

Abstract
Small-cell lung cancer (SCLC) is an aggressive lung tumor subtype with poor survival1–3. We sequenced 29 SCLC exomes, two genomes and 15 transcriptomes and found an extremely high mutation rate of 7.4±1 protein-changing mutations per million basepairs. Therefore, we conducted integrated analyses of the various data sets to identify pathogenetically relevant mutated genes. In all cases we found evidence for inactivation of TP53 and RB1 and identified recurrent mutations in histone-modifying genes, CREBBP, EP300, and MLL. Furthermore, we observed mutations in PTEN, in SLIT2, and EPHA7, as well as focal amplifications of the FGFR1 tyrosine kinase gene. Finally, we detected many of the alterations found in humans in SCLC tumors from p53/Rb1-deficient mice4. Our study implicates histone modification as a major feature of SCLC, reveals potentially therapeutically tractable genome alterations, and provides a generalizable framework for identification of biologically relevant genes in the context of high mutational background.
Keywords: small-cell lung cancer, cancer genome, integrated analysis
Small-cell lung cancer (SCLC; ~15% of all lung cancer cases) typically occurs in heavy smokers and is characterized by aggressive growth, frequent metastases and early death1,2,5. Unfortunately, no single molecularly targeted drug has yet shown any clinical activity in SCLC6. Genomic analyses have revealed genetically altered therapeutic targets in lung adenocarcinoma7–16 and in squamous-cell lung carcinoma17–19. By contrast, little is known about the molecular events causing SCLC beyond the high prevalence of mutations in TP53 and RB13. Systematic genomic analyses in SCLC are challenging because these tumors are rarely treated by surgery resulting in a lack of suitable fresh-frozen tumor specimens.
We have established a global lung cancer genome research consortium19, giving us access to approximately 6,600 surgically resected lung cancer specimens, out of which we retrieved 99 SCLC specimens. We conducted 6.0 SNP array analyses on 63 tumors, exome sequencing of 27 tumors and two cell lines, transcriptome sequencing of 15 tumors, and genome sequencing of two tumors (Supplementary Tab. 1).
We applied a novel algorithm in order to identify significant broad (Supplementary Fig. 1a) and focal copy number alterations (CNAs) (Fig. 1a, Supplementary Tab. 2) and observed almost universal deletions affecting 3p and 13q (containing RB1), frequent gains of 3q, 5p, and losses of 17p (containing TP53) (Supplementary Fig. 1a). Gains of 3q affected the region containing SOX2, recently shown to be focally amplified in squamous-cell lung cancer19,20. However, 3q gains in SCLC were less focal than those in squamous-cell lung cancer (Supplementary Fig. 1b). Focal amplifications affected MYCL1 (5/63 cases) and MYCN (4/63 cases)21,22 (Fig. 1a). A single case harbored a focal amplification of MYC. All MYC family member amplifications (16% of cases) were mutually exclusive suggesting genetic epistasis21–23. Focal amplifications affected 8p12 including FGFR1 (6% with copy number ≥3.5; Fig. 1b) and 19q12 containing CCNE124. Fluorescent in-situ hybridization analyses in 51 independent specimens validated the occurrence of FGFR1 amplifications in SCLC (n=3, 6%, Fig. 1c). We and others have recently reported focal FGFR1 amplifications in squamous-cell lung cancer; FGFR inhibitors are currently being tested in such patients17,19,25. Thus, FGFR1-amplified SCLC might benefit from FGFR inhibition. The only significant focal deletion involved FHIT26 (Fig. 1a, Supplementary Tab. 2).
Figure 1.
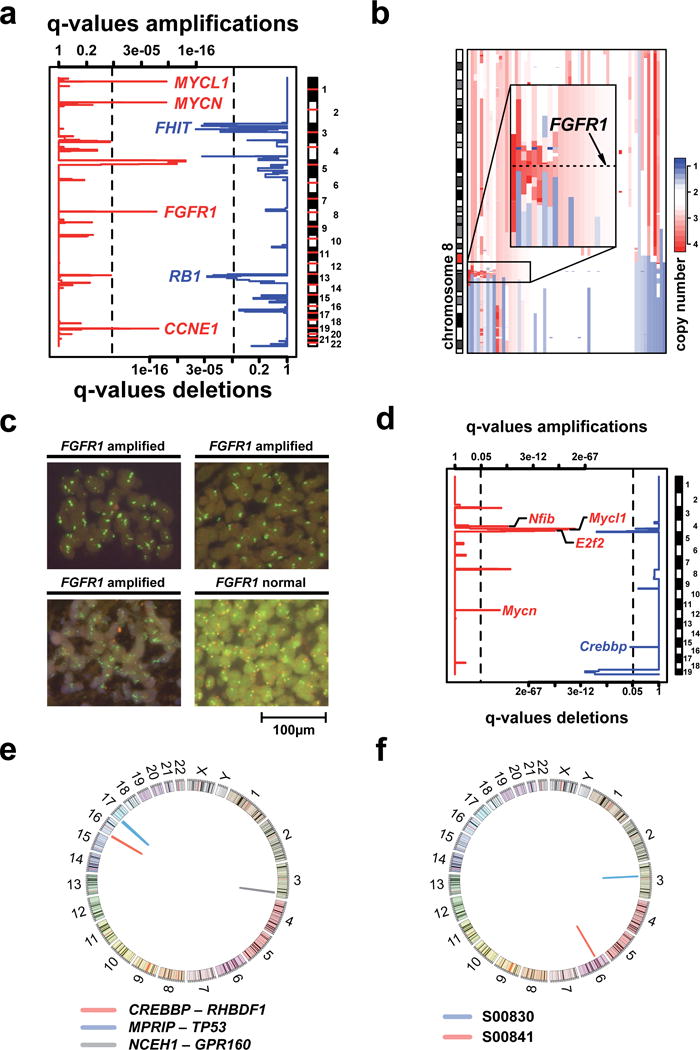
a) Copy number analysis to detect significantly altered regions across 63 tumors. Statistical significance, expressed by q-values (x-axes), is computed for each genomic location (y-axis) (Supplementary Information). Deletions (blue lines, lower scale) and amplifications (red lines, upper scale) are analyzed independently and vertical dashed black lines indicate the significance threshold of 1%. Focally amplified and deleted regions were identified using narrow thresholds (upper quantile: 10%; lower quantile: 15%) to resolve CNAs down to candidate driver genes. b) CNAs of chromosome 8 containing FGFR1 (8p12). Samples are sorted according to the amplitude of FGFR1 amplification. c) FISH analysis to screen for FGFR1 amplifications in an independent set of 51 tumors. Quantification of green signals (FGFR1 specific probe) in comparison to red signals (centromere 8 probe) reveals three FGFR1 amplified samples. d) Copy number analysis based on array-CGH data of 20 SCLC tumors derived from p53/Rb1-deficient mice. Data was analyzed similar to the analysis presented in a). Due to the small sample size, we used a significance threshold of 5% (vertical dashed lines). e) Circos plot of all validated chimeric transcripts detected by transcriptome sequencing. f) Circos plot of validated genomic rearrangements obtained from whole genome sequencing. Both rearrangements affect only portions of the genome smaller than 500kbp. While the structural variant in sample S00841 affects non-coding DNA, the rearrangement in S00830 leads to a loss of exon 7 to 11 of the gene FOXP1.
