

Abstract
Deficiency of Asparagine Synthetase (ASNSD, MIM 615574) is a very rare autosomal recessive disorder presenting with some brain abnormalities. Affected individuals have congenital microcephaly and progressive encephalopathy associated with severe intellectual disability and intractable seizures. The loss of function of the asparagine synthetase (ASNS, EC 6.3.5.4), particularly in the brain, is the major cause of this particular congenital microcephaly. In this study, we clinically evaluated an affected child from a consanguineous Emirati family presenting with congenital microcephaly and epileptic encephalopathy. In addition, whole-exome sequencing revealed a novel homozygous substitution mutation (c.1193A>C) in the ASNS gene. This mutation resulted in the substitution of highly conserved tyrosine residue by cysteine (p.Y398C). Molecular modeling analysis predicts hypomorphic and damaging effects of this mutation on the protein structure and altering its enzymatic activity. Therefore, we conclude that the loss of ASNS function is most likely the cause of this condition in the studied family. This report brings the number of reported families with this very rare disorder to five and the number of pathogenic mutations in the ASNS gene to four. This finding extends the ASNS pathogenic mutations spectrum and highlights the utility of whole-exome sequencing in elucidation the causes of rare recessive disorders that are heterogeneous and/or overlap with other conditions.
Keywords: Asparagine synthetase deficiency, ASNS, WES, Microcephaly, Epileptic encephalopathy, Hypomorphic mutation
Introduction
Asparagine synthetase deficiency (ASNSD, MIM 615574) is a very rare severe neuro developmental disorder shown to be caused by mutations in ASNS gene. It has been suggested to classify ASNSD as an inborn error of non-essential amino acid synthesis (Ruzzo et al. 2013). This condition is characterized by congenital microcephaly, severely delayed psychomotor development, progressive encephalopathy, and cortical atrophy, associated with seizure or hyperekplexic activity (Ruzzo et al. 2013). Additional variable dysmorphic features include micrognathia, receding forehead and large ears. Axial hypotonia and cortical blindness can be seen as well (Ruzzo et al. 2013). To date, most of the reported patients had feeding difficulties and respiratory insufficiency, which led to early death in infancy (Ruzzo et al. 2013). The symptoms associated with this condition overlap with numerous other intellectual disability disorders such as serine deficiency syndromes, acute mitochondrial encephalopathy, neuronal ceroid–lipofuscinosis, and pyridoxine-dependent seizure (Samson et al. 1994; Haltia 2003; de Koning and Klomp 2004; Stockler et al. 2011). A gene-trap mouse model for asparagine synthetase deficiency showed a milder phenotype compared to humans (Ruzzo et al. 2013). The overlapping clinical presentation includes learning and memory deficiency with structural brain abnormalities including reduced cortical thickness and enlarged ventricles. However, mutant mice do not have any motor abnormalities or seizures. Though asparagine is a non-essential amino acid, the existence of the syndrome suggests its deficiency is pathogenic, particularly for brain function.
The ASNS gene maps to the cytogenetic band 7q21.3 and has a total of 14 spliced coding transcript variants with most variation occurring in the 5′ UTR regions. The gene encodes an asparagine synthetase enzyme (EC 6.3.5.4) involved in the synthesis of asparagine (Zhang et al. 1989; Ruzzo et al. 2013). ASNS enzyme catalyzes the transfer of ammonia from glutamine to aspartic acid to form asparagine (Zhang et al. 1989; Ruzzo et al. 2013). It has an approximate molecular weight of 64 KDa for most common transcripts. The asparagine synthetase is expressed in all mammalian tissues with particularly high expression levels in the adult brain (Ruzzo et al. 2013).
So far, only three pathogenic mutations are known to be responsible for asparagine synthetase deficiency, all from a single report on the role of human ASNS gene (Fig. 2. A–C) (Ruzzo et al. 2013). In the present study, we report the use of whole-exome sequencing to identify a novel homozygous mutation (c.1193A>C/p.Y398C) in an Emirati child exhibiting congenital microcephaly with epileptic encephalopathy and psychomotor delay, consistent with the diagnosis of asparagine synthetase deficiency.
Fig. 2.
