

Abstract
Two novel pyridine pendant-armed macrocycles structurally reinforced by an ethyl bridge, either between adjacent nitrogens (for side-bridged) or non-adjacent nitrogens (for cross-bridged), have been synthesized and complexed with a range of transition metal ions (Co2+, Ni2+, Cu2+ and Zn2+). X-ray crystal structures of selected cross-bridged complexes were obtained which showed the characteristic cis-V configuration with potential labile cis binding sites. The complexes have been characterized by their electronic spectra and magnetic moments, which show the expected high spin divalent metal complex in most cases. Exceptions are the nickel side-bridged complex, which shows a mixture of high-spin and low spin, and the cobalt cross-bridged complex which has oxidized to cobalt(III). Cyclic voltammetry in acetonitrile was carried out to assess the potential future use of these complexes in oxidation catalysis. Selected complexes offer significant catalytic potential enhanced by the addition of the pyridyl arm to a reinforced cyclen backbone.
Keywords: pendant-armed, pyridyl, reinforced macrocycles, tetraazamacrocycles, cyclen, cross-bridged
Synopsis
Mono-pyridyl pendant armed side-bridged and cross-bridged cyclen ligands have been synthesized and complexed to Co, Ni, Cu, and Zn. Their electronic, electrochemical, and structural characterization suggests that they may be useful as catalysts.
1. Introduction
Tetraazamacrocycles form complexes of high thermodynamic and kinetic stability with transition metals. [1] Depending on the chelated metal, tetraazamacrocyclic complexes have found use in a range of applications, including medical imaging, [2] [3] [4] [5] protein binding agents, [6] [7] and as anti-malarial drugs. [8] [9] Recently we have been interested in the use of this complex type as oxidation catalysts. [10] [11] [12] A recent papers by Que, et al, has shown that addition of a pyridyl pendant arm to a tetraazamacrocycle can result in new and improved oxidation catalysis. [13] The literature contains numerous other pyridyl pendant armed tetraazamacrocycles, indicating a general interest in the community and known synthetic techniques for adding pendant pyridyl donors. [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24]
Our specific exploration is aimed at forming catalysts which can perform under a range of aqueous conditions. Transition metal complexes of tetraazamacrocycles reinforced with additional ethylene bridges have produced oxidation catalysts with high kinetic stability under such harsh conditions. [25] [26] [27] [28] [29] Transition metal complexes of macrocycles of this type are known to be stable to a range of pH’s and temperatures. A cross-bridged cyclen ligand with two pyridyl pendant arms (CB-Py2Cyclen, Figure 1) has been reported [30], but its hexadentate nature coordinatively saturates its complexed metal ions, while we wished to leave at least one coordination site available for interaction with oxidant and/or substrate. Our first communications regarding complexes incorporating a single pyridyl labile coordinating group have described reinforced cyclam derivatives with modified steric and electronic properties to cause improved properties as oxidation catalysts, [10] [12] see Figure 1. Herein, we report the synthesis and characterization of analogous smaller cyclen derivatives as potential future oxidation catalysts. Comparisons between our cyclam and cyclen derivatives, as well as with other closely related cyclen-pyridyl ligands (Figure 1) will be presented.
Figure 1.

Ligands discussed within this work; SB-PyCyclam [10] PyMeEBC [12] Me2BCyclen, [31] PyCyclen [21] [22] CB-Py2Cyclen [30]; alongside SB-PyCyclen and CB-MePyCyclen first described in this study.
2. Results and discussion
2.1. Synthesis
We have recently demonstrated that alkylation of tetraazamacrocycle-glyoxal condensates using typical acetonitrile conditions described by Weisman et al. [32] [33] [34] and Handel et al. [35] are not ideal when using pyridyl derived alkyl halides and nonpolar chlorinated solvents are preferred. [10] [12] Analogous conditions were used for the formation of 1. Using dichloromethane gave the desired product in relatively high yields (~80%), see Scheme 1. Methylation of the non-adjacent nitrogen atom with methyl iodide gives a bis-quaternary ammonium salt 2 by what are now routine methods for cyclam, [12] [36] although this is the first reported use of methylation to form unsymmetrical ethyl bridged cyclen derivatives. Reduction of mono (1) and bis (2) quaternary ammonium salts is carried out with sodium borohydride to form ethyl side-bridged (SB-PyCyclen) 3 and cross-bridged (CB-MePyCyclen) 4 derivatives respectively using well described analogous reactions. [32] [33] [34]
Scheme 1.

Ligand synthesis. (a) (i) CH2Cl2, 2-picolyl chloride hydrochloride, NaHCO3 (ii) Decahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene, KI. (b) MeCN, CH3I. (c) EtOH, NaBH4. (d) EtOH, NaBH4.
Metal complexes have been formed with both SB 3 and CB 4 ligands with a range of divalent transition metals (Co2+, Ni2+, Cu2+ and Zn2+). Due to the proton sponge nature of cross-bridged tetraazamacrocyclic ligands, complexation reactions were carried out in an inert atmosphere glovebox using anhydrous metal salts in acetonitrile (with additional DMF for acetonitrile-insoluble NiCl2). In a similar manner to our cyclam analogues, [10] [12] the reactions did not require heating and the complexes did not precipitate from the reaction solution. After drying the crude solutions the complexes were purified by changing the counter ion from chloride to hexafluorophosphate in methanol, with the conversion causing a reduction in solubility and precipitation of the pure complex salt.
According to elemental analysis, the copper complexes of both ligands, as well as the Ni2+, and Zn2+ complex of 3, replace both chloride counter ions with hexaflurophosphates, indicating no bound chloro ligands and five-coordinate geometries. In contrast, the other metal ion complexes show replacement of only one chloro counter ion, indicating a six-coordinate complexes with a coordinated chloride anion (vide infra). The five-coordinate behavior of the side-bridged ligand is consistent with the complexation of its cyclam analogue SB-PyCyclam, which coordinates Fe2+ and Cu2+ in a 5-coordinate manner. [10] Similarly, the 6-coordination (in combination with a chloro ligand) of the cross-bridged 4 is consistent with its cyclam analogue PyMeEBC with Mn2+, Fe2+, Co2+, Ni2+, and Zn2+. PyMeEBC is only 5-coordinate, as seen for 4 here, with Cu2+. [12] Uniquely, the cobalt complex of 4 has oxidized to Co3+ upon its workup in air. Its elemental analysis shows one chloro ligand completing its six-coordination, but the presence of two additional hexafluorophosphate counter ions. This behavior has been observed for other cross-bridged cyclen ligand analogues with cobalt. [30] [37]
2.2. X-ray crystal structures
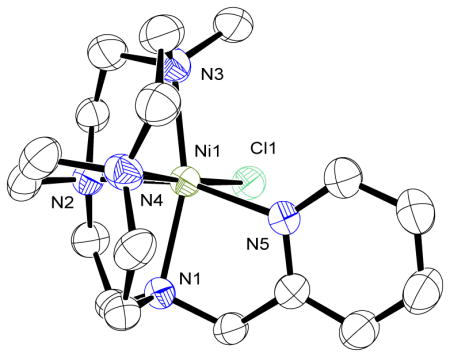
X-ray crystal structures could be determined for the Ni2+, Cu2+ and Zn2+ complexes of CB-MePyCyclen (4), see Figure 2. These solid state structures are consistent with the elemental analyses in which Ni(4)Cl+ and Zn(4)Cl+ retain a chloro ligand, but Cu(4)2+ displaces both. All three complexes crystalized in the monoclinic P21 space group. Ni(4)Cl+ and Zn(4)Cl+ are isostructural and will be discussed collectively, with Cu(4)2+ crystalizing differently.
Figure 2.

X-ray crystal structures of Zn(4)Cl+ (left) Ni(4)Cl+ (middle) and Cu(4)2+ (right) showing 50% ellipsoids.