Mice with conditional deletion of Rb1 and p53 develop SCLC4,27–31 bearing amplifications of Mycl1, Mycn, and Nfib, which were subsequently also found in human SCLC28. We analyzed CNAs in 20 SCLC tumors (15 primary tumors and 5 metastases) from p53/Rb1 conditional knockout mice4 in order to identify alterations shared by both human and mouse tumors. We found significant amplifications of Mycl1, Mycn, and of Nfib (Fig. 1d). In the 15 primary tumors (Supplementary Fig. 2), Nfib did not reach statistical significance, suggesting that Nfib amplifications occur later in tumor evolution. While NFIB was not significantly amplified in the human tumors, three samples exhibited copy number gain at this locus (data not shown). Furthermore, we identified significant amplifications affecting E2f2, a mediator of RB1 function32 and deletions of the histone acetyl transferase gene Crebbp in two mouse tumors (Fig. 1d).
By analyzing transcriptome sequencing data of 15 human tumors we next identified and validated three chimeric transcripts (Fig. 1e, Supplementary Tab. 3). Two contained a fusion partner that was also mutated, MPRIP-TP53 and CREBBP-RHBDF1 (Fig. 1e); both of which are predicted to create a loss-of-function of the genes involved (Supplementary Fig. 3a, b). Similarly, we also found a low genomic rearrangement frequency by reconstruction from paired-end whole genome sequencing data of two specimens (Fig. 1f). This low frequency is in accordance with the spectrum of CNAs in these samples exhibiting almost exclusively arm-level events (Supplementary Fig. 4a).
In order to identify possible differences in the overall genomic architecture between surgically resected (i.e., early stage) samples (n=17) and samples obtained by autopsy (i.e., late stage, n=10) we compared the spectrum of broad CNAs in these two sets. We computed absolute copy numbers from sequencing data in order to correct for admixture of nontumoral cells and for ploidy (Supplementary Note, Supplementary Fig. 4b), but found no significant difference between resected and autopsy cases (Fig. 2a). Furthermore, there was no difference in the total mutation frequency (Fig. 2b) and no segregation between resected and autopsy cases in an analysis of mutated “driver” genes (Fig. 2c, d). We further identified 5 triploid and 2 near-tetraploid cases (n=29) and found no statistical significant overrepresentation of samples with ploidy >2 between resected and autopsy cases (p=0.15). On average we observed a ploidy of 2.3, in line with previously reported studies based on DNA cytometry5. Thus, resected early-stage tumors and late-stage tumors are genomically similar, underscoring the representative nature of our analysis.
Figure 2.