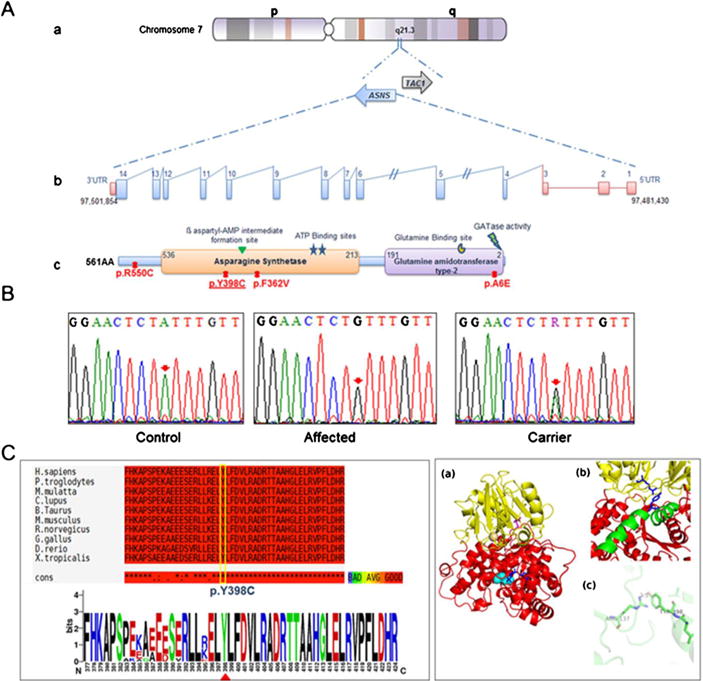
Schematic representation and molecular analysis of ASNS gene. a-(a to b) genomic organization of the ASNS gene. a to c. Protein representation of functional domain of the asparagine synthetase enzyme along with the corresponding position of all reported mutations. The identified mutation in this study is underlined b. Chromatograms of DNA sequence changes in the ASNS gene. A homozygous c.1193A>C mutation was identified in the affected child, which results in the substitution of p.Y398C. Both parents are carrier for this mutation. c. Schematic representation of amino acid sequence alignment of p.Y398C in different orthologues. c to a Alignment conversation was compiled using T-coffee online website (http://tcoffee.crg.cat/). C to b Logo was generated using WebLogo (weblogo.berkeley.edu) (Crooks et al. 2004). d. Humanized model of ASNS protein. a The cartoon representation shows that the protein is divided into distinct N-terminal domain (represented in yellow color) and C-terminal domain (represented in red color). The glutamine (represented as sticks in magenta color) is bound in N-terminal domain while AMP (represented as sticks in blue color) is bound in C-terminal domain. The cyan spheres represent the site for cation binding. b The protein consists of a long helix (represented in green color) which is distorted in the middle. Y398 (blue sticks) lies on this long helix and is at the interface of N-and C-terminal domain. R137 (blue sticks) lies in N-terminal domain and is within hydrogen bonding distance of Y398. c Representation of R137 and Y398 hydrogen bonding indicating that it is a key residue between the domain-domain interactions which contains the two active sites of the enzyme
Patient and methods
Ethics statement
This study has been approved by Al-Ain Medical Human Research Ethics Committee according to the national regulations (protocol number 10/09). The parents in this family provided an informed written consent form prior to research and publication.
Patient
We ascertained one consanguineous family from the United Arab Emirates (UAE) with one affected child (Fig. 1A. Patient V-1) exhibiting congenital microcephaly with epileptic encephalopathy and severe psychomotor delay. Peripheral blood samples were collected from members of this family in EDTA tubes after obtaining informed written consent. Genomic DNA was extracted using the Flexigene DNA extraction kit (Qiagen Gmbh, Germany) following the manufacturer’s instructions, and was available on the proband and the mother.
Fig. 1.
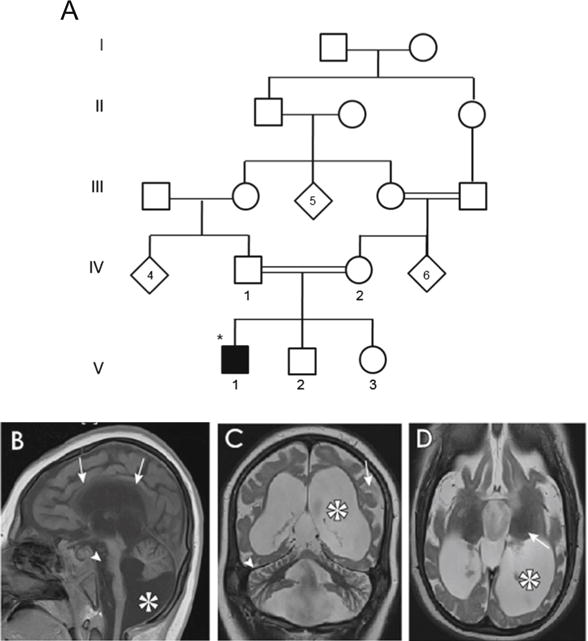
Pedigree and MRI pictures of a patient with asparagine synthetase deficiency. a. Pedigree showing one affected Emirati patient with asparagine synthetase deficiency. Circles and squares denote females and males respectively, filled symbols represent affected members, diagonal symbols indicate individuals with undesignated sex, double lines denote consanguineous marriage, Roman numbers indicate the generation number until their offspring, Arabic numbers depict participants in this study, star symbols represent the index patient. b to d. MRI pictures showing decreased cerebral volume and atrophy/hypoplasia. b. Midline sagittal with thin corpus callosum (arrows), reduced volume of pons (arrowhead), cerebellar hypoplasia and Blake’s pouch cyst (asterisk). c. Simplified gyral pattern (arrow), massive ventriculomegaly (asterisk, and cerebellar folia atrophy (arrowhead). d. Decreased cerebral volume and ventriculomegaly (asterisk). Arrow in 2D highlights that thalamus, which is relatively dark suggesting lack of edema
Whole-exome sequencing and variant analysis