Ni(4)Cl+ and Zn(4)Cl+ have the metal ion in a slightly distorted octahedral environment, coordinating with the four nitrogen atoms of the folded macrocycle along with the pyridyl arm and a chloride counter ion located cis to each other. Cu(4)2+ has displaced the chloride ion with a non-coordinating hexafluorophosphate and is subsequently square based pyramidal. The Addison Parameter [38] (τ) has been used to quantify the geometry of 5-coordinate structures, with τ = 0 for a perfect square pyramidal complex, and τ = 1 for a perfect trigonal bipyramidal one. For Cu(4)2+ τ = 0.09, which confirms our square pyramidal designation. Of note for Cu(4)2+, The “empty” coordination position on the square pyramidal Cu2+ ion is occupied in the solid state structure by one of the PF6− anions, with the Cu(1)-F(1) distance at 2.787 Å (see Figure 2c). This Cu-F distance is rather typical for similar interactions where copper is coordinated by four or five nitrogen atoms. [39]
The cross-bridged tetraazamacrocycle complexes show the well-recognized “cis-V” configuration, [40] with the metal ion being slightly pushed out of the macrocyclic plane. The degree of distortion from octahedral can be quantified by looking at Nax-M-Nax and Neq-M-Neq bond angles, where Nax are the non-cross-bridged nitrogen atoms of the cyclen ring, while Neq are the cross-bridged nitrogen atoms. For comparison (Table 1), the metrical parameters of the complexes of the two most relevant ligand analogues, Me2Bcyclen and PyMeEBC, with the same metal ions and in the same coordination geometries are provided.
Table 1.
Structural data for transition metal complexes
| Metal ion | Ionic radiusa [41] | Me2BCyclen | Ref | PyMeEBC | Ref | CB-MePyCyclen | |||
|---|---|---|---|---|---|---|---|---|---|
| Nax-M-Nax | Neq-M-Neq | Nax-M-Nax | Neq-M-Neq | Nax-M-Nax | Neq-M-Neq | ||||
| Zn2+ | 88 | 150.75 | 78.32 | [31] | 168.66 | 81.84 | [12] | 153.69(10) | 81.26(11) |
|
| |||||||||
| Ni2+ | 83 | 161.58 | 85.46 | [42] | 173.56 | 84.85 | [12] | 160.8(2) | 84.46(19) |
|
| |||||||||
| Cu2+ | 79 | 164.06 | 85.95 | [37] | 175.66 | 85.24 | [12] | 170.6(2) | 83.0(2) |
High spin 6-coordinate (Zn2+/Ni2+) or 5-coordinate (Cu2+)
The first trend that is apparent from this data is that the Nax-M-Nax and Neq-M-Neq bond angles increase (becoming less distorted) as the ionic radius of the metal ion decreases. For example, the smaller Cu2+ ion fits into the CB-MePyCyclen cavity (Nax-M-Nax = 170.6° and Neq-M-Neq = 83.0°) with less distortion of the ideal 180° and 90° expected bond angles than the larger Zn2+ ion (Nax-M-Nax = 153.69° and Neq-M-Neq = 81.26°). This trend is also followed by analogous bridged ligands Me2Bcyclen and PyMeEBC.
Another interesting comparison is between the larger PyMeEBC complexes and the small CB-MePyCyclen complexes. From the bond angles it is obvious the 14-membered cyclam ring of PyMeEBC better accommodates these metal ions with less distortion from the expected bond angles than does the 12-membered cyclen ring of CB-MePyCyclen. For example, the Zn2+ ion is difficult for CB-MePyCyclen to hold in an undistorted octahedral geometry (Nax-M-Nax = 153.69° and Neq-M-Neq = 81.26°) while the larger PyMeEBC accommodates Zn2+ with much less distortion of the octahedral geometry (Nax-M-Nax = 168.66° and Neq-M-Neq = 81.84°).
Finally, the effect of the pyridine pendant arm on the coordination geometry can be analyzed by comparing the coordination geometries of the CB-MePyCyclen complexes with the pendant-arm-lacking Me2BCyclen complexes. In each ion’s case, addition of the pendant arm allows the metal ion to be better engulfed by the bridged macrocycle with less geometric distortion than for Me2Bcyclen, at least judged by the Nax-M-Nax angles. For example Cu(Me2Bcyclen)Cl+ has Nax-M-Nax = 164.06°, while Cu(CB-MePyCyclen)2+ has Nax-M-Nax = 170.6°. Interestingly, the trend towards larger bond angles for CB-MePyCyclen complexes does not always extend to the Neq-M-Neq angles, which are smaller for Cu2+ and Ni2+ (see Table 1), but larger for Zn2+.
Structural comparisons with CB-Py2Cyclen [30] and PyCyclen [21] [22] (see Figure 1) are also desirable. Unfortunately, although Co3+, Cu2+, and Zn2+ complexes with CB-Py2Cyclen, the most direct analogue including an ethylene cross-bridge, have been synthesized and characterized, no X-ray crystal structures have been published. However, [Ni(PyCyclen)(CH3CN)]2+ [22] and [Cu(PyCyclen)]2+ [21] have published crystal structures.
Comparison between octahedral Ni(4)Cl+ and octahedral [Ni(PyCyclen)(CH3CN)]2+ [22] begins by recognition that the pyridine donor of Ni(4)Cl+ is forced by the short cross-bridge to be located on an axially positioned nitrogen of the coordinated folded macrocycle, positioning the pyridine donor on the equator of the distorted octahedral complex with the pyridine ring perpendicular to the equator. However, in [Ni(PyCyclen)(CH3CN)]2+, although the unbridged cyclen is similarly folded, the pyridyl donor is attached to an equatorial positioned cyclen nitrogen with the pyridine ring parallel to the equator. For comparison to Table 1, the Nax-Ni-Nax and Neq-Ni-Neq bond angles for in [Ni(PyCyclen)(CH3CN)]2+ are 156.00° and 104.01° respectively. These values are 160.8° and 84.46° for Ni(4)Cl+. Clearly, the most significant difference is the ~20 degree larger Neq-Ni-Neq bond angle for the unbridged PyCyclen complex, demonstrating the severe effect of the short ethylene cross-bridge on complex coordination geometry. Very little difference in Ni-N bond lengths are noted, with the Ni-Npy bond lengths nearly identical at 2.085 and 2.083 Å, respectively.
Comparison of Cu(4)2+ and Cu(PyCyclen)2+ can begin with their Addison Parameters [38] which are t = 0.09 for both compounds, indicating similar square pyramidal coordination geometries. However, the axial position is occupied by a macrocycle nitrogen in cross-bridged Cu(4)2+, while the axial position is occupied by the pyridine nitrogen in unbridged Cu(PyCyclen)2+. Due to the Jahn-Teller distortion of d9 Cu2+, the axial bond is typically lengthened and weakened. Here, that results in a long Cu-Npy bond of 2.164 Å for Cu(PyCyclen)2+, while the equatorial pyridine of Cu(4)2+ has a Cu-Npy bond length of 2.012 Å. The short, strong, equatorial coordination of the non-macrocycle donor has been credited with the more effective CXCR4 antagonism and longer residence time of Cu2+ cross-bridged tetraazamacrocyle CXCR4 antagonists. [43] The Neq-Ni-Neq bond angle for Cu(4)2+ is 83.0° and is this small because of the short ethylene cross-bridge. Cu(PyCyclen)2+ does not have a completely analogous bond angle, because the macrocycle occupies the base of the square pyramid, rather than having on macrocycle nitrogen at the axial position. The smallest nonadjacent Ncyclen-Cu-Ncyclen bond angle in Cu(PyCyclen)2+ is 146.94°, reflecting the very large structural difference with Cu(4)2+.
2.3. Electronic structure
The magnetic moments and UV-vis spectra were recorded on all paramagnetic ions in an attempt to glean structural information about the complexes, see Table 4. The elemental analysis of Ni(3)2+ shows no bound chloride, making it at most 5-coordinate. However, the electronic spectrum appears to indicate a square-planar 4-coordinate structure. Square planar Ni2+ usually has only one absorption near 460 nm, [44] in this case, it is at 441 nm with an extinction coefficient of 234 M−1cm−1, indicating that the pyridine pendant arm does not bind to the metal center in this complex. The magnetic moment of this complex is 1.13 BM, which is well below the expected value for a high spin Ni2+ ion of 2.80–3.50. [45] This low value indicates that there may be a mixture of high and low spin Ni2+ complexes in the solid state, which is in agreement with the low-spin square planar electronic spectrum in an acetonitrile solution.
Table 4.