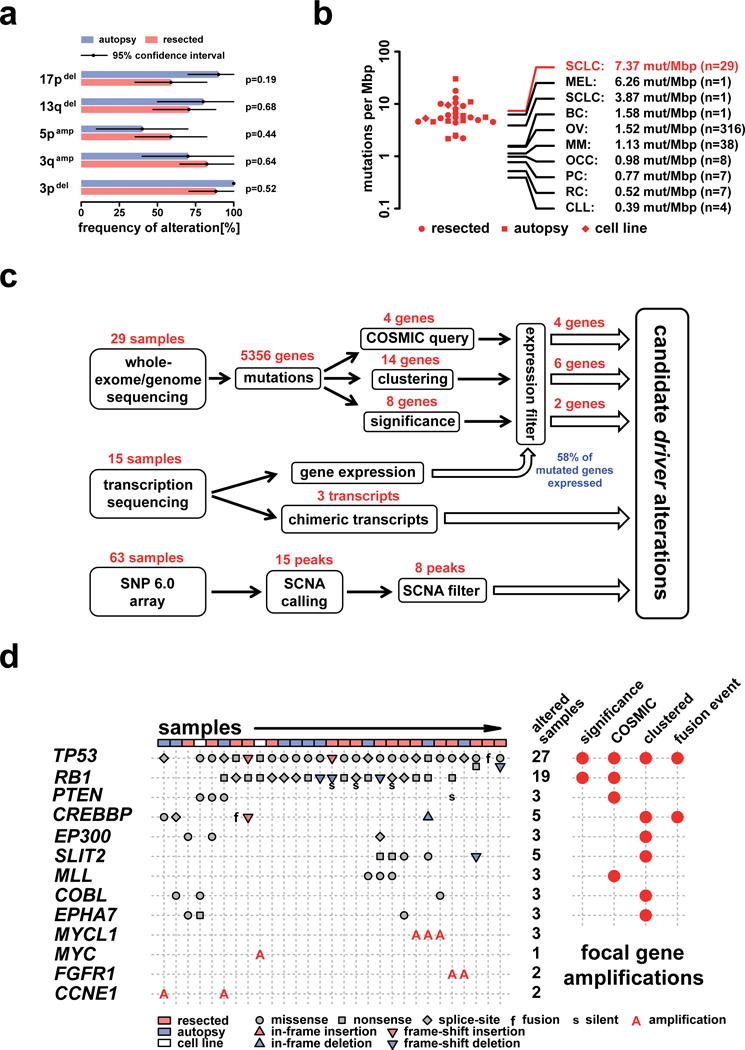
a) Comparison of broad structural genome alterations between surgically resected and autopsy samples. The analysis is based on absolute copy numbers determined using a reconstruction of the allelic state (Supplementary Note). A broad alteration is assigned to be present if 1/4 of the chromosome arm is altered accordingly. Difference between resected and autopsy samples of broad CNAs in 3p, 3q, 5p, 13q, and 17p were statistically tested by a Fisher’s exact test. b) Distribution of the mutation frequency observed in SCLC (points: resected cases; squares: autopsy samples; diamonds: cell lines). The average of the mutation frequency in SCLC (red lines and label) is compared to various tumor types taken from recent large-scale sequencing studies of: melanoma (MEL)37, SCLC38, breast cancer (BC)35, ovarian cancer (OC)40, multiple myeloma (MM)34, ovarian clear cell carcinoma (OCC)36, prostate cancer (PC)33, renal cell cancer (RC)41, and chronic lymphocytic leukemia (CLL)39. c) A schema showing the various steps of our integrated analysis and filtering procedures. All candidate driver genes extracted from sequencing are filtered against gene expression derived from transcription sequencing. CNAs are identified from SNP arrays and candidate CNA regions that are entirely driven by a single SCLC sample were subsequently removed. d) Candidate driver genes identified by significance analysis, presence in the COSMIC database, clustered mutations, and genes that are also involved in fusion events. The type of each mutation is shown for every sample including the gene specific total number of mutated samples.
Compared to global sequencing studies of other tumor types33–41, SCLC exhibits an extremely high mutation rate of 7.4 protein-changing mutations per million basepairs (Fig. 2b, Supplementary Fig. 5a). This high mutation rate is likely linked to tobacco carcinogens, reflected by an elevated rate of C:G>A:T transversions compared to the neutral mutation rate observed in evolution (Supplementary Fig. 5b)38,42–44. In order to identify pathogenetically relevant driver genes in the context of frequent background mutations we applied several filters, including analyses of a signature of mutational selection and of gene expression (Fig. 2c, Supplementary Note). In particular, significantly mutated genes showing an expression level lower than 1 FPKM (fractions per kilobase of exon per million fragments mapped) in more than half of the 15 transcriptomes were removed. Using these adjustments only two genes had q-values of ≤ 0.1: TP53 and RB1 (Fig. 2d)22,29,30,45,46. Remarkably, many of the significant genes were actually not expressed (Supplementary Tab. 4) and none of these mutations were called in the transcriptomes. By contrast, all known tumor suppressors exhibited expression in the upper part of the overall distribution (Supplementary Fig. 6) supporting our strategy for elimination of passenger mutations. Additional filters included an analysis of regional clustering of mutations in a given gene (defining a mutational hotspot) and integration with orthogonal datasets and databases (Fig. 2c)47. Similar to the analysis of significantly mutated genes, we discarded genes that were enriched for silent mutations. Together, these filters yielded a list of likely driver genes in SCLC: TP53, RB1, PTEN, CREBBP, EP300, SLIT2, MLL, COBL, and EPHA7 (Fig. 2d).
SLIT2 showed a pronounced clustering of mutations (5 of 29 cases). The observed mutation spectrum (2 nonsense, 1 frame-shift deletion, 2 missense) (Fig. 3a) together with frequent genomic losses (Supplementary Fig. 7a) suggests that SLIT2 may be a novel tumor suppressor gene in SCLC. We sequenced SLIT2 in 26 additional tumors and 34 cell lines and found an overall mutation frequency of 10% (n=89). Slit proteins are secreted ligands for Robo receptors involved in axon guidance and cellular migration48,49. Supporting a tumor suppressive function of SLIT2/ROBO1 in the lung, Robo1 knockout mice fail to develop normal lungs; surviving mice exhibit bronchial hyperplasia50. Accordingly, a tumor suppressive role for SLIT2 has recently been implied in lung cancer cell lines51. Furthermore, ROBO1 was recently found to be a specific serum biomarker of SCLC52. EPHA7 was recently described as a tumor suppressor gene frequently lost in lymphomas53. Given its role in embryonic development and neural tube closure54, EPHA7 mutations may contribute to the invasive phenotype of SCLC.
Figure 3.
