

Abstract
Arginase 1 deficiency is a urea cycle disorder associated with hyperargininemia, spastic diplegia, loss of ambulation, intellectual disability, and seizures. To gain insight on how loss of arginase expression affects the excitability and synaptic connectivity of the cortical neurons in the developing brain, we used anatomical, ultrastructural, and electrophysiological techniques to determine how single-copy and double-copy arginase deletion affects cortical circuits in mice. We find that the loss of arginase 1 expression results in decreased dendritic complexity, decreased excitatory and inhibitory synapse numbers, decreased intrinsic excitability, and altered synaptic transmission in layer 5 motor cortical neurons. Hepatic arginase 1 gene therapy using adeno-associated virus rescued nearly all these abnormalities when administered to neonatal homozygous knock-out animals. Therefore, gene therapeutic strategies can reverse physiological and anatomical markers of arginase 1 deficiency and therefore may be of therapeutic benefit for the neurological disabilities in this syndrome.
SIGNIFICANCE STATEMENT These studies are one of the few investigations to try to understand the underlying neurological dysfunction that occurs in urea cycle disorders and the only to examine arginase deficiency. We have demonstrated by multiple modalities that, in murine layer 5 cortical neurons, a gradation of abnormalities exists based on the functional copy number of arginase: intrinsic excitability is altered, there is decreased density in asymmetrical and perisomatic synapses, and analysis of the dendritic complexity is lowest in the homozygous knock-out. With neonatal administration of adeno-associated virus expressing arginase, there is near-total recovery of the abnormalities in neurons and cortical circuits, supporting the concept that neonatal gene therapy may prevent the functional abnormalities that occur in arginase deficiency.
Keywords: adeno-associated virus, arginase deficiency, electrophysiology, gene therapy, motor cortex, synapses
Introduction
Urea cycle disorders result from a deficiency of one of six enzymes or one of the two transporter systems necessary for metabolism of ammonia. In general, the prognosis can be mild encephalopathy to substantial developmental disability, often depending on the severity, duration, and the frequency of episodes of hyperammonemia (Helman et al., 2014). Despite intensive study in the clinic, little is known about the mechanisms of neurological dysfunction. Several hypotheses have been formulated based on biochemical and structural changes (Summar and Tuchman, 2001) in part related to hyperammonemia. Ammonia is freely diffusible across the blood–brain barrier, and, although astroglial cells can provide short-term buffering, osmotically active glutamine leads to edema in addition to alteration of neurotransmitter systems and signal transduction pathways, energy metabolism, and oxidative stress (Butterworth et al., 1987; Braissant et al., 2013; Helman et al., 2014). Ammonia has been shown to compromise potassium buffering of astrocytic membranes and cause clinical neurological abnormalities by impairing cortical inhibition (Rangroo Thrane et al., 2013).
Arginase deficiency results from a loss of arginase 1 (Arg1), the final enzyme in the urea cycle (Brusilow and Horwich, 2001; Lee et al., 2012, 2013), resulting in hyperargininemia (Fig. 1A). It usually presents later in life beginning in late infancy to the second year with microcephaly, spasticity, seizures, loss of ambulation, progressive mental impairment, growth retardation, periodic episodes of hyperammonemia, and failure to thrive (Prasad et al., 1997). Spastic diplegia, a unique feature of arginase deficiency, is not typical of any other urea cycle disorder (Prasad et al., 1997). Although the etiology of this neurologic phenotype is unknown, it appears that the origin is in upper motor neuron function affecting the motor cortex, the basal ganglia, and the corticospinal tract.
Figure 1.