Electronic structural data for transition metal complexes
| Electronic Spectra | Magnetic moment | |||
|---|---|---|---|---|
|
| ||||
| Compound | λmax (nm) | ε (M−1cm−1) | Literature range (BM) | Experimental value (BM) |
|
| ||||
| Co(3)Cl+ | 219 | 35100 | 4.30–5.20 | 4.10 |
| 521 | 100.3 | |||
| 263(sh) | 5760 | |||
|
| ||||
| Ni(3)2+ | 240 | 4820 | 2.80–3.50 | 1.13 |
| 441 | 230 | |||
|
| ||||
| Cu(3)2+ | 612 | 210 | 1.70–2.20 | 1.92 |
| 289 | 6140 | |||
| 263.5 | 8520 | |||
| 216 | 34460 | |||
|
| ||||
| Co(4)Cl2+ | 227 | 44500 | 0 | 0 |
| 499 | 280 | |||
| 245(sh) | 2310 | |||
|
| ||||
| Ni(4)Cl+ | 847 | 31 | 2.80–3.50 | 3.00 |
| 538 | 26 | |||
| 233 | 440 | |||
| 332(sh) | 150 | |||
|
| ||||
| Cu(4)2+ | 216 | 33600 | 1.70–2.20 | 1.91 |
| 262 | 5700 | |||
| 295 | 3220 | |||
| 559 | 390 | |||
The elemental analysis of the cross bridged Ni(4)Cl+ points to a bound chloride ligand, while its electronic spectrum supports an octahedral Ni2+ ion. Such an ion generally exhibits three weak absorptions from 300–1100 nm. [44] In this case, these absorptions are at 332, 538, and 847 nm. The electronic spectra of octahedral Ni2+ complexes can be used to determine the ligand field strength of the ligands, [46] equivalent to the energy of the lowest energy absorption. In this case Δo = 11,800 cm−1. This can be compared to the bridged cyclen ligand having two methyl groups and no pyridine donor, [Ni(Me2BCyclen)Cl2] which has Δo = 9,840 cm−1. [42] The pyridine donor and 5-coordinate ligand in the present case greatly increases the ligand field strength. Pyridine is a strong field ligand in the spectrochemical series [45] as its empty π orbitals allow for back bonding, while Cl− is a weak field ligand because its filled p-orbitals reduce Δo. This trend is clearly reflected in the Δo values observed. Also, comparing with ligand field strength of the very similar [Ni(PyMeEBC)Cl2] complex (Δo = 11,223 cm−1) [12] shows a slight increase for Ni(4)Cl+. Additionally, the magnetic moment for Ni(4)Cl+ (3.00 BM) is consistent with high-spin Ni2+, which generally ranges from 2.80–3.50 BM. [45]The elemental analysis, electronic spectrum, and magnetic moment all point to an octahedral, high spin, Ni2+ complex in this case, in contrast to the low spin, square planar complex of Ni(3)2+. This result is expected, as the short 2-carbon cross bridge allows only for a folded, cis configuration of the cyclen ring, which leads only to the distorted octahedral structure and a high spin Ni2+ cation. The electronic data for Ni(4)Cl+ is in general very similar to the slightly larger cyclam analogue. [12]
The side-bridged complex Co(3)Cl+ has an elemental analysis matching a Co2+ ion with one bound chloride and a PF6− anion. Its electronic spectra has a weak absorbance at 521 nm (ε = 100 M−1cm−1). This is consistent with high spin cobalt(II), which generally has a single band centered between 500–600 nm. [44] This observation is in agreement with the magnetic moment of the compound, 4.10 BM, which is also consistent with high spin cobalt(II). [45]
However, the cross-bridged complex Co(4)Cl2+ has an elemental analysis that indicates an oxidized Co3+, having one bound chloride ligand and two PF6− anions. This behavior was not noted with the cross-bridged cyclam analogue. [12] This compound also gave a diamagnetic magnetic moment, consistent with low spin d6 Co3+ in an octahedral environment. Its electronic spectrum is similar to Co(3)Cl+, yet the absorbances are stronger and there are two peaks plus a shoulder: 499 (281), 387 (281), and 252 nm (2307 M−1cm−1). These bands are consistent with the di-methyl ligand complex [47] [Co(Me2EBCyclen)Cl2]+: 577 nm (250), 415(210), and 346(950). This cobalt(III) complex had three bands with similar intensities and included a shoulder for the middle absorbance. The major difference between the pyridine-armed and unarmed complexes is the increase in energy of each of the bands for the pyridine armed complex. This can be quantified by calculating Δo for the pyridine-armed complex, which is accomplished by adding the Racah parameter, 3800cm−1 to the energy of the lowest energy band for an octahedral Co3+ complex. [48] In this case Δo = 23,840 cm−1. This compares to the Δo = 21,130 cm−1 value for the [Co(Me2EBCyclen)Cl2]+ complex. As in the case with nickel(II), a clear increase in ligand field strength is observed, which makes sense as we have added a strong field pyridine ligand in place of a weak field chloride anion.
The electronic spectra of the Cu2+ complexes in acetonitrile demonstrate the expected ligand field transitions [45] for d9 Cu2+. A number of high-intensity bands are present in the UV, which are likely ligand to metal charge transfer bands. Each complex has one d-d absorption: at 559 nm (390 M−1cm−1) for Cu(3)2+ and at 612 nm (210 M−1cm−1) for Cu(4)2+. Cu2+ electronic spectra generally exhibit one such d-d band, which has an energy equivalent to Δo. Thus, Δo = 17,900 cm−1 for Cu(3)2+ and Δo =16,300 cm−1 for Cu(4)2+. The oxidation state is confirmed by the magnetic moments, Cu(3)2+ μ = 1.91 BM and Cu(4)2+ μ = 1.92, both consistent with the usual Cu2+ range of 1.70–2.20 BM, [31] and similar to that of the cross-bridged cyclam analogue. [12]
2.4. Electrochemical Studies
After our encouraging results using transition metal complexes of SB-PyCyclam [10] and PyMeEBC [12] in oxidation catalysis, we wished to determine if a reduction in macrocyclic size would result in beneficial shifts in the red-ox potentials of their transition metal complexes and also determine the degree of influence achieved by addition of the pyridyl arm compared to the analogous non-pyridyl complexes [M(Me2BCyclen)Cl2]. [31] [42] [42] The electrochemical properties were assessed via cyclic voltammetry in acetonitrile; all redox potentials given are vs. SHE.
[Ni(Me2BCyclen)Cl2] in acetonitrile has an irreversible reduction to Ni+ with Ered = −2.036 V, a reversible Ni2+/3+ couple at E1/2 = +0.863 V (ΔE = 68 mV), and an additional unassigned irreversible oxidation at Eox = +1.450 V that is most likely due to oxidation of a bound chloride ligand. [42] Ni(3)2+ has only an irreversible reduction at Ered = −1.287 V, likely from Ni2+ to Ni+, and an irreversible oxidation at Eox = +1.375 V, likely from Ni2+ to Ni3+. In comparison to [Ni(Me2EBCyclen)Cl2], the reduction to Ni+ is substantially (749 mV) easier for this side-bridged complex with a pyridine arm. Recall that this complex appears to be low spin, d8, square planar Ni2+ in its electronic structure, with no bound chloride. Reduction would result in a d9 Ni+ ion, which has a preference for 5-coordinate structures. [48] Perhaps the easier reduction is due to lack of a negatively charged chloro ligand combined with the ready presence of a fifth donor in the form of the pyridine pendant arm. The irreversibility of this reduction would be explained by the change in structure if the pyridine donor does indeed bind upon reduction to Ni+. The lack of a chloro donor would also explain the missing second oxidation, which was assigned to oxidation of a bound chloro ligand in [Ni(Me2EBCyclen)Cl2]. Finally, the oxidation that is present at +1.375 V (likely Ni2+ to Ni3+) is at a much higher potential compared to this reversible oxidation in [Ni(Me2EBCyclen)Cl2], which is at +0.863 V. Again, lack of negatively charged chloro ligands may explain this difference, as the Ni3+ ion would not be as stabilized in Ni(3)2+. The lack of reversibility of this oxidation may also be explained by the Ni3+ ion binding to the pyridine pendant arm as it would require more electron donation to be stabilized.
Ni(4)Cl+ is quite different from either Ni(3)2+ or [Ni(Me2EBCyclen)Cl2. This complex gave no reduction wave in acetonitrile out to approximately −2.0 V which is very different from the reduction of the analogous Ni(PyMeEBC)Cl+ (−1.026 V). Ni(4)Cl+ does demonstrate a reversible oxidation at E1/2 = +1.034 V (ΔE = 76 mV) and another quasi-reversible oxidation at E1/2 = +1.313 V (ΔE = 87 mV). The first oxidation is likely from Ni2+ to Ni3+. Recall that the electronic structure of this complex, from its magnetic moment and electronic spectrum in acetonitrile appears to be octahedral Ni2+, which along with its elemental analysis implies binding of the pyridine pendant arm and one chloro ligand. This structure is apparently maintained upon oxidation, with a very reversible reduction wave present. This is similar to the [Ni(Me2BCyclen)Cl2] complex, where this Ni2+/3+ couples occurs at +0.863 V. The increase in oxidation potential for the pendant arm complex could be explained by the presence of only one negatively charged chloro ligand, which makes it more challenging to reach Ni3+ than in [Ni(Me2EBCyclen)Cl2], which has two negatively charged chloro ligands and would make it easier to oxidize. When compared with the analogous Ni(PyMeEBC)Cl+ complex, we see a similar (+1.290 V) oxidation potential to Ni3+ which is slightly (0.171 V) stabilized compared with Ni(4)Cl+. The identity of the second quasi-reversible wave, which is less intense than the first one, could be assigned, as in [Ni(Me2BCyclen)Cl2] to oxidation of a chloro ligand. Although the quasi-reversible nature of this oxidation would not be expected for oxidation of a chloro ligand, which would not likely stay bound upon oxidation. An alternative explanation could be the presence of a second form of the complex in which the pyridine arm is not bound to the nickel ion. This form would have less electron density to stabilize Ni3+ and would therefore likely require a larger voltage to reach this oxidation state.