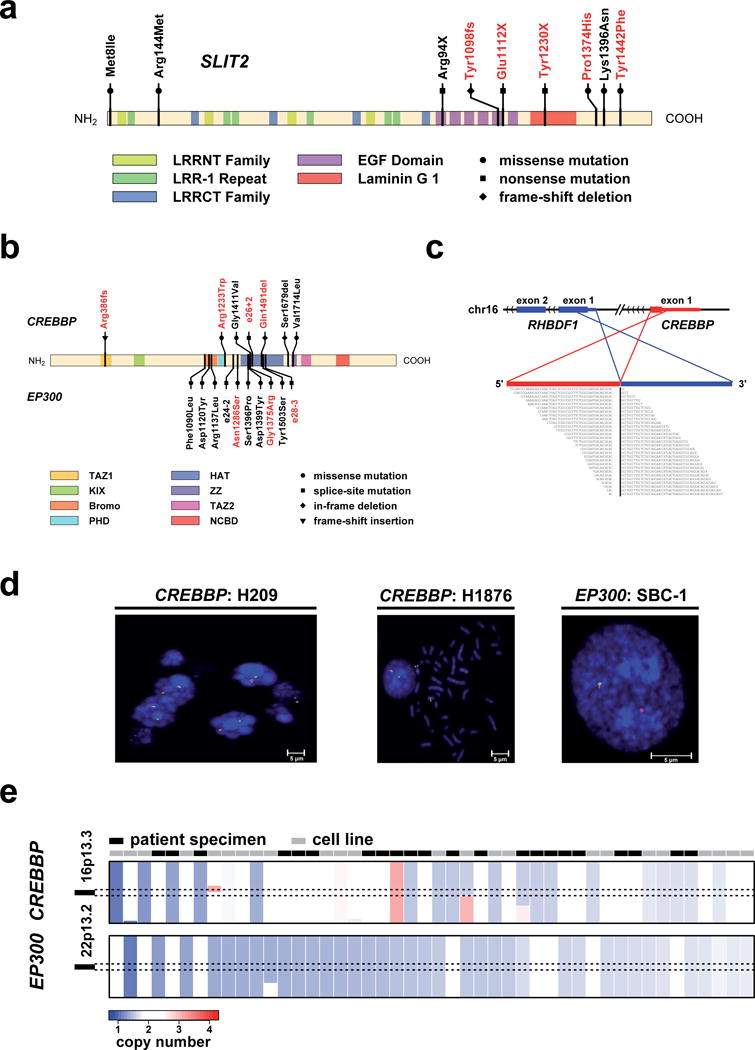
a) The spectrum of mutations affecting SLIT2. Red mutation labels indicate mutations detected by exome sequencing and black labels indicate the results of the extended screen using 454 sequencing. b) Mutations in CREBBP and EP300. Similar to a), red mutation labels indicate mutations discovered by whole exome sequencing, whereas black mutation labels show the results from the extended sequencing around the HAT domain. c) The structure of the chimeric transcript affecting RHBDF1 and CREBPP is shown. Note, that the genomic scale has been adapted to accommodate exons from both genes (axis break, dashes). Chimeric reads are shown below. d) Cell lines that show abnormal signals in the break-apart FISH assay of CREBBP/EP300. In case of CREBBP, both cell lines are showing a loss of the telomeric signal (red signal). For EP300 one cell line also showed a loss of the telomeric signal (here green signal). Break-apart FISH results for CREBBP in H209 are shown as a control38. e) Copy number status for CREBBP and EP300 of all samples that show a deletion in one of the two genes (copy numbers ≤ 1.6 are considered as being deleted). Copy numbers are sorted with respect to the minimal copy number between CREBBP and EP300.
Mutations in CREBBP and EP300 were significantly clustered around the histone acetyltransferase (HAT) domain (Fig. 3b). Of these, mutations affecting the homologous Asp1399 (EP300) and Asp1435 (CREBBP) residues both affect acetylase activity in vitro55–57. Furthermore, Gly1411Glu in CREBBP has been previously identified in lung cancer58 and follicular lymphoma59 and Gly1411Val as well as Asp1435Gly were found in relapsed acute lymphoblastic leukemia60, suggesting a mutational hotspot. By contrast, the Arg386fs mutation and the CREBBP–RHBDF1 gene fusion truncate the open reading frame in the amino terminus (Fig. 3c, Supplementary Fig. 3a). Together with the observation of Crebbp deletions in mouse SCLC (Fig. 1d) and the recently described CREBBP–BTBD12 gene fusion in the NCI-H209 SCLC cell line38, inactivation of CREBBP and EP300 likely plays a major role in SCLC. Focused sequencing of the HAT domain-encoding exons of CREBBP and EP300 in a validation set of 26 additional SCLC tumor specimens and 45 cell lines as well as break-apart FISH performed in 34 SCLC cell lines, confirmed an overall mutation frequency of 18% (point-mutations, indels, and gene rearrangements) (Fig. 3b, c, d). CREBBP/EP300 mutations have recently been described in relapsed acute lymphoblastic leukemia and B-cell lymphoma57,61 but have not been observed at such high frequency in solid tumors so far. Furthermore, all mutations and most of the deletions of CREBBP and EP300 occurred in mutually exclusive fashion in the total set of 101 samples analyzed suggesting epistasis (Fig. 3e). The observed alterations are predominantly heterozygous supporting haploinsufficiency57,62. Thus, even hemizygous deletions occurring in at least 10% of non-mutant samples (Fig. 3e; Supplementary Fig. 7b) may be considered inactivating.