The DNA samples of both the mother and the affected child were investigated by whole-exome sequencing (WES). The Agilent SureSelect Human All Exome 50 Mb Kit was used as exome enrichment probe sets according to the manufacturer’s protocol (Agilent, Inc. USA). Genomic DNA libraries were prepared according to the manufacturer’s instructions (Illumina, San Diego, CA). Briefly, genomic DNA was fragmented in a Bioruptor (Diagenode). These fragments were ligated to the Illumina multi-PE-adaptor followed by PCR amplification. Captured DNA libraries, of 100–base pair sequencing length, were sequenced with HiSeq2000 instrument (Gnirke et al. 2009). The exome reads obtained were aligned to the reference human genome (hg19) and annotated SNPs were mapped to dbSNP v131, v132 and 1,000 Genomes. Depth of sequencing was 60X±16 (SD) per exome. Genetic variants were delineated with the GATK software for both SNPs and INDELs (DePristo et al. 2011). Each aligned exome was assessed for sufficient quality with the following parameters: number of SNPs called in each lane (average~26,000), accuracy, depth of coverage, and error rate per read position. Only exomes of high quality were analyzed further using simultaneously our locally “sequencing analysis pipeline” and “potential disease-causing variants filtering and prioritization pipeline” (Dixon-Salazar et al. 2012). Based on autosomal recessive inheritance, SNPs and INDELs present in stretches of homozygosity, as determined by either linkage analysis or publicly available homozygosity mapping software (Homozygosity Mapper) were identified (Seelow et al. 2009). Selected variants were cross compared to the Genome Variation Server (http://gvs.gs.washington.edu/GVS/) and SIFT (http://sift.jcvi.org/) databases to determine single-nucleotide evolutionarily constraints and conservation scores (GERP, PhastCons), protein damage prediction determinations (Polyphen2), and relationship of variants to OMIM classifications (Cooper et al. 2010). Potential disease-causing variants were validated using in-house “potential disease-causing variants validation pipeline” (Dixon-Salazar et al. 2012).
Sanger DNA sequencing
Probable disease-causing variants detected by WES were confirmed using Sanger sequencing method. Primers for potential variants were designed using Primer3 version 0.4.0 (http://primer3.ut.ee/). Direct sequencing was carried out to validate the segregation of these variants within this family and to further confirm the absence of these variants in large cohort of healthy, ethnically matched control individuals. Amplified PCR products were purified and sequenced using the BigDye Terminator kit v3.1 (Applied Biosystems, USA) on a 3130xl Genetic Analyzer System (Applied Biosystems, USA). DNA chromatograms were inspected and analyzed based on cDNA sequence in accordance with the GenBank entry NM_133436.3 using the Sequencing Analysis® 5.3 software (Applied Biosystems, USA) and ClustalW2 algorithms (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Protein structure modelling analysis
To evaluate the effect of p.Y398C mutation on the protein structure and/or function, homology model of ASNS was constructed by MODELLER program version 9.10 (Eswar et al. 2006). The amino acid sequence of human ASNS protein was obtained from UniProt (accession # P08243) and structure closest to the sequence was searched using PDBe webserver (https://www.ebi.ac.uk/pdbe/). The crystal structure of asparagine synthetase B of E. coli (Protein Data Bank code: 1CT9) showed best sequence identity and was used as template for the construction of human ASNS protein model (Larsen et al. 1999). The alignment of human ASNS and E.coli ASNB was carried out by Clustalw2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The model which had lowest energy and least Cα RMSD value with respect to the template structure was selected. The stereo-chemical properties of this model were evaluated using PROCHECK program version 3.5.4 (Laskowski et al. 1993). Evaluation of steric clashes in the side chains was performed. The ligand binding sites on ASNS model were identified by superimposing the model and the template using the software Chimera (Yang et al. 2012). Residue Y398 was mutated to cysteine in the ASNS model using mutagenesis wizard of PYMOL software (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC). All graphic representations of ASNS model and corresponding mutated protein were generated using PYMOL software.