[Co(Me2BCyclen)Cl2] in acetonitrile has an irreversible reduction to Co+ with Ered = −2.202 V, a quasi-reversible Co2+/3+ couple at E1/2 = −0.157 V (ΔE = 288 mV), and an additional unassigned irreversible oxidation at Eox = +0.983 V that is most likely due to oxidation of a bound chloride ligand. Co(3)Cl+, has a voltammogram much like [Co(Me2EBCyclen)Cl2]. Similarly, there is an irreversible reduction to Co+ with Ered = −1.884 V, compared to −2.202 volts for [Co(Me2BCyclen)Cl2]. Since there is only one negatively charged chloride ligand in Co(3)Cl+, it makes sense that it is easier to reduce this complex to Co+ than for [Co(Me2EBCyclen)Cl2]. The irreversible nature of both complexes upon reduction is probably due to loss of a ligand, either chloride or the pendant arm pyridine. The oxidation behavior of both complexes differs more than the reduction. [Co(Me2EBCyclen)Cl2] has a reversible oxidation to Co3+ at a mild potential of −0.157 V, whereas Co(3)Cl+ doesn’t oxidize to Co3+ until a potential of +1.121 V is reached. Again, having only one bound chloro ligand may make it less favorable to oxidize to Co3+ in this case. [Co(Me2BCyclen)Cl2] shows reversible behavior for this oxidation, while Co(3)Cl+ does not. Perhaps the pendant arm pyridine is dissociated or modified causing the irreversible behavior, where this pathway is not available to [Co(Me2BCyclen)Cl2]. Another similarity is that [Co(Me2BCyclen)Cl2] has a second oxidation, probably of bound chloro ligand at +0.983 V while Co(3)Cl+ also has a second irreversible oxidation at +1.411 V.
The cross-bridged pyridine pendant arm complex Co(4)Cl2+ differs significantly from [Co(Me2BCyclen)Cl2] or Co(PyMeEBC)Cl+, as might be expected since it appears to be a diamagnetic, octahedral Co3+ complex, where the others are paramagnetic Co2+ complexes. Starting as Co3+, there is no observed oxidation for this complex, even of the irreversible variety of a bound chloro ligand. Reduction to Co2+, however is observed, and is quasi-reversible: E1/2 = −0.127 V (ΔE = 107 mV). Apparently both 3+ and 2+ oxidation states are stable with the bound chloride and, presumably, the bound pyridine pendant arm. Unlike the other two complexes, no further reduction to Co+ is observed. Perhaps the bound pyridine and chloro ligands particularly stabilize Co2+ in this case, or provide too much electron density to allow for reduction to Co+ in the observed region (out to ~ −2.0 V). The difference in the Co2+/3+ reversible couple for Co(4)Cl2+ and Co(PyMeEBC)Cl+ is instructive. The smaller cyclen-based ligand 4 clearly selects for the smaller Co3+ ion with an easily obtained oxidation to Co3+ at −0.127 V. This is likely the reason this complex oxidized in air to Co3+ during workup. In contrast, Co(PyMeEBC)Cl+ is much more difficult to oxidized (+0.657 V), likely because the larger cyclam-based ligand prefers the larger Co2+ ion.
A copper complex serving as the basis for comparison is [Cu(Me2BCyclen)(CH3CN)2](PF6)2. [31] Interestingly, this complex in acetonitrile does not bind chloride, but prefers acetonitrile, as evidenced by a crystal structure grown from acetonitrile and molar conductance data. [31] The explanation for this behavior is the strained, distorted octahedral structure of this complex due to the small cavity of the bridged cyclen ligand. Cu(3)2+, like [Cu(Me2BCyclen)(CH3CN)2](PF6)2 has no bound chloride, according to its elemental analysis. This is to be expected if the pyridine pendant arm is bound, as Cu2+ prefers 5-coordinate geometries. [48] Cu(3)2+ has an irreversible reduction to Cu+ at Ered = −0.406 V, which occurs about 250 mV less negative than the same reduction in [Cu(Me2EBCyclen)(CH3CN)2](PF6)2. Perhaps the bound pendant arm helps to stabilize the large Cu+ ion, making reduction easier. Cu(3)2+ also has an irreversible oxidation at Eox ~ +1.75 V (solvent oxidation prevents exact measurement of the peak), this oxidation is not seen in [Cu(Me2BCyclen)(CH3CN)2](PF6)2. The explanation for the lack of observation of Cu3+ in the Me2BCyclen complex is that strong bonds are required to stabilize the reactive Cu3+ ion, which are not possible because of the small cavity size and distorted geometry of the complex. [31] Perhaps the pyridine pendant arm provides enough additional bonding strength to allow the formation of Cu3+ in this case.
Similar behavior is seen in Cu(4)2+. One difference is that the reduction to Cu+ is now quasi-reversible with E1/2 = −0.341 V (ΔE = 86 mV). The 5-coordinate ligand, including the pyridine pendant arm can obviously stabilize the Cu+ oxidation state, which is reached at a milder potential than either of the other two compared complexes. Finally, an oxidation to Cu3+ is also observed for Cu(4)2+ with Eox ~ 1.86 V (again solvent oxidation prevents exact measurement of the peak). This is about 100mV more positive than for Cu(3)2+. Due to their expected structural similarities as distorted 5-coordinate complexes, there is very little difference in reduction potentials between all four analogous compounds (Cu(3)2+, Cu(4)2+, Cu(SB-PyCyclam)2+ and Cu(PyMeEBC)2+ being in the range −0.3 to −0.65 V.
3. Conclusion
Two novel topologically constrained cyclen chelators bearing pyridyl arms were described which were synthesized in a simple and scalable fashion. Access to unsymmetrically substituted tetraazamacrocycles is valuable and was achieved here by controlling the reactivity of the pyridyl alkyl halide by selection of an appropriate solvent. Transition metal complexes were formed with both ligands using Zn2+, Ni2+, Co2+ and Cu2+ which were characterized using UV-vis spectroscopy and magnetic moments to gain insights into their electronic structure along with X-ray crystal structure determination for selected cross-bridge complexes. The magnetic and electronic spectral studies revealed both five- and six-coordinate structures controlled by selection of the appropriate side- or cross-bridged ligand. The X-ray crystal structures were limited to the cross-bridged ligand complexes only, yet revealed interesting trends in geometric parameters based on metal ionic radius, macrocyclic ring size, and the effect of the additional pendant pyridine donor, especially in comparison to other known structural analogues. Electrochemical studies were carried out using cyclic voltammetry to determine oxidation and reduction potentials and give indications for their future potential as oxidation catalysis. Our current model relies on forming simple to work with divalent metal complexes (such as those used in this study) as a more rapid pre-screening of ligands which could be useful for oxidation catalysis. Indications from this study, particularly with respect to reversible access to multiple oxidation states stabilized by ligands 3 and 4, encourage us to subsequently form the Mn2+ and Fe2+ complexes which have shown oxidation catalytic promise previously and assess their potential for this application.
