]
- Ong S.E., Schenone M., Margolin A.A., Li X., Do K., Doud M.K., Mani D.R., Kuai L., Wang X., Wood J.L. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4617–4622. doi: 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Hoppe S., Saher G., Krijnse-Locker J., Bartenschlager R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013;87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Schwartz D., Elias J.E., Thoreen C.C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Kandasamy K., Alvisi G., Mulhern O., Sacco R., Habjan M., Binder M., Stefanovic A., Eberle C.A., Goncalves A. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature. 2012;487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- Radoshitzky S.R., Abraham J., Spiropoulou C.F., Kuhn J.H., Nguyen D., Li W., Nagel J., Schmidt P.J., Nunberg J.H., Andrews N.C. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiyaah J., Tan C.P., Fletcher A.J., Price A.J., Blondeau C., Hilditch L., Jacques D.A., Selwood D.L., James L.C., Noursadeghi M. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stencel-Baerenwald J.E., Reiss K., Reiter D.M., Stehle T., Dermody T.S. The sweet spot: defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014;12:739–749. doi: 10.1038/nrmicro3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q., Li C., Yuan P., Cai C., Zhang L., Luo G.G., Wei W. A Dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci. Rep. 2015;5:8865. doi: 10.1038/srep08865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea J.M., Diwan C.A., Molinaro R.J. Mass spectrometry-coupled techniques for viral-related disease biomarker identification. Biomark. Med. 2010;4:859–870. doi: 10.2217/bmm.10.110. [DOI] [PubMed] [Google Scholar]
- Rocha-Perugini V., Montpellier C., Delgrange D., Wychowski C., Helle F., Pillez A., Drobecq H., Le Naour F., Charrin S., Levy S. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One. 2008;3:e1866. doi: 10.1371/journal.pone.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M.J., Lakadamyali M., Zhang F., Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004;11:567–573. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser H.J., Levy E.M., Mandel R., DiSciullo G.J. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. U. S. A. 1994;91:4559–4563. doi: 10.1073/pnas.91.10.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R.M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Schaulies J., Dunster L.M., Schwartz-Albiez R., Krohne G., ter Meulen V. Physical association of moesin and CD46 as a receptor complex for measles virus. J. Virol. 1995;69:2248–2256. doi: 10.1128/jvi.69.4.2248-2256.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N.R., Mateu G., Dreux M., Grakoui A., Cosset F.L., Melikyan G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011;286:30361–30376. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima M., Stroher U., Ebihara H., Feldmann H., Kawaoka Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 2012;86:2067–2078. doi: 10.1128/JVI.06451-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.L., Lidke D.S., Ozbun M.A. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology. 2008;381:16–21. doi: 10.1016/j.virol.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stantchev T.S., Paciga M., Lankford C.R., Schwartzkopff F., Broder C.C., Clouse K.A. Cell-type specific requirements for thiol/disulfide exchange during HIV-1 entry and infection. Retrovirology. 2012;9:97. doi: 10.1186/1742-4690-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf M.P., Thorne T., de Silva E., Stewart R., An H.J., Lappe M., Wiuf C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W.C., Chen Y.C., Tseng C.H., Hsu P.W., Tung K.F., Jeng K.S., Lai M.M. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. U. S. A. 2013;110:17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze Z., Sterk R.T., DeHaro S.A., Ozbun M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013;87:2508–2517. doi: 10.1128/JVI.02319-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P., Davey N.E., Gibson T.J., Babu M.M. A million peptide motifs for the molecular biologist. Mol. Cell. 2014;55:161–169. doi: 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L. Resolving protein interactions and complexes by affinity purification followed by label-based quantitative mass spectrometry. Proteomics. 2012;12:1623–1638. doi: 10.1002/pmic.201100438. [DOI] [PubMed] [Google Scholar]
- Ueki I.F., Min-Oo G., Kalinowski A., Ballon-Landa E., Lanier L.L., Nadel J.A., Koff J.L. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon lambda and antiviral defense in airway epithelium. J. Exp. Med. 2013;210:1929–1936. doi: 10.1084/jem.20121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Almeida T.O., Salas-Benito M., De Nova-Ocampo M.A., Del Angel R.M., Salas-Benito J.S. Surface proteins of C6/36 cells involved in dengue virus 4 binding and entry. Arch. Virol. 2013;158:1189–1207. doi: 10.1007/s00705-012-1596-0. [DOI] [PubMed] [Google Scholar]