The urea cycle removes excess nitrogen from terrestrial mammals. A, The urea cycle metabolizes excess ammonia with the production of urea; in arginase deficiency, the last step is blocked and hyperargininemia results. B, At 14 d of life, WT and HET mice are indistinguishable, whereas homozygous KO mice are substantially smaller. C, AAV was produced with the thyroxine-binding globulin (TBG) promoter, codon-optimized murine arginase 1 cDNA (co ARG1), human growth hormone polyadenylation signal (hGH PA), and inverted terminal repeats (ITR) at the 5′ and 3′ ends. D, Plasma arginine is markedly elevated in KO animals at day 14, whereas the Treated KO has a substantial reduction [WT, 316.068 ± 14.2 μm, n = 5 mice; HET, 438.4 ± 24.49 μm, n = 5 mice; KO, 2789.17 ± 498.14 μm, n = 9 mice; treated KO, 847.934 ± 30.4 μm, n = 5 mice; one-way ANOVA Kruskal–Wallis test, p ≤ 0.001; Dunn's post hoc test (all pairwise), p < 0.05]. E, With AAV gene therapy, note that the plasma ammonia is equivalent between the Treated KO, HET, and WT [WT, 115.01 ± 22.99 μm; n = 5 mice; HET, 142.27 ± 20.8 μm, n = 5 mice; KO, 973.5 ± 70.5 μm, n = 5 mice; Treated KO, 136.3 ± 19.02 μm, n = 5 mice; one-way ANOVA Kruskal–Wallis test, p = 0.01; Student–Newman–Keuls (SNK) post hoc test (all pairwise), p < 0.05]. F, Plasma taurine is increased in KO animals when compared with WT (WT, 568.6 ± 84.3 μm, n = 5 mice; HET, 902.3 ± 142.16 μm, n = 5 mice; KO, 1271.57 ± 186.9 μm, n = 9 mice; Treated KO, 670.08 ± 80.34 mm, n = 5 mice; one-way ANOVA, p = 0.018; Bonferroni's post hoc test (all pairwise), p < 0.05]. All values are mean ± SEM.
Because Arg1-deficient patients usually avoid the catastrophic hyperammonemic crises characteristic of the other urea cycle disorders and tend to survive much longer (Iyer et al., 1998), their course tends to be one of slowly progressive neurological impairment and intellectual disability. It is not known what the proximate toxin is but, because the neurologic picture is very different from those patients who suffer from severe hyperammonemia of other urea cycle disorders, it is believed to be arginine or a product produced directly from it. To develop a better understanding of the causes of neurological dysfunction in arginase deficiency, we performed anatomical, ultrastructural, and electrophysiological studies of layer 5 (L5) motor cortical neurons with single-copy (heterozygote) or double-copy [homozygous knock-out (KO)] loss of Arg1. We find decreased dendritic complexity, decreased synapse number, decreased intrinsic excitability, and functional synaptic deficits consistent with the anatomical changes observed with an unexpected gradation of abnormalities based on whether this is a single-copy or double-copy loss of Arg1. Strikingly, gene therapy with Arg1 at neonatal stages could rescue nearly all of these abnormalities.
Materials and Methods
Mouse procedures.
Exon 4 of the Arg1 allele was replaced by the neomycin resistance gene to generate the Arg1 heterozygous KO mouse (Iyer et al., 2002), which was backcrossed to achieve a homogeneous NIH–Swiss strain background as described previously (Lee et al., 2012). All mice were housed under specific pathogen-free conditions with food and water provided ad libitum. Mice were kept according to the National Institutes of Health guidelines, and all experimental procedures were conducted in accordance with guidelines for the care and use of research animals at our institution. To generate the gene therapy mice (Treated KO), arginase-deficient pups on postnatal day 2 (determined by PCR genotyping; Gau et al., 2009; Lee et al., 2012) were injected with 1.0 × 1014 genome copies kg−1 of AAVrh10 with thyroxine-binding globulin promoter driving murine Arg1 (codon-optimized) diluted in pharmaceutical-grade saline administered by the superficial facial vein as described previously (Hu et al., 2014). The injections were performed in a total volume of 50 μl with a 28 gauge needle. Mice aged 13–16 d were studied. Mice underwent genotyping by PCR a second time at the time of study to ensure that the initial genotype was correct; this was performed with a second collected tissue sample. Mice were fed standard mouse chow (Picolab Rodent diet 20, catalog #5053; Labdiet/PMI Nutrition International). We attempted to include an equal number of male and female mice.
PCR genotyping.
Genomic DNA was prepared from tail tip by using 5 PRIME ArchivePure DNA Purification (catalog #FP2300860). The genotyping was performed by standard PCR amplification as described previously (Gau et al., 2009; Lee et al., 2012).
Biochemical analysis of plasma.
Plasma amino acid analysis was performed on a Biochrom 30 HPLC amino acid analyzer in a blinded manner. In brief, 30–50 μl of plasma was mixed with equal volumes of Biochrom Seraprep and Lithium dilution buffer. Protein was precipitated by centrifugation, and 10 μl supernatant was injected into the analyzer. Physiological amino acid standards (Sigma-Aldrich) were used to calibrate and determine analyte concentration. Analysis of results was performed using EZchrom Elite software (Agilent Technologies). Results are presented as mean ± SEM.
Ammonia analysis of plasma.
Ammonia analysis was performed using a colorimetric commercial assay kit from Abcam (catalog #ab83360). We followed the protocol recommended by the manufacturer. With this assay, ammonia or ammonium is converted to a product that reacts with the OxiRed probe to generate color (λmax = 570 nm), which can be easily quantified using a plate reader to compare with the values obtained from a standard curve. Five microliters of plasma was used per replicate for each sample tested that was performed in duplicate. Results are presented as mean ± SEM.