4. Experimental
Materials and methods
1,4,7,10-tetraazatetradodecane (Cyclen), 98%, was purchased from CheMatech. Glyoxal (40% wt in water), methyl iodide (99%), and sodium borohydride (98%) were purchased from Sigma Aldrich. Elemental analyses were performed by Quantitative Technologies Inc. Electrospray Mass spectra were collected on a Shimadzu LCMS 2020 Electrospray Mass Spectrometer. NMR spectra were obtained on a Varian Bruker AVANCE II 300 MHz NMR Spectrometer instrument. Electronic spectra were recorded using a Shimadzu UV-240 UV-Vis Spectrometer. Conductance measurements were obtained with an Oakton CON510 Bench Conductivity/TDS Meter on 0.001 M solutions at room temperature. Magnetic moments were obtained on finely ground solid samples at ambient temperatures using a Johnson Matthey MSB Auto magnetic susceptibility balance. Electrochemical experiments were performed on a BAS100B Electrochemical Analyzer. A button Pt electrode was used as the working electrode with a Pt-wire counter electrode and a Ag-wire pseudo-reference electrode. Scans were taken at 200 mV/s. Acetonitrile solutions of the complexes (1 mM) with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte were used. The measured potentials were referenced to SHE using ferrocene (+0.400 V versus SHE) as an internal standard. All electrochemical measurements were carried out under N2. The Co2+ complexes were analyzed using tetrabutylammonium chloride (0.1 M) as supporting electrolytes due to the simplification of the voltammograms in the latter. Partial replacement of chloro ligands by solvent in the former complicated the voltammograms. Synthesis of decahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene was carried out via literature methods. [11]
Crystal Structure Analysis
Single crystal X-ray diffraction data were collected in series of ω-scans using a Stoe IPSD2 image plate diffractometer utilizing monochromated Mo radiation (λ = 0.71073 Å). Standard procedures were employed for the integration and processing of the data using X-RED. [49] Samples were coated in a thin film of perfluoropolyether oil and mounted at the tip of a glass fibre located on a goniometer. Data were collected from crystals held at 150 K in an Oxford Cryosystems nitrogen gas cryostream.
Crystal structures were solved using routine automatic direct methods implemented within SHELXS-97. [50] Completion of structures was achieved by performing least squares refinement against all unique F2 values using SHELXL-97. [50] All non-H atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed using a riding model. Where the location of hydrogen atoms was obvious from difference Fourier maps, C-H bond lengths were refined subject to chemically sensible restraints.
Synthesis of 6a-(pyridin-2-ylmethyl)decahydro-5H-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylen-6a-ium chloride (1)
Sodium bicarbonate (8.7 g, 0.103 ml) was added to 2-picolyl chloride hydrochloride (8.5 g, 0.052 mol) in dichloromethane (500 mL) and stirred at room temperature for 1 h. The tan, solid precipitate was filtered and discarded. Decahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene (5.2 g, 0.027 mol) was added to the light yellow liquid filtrate along with potassium iodide (0.854 g, 0.00514 mol). The reaction was heated at reflux under nitrogen for 24 hours. The solution was allowed to cool to room temperature and filtered. The filtrate was also precipitated with ether (1.5 L) and more product was collected via filtration. Yield 6.818 g (79.43%) of 1. 1H-NMR (300 MHz, D2O): δ 2.62 (m, 2H), 2.89 (m, 3H), 3.03 (m, 1H), 3.23 (m, 2H), 3.39 (m, 3H), 3.57 (m, 1H), 3.72 (m, 2H), 3.85 (m, 2H), 4.17 (m, 1H), 4.45 (td, 1H), 4.87 (m, 1H), 5.06 (m, 1H), 7.68 (m, 1H), 7.81 (d, 1H), 8.09 (m, 1H), 8.76 (d, 1H). 13C{1H} NMR (75.6 MHz, D2O): δ 43.91, 47.64, 47.74, 48.20, 48.33, 51.32, 58.06, 61.81, 62.08, 83.11, 125.92, 128.21, 138.98, 147.23, 150.33. MS (ESI) 286.2 (M-Cl−)+. Anal calc. for C15H24N5Cl•0.38CH2Cl2 calc: C 55.56%, H 7.05%, N 19.78%; Found: C 55.36%, H 6.95%, N 20.02%.
Synthesis of mono(2a-methyl-6a-(pyridin-2-ylmethyl)dodecahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene-2a,6a-diium) diiodide (2)
Methyl iodide (0.306 moles, 19 mL) was added to 6a-(pyridin-2-ylmethyl)decahydro-5H-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylen-6a-ium chloride (1, 6.56 g, 0.02 moles) in dry acetonitrile (250 mL) and stirred at room temperature for 3 days. After which, ether (500 mL) was added and the precipitate was filtered and washed with acetonitrile (200 mL) and ether (200 mL). Yield 9.41 g (84.70%) of 2. 1H-NMR (300 MHz, D2O): δ 2.49 (m, 1H), 2.88 (m, 1H), 3.00 (m, 2H), 3.34 (m, 8H), 3.81 (m, 7H), 4.32 (m, 1H), 4.68 (m, 1H), 4.84 (m, 1H), 5.08 (m, 1H), 7.56 (m, 1H), 7.76 (d, 1H), 8.02 (m, 1H), 8.73 (d, 1H). 13C{1H} NMR (75.6 MHz, D2O): δ 42.36, 42.74, 46.31, 46.50, 46.72, 56.43, 58.33, 60.38, 61.84, 64.62, 76.94, 77.19, 125.14, 127.33, 138.15, 148.68, 150.58. MS (ESI) 428.1 (M-I−)+. Anal calc. for C17H27N5I2 calc: C 36.77%, H 4.90%, N 12.61%; Found: C 36.79%, H 4.69%, N 12.69%.
Synthesis of 4-(pyridin-2-ylmethyl)-1,4,7,10-tetraazabicyclo[8.2.2]tetradecane (SB-PyCyclen, 3)
Sodium borohydride (1.30 g, 0.0345 mol) was added slowly to a solution of 1 (2.22 g, 0.0069 mol) in ethanol (100 mL) and the reaction was heated under reflux for 1 hour. After cooling to room temperature, 6 M hydrochloric acid was added drop wise until a pH of 1–2 was reached. The ethanol was removed via rotary evaporation. Water (50 mL) and 30% w/w potassium hydroxide solution was added until a pH of 14 was reached. The product was extracted into benzene, dried (NaSO4) and reduced under vacuum and subsequently purified via short-path distillation. Yield 1.89 g (94.49%) of 3. 1H-NMR (300 MHz, CDCl3): δ 2.42 (m, 4H), 2.59 (m, 2H), 2.72 (m, 5H), 2.90 (m, 4H), 3.11 (m, 4H), 3.23 (m, 2H), 3.83 (m, 2H), 7.15 (m, 1H), 7.56 (d, 1H), 7.67 (m, 1H), 8.51 (d, 1H). 13C{1H} NMR (75.6 MHz, CDCl3): δ 45.72, 47.38, 48.88, 48.92, 50.77, 52.53, 52.74, 52.80, 56.88, 57.72, 61.76, 121.91, 122.48, 128.42, 136.76, 148.87. MS (ESI) 290.1 (M+H+)+. Anal calc. for C16H27N5: C 66.40%, H 9.40%, N 24.20%; Found: C 66.15%, H 9.61%, N 24.20%.
Synthesis of 4-methyl-10-(pyridin-2-ylmethyl)-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane (CB-MePyCyclen, 4)
Sodium borohydride (2.43 g, 0.064 mol) was added slowly to a solution of 2 (2.35 g, 0.0042 mol) in 95% ethanol (180 mL) and the reaction was stirred for 5 days under nitrogen. 6 M hydrochloric acid was added drop wise until a pH of 1–2 was reached. The ethanol was removed via rotary evaporation. 30% w/w potassium hydroxide solution was added until a pH of 14 was reached. The product was extracted into benzene, dried (NaSO4) and reduced under vacuum. Yield 0.96 g (68.3%) of 4. 1H-NMR (300 MHz, CDCl3): δ 2.43 (m, 3H), 2.70 (m, 16H), 2.97 (m, 3H), 3.82 (s, 2H), 7.27 (td, 1H), 7.41 (d, 1H), 7.77 (td, 1H), 8.34 (d, 1H). 13C{1H} NMR (75.6 MHz, CDCl3): δ 42.82, 47.04, 51.78, 52.39, 54.38, 55.07, 59.72, 123.10, 123.94, 138.02, 148.16, 158.16. MS (ESI) 304.2 (M+H+)+. C17H29N5•0.35H2O: C 65.92%, H 9.66%, N 22.61%; Found: C 66.04%, H 9.61%, N 22.22%.
General method for synthesis of transition metal complexes (with below exceptions)
In a glove-box 3 (1 mmol, 0.289 g) or 4 (1 mmol, 0.303 g) was dissolved in 10 mL of acetonitrile. 1 mmol of zinc chloride, nickel chloride, copper chloride, and cobalt chloride were added in separate vials containing the ligand. These reactions were left to stir for 2 days at room temperature. The solutions were gravity filtered to remove any un-reacted metal salt. The reaction vials were removed from the glove-box and the acetonitrile removed on a rotary evaporator. The oily residues were dissolved in a minimum of methanol. A solution ammonium hexafluorophosphate (5 mmol, 0.815 g) in methanol was added to each reaction. The metal complex solutions were placed in the freezer to help promote precipitation. All solid precipitates were filtered on glass frits, washed with cold methanol, then ether, and dried prior to characterization. NiCl2 is not completely soluble in acetonitrile so these reactions were carried out with additional DMF (15 mL) and left to react for an additional 2 days.