Results
Clinical data
We evaluated a five year old male child (Fig. 1A. individual V-1), from a consanguineous Emirati family, who suffered from primary microcephaly, severe psychomotor delay, cortical blindness and intractable seizures (Fig. 1B to D). The healthy parents were first cousins (Fig. 1A). The child was born at 39 weeks gestation by spontaneous vaginal delivery after an uncomplicated pregnancy. His birth weight, length and head circumference were 3.1 kg (<10 centile), 51 cm (<10 centile) and 29.5 cm (−10SD), respectively (Table 1). On the first day of life he developed myoclonic movements. The seizures responded well to conventional treatment (Phenobarbitone). EEG showed abnormal background activity with low amplitude and frequent interictal bilateral multifocal spike slow wave activity. During his early infancy he was noticed to have spasticity with exaggerated deep tendon reflexes of both upper and lower extremities, severe microcephaly, with no dysmorphic features and unremarkable other systemic examination. He did not acquire any developmental milestones, never recognized his mother and never vocalized. His parents noted lack of response to any visual stimuli since birth and ophthalmological examination revealed bilateral pale optic discs. His lack of visual response was attributed to cortical blindness. Metabolic screening revealed normal results including liver and renal function tests, complete blood count, blood gas analysis, ammonia, serum lactate, plasma urea, electrolytes, minerals including calcium, magnesium and phosphate, urine amino acids and urine organic acids. Plasma amino acids were normal, including the aspargine level. Cranial MRI showed decreased cerebral volume and atrophy/hypoplasia with thin corpus callosum, simplified gyral pattern with massive ventriculomegaly. In addition, there was reduced volume of pons, cerebellar hypoplasia, Blake’s pouch cyst, and cerebellar folia atrophy (Fig. 1. B–D). Currently, the patient is admitted in long-term care institute, bedridden in encephalopathic state, on tracheostomy ventilation, percutaneous endoscopic gastrostomy (PEG) tube feeding, and multiple anti-convulsion medications.
Table 1.
Clinical features of cases with Asparagine synthestase deficiency
| Family A | Family B | Family C | Family D | Family E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cases | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 3 | 1 |
| Consanguinity | Yes | No | Yes | No | Yes | |||||
| Ethnic origin | Iranian Jews | Iranian Jews | Bangladeshi | French Canadian | Emirati | |||||
| Gender | Male | Male | Female | Male | Male | Male | Male | Male | Male | Male |
| Age | 14Y | 14Y | 12Y | 4 M | 3 M | 6 M | 9D | 11 M | 12 M | 5Y |
| HC* at birth | 31.5 cm | 31 cm | 31 cm | 30.5 cm | 33 cm | 32 cm | 31.5 cm | 31 cm | 28.5 cm | 29.5 cm |
| Progressive microcephaly | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Epilepsy (Age of onset) | 1 M | 2 W | 3 W | ND | ND | ND | 4D | 9 M | 8D | 1stD |
| Psychomotor delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Spasticity | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hyperreflexia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Deceased | No | No | No | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Brain MRI | Reduced cerebral volume | Reduced cerebral volume | Reduced cerebral volume, reduced size of Pons and simplified gyri | Reduced cerebral volume, reduced size of Pons and simplified gyri | Ventricular system dilatation, cerebral and cerebellar atrophy | |||||
| DNA change | Homo | Homo | Homo | Comp. Hetero | Homo | |||||
| C.1084 T>G | c.1084 T>G | c.1648 C>T | c.1648C>T/c.17C>A | c.1193A>C | ||||||
| Protein Change | F362V | F362V | R550C | A6E/R550C | Y398C | |||||
| (Ruzzo et al. 2013) | This study | |||||||||
Y, years; M, months; D, days; W, weeks; HC, head circumference; ND, Not determined; Homo, Homozygous; Comp. Hetero, Compound heterozygous
Whole-exome sequencing revealed a homozygous missense mutation in ASNS gene
Whole-exome sequencing has revealed a total of 3 potential disease-causing variants as illustrated in Table 2. One of the detected variants was a homozygous single nucleotide substitution (c.1193A>C) in exon 11 of the ASNS gene (Table 2). The depth of coverage for this mutation was showed high reliability of sequencing. Sanger sequencing validated the segregation of this substitution within the studied family showing that both parents and siblings were carriers of c.1193A>C substitution (Fig. 2B). This substitution leads to a novel missense mutation changing a tyrosine residue at position 398 by cysteine (p.Y398C). The tyrosine residue (Y398) is highly conserved across different species (Fig. 2C). In addition, prediction analyses using (SIFT, polyphen2, mutation taster) showed that this substitution is probably damaging (SIFT score 0, polyphen2 score 0.997 and mutation taster score 0.999). Moreover, the c.1193A>C/p.Y398C was not detected in large controls samples from the same ethnic group nor reported in the exome variant database (http://gvs.gs.washington.edu/GVS/).
Table 2.