Further supporting the relevance of CREBBP/EP300 mutations in SCLC, all but one (Asn1286Ser in EP300) of the missense mutations were classified as being damaging by computational analyses63. Furthermore, all HAT domain mutations were located at the interface of substrate binding56 (Fig. 4a), thus supporting the notion that they may impact catalytic activity. We assessed the functional impact on histone acetylation of the Gly1411Arg, Asp1435Tyr, and Ser1432Pro CREBBP mutations (homologous to Gly1375Arg, Asp1399Tyr, and Ser1396Pro in EP300) in reconstitution experiments in CrebbpΔflox/Δflox, Ep300Δflox/Δflox (Crebbp/Ep300 Cre-deleted double knockout, or dKO) murine embryonic fibroblasts (MEFs)64–66. All three mutations significantly reduced acetylation of histone 3 lysine 18 (H3K18) (Fig. 4b, c). Specifically, Asp1435Tyr induced complete, Gly1411Arg pronounced, and Ser1432Tyr moderate loss of H3K18 acetylation. Furthermore, knockdown of CREBBP in the cell line DMS114 that lacks CREBBP HAT domain mutations resulted in a moderate but significant increase of cell proliferation (Fig. 4d, e). Tumors with mutations and hemizygous deletions in CREBBP/EP300 did not exhibit a significantly different pattern of gene expression as compared to wild-type tumors after correcting for multiple hypothesis testing (data not shown), suggesting that global changes in gene expression are not the predominant mechanism by which loss of HAT activity contributes to SCLC pathogenesis. Together, these results support a role for loss of CREBBP/EP300 function in the biology of SCLC.
Figure 4.
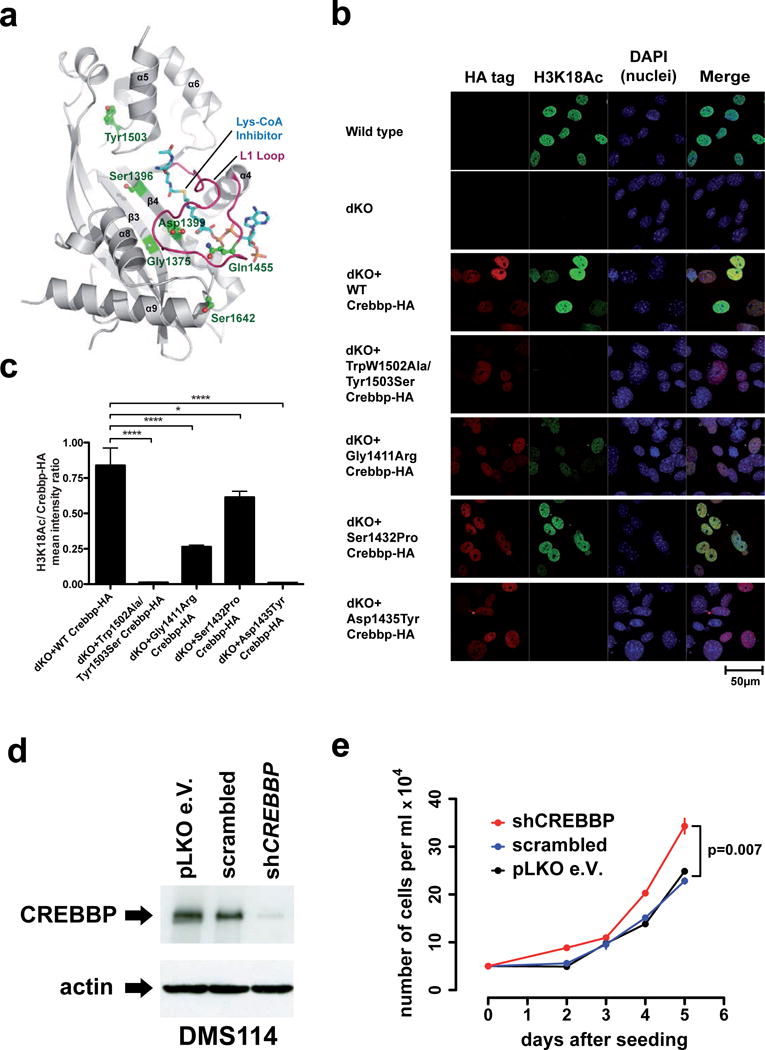
a) CREBBP/EP300 mutations mapped to the crystal structure of the EP300 HAT domain56. All mutations are positioned at the molecular interface involved in Lys-CoA inhibitor binding. In particular, Asp1399 and Gln1455 (equivalent to CREBBP Asp1435 and Gln1491) are located on the substrate-binding loop L1 (red). b) Immunofluorescence was applied to measure levels of acetylated lysine 18 on histone H3 (H3K18Ac) in wild-type MEFs, Crebbp/Ep300 dKO MEFs and dKO MEFs transduced with retroviruses expressing wild-type or SCLC-derived mutants of mouse Crebbp. Human mutations were made at the equivalent murine amino acid, but human numbering is shown in labels. Crebbp-HA signal, red (CY3); H3K18Ac, green (Alexa 488); nuclei, blue (DAPI). The functionally defective Trp1502Ala/Tyr1503Ser81 was included as a control. c) Quantification of H3K18Ac mean signal intensity per nucleus relative to the HA-tagged Crebbp mean signal intensity. P-values shown are from Bonferroni post test of one way ANOVA. * P<0.05, **** P<0.0001. d) Whole cell lysates of DMS114 cells stably infected with lentiviruses expressing shRNAs targeting CREBBP were analyzed for CREBBP protein levels by immunoblotting. e) DMS114 cells stably infected with lentiviral shRNAs targeting CREBBP or the indicated control cells were seeded in 6-well plates and counted as triplicates at the indicated time points (x-axis). Absolute numbers are given on the y-axis and error bars are showing one standard deviation of the mean.
Another histone-modifying enzyme mutated in SCLC was the methyltransferase gene MLL, which was recurrently mutated at isoleucin 960 (Ile960Met)47. MLL rearrangements occur in acute leukemia67,68. Similarly, recurrent genetic alterations in histone modifying genes appear to be a novel hallmark feature of SCLC.
Confirming previous reports69, we found mutations in PTEN (3 of 29 cases), all of which and are likely (Gly165Glu) or proven (His61Arg, Arg130Gly) to affect phosphatase activity70, thereby activating the PI3-kinase pathway. We did not observe any mutations in PIK3CA71.