The complexation of 3 with CuCl2 was stirred for only 15 minutes. Leaving the reaction longer resulted in a reduced copper metal precipitation. From this reaction, and other unpublished results, [37] side-bridged macrocycle ligand complexation reactions in acetonitrile with CuCl2 are susceptible to some kind of uncharacterized redox process whereby the complex is destroyed, and this is accompanied by the production of metallic copper. The secondary NH of the side-bridged ligand is the main difference from cross-bridged, all-tertiary ligands, where this process does not occur. The process can be limited or avoided with SB ligands by using only short reactions times. We have not investigated the specific deleterious reaction, so can only speculate on its mechanism. Since we have overcome this problem with short reaction time, we have not chosen to focus on this mechanism.
The zinc complex of 3 gave a low yield upon precipitation with NH4PF6, apparently because the complex is much more soluble in MeOH than its analogues with other metal ions. Since a sufficient amount of product was obtained for characterization and was analytically pure, we have not optimized this reaction.”
[Zn3](PF6)2
Yield: 0.025 g (4%) of a beige powder. MS (ESI) 388 (M-2PF6−+Cl−)+. Anal calc. for [Zn(C16H27N5)](PF6)2: C 29.81%, H 4.22%, N 10.86%; Found: C 30.07%, H 4.10%, N 10.80%.
[Ni3](PF6)2
Yield: 0.337 g (53%) of a rust orange powder. MS (ESI) 346 (M-2PF6−)+. Anal calc. for [Ni(C16H27N5)](PF6)2 • 0.2 (NH4PF6): C 28.66%, H 4.18%, N 10.86%; Found: C 28.43%, H 3.79%, N 10.75%.
[Co3Cl](PF6)
Yield: 0.212 g (40%) of a brick red powder. MS (ESI) 383 (M-2PF6−+Cl−)+. Anal calc. for [Co(C16H27N5)Cl]PF6•0.5H2O: C 35.74%, H 5.25%, N 13.02%; Found: C 35.56%, H 5.10%, N 12.99%.
[Cu3](PF6)2
Yield: 0.625 g (97%) of a violet powder. MS (ESI) 387 (M-2PF6−+Cl−)+; 351 (M-2PF6−)+. Anal calc. for [Cu(C16H27N5)](Cl0.1)(PF6)1.9: C 30.41%, H 4.31%, N 11.08%; Found: C 30.37%, H 4.07%, N 10.70%.
[Zn4Cl](PF6)
Yield: 0.540 g (98%) of a beige powder. MS (ESI) 402 (M-2PF6−+Cl−)+. Anal calc. for [Zn(C17H29N5)Cl]PF6: C 37.18%, H 5.32%, N 12.75%; Found: C 36.98%, H 5.20%, N 12.41%.
Ni4(PF6)2
Yield: 0.429 g (79%) of a purple powder. MS (ESI) 396 (M-2PF6−+Cl−)+. Anal calc. for [Ni(C17H29N5)Cl]PF6•0.5H2O: C 37.02%, H 5.48%, N 12.70%; Found: C 36.85%, H 5.27%, N 12.36%.
[Co4Cl](PF6)2
Yield: 0.532 g (77%) of a pink powder. MS (ESI) 396 (M-2PF6−+Cl−)+. Anal calc. for [Co(C17H29N5)Cl](PF6)2: C 29.69%, H 4.25%, N 10.18%; Found: C 29.59%, H 4.23%, N 10.04%.
[Cu4](PF6)2
Yield: 0.442 g (67%) of a blue powder. MS (ESI) 401 (M-2PF6−+Cl−)+. Anal calc. for [Cu(C17H29N5)](PF6)2: C 31.08%, H 4.45%, N 10.66%; Found: C 30.73%, H 4.33%, N 10.52%.
Supplementary Material
Figure 3.

Cyclic voltammograms of Ni(4)Cl+ and Ni(3)2+.
Figure 4.
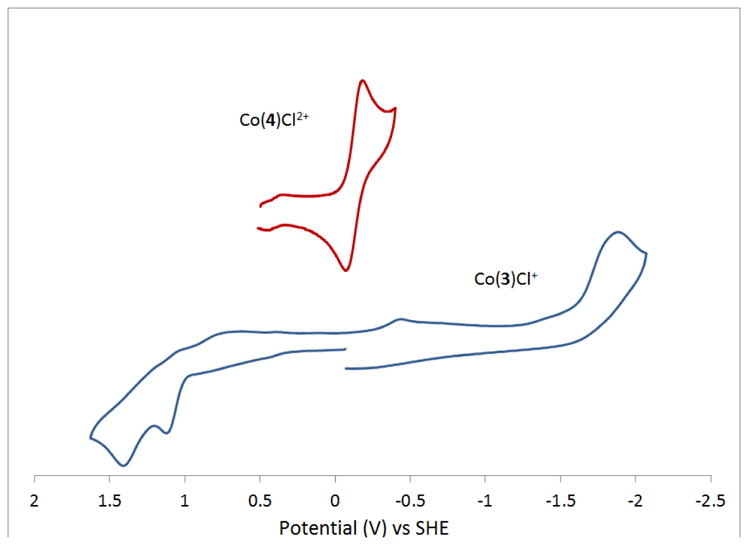
Cyclic voltammograms of Co(4)Cl2+ and Co(3)Cl2+.
Figure 5.
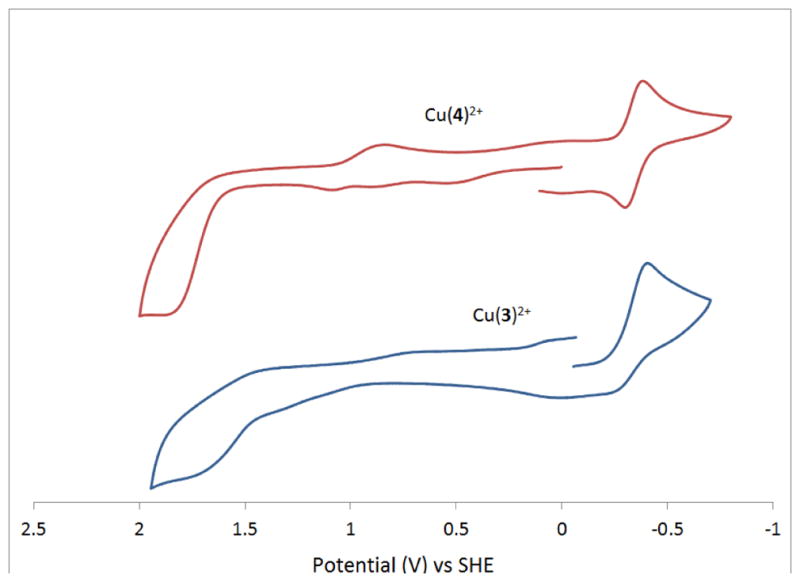
Cyclic voltammograms of Cu(4)2+ and Cu(3)2+.
Table 2.
Crystallographic data. L = PyMeEBC
| [Ni(4)Cl]PF6 sja4_12 |
[Cu(4)](PF6)2 sja26_12 |
[Zn(4)Cl]PF6 sja3_12 |
|
|---|---|---|---|
| Chemical Formula | C17 H29 Cl F6 N5 Ni P | C17 H29 Cu F12 N5 P2 | C17 H29 Cl F6 N5 P Zn |
| a = …(esd) Å | 11.6458(10) | 11.4577(9) | 11.584(2) |
| b = …(esd) Å | 11.6930(12) | 12.2495(14) | 11.729(3) |
| c = …(esd) Å | 15.6867(13) | 17.1539(14) | 15.730(3) |
| α = …(esd) degrees | 90 | 90 | 90 |
| β = …(esd) degrees | 97.680(7) | 95.837(6) | 96.95(2) |
| γ = …(esd) degrees | 90 | 90 | 90 |
| V = … Å3 | 2117.0(3) | 2395.1(4) | 2121.6(9) |
| Z = | 4 | 4 | 4 |
| Formula Weight | 542.58 | 656.93 | 549.24 |
| Space Group | P21/c | P21/c | P21/c |
| T = … K | 150(2) | 150(2) | 150(2) |
| λ = … Å | 0.71073 | 0.71073 | 0.71073 |
| Dcalcd = … g cm−3 | 1.702 | 1.822 | 1.720 |
| μ = … mm−1 | 1.186 | 1.157 | 1.427 |
| R1(Fo2) = | 0.1144 | 0.1125 | 0.0598 |
| wR2 (Fo2) = | 0.1980 | 0.1389 | 0.0660 |
Table 3.