Variants detected by whole exome sequencing
| Chr. | Pos. | Ref. | Mut | gene | Fct. GVS | cDNA | AA_Charge | AA_Pos | OMIM | accession | PolyPhen2 | % Hom |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 97483937 | T | C | ASNS | missense | 1193 | TYR/CYS | 398/562 | Asparagine synthetase deficiency, 615574 (3) | NM_183356.3 | probably damaging | 0.876 |
| 20 | 62729793 | C | T | OPRL1 | stop-gained | 754 | ARG/stop | 252/371 | NM_182647.2 | 0.906 | ||
| 1 | 207112648 | G | C | PIGR | missense | 204 | ILE/MET | 68/765 | NM_002644.3 | possibly damaging | 0.857 |
Chr, chromosome; Pos, position; Ref, reference; Mut, mutant; Fct, function; AA, Amino acid; %, percentage
Filtering for rare homozygous potentially deleterious variants within blocks of homozygosity revealed five variants that were homozygous in the child and heterozygous in the mother. Of these, only the ASNS mutation occurred within a conserved residue and was predicted to be probably damaging and was known to lead to a condition nearly identical to the clinical presentation. Other variants included a stop-gained in the OPRL1 gene, encoding opioid receptor-like 1, a missense in the PIGR gene, encoding polymeric immunoglobulin receptor
Molecular modeling suggested that the mutation will result in dramatic changes to the structure and enzymatic activity of the asparagine synthetase
We performed computational modeling to predict the structure of ASNS protein and evaluate the effect of Y398C mutation. From all the structures in RCSB protein data bank, the crystal structure of asparagine synthetase B (ASNB) of E. coli showed the highest E value (1e−70) with 37.8 % sequence identity. Moreover, the protein showed a complete overlap on human ASNS protein sequence. Therefore, we selected this structure to be used as template for the construction of the present model. The overlap of model and template using chimera revealed the corresponding AMP and glutamine binding site on ASNS protein. In E. coli residues Arg49, Asn74, Glu76, and Asp98 form the glutamine binding pocket and Val 272, Leu 232, Ser346 and Gly 347 bind the AMP moiety (Larsen et al. 1999). The ClustalW2 alignment of E. coli ASNB and ASNS shows that glutamine and AMP binding residues are highly conserved (Sup Fig. S1). Among all the ligand binding residues, only residue Val232 of the AMP binding site in ASNB from E.coli was substituted by Isoleucine in corresponding position (residue 287) in human ASNS model (Sup Fig. S1). The model revealed that similar to E. coli ASNB structure, human ASNS protein is divided into distinct N and C-terminal regions (Fig. 2D-a). The N-terminal is rich in beta-strands and has glutamine binding site while C-terminal possesses AMP-binding site. The two active sites are separated by a distance of 19 Å. It also has a long helix of 29 residues (384–413) similar to the 27 residue long helix in E. coli ASNB protein. This helix is slightly distorted at residues 397–400 (LYLF) (Fig. 2D-b). Y398 lies in the interface of N and C-terminal motif of the protein and forms hydrogen bonds with R137 of N-terminal domain (Fig. 2D-c). The identified mutation (p.Y398C) will disrupt this hydrogen bonding interaction which is important for domain-domain interaction thereby impairing the catalytic function of ASNS protein.
Discussion
A recent report has implicated ASNS gene in an autosomal recessive form of a neuro developmental condition (Ruzzo et al. 2013). The manifestation of this condition is exclusively neurological abnormalities and includes congenital microcephaly, delayed psychomotor development, progressive encephalopathy and early onset of seizure (Ruzzo et al. 2013). As Mendelian pathogenic mutations are frequently exonic, it has been shown that exome-sequencing is an important tool in genetic research by screening efficiently many coding regions (Biesecker et al. 2011). In this study, we report the use of whole-exome sequencing to identify a novel missense mutation affecting the ASNS gene as responsible for asparagine synthetase deficiency in an Emirati child with early onset encephalopathy. Congenital microcephaly was apparent at birth, intractable seizures developed in the first few weeks of life and the child had severe psychomotor delay. All investigations including full metabolic work up were normal, including normal asparagine levels. Lumbar puncture for CSF asparginase levels is technically impossible because of the severe scoliosis resulting from his neurological condition. The diagnosis was eventually established by WES identifying the mutation in the ASNS gene. This was also the case with the previously reported patients (Ruzzo et al. 2013), so that at the present time molecular diagnosis is the best approach for this condition diagnosis. Screening for mutations in the ASNS gene should be included in the investigations of a child with congenital microcephaly and early onset encephalopathy. Our patient shares most of the clinical features with the reported cases such as progressive microcephaly, epileptic encephalopathy and psychomotor delay as summarized in Table 1. Interestingly, these neurological manifestations was observed in cases with serine and glutamine deficiencies diseases (Häberle et al. 2012; van der Crabben et al. 2013). It has been shown that an oral supplementation of L-serine in cases with serine deficiency seems to be an effective treatment of intractable seizures by improving the white matter volume on MRI in these patients (de Koning et al. 2000; De Koning et al. 2002; de Koning 2006). Therefore, dietary asparagine supplementation, particularly at early stage, might help to alleviate the neurological complications by preventing neuronal damage and provide the brain with the deficient amino acid.