We developed a mathematical model that gives insight into the allelic state of each tumor and yields estimates of tumor heterogeneity (Supplementary Note). On average, we observed a rather low heterogeneity of about 6.5% (Supplemetary Tab. 5). Using the reconstructed allelic states of each tumor, we found that copy-neutral loss of heterozygosity (CNLOH) events (i.e., complete loss of one allele at a given locus combined with a match of the absolute copy number at that locus with the overall ploidy of the sample) were enriched at the TP53 and RB1 loci (Fig. 5a, b). Furthermore, all TP53 and RB1 mutations in CNLOH regions were early events (Fig. 5b) as their allelic fractions were compatible with the tumor purity. By integrating the different datasets we found that at least one allele of TP53 and RB1 was affected by any genomic event (i.e., mutation (including rearrangements), or hemizygous deletion, LOH) in all cases (Fig. 5c). Thus, similar to genetically manipulated mouse models of SCLC, inactivation of TP53 and RB1 are early and necessary events in the development of SCLC in humans as well4,27–31. Finally, we identified one case, in which the patient had undergone surgery for lung adenocarcinoma three years prior diagnosis of SCLC. While both tumors contained the identical TP53 mutation (Val73fs), the RB1 mutation (Arg251X) was restricted to the SCLC tumor (Supplementary Fig. 8), compatible with trans-differentiation of adenocarcinoma cells to SCLC cells, mediated in part through loss of RB1. Acquired resistance of EGFR-mutant lung adenocarcinomas to EGFR inhibition has been linked with trans-differentiation to SCLC72,73. It is tempting to speculate that loss of RB1 may be mechanistically involved in such cases of acquired resistance as well.
Figure 5.
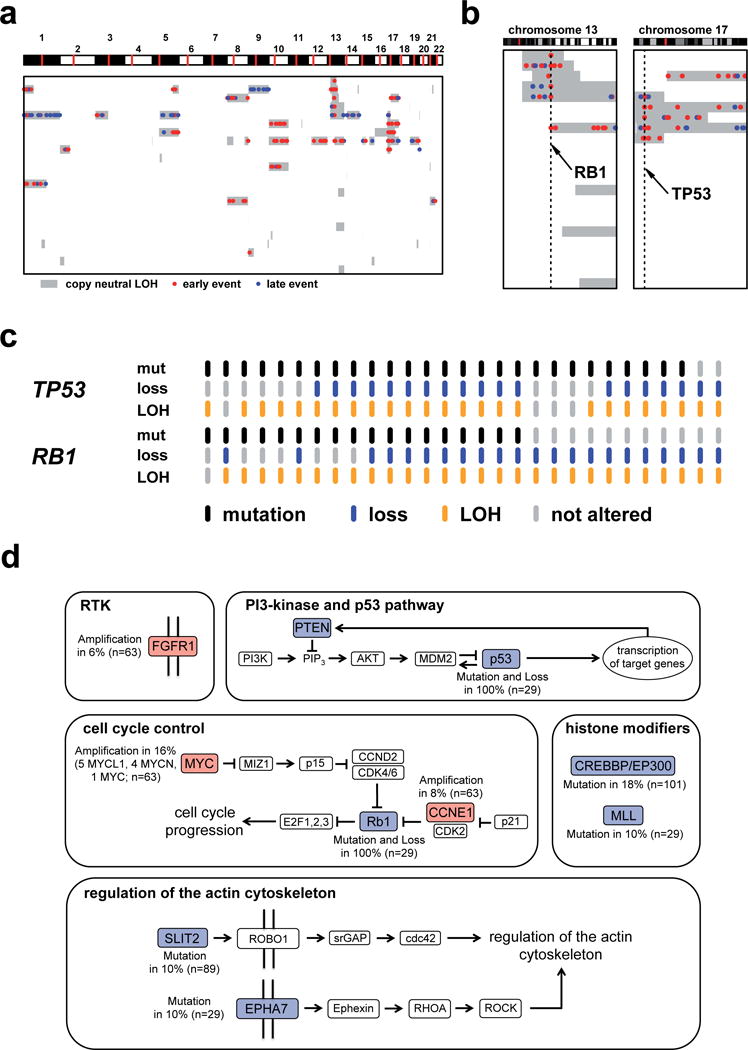
a) Analysis of copy-neutral LOH events (CNLOH) in SCLC. The allelic state of each exome-sequenced sample was reconstructed by applying a detailed mathematical model (Supplementary Information). Genomic portions that showed a loss of heterozygosity (LOH) and an absolute copy number equal to the overall samples’ ploidy are classified CNLOH events. Only samples showing at least one CNLOH event are shown. An analysis of the allelic fraction of somatic mutations in CNLOH regions yields information about the timing of these mutational events. b) TP53 and RB1 mutations in CNLOH regions. c) Distribution of mutations (including rearrangements), hemizygous deletions, and LOH affecting TP53 and RB1 across all exome-sequenced samples. d) SCLC driver genes and their mutation frequency mapped to signaling pathways. We classified the occurring mutations into 5 major groups: receptor tyrosine kinase (RTK) alterations, PI3-kinase and p53 pathway, cell cycle control, histone modifiers, and regulation of actin cytoskeleton.