Selected Bond Lengths [Å] and angles [°].
| [Ni(4)Cl]PF6 | [Cu(4)](PF6)2 | [Zn(4)Cl]PF6 | |||
|---|---|---|---|---|---|
|
| |||||
| N(1)-Ni(1) | 2.117(4) | N(1)-Cu(1) | 1.998(6) | N(1)-Zn(1) | 2.255(3) |
|
|
|
|
|||
| N(2)-Ni(1) | 2.057(4) | N(2)-Cu(1) | 2.042(5) | N(2)-Zn(1) | 2.185(2) |
|
|
|
|
|||
| N(3)-Ni(1) | 2.153(5) | N(3)-Cu(1) | 2.014(6) | N(3)-Zn(1) | 2.202(3) |
|
|
|
|
|||
| N(4)-Ni(1) | 2.077(5) | N(4)-Cu(1) | 2.165(7) | N(4)-Zn(1) | 2.142(3) |
|
|
|
|
|||
| N(5)-Ni(1) | 2.085(5) | N(5)-Cu(1) | 2.012(5) | N(5)-Zn(1) | 2.118(3) |
|
|
|
|
|||
| Cl(1)-Ni(1) | 2.4302(16) | N(1)-Cu(1)-N(5) | 83.9(2) | Cl(1)-Zn(1) | 2.3870(8) |
|
|
|
|
|||
| N(2)-Ni(1)-N(4) | 84.4(2) | N(1)-Cu(1)-N(3) | 170.6(2) | N(5)-Zn(1)-N(4) | 156.13(11) |
|
|
|
|
|||
| N(2)-Ni(1)-N(5) | 162.85(18) | N(5)-Cu(1)-N(3) | 105.5(2) | N(5)-Zn(1)-N(2) | 94.28(11) |
|
|
|
|
|||
| N(4)-Ni(1)-N(5) | 94.1(2) | N(1)-Cu(1)-N(2) | 84.4(2) | N(4)-Zn(1)-N(2) | 81.26(11) |
|
|
|
|
|||
| N(2)-Ni(1)-N(1) | 81.32(16) | N(5)-Cu(1)-N(2) | 164.9(3) | N(5)-Zn(1)-N(3) | 118.85(10) |
|
|
|
|
|||
| N(4)-Ni(1)-N(1) | 84.46(19) | N(3)-Cu(1)-N(2) | 86.5(2) | N(4)-Zn(1)-N(3) | 83.49(11) |
|
|
|
|
|||
| N(5)-Ni(1)-N(1) | 81.54(17) | N(1)-Cu(1)-N(4) | 88.2(2) | N(2)-Zn(1)-N(3) | 78.88(11) |
|
|
|
|
|||
| N(2)-Ni(1)-N(3) | 84.06(18) | N(5)-Cu(1)-N(4) | 106.0(2) | N(5)-Zn(1)-N(1) | 78.58(11) |
|
|
|
|
|||
| N(4)-Ni(1)-N(3) | 81.8(2) | N(3)-Cu(1)-N(4) | 88.5(2) | N(4)-Zn(1)-N(1) | 77.55(9) |
|
|
|
|
|||
| N(5)-Ni(1)-N(3) | 112.67(18) | N(2)-Cu(1)-N(4) | 83.0(2) | N(2)-Zn(1)-N(1) | 80.34(10) |
|
|
|
|
|||
| N(1)-Ni(1)-N(3) | 160.8(2) | N(3)-Zn(1)-N(1) | 153.69(10) | ||
|
|
|
|
|||
| N(2)-Ni(1)-Cl(1) | 96.69(15) | N(5)-Zn(1)-Cl(1) | 87.75(8) | ||
|
|
|
|
|||
| N(4)-Ni(1)-Cl(1) | 176.36(13) | N(4)-Zn(1)-Cl(1) | 98.00(8) | ||
|
|
|
|
|||
| N(5)-Ni(1)-Cl(1) | 85.95(15) | N(2)-Zn(1)-Cl(1) | 176.51(9) | ||
|
|
|
|
|||
| N(1)-Ni(1)-Cl(1) | 99.13(14) | N(3)-Zn(1)-Cl(1) | 97.66(8) | ||
|
|
|
|
|||
| N(3)-Ni(1)-Cl(1) | 94.86(17) | N(1)-Zn(1)-Cl(1) | 102.86(8) | ||
|
|
|
|
|||
Table 5.
Redox potentials (vs SHE) with peak separations for complexes of 3, 4, Me2BCyclen, SB-PyCyclen, and PyMeEBC.
| Complex | Redox Process | Potential (V) | Peak Separation (mV) | Ref |
|---|---|---|---|---|
| Ni(Me2Bcyclen)Cl2 | Ni2+ → Ni+ | Ered = −2.036 | --- | [42] |
| Ni2+ / Ni3+ | E1/2 = +0.863 | 68 | ||
| ???? | Eox = +1.450 | --- | ||
| Ni(3)2+ | Ni2+ → Ni+ | Ered = −1.287 | --- | |
| Ni2+ → Ni3+ | Eox = +1.375 | --- | ||
| Ni(4)Cl+ | Ni2+ / Ni3+ | E1/2 = +1.034 | 76 | |
| ???? | E1/2 = +1.313 | 87 | ||
| Ni(PyMeEBC)Cl+ | Ni2+ → Ni+ | Ered = −1.026 | --- | [12] |
| Ni2+ → Ni3+ | Eox = +1.290 | --- | ||
| Co(Me2BCyclen)Cl2 | Co2+ → Co+ | Ered = −2.202 | --- | [47] |
| Co2+ / Co3+ | E1/2 = −0.157 | 288 | ||
| ???? | Eox = +0.983 | --- | ||
| Co(3)Cl+ | Co2+ → Co+ | Ered = −1.884 | --- | |
| Co2+ → Co3+ | Eox = +1.121 | --- | ||
| ???? | Eox = +1.411 | --- | ||
| Co(4)Cl2+ | Co3+ → Co2+ | E1/2 = −0.127 | 107 | |
| Co(PyMeEBC)Cl+ | Co2+ / Co+ | E1/2 = −1.386 | 143 | [12] |
| Co2+ / Co3+ | E1/2 = +0.657 | 154 | ||
| Cu(Me2BCyclen)2+ | Cu2+ → Cu+ | Ered = −0.651 | --- | [31] |
| Cu(3)2+ | Cu2+ → Cu+ | Ered = −0.406 | --- | |
| Cu2+ → Cu3+ | Eox ~ +1.75 | --- | ||
| Cu(4)2+ | Cu2+ / Cu+ | E1/2 = −0.341 | 86 | |
| Cu2+ → Cu3+ | Eox ~ +1.86 | --- | ||
| Cu(SB-PyCyclam)2+ | Cu2+ / Cu+ | E1/2 = −0.586 | 77 | [10] |
| Cu(PyMeEBC)2+ | Cu2+ / Cu+ | E1/2 = −0.402 | 80 | [12] |
Acknowledgments
TJH acknowledges Southwestern Oklahoma State University for internal funding through a Proposal Development Award. TJH acknowledges the Donors of the American Chemical Society Petroleum Research Fund; Health Research award for project number HR13-157, from the Oklahoma Center for the Advancement of Science and Technology; and Grant Number P20RR016478 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) for partial support of this research. TJH also acknowledges the Henry Dreyfus Teacher-Scholar Awards Program for support of this work.