At the molecular level, only three variants were predicted to be deleterious after exome variants prioritization (Table 2). The first variant was detected in the OPRL1 gene (c.754C>T) leading to premature termination codon p.R252*. Knockdown of this gene in mice causes onset and persistence of posttraumatic stress disorder (Andero et al. 2013). In human, two variants in this gene were associated with vulnerability to develop opiate addiction among Caucasians (Briant et al. 2010). A frame shift variant (p.A189Pfs*39) in this gene was listed in the exome variant Server database with a frequency of 3/1988 for homozygous alleles and 5/1988 for heterozygous alleles. Another variant (p.Val271Glyfs*55) has been reported in 1,000 genomes database (rs147012148) but no frequency was determined. Therefore, the nonsense mutation detected in our patient is less likely to be pathogenic. The second variant is a missense mutation c.204G>C in the PIGR gene which was predicted to be possibly pathogenic. This gene is a member of the immunoglobulin superfamily that has been associated with nephropathy in Japanese patients (Obara et al. 2003). Hence, the PIGR gene is less likely the cause of this condition based on its function in inflammation and tumour genesis (Ai et al. 2011; Ocak et al. 2012). The third variant is an additional homozygous missense mutation identified in exon 11 of the ASNS gene. This mutation affects a tyrosine residue that is highly conserved among different species (Fig. 2C). To date, only three homozygous missense mutations have been reported in four families from different ethnic backgrounds (Fig. 2A-c) (Ruzzo et al. 2013). Molecular modeling suggested that this mutation might alter the function/or structure of the asparagine synthetase domain (Fig. 2A-c). This conclusion was based on modeling the structure of the human ASNS mutant protein using as template the closest homologue asparagine synthetase (ASNB) from E.coli. This showed a complete overlap over the core regions, while variations at the extremities can be observed. The protein is highly conserved and the present model is appropriate for the analysis of p.Y398C mutation. The generated model showed that residue Y398 forms hydrogen bonds with R137 which mediates the N and C-domain interaction of the protein (Fig. 2D-b,c). ASNS catalyzes ammonia transfer from glutamine to aspartic acid via a β-aspartyl-AMP intermediate. In the present model, the glutamine binding site in the N-terminal region and the AMP binding site in the C-terminal region are separated by approximately 19 Å. Therefore the coordination between the two sites is brought about by tunnel/channel formed by residues within the protein. Thereby, the interaction of residue Y398 with R137 forms a direct link between C-terminal and N-terminal domains and may form part of the tunnel that bridges the N and C-terminal of ASNS protein. Consequently, mutation of residue Y398 by cysteine will disrupt this interface and may impede the connection between the two active sites. As a result, this substitution will lead to the loss of ASNS activity and therefore is most likely the cause of asparagine synthetase deficiency in the studied patient. Furthermore, it has been shown that the loss of the ASNS function results in accumulation of aspartate and glutamate in the brain leading to in increased excitability, seizure activity, and neuronal damage (Ruzzo et al. 2013). The discovery of pathogenic mutations is important for adopting effective prevention and therapeutic approaches that might minimize the burden of genetic conditions particularly in Arab population (Al-Gazali and Ali 2010; Schuurs-Hoeijmakers et al. 2012; Akawi et al. 2013; Reiff et al. 2014).
In conclusion, we report a novel pathogenic cause of asparagine deficiency in a child with congenital microcephaly and early onset encephalopathy which expands the spectrum of ASNS mutations and supports the relevance of next-generation sequencing diagnosis approaches for autosomal recessive conditions.
Supplementary Material
Acknowledgments
We are indebted the family members for their participation in this study. We are also grateful to Sheikh Hamdan Bin Rashid Al-Maktoum Award for Medical Sciences for their financial support. Molecular work was funded by NIH grants 1P01HD070494 and R01 NS048453.
Footnotes
Conflict of interest
All authors have declared that no competing interests exist.
Electronic supplementary material The online version of this article (doi:10.1007/s11011-014-9618-0) contains supplementary material, which is available to authorized users.
Contributor Information
Salma Ben-Salem, Email: salmabs@uaeu.ac.ae, salma81bs@yahoo.fr, Department of Pathology, College of Medicine and Heath Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates.
Joseph G. Gleeson, Email: jogleeson@ucsd.edu, Department of Neuroscience and Pediatrics, Neurogenetics Laboratory, Howard Hughes Medical Institute, University of California, San Diego, CA, USA.