Despite methodological challenges (limited sample set, high mutation frequency), integrative genome analyses of human and mouse SCLC afforded sketching a molecular map of this tumor type, condensed in 5 categories (Fig. 5d). The tumor suppressive functions of p53 rely on its acetylation by CREBBP or EP30074-79. However, given the universal loss of p53 function in SCLC, the tumor suppressive functions of CREBBP that we observed are likely independent of p53. One of the best-studied functions of SLIT2 is its involvement in actin polymerization mediated by Cdc4280. We speculate that this property might enhance invasive capabilities and thus contribute to the aggressiveness of SCLC. The reported functions of EPHA753,54 may also contribute to this phenotype. Beyond universal losses of TP53 and RB1 and amplifications of MYCL1, MYCN, MYC, we present PTEN mutations and FGFR1 amplifications as potentially therapeutically tractable genome alterations. Finally, we define genomic alterations affecting histone modifying enzymes CREBBP, EP300, and MLL as the second most frequently mutated class of genes in SCLC. In summary, our study represents a significant extension of the current molecular concept of SCLC and, more broadly, provides an example of how integrative computational genome analyses can provide functionally tractable information in the context of a highly mutated cancer genome.
Supplementary Material
Acknowledgments
We are indebted to the patients donating their tumor specimens as part of the Clinical Lung Cancer Genome Project initiative. Additional biospecimens for this study were obtained from Victorian Cancer Biobank, Melbourne, Australia. The Institutional Review Board (IRB) of each participating institution approved collection and use of all patient specimens in this study. We also thank our colleagues of The Cancer Genome Atlas Research Network (TCGA) and Andrew L. Kung (Dana-Farber Cancer Institute, Children’s Hospital, Boston, MA) for invaluable discussion and many helpful comments. This work was supported by the German Ministry of Science and Education (BMBF) as part of the NGFNplus program (grant 01GS08100 to RKT and 01GS08101 to JW and PN), by the Max Planck Society (M.I.F.A.NEUR8061 to RKT), by the Deutsche Forschungsgemeinschaft (DFG) through SFB832 (TP6 to RKT and RTU; TP5 and Z1 to LH and RB) and TH1386/3-1 (to RKT and MLS), by the EU-Framework Programme CURELUNG (HEALTH-F2-2010-258677) (to RKT, JF, EB, CB, SL, BB, and JW), Stand Up To Cancer-American Association for Cancer Research Innovative Research Grant (SU2C-AACR-IR60109) (to RKT and WP), by the Behrens-Weise Foundation (to RKT) and by an anonymous foundation to RKT. MLS is a fellow of the International Association for the Study of Lung Cancer (IASLC). PB and LK thank the St. Jude Cell and Tissue Imaging facility, and support from NIH Cancer Center grant P30 CA021765 and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital. RKT reports the following potential sources of conflict of interest: consulting and lecture fees (Sanofi-Aventis, Merck KGaA, Bayer, Lilly, Roche, Boehringer Ingelheim, Johnson&Johnson, AstraZeneca, Atlas-Biolabs, Daiichi-Sankyo); research support (AstraZeneca, Merck, EOS).
Footnotes
Data Access
Binary sequence alignment data of 300bp regions around all identified somatic mutations and SNP array data were deposited in the European Genome-Phenome Archive (EGA; EGAS00001000299).
Author Contributions
MP, LFC contributed equally. MP, LFC, MLS, JG, DS, LHK, FL, RM, JV, PS, JS, RS, RB, SP, LH, PKB, RKT conceived and designed the experiments. LFC, MLS, JG, DS, LHK, DP, RM, MK, ID, CM, VDC, HUS, JA, IB, CB, BDW, DB, FG, IW, SH, JH preformed experiments. MP, LFC, MLS, JG, DS, LHK, DP, FL, RS, TZ, RM, VDC, BDW, JV, XL, WP, MLW, JS, RS, SP, LH, PKB, SH, RKT analyzed the data. MP, RS, TZ, SA, SLC, KC, SB, GG, KSP, DR, CG, MF, LP, GW, ZW, PR, IP, YC, ES, CL, PS, HH, TM, MB WER, LAM, VMF, HG, WT, HS, ET, ES, DAMH, PJFS, FC, CL, SD, JF, SS, OTB, MLI, JS, JHC, AS, HM, WW, BS, JCS, BB, EB, CB, SL, MH, JS, JW, PN, LH, PKB, SH contributed reagents/materials/analysis tools. MP, LFC, RKT wrote the paper.
References
- 1.Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113:5–21. doi: 10.1002/cncr.23542. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD. Lung tumours with neuroendocrine differentiation. European journal of cancer. 2009;45(Suppl 1):251–66. doi: 10.1016/S0959-8049(09)70040-1. [DOI] [PubMed] [Google Scholar]
- 3.van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741–55. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- 4.Park KS, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nature medicine. 2011;17:1504–8. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Travis WD, World Health Organization., International Agency for Research on Cancer., International Association for the Study of Lung Cancer & International Academy of Pathology . Pathology and genetics of tumours of the lung, pleura, thymus and heart. IARC Press, Oxford University Press (distributor); Lyon, Oxford: 2004. p. 344. [Google Scholar]
- 6.Tiseo M, Ardizzoni A. Current status of second-line treatment and novel therapies for small cell lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2007;2:764–72. doi: 10.1097/JTO.0b013e3180986262. [DOI] [PubMed] [Google Scholar]
- 7.Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 9.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 10.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nature reviews– Cancer. 2010;10:760–74. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 13.Bergethon K, et al. ROS1 rearrangements define a unique molecular class of lung cancers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohno T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nature medicine. 2012;18:375–7. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipson D, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nature medicine. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi K, et al. RET, ROS1 and ALK fusions in lung cancer. Nature medicine. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 17.Dutt A, et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PloS one. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammerman PS, et al. Mutations in the DDR2 Kinase Gene identify a Novel therapeutic target in squamous cell lung cancer. Cancer Discovery. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss J, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science translational medicine. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bass AJ, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics. 2009;41:1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YH, et al. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene. 2006;25:130–8. doi: 10.1038/sj.onc.1208997. [DOI] [PubMed] [Google Scholar]
- 22.Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Seminars in oncology. 2001;28:3–13. [PubMed] [Google Scholar]
- 23.Gazzeri S, et al. Activation of myc gene family in human lung carcinomas and during heterotransplantation into nude mice. Cancer research. 1991;51:2566–71. [PubMed] [Google Scholar]