Appendix A: Supplementary data
CCDC 1428920, 1428921, and 1428922 contain the supplementary crystallographic data for [Zn(4)Cl]PF6, [Ni(4)Cl]PF6, and [Cu(4)](PF6)2, respectively. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Busch DH. Chem Rev. 1993;93:847–860. [Google Scholar]
- 2.Silversides JD, Burke BP, Archibald SJ. C R Chemie. 2013;16:524–530. [Google Scholar]
- 3.Silversides JD, Allan CC, Archibald SJ. Dalton Trans. 2007:971. doi: 10.1039/b615329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL, Anderson CJ. J Med Chem. 2004;47:1465–1474. doi: 10.1021/jm030383m. [DOI] [PubMed] [Google Scholar]
- 5.Sun X, Wuest M, Weisman GR, Wong EH, Reed DP, Boswell CA, Motekaitis R, Martell AE, Welch MJ, Anderson CJ. J Med Chem. 2002;45 doi: 10.1021/jm0103817. [DOI] [PubMed] [Google Scholar]
- 6.Smith R, Huskens D, Daelemans D, Mewis RE, Garcia CD, Cain AN, Carder Freeman TN, Pannecouque C, De Clercq E, Schols D, Hubin TJ, Archibald SJ. Dalton Trans. 2012;41:11369–11377. doi: 10.1039/c2dt31137b. [DOI] [PubMed] [Google Scholar]
- 7.Khan A, Silversides JD, Madden L, Greenman J, Archibald SJ. Chem Commun. 2007:416. doi: 10.1039/b614557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubin TJ, Amoyaw PN-A, Roewe KD, Simpson NC, Maples RD, Carder Freeman TN, Cain AN, Le JG, Archibald SJ, Khan SI, Tekwani BL, Khan MOF. Bioorg & Med Chem. 2014;22:3239–3244. doi: 10.1016/j.bmc.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudraraju AV, Amoyaw PNA, Hubin TJ, Khan MOF. Pharmazie. 2014;69:655–662. [PubMed] [Google Scholar]
- 10.Shircliff AD, Wilson kR, Cannon-Smith DJ, Jones DG, Zhang Z, Chen Z, Yin G, Prior TJ, Hubin TJ. Inorg Chem Commun. 2015;59:71. doi: 10.1016/j.inoche.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matz DL, Jones DG, Roewe KD, Gorbet MJ, Zhang Z, Chen Z, Prior TJ, Archibald SJ, Yin G, Hubin TJ. Dalton Trans. 2015;44:1220–12224. doi: 10.1039/c5dt00742a. [DOI] [PubMed] [Google Scholar]
- 12.Jones DG, Wilson KR, Cannon-Smith DJ, Shircliff AD, Zhang Z, Chen Z, Prior TJ, Yin TJ, Hubin TJ. Inorg Chem. 2015;54:2221–2234. doi: 10.1021/ic502699m. [DOI] [PubMed] [Google Scholar]
- 13.Thibon A, England J, Martinho M, Young VGJ, Frisch JR, Guillot R, Girerd JJ, Munck E, Que LJ, Banse F. Angew Chem Int Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcock NW, Balakrishnan KP, Moore P. J Chem Soc Dalton Trans. 1986:1743–1745. [Google Scholar]
- 15.Asato E, Hashimoto S, Matsumoto N, Kida S. J Chem Soc Dalton Trans. 1990:1741–1746. [Google Scholar]
- 16.Vuckovic G, Asato E, Matsumoto N, Kida S. Inorg Chim Acta. 1990;171:45–52. [Google Scholar]
- 17.Royal G, Dahaoui-Gindrey V, Dahaoui S, Tabard A, Guilard R, Pullumbi P, Lecomte C. Eur J Org Chem. 1998:1971–1975. [Google Scholar]
- 18.Bucher C, Royal G, Barbe JM, Guilard R. Tetrahedron Lett. 1999;40:2315–2318. [Google Scholar]
- 19.Goeta AE, Howard JAK, Maffeo D, Puschmann H, Williams JAG, Yufit DS. J Chem Soc, Dalton Trans. 2000:1873–1880. [Google Scholar]
- 20.Batsanov AS, Goeta AE, Howard JAK, Maffeo D, Puschmann H, Williams JAG. Polyhedron. 2001;20:981–986. [Google Scholar]
- 21.El Ghachtouli S, Cadiou C, Dechamps-Olivier I, Chuburu F, Aplincourt M, Roisnel T. Eur J Inorg Chem. 2006:3472–3481. [Google Scholar]
- 22.El Ghachtouli S, Cadiou C, Dechamps-Olivier I, Chuburu F, Aplincourt M, Patinec V, Le Baccon M, Handel H, Roisnel T. New J Chem. 2006;30:392–398. [Google Scholar]
- 23.Narayanan J, Solano-Peralta A, Ugalde-Salvidar VM, Escudero R, Hopfl H, Sosa-Torres ME. Inorg Chim Acta. 2008;361:2747–2758. [Google Scholar]
- 24.Morfin JF, Tripier R, Le Baccon M, Handel H. Polyhedron. 2009;28:3691–3698. [Google Scholar]
- 25.Hubin TJ, McCormick JM, Collinson SR, Alcock NW, Busch DH. Chem Commun. 1998:1675–1676. [Google Scholar]
- 26.Hubin TJ, McCormick JM, Collinson SR, Buchalova M, Perkins CM, Alcock NW, Kahol PK, Raghunathan A, Busch DH. J Am Chem Soc. 2000;122:2512–2522. [Google Scholar]
- 27.Hubin TJ, McCormick JM, Alcock NW, Busch DH. Inorg Chem. 2001;40:435–444. doi: 10.1021/ic9912225. [DOI] [PubMed] [Google Scholar]
- 28.Collinson SR, Alcock NW, Hubin TJ, Busch DH. Journal of Coordination Chemistry. 2001:317–331. [Google Scholar]
- 29.Hubin TJ. Coord Chem Rev. 2003;241:27–46. [Google Scholar]
- 30.Bernier N, Costa J, Delgado R, Felix V, Royal G, Tripier R. Dalton Trans. 2011;40:4514–4526. doi: 10.1039/c0dt01191f. [DOI] [PubMed] [Google Scholar]
- 31.Hubin TJ, Alcock NW, Morton MD, Busch DH. Inorg Chim Acta. 2003;348:33–40. [Google Scholar]
- 32.Wong EH, Weisman GR, Hill DC, Reed DP, Rogers ME, Condon JP, Fagan MA, Calabrese JC, Lam KC, Guzei IA, Rheingold AL. J Am Chem Soc. 2000;122:10561. [Google Scholar]
- 33.Weisman GR, Wong EH, Hill DC, Rogers ME, Reed DP, Calabrese JC. Chem Commun. 1996:947–948. [Google Scholar]
- 34.Weisman GR, Rogers ME, Wong EH, Jasinski JP, Paight ES. J Am Chem Soc. 1990;112:8604–8605. [Google Scholar]
- 35.Le Baccon M, Chuburu F, Toupet L, Handel H, Soibinet M, Deschamps-Olivier I, Barbier JP, Aplincourt M. New J Chem. 2001;25:1168–1174. [Google Scholar]
- 36.Silversides JD, Smith R, Archibald SJ. Dalton Trans. 2011;40:6289–6297. doi: 10.1039/c0dt01395a. [DOI] [PubMed] [Google Scholar]
- 37.Hubin TJ. 2015 unpublished data. [Google Scholar]
- 38.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. J Chem Soc Dalton Trans. 1984:1349–1356. [Google Scholar]
- 39.Groom CR, Allen FH. Angew Chem Int Ed. 2014;53:662–671. doi: 10.1002/anie.201306438. [DOI] [PubMed] [Google Scholar]
- 40.Bosnich B, Poon CK, Tobe ML. Inorg Chem. 1965;4:1102–1108. [Google Scholar]
- 41.Shannon RD. Acta Crystallogr. 1976;A32 [Google Scholar]
- 42.Hubin TJ, Alcock NW, Clase HJ, Busch DH. Supramolec Chem. 2001;13:261–276. [Google Scholar]
- 43.Khan A, Nicholson G, Greenman J, Madden L, McRobbie G, Pannecouque C, De Clercq E, Silversides JD, Ullom R, Maples DL, Maples RD, Hubin TJ, Archibald SJ. J Am Chem Soc. 2009;131:3416–3417. doi: 10.1021/ja807921k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lever ABP. Inorganic Electronic Spectroscopy. 2. Amsterdam: Elsevier; 1984. [Google Scholar]
- 45.Huheey JE, Keiter EA, Keiter RL. Inorganic Chemistry: Principles of Structure and Reactivity. 4. New York: HarperCollins; 1993. [Google Scholar]
- 46.Drago RS. Physical Methods for Chemists. 2. Ft. Worth: Saunders College Publishing-Harcourt Brace Jovanovich; 1992. [Google Scholar]
- 47.Hubin TJ, Alcock NW, Clase HJ, Seib LL, Busch DH. Inorg Chim Acta. 2002;337:91–102. [Google Scholar]
- 48.Cotton FA, Wilkinson G. Advanced Inorganic Chemistry. 5. New York: Wiley & Sons; 1988. [Google Scholar]
- 49.X-AREA v 1.64. Darmstadt: STOE & Cie GmbH; 2012. [Google Scholar]
- 50.Sheldrick G. Acta Crystallogr Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.