Aisha M. Al-Shamsi, Email: aishamsi@tawamhospital.ae, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates, Al-Ain, United Arab Emirates.
Barira Islam, Email: barira.khan@gmail.com, Department of Pediatrics, College of Medicine and Heath Sciences, United Arab Emirates University, P.O. Box 17666, Al-Ain, United Arab Emirates.
Jozef Hertecant, Email: jhertecant@tawamhospital.ae, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates, Al-Ain, United Arab Emirates; Department of Pediatrics, College of Medicine and Heath Sciences, United Arab Emirates University, P.O. Box 17666, Al-Ain, United Arab Emirates.
Bassam R. Ali, Email: bassam.ali@uaeu.ac.ae, Department of Pathology, College of Medicine and Heath Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates.
Lihadh Al-Gazali, Email: l.algazali@uaeu.ac.ae, Department of Pediatrics, College of Medicine and Heath Sciences, United Arab Emirates University, P.O. Box 17666, Al-Ain, United Arab Emirates.
References
- Ai J, Tang Q, Wu Y, Xu Y, Feng T, Zhou R, Chen Y, Gao X, Zhu Q, Yue X, Pan Q, Xu S, Li J, Huang M, Daugherty-Holtrop J, He Y, Xu HE, Fan J, Ding J, Geng M. The role of polymeric immunoglobulin receptor in inflammation-induced tumor metastasis of human hepatocellular carcinoma. J Natl Cancer Inst. 2011;103:1696–1712. doi: 10.1093/jnci/djr360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akawi NA, Al-Jasmi F, Al-Shamsi AM, Ali BR, Al-Gazali L. LINS, a modulator of the WNT signaling pathway, is involved in human cognition. Orphanet J Rare Dis. 2013;8:87. doi: 10.1186/1750-1172-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gazali L, Ali BR. Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE) Hum Mutat. 2010;31:505–520. doi: 10.1002/humu.21232. [DOI] [PubMed] [Google Scholar]
- Andero R, Brothers SP, Jovanovic T, Chen YT, Salah-Uddin H, Cameron M, Bannister TD, Almli L, Stevens JS, Bradley B, Binder EB, Wahlestedt C, Ressler KJ. Amygdala-dependent fear is regulated by Oprl1 in mice and humans with PTSD. Sci Transl Med. 2013;5(188):188ra73. doi: 10.1126/scitranslmed.3005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Shianna KV, Mullikin JC. Exome sequencing: the expert view. Genome Biol. 2011;12:128. doi: 10.1186/gb-2011-12-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant JA, Nielsen DA, Proudnikov D, Londono D, Ho A, Ott J, Kreek MJ. Evidence for association of two variants of the nociceptin/orphanin FQ receptor gene OPRL1 with vulnerability to develop opiate addiction in Caucasians. Psychiatr Genet. 2010;20(2):65–72. doi: 10.1097/YPG.0b013e32833511f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, Nickerson DA. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning TJ. Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis. 2006;29:347–351. doi: 10.1007/s10545-006-0269-0. [DOI] [PubMed] [Google Scholar]
- de Koning TJ, Klomp LW. Serine-deficiency syndromes. Curr Opin Neurol. 2004;17:197–204. doi: 10.1097/00019052-200404000-00019. [DOI] [PubMed] [Google Scholar]
- de Koning TJ, Jaeken J, Pineda M, Van Maldergem L, Poll-The BT, van der Knaap MS. Hypomyelination and reversible white matter attenuation in 3-phosphoglycerate dehydrogenase deficiency. Neuropediatrics. 2000;31:287–292. doi: 10.1055/s-2000-12944. [DOI] [PubMed] [Google Scholar]
- De Koning TJ, Duran M, Van Maldergem L, Pineda M, Dorland L, Gooskens R, Jaeken J, Poll-The BT. Congenital microcephaly and seizures due to 3-phosphoglycerate dehydrogenase deficiency: outcome of treatment with amino acids. J Inherit Metab Dis. 2002;25:119–125. doi: 10.1023/a:1015624726822. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, Olvera J, Bafna V, Zaki MS, Abdel-Salam GH, Mansour LA, Selim L, Abdel-Hadi S, Marzouki N, Ben-Omran T, Al-Saana NA, Sonmez FM, Celep F, Azam M, Hill KJ, Collazo A, Fenstermaker AG, Novarino G, Akizu N, Garimella KV, Sougnez C, Russ C, Gabriel SB, Gleeson JG. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78. doi: 10.1126/scitranslmed.3003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom M, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5 Unit 5.6. 2006 doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häberle J, Shahbeck N, Ibrahim K, Schmitt B, Scheer I, O’Gorman R, Chaudhry FA, Ben-Omran T. Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet J Rare Dis. 2012;7:48. doi: 10.1186/1750-1172-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia M. The neuronal ceroid-lipofuscinosis. J Neuropathol Exp Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- Larsen TM, Boehlein SK, Schuster SM, Richards NG, Thoden JB, Holden HM, Rayment I. Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry. 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Obara W, Iida A, Suzuki Y, Tanaka T, Akiyama F, Maeda S, Ohnishi Y, Yamada R, Tsunoda T, Takei T, Ito K, Honda K, Uchida K, Tsuchiya K, Yumura W, Ujiie T, Nagane Y, Nitta K, Miyano S, Narita I, Gejyo F, Nihei H, Fujioka T, Nakamura Y. Association of single-nucleotide polymorphisms in the polymeric immunoglobulin receptor gene with immunoglobulin A nephropathy (IgAN) in Japanese patients. J Hum Genet. 2003;48:293–299. doi: 10.1007/s10038-003-0027-1. [DOI] [PubMed] [Google Scholar]
- Ocak S, Pedchenko TV, Chen H, Harris FT, Qian J, Polosukhin V, Pilette C, Sibille Y, Gonzalez AL, Massion PP. Loss of polymeric immunoglobulin receptor expression is associated with lung tumour genesis. Eur Respir J. 2012;39:1171–1180. doi: 10.1183/09031936.00184410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff RE, Ali BR, Baron B, Yu TW, Ben-Salem S, Coulter ME, Schubert CR, Hill RS, Akawi NA, Al-Younes B, Kaya N, Evrony GD, Al-Saffar M, Felie JM, Partlow JN, Sunu CM, Schembri-Wismayer P, Alkuraya FS, Meyer BF, Walsh CA, Al-Gazali L, Mochida GH. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum Mol Genet. 2014;23(13):3456–3466. doi: 10.1093/hmg/ddu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzo EK, Capo-Chichi JM, Ben-Zeev B, Chitayat D, Mao H, Pappas AL, Hitomi Y, Lu YF, Yao X, Hamdan FF, Pelak K, Reznik-Wolf H, Bar-Joseph I, Oz-Levi D, Lev D, Lerman-Sagie T, Leshinsky-Silver E, Anikster Y, Ben-Asher E, Olender T, Colleaux L, Décarie JC, Blaser S, Banwell B, Joshi RB, He XP, Patry L, Silver RJ, Dobrzeniecka S, Islam MS, Hasnat A, Samuels ME, Aryal DK, Rodriguiz RM, Jiang YH, Wetsel WC, McNamara JO, Rouleau GA, Silver DL, Lancet D, Pras E, Mitchell GA, Michaud JL, Goldstein DB. Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron. 2013;80:429–441. doi: 10.1016/j.neuron.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson JF, Barth PG, de Vries JI, Menko FH, Ruitenbeek W, van Oost BA, Jakobs C. Familial mitochondrial encephalopathy with fetal ultrasonographic ventriculomegaly and intracerebral calcifications. Eur J Pediatr. 1994;153:510–516. doi: 10.1007/BF01957007. [DOI] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B, van de Vondervoort II, van der Graaf M, Nobau AC, Otte-Höller I, Vermeer S, Smith AC, Humphreys P, Schwartzentruber J, Ali BR, Al-Yahyaee SA, Tariq S, Pramathan T, Bayoumi R, Kremer HP, van de Warrenburg BP, van den Akker WM, Gilissen C, Veltman JA, Janssen IM, Vulto-van Silfhout AT, van der Velde-Visser S, Lefeber DJ, Diekstra A, Erasmus CE, Willemsen MA, Vissers LE, Lammens M, van Bokhoven H, Brunner HG, Wevers RA, Schenck A, Al-Gazali L, de Vries BB, de Brouwer AP, Consortium FC. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockler S, Plecko B, Gospe SM, Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, Tein I, Jakobs C, Clayton P, Van Hove JL. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104:48–60. doi: 10.1016/j.ymgme.2011.05.014. [DOI] [PubMed] [Google Scholar]
- van der Crabben SN, Verhoeven-Duif NM, Brilstra EH, Van Maldergem L, Coskun T, Rubio-Gozalbo E, Berger R, de Koning TJ. An update on serine deficiency disorders. J Inherit Metab Dis. 2013;36:613–619. doi: 10.1007/s10545-013-9592-4. [DOI] [PubMed] [Google Scholar]
- Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF, Goddard TD, Meng EC, Sali A, Ferrin TE. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol. 2012;179:269–278. doi: 10.1016/j.jsb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Lambert MA, Cairney AE, Wills D, Ray PN, Andrulis IL. Molecular structure of the human asparagine synthetase gene. Genomics. 1989;4:259–265. doi: 10.1016/0888-7543(89)90329-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.