- 24.Zhao X, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer research. 2005;65:5561–70. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 25.Voortman J, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13040–5. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassan MI, Naiyer A, Ahmad F. Fragile histidine triad protein: structure, function, and its association with tumorogenesis. Journal of cancer research and clinical oncology. 2010;136:333–50. doi: 10.1007/s00432-009-0751-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calbo J, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer cell. 2011;19:244–56. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Dooley AL, et al. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes & development. 2011;25:1470–5. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meuwissen R, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer cell. 2003;4:181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 30.Schaffer BE, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer research. 2010;70:3877–83. doi: 10.1158/0008-5472.CAN-09-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutherland KD, et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer cell. 2011;19:754–64. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 32.Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Current opinion in cell biology. 2007;19:649–57. doi: 10.1016/j.ceb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–90. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karro JE, Peifer M, Hardison RC, Kollmann M, von Grunberg HH. Exponential decay of GC content detected by strand-symmetric substitution rates influences the evolution of isochore structure. Molecular biology and evolution. 2008;25:362–74. doi: 10.1093/molbev/msm261. [DOI] [PubMed] [Google Scholar]
- 43.Hecht SS. Progress and challenges in selected areas of tobacco carcinogenesis. Chemical research in toxicology. 2008;21:160–71. doi: 10.1021/tx7002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Seminars in cancer biology. 2005;15:103–12. doi: 10.1016/j.semcancer.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Horowitz JM, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:2775–9. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mori N, et al. Variable mutations of the RB gene in small-cell lung carcinoma. Oncogene. 1990;5:1713–7. [PubMed] [Google Scholar]
- 47.Bamford S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British journal of cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong K, Park HT, Wu JY, Rao Y. Slit proteins: molecular guidance cues for cells ranging from neurons to leukocytes. Current opinion in genetics & development. 2002;12:583–91. doi: 10.1016/s0959-437x(02)00343-x. [DOI] [PubMed] [Google Scholar]
- 49.Zhou WJ, et al. Slit-Robo signaling induces malignant transformation through Hakai-mediated E-cadherin degradation during colorectal epithelial cell carcinogenesis. Cell research. 2011;21:609–26. doi: 10.1038/cr.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xian J, et al. Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15062–6. doi: 10.1073/pnas.251407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tseng RC, et al. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer research. 2010;70:543–51. doi: 10.1158/0008-5472.CAN-09-2084. [DOI] [PubMed] [Google Scholar]
- 52.Taguchi A, et al. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer cell. 2011;20:289–99. doi: 10.1016/j.ccr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oricchio E, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147:554–64. doi: 10.1016/j.cell.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holmberg J, Clarke DL, Frisen J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 2000;408:203–6. doi: 10.1038/35041577. [DOI] [PubMed] [Google Scholar]
- 55.Muraoka M, et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–9. [PubMed] [Google Scholar]
- 56.Liu X, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–50. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 57.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kishimoto M, et al. Mutations and deletions of the CBP gene in human lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:512–9. [PubMed] [Google Scholar]
- 59.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inthal A, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2012 doi: 10.1038/leu.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tillinghast GW, et al. Analysis of genetic stability at the EP300 and CREBBP loci in a panel of cancer cell lines. Genes, chromosomes & cancer. 2003;37:121–31. doi: 10.1002/gcc.10195. [DOI] [PubMed] [Google Scholar]
- 63.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang-Decker N, et al. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer cell. 2004;5:177–89. doi: 10.1016/s1535-6108(04)00022-4. [DOI] [PubMed] [Google Scholar]
- 65.Kasper LH, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Molecular and cellular biology. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kasper LH, et al. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. The EMBO journal. 2010;29:3660–72. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thirman MJ, et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. The New England journal of medicine. 1993;329:909–14. doi: 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- 68.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic acids research. 2004;32:959–76. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yokomizo A, et al. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene. 1998;17:475–9. doi: 10.1038/sj.onc.1201956. [DOI] [PubMed] [Google Scholar]
- 70.Han SY, et al. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer research. 2000;60:3147–51. [PubMed] [Google Scholar]
- 71.Yamamoto H, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer research. 2008;68:6913–21. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zakowski MF, Ladanyi M, Kris MG. EGFR mutations in small-cell lung cancers in patients who have never smoked. The New England journal of medicine. 2006;355:213–5. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 73.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819–23. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 75.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Current opinion in cell biology. 2003;15:164–71. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 76.Sakaguchi K, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes & development. 1998;12:2831–41. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grossman SR, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–4. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- 79.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–7. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 80.Wong K, et al. Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107:209–21. doi: 10.1016/s0092-8674(01)00530-x. [DOI] [PubMed] [Google Scholar]
- 81.Bordoli L, et al. Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic acids research. 2001;29:4462–71. doi: 10.1093/nar/29.21.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome research. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scheble VJ, et al. ERG rearrangement is specific to prostate cancer and does not occur in any other common tumor. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:1061–7. doi: 10.1038/modpathol.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Laframboise T, Harrington D, Weir BA. PLASQ: a generalized linear model-based procedure to determine allelic dosage in cancer cells from SNP array data. Biostatistics. 2007;8:323–36. doi: 10.1093/biostatistics/kxl012. [DOI] [PubMed] [Google Scholar]
- 87.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 88.Sos ML, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. The Journal of clinical investigation. 2009;119:1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Querings S, et al. Benchmarking of mutation diagnostics in clinical lung cancer specimens. PloS one. 2011;6:e19601. doi: 10.1371/journal.pone.0019601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.