- Venkatesan K., Rual J.F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K.I. An empirical framework for binary interactome mapping. Nat. Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M., Hubner N.C., Manns M. High confidence determination of specific protein–protein interactions using quantitative mass spectrometry. Curr. Opin. Biotechnol. 2008;19:331–337. doi: 10.1016/j.copbio.2008.06.001. [DOI] [PubMed] [Google Scholar]
- von Hahn T., Ciesek S., Manns M.P. Arrest all accessories–inhibition of hepatitis C virus by compounds that target host factors. Discov. Med. 2011;12:237–244. [PubMed] [Google Scholar]
- Wang X., Huang D.Y., Huong S.M., Huang E.S. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 2005;11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes M.P., Tomasec P., Huttlin E.L., Fielding C.A., Nusinow D., Stanton R.J., Wang E.C., Aicheler R., Murrell I., Wilkinson G.W. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell. 2014;157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild C., Greenwell T., Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell–cell fusion. AIDS Res. Hum. Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- Wisniewski J.R., Hein M.Y., Cox J., Mann M. A proteomic ruler for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics. 2014;13:3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcechowskyj J.A., Didigu C.A., Lee J.Y., Parrish N.F., Sinha R., Hahn B.H., Bushman F.D., Jensen S.T., Seeholzer S.H., Doms R.W. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe. 2013;13:613–623. doi: 10.1016/j.chom.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N., Huang K., Zhang T., Lei Y., Liu R., Wang K., Zhou S., Li J., Wu J., Wu H. Comprehensive proteomic analysis of host cell lipid rafts modified by HBV infection. J. Proteomics. 2012;75:725–739. doi: 10.1016/j.jprot.2011.09.011. [DOI] [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough M.L., Mata M.A., Sakthivel R., Fontoura B.M. Viral subversion of nucleocytoplasmic trafficking. Traffic. 2014;15:127–140. doi: 10.1111/tra.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z., Fang C., Pan T., Wang J., Yang P., Yuan Z. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem. Biophys. Res. Commun. 2006;350:174–178. doi: 10.1016/j.bbrc.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Yi Z., Pan T., Wu X., Song W., Wang S., Xu Y., Rice C.M., Macdonald M.R., Yuan Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 2011;85:6996–7004. doi: 10.1128/JVI.00013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A., Yu D., Dong L., Iyer S.R., Xu X., Kelly J., Liu J., Wang W., Vorster P.J., Agulto L. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanivan S., Krueger M., Mann M. In vivo quantitative proteomics: the SILAC mouse. Methods Mol. Biol. 2012;757:435–450. doi: 10.1007/978-1-61779-166-6_25. [DOI] [PubMed] [Google Scholar]
- Zanivan S., Maione F., Hein M.Y., Hernandez-Fernaud J.R., Ostasiewicz P., Giraudo E., Mann M. SILAC-based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Mol. Cell. Proteomics. 2013;12:3599–3611. doi: 10.1074/mcp.M113.031344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Lu Z., Liu H., Xiao X., Zhao Z., Bao G., Han J., Jing T., Chen G. Incidence of Japanese encephalitis, visceral leishmaniasis and malaria before and after the Wenchuan earthquake, in China. Acta Trop. 2013;128:85–89. doi: 10.1016/j.actatropica.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Zhang S., Sodroski J. Efficient human immunodeficiency virus (HIV-1) infection of cells lacking PDZD8. Virology. 2015;481:73–78. doi: 10.1016/j.virol.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K., Xiang Y., Wang X., Wang Q., Zhong M., Wang S., Wang X., Fan J., Kitazato K., Wang Y. Epidermal growth factor receptor-PI3K signaling controls cofilin activity to facilitate herpes simplex virus 1 entry into neuronal cells. mBio. 2014;5:e00958–00913. doi: 10.1128/mBio.00958-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.Z., Luo Y., Cao M.M., Liu Y., Liu X.Q., Wang W., Wu D.G., Guan M., Xu Q.Q., Ren H. Significance of palmitoylation of CD81 on its association with tetraspanin-enriched microdomains and mediating hepatitis C virus cell entry. Virology. 2012;429:112–123. doi: 10.1016/j.virol.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Zona L., Lupberger J., Sidahmed-Adrar N., Thumann C., Harris H.J., Barnes A., Florentin J., Tawar R.G., Xiao F., Turek M. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe. 2013;13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]