Electrophysiology.
All recordings were made from 13- to 16-d-old mice. The brain was removed and placed in ice-cold cutting solution (in mm): 222 sucrose, 11 d-glucose, 26 NaHCO3, 1 NaH2PO4, 3 KCl, 7 MgCl2, and 0.5 CaCl2 (aerated with 95% O2, 5% CO2). Three hundred micrometer coronal slices were cut with a Leica VT1000S Vibratome. Slices were allowed to recover at 37°C in artificial CSF (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 d-glucose, 4 sucrose, 2.5 CaCl2, and 2 MgCl2 (aerated with 95% O2, 5% CO2). To analyze the intrinsic excitability and intrinsic properties, whole-cell recordings of L5 neurons were made in current-clamp mode using a Multiclamp (Molecular Devices) patch-clamp amplifier under visual guidance using differential interference contrast optics with an Olympus BX51 microscope. Traces were low-pass filtered at 10 kHz and acquired at 20 kHz, using NIDAQ cards (National Instruments) running under the WinWCP Strathclyde Electrophysiology Software version 4.8.6. All recordings were performed at 32–35°C. The internal solution contained the following (in mm): 115 K-gluconate, 20 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP-Mg2+, and 0.3 GTP-Na+. Micropipettes were 2–5 MΩ in resistance for all recordings. Series resistance was below 30 MΩ and fully compensated in current-clamp mode. To analyze all action potential (AP) parameters, we injected the minimum current intensity to evoke one AP. Rise time was calculated as the slope between 10 and 90% of the AP amplitude. AP width was measured at 50% of the peak amplitude. Input resistance was calculated as the slope of the linear fit of the voltage–current plot generated from a family of positive and negative current injections (16 steps of 50 pA, 500 ms duration). For all analyses, we used Axograph X software (Molecular Devices).
To measure and analyze the miniature EPSCs (mEPSCs) and the GABA receptor miniature IPSCs (mIPSCs), whole-cell recordings were performed using the Multiclamp (Molecular Devices) patch-clamp amplifier. Data were low-pass filtered at 4 kHz and acquired at 12 kHz using Win EDR Strathclyde Electrophysiology Software version 3.5.2. Cells were voltage clamped at −70 mV. The internal solution for mEPSC recordings was the same that was used previously for current-clamp recordings. These recordings were usually performed for at least 120 s in the presence of 1 μm tetrodotoxin and 50 μm picrotoxin. In the case of mIPSCs recordings, the internal pipette solution contained the following (in mm): 120 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP-Mg2+, and 0.3 GTP-Na+. Recordings were performed for at least 120 s in the presence of 1 μm tetrodotoxin, 10 μm CNQX, and 50 μm AP-5. All miniature postsynaptic currents were analyzed with Axograph X software (Molecular Devices) using a very sensitive method based on an automated search of putative synaptic events by comparison with a template selected by the user (Clements and Bekkers, 1997). Recordings were first digitally filtered at 1 kHz. A few (eight) random events were handpicked from every recording (120 s duration) and averaged to get a representative template. The program used that template to adjust the values of a function that generated a new template with a linear basal region, followed by an idealized synaptic time course. The program used the new template for the detection of synaptic events. Threshold for detection was set to 2.5 times the baseline SD for mEPSCs and 3.2 times the baseline SD for mIPSCs. Captured miniature currents of individual cells were averaged to determine mean amplitude and waveform per cell.
Electron microscopy.
A total of three mice per genotype were included in the electron microscopic (EM) study. The mice were anesthetized and perfused with 1× PBS, followed by cold 4% paraformaldehyde plus 2.5% glutaraldehyde diluted in 1× PBS, pH 7.4. Brains were removed and stored in the fixative for 1–2 d at 4°C. Brains were sliced as coronal sections at ∼200–300 μm in thickness, and the sections were collected in cold 0.1 m phosphate buffer. Sections were dehydrated in graded ethanol and pure acetone and embedded into Araldite based on previous publications (Glynn et al., 2011; Liu and Schumann, 2014). Embedded sections were identified with a light microscope. Sigmacote-treated coverslips (Sigma-Aldrich) were removed, and the portion of cerebral cortex including the whole cortical layers were dissected out and glued to a blank resin block. The blocks were trimmed, and the semithin sections at 0.5–1 μm were cut on an ultramicrotome (Leica EM UC7). Sections were collected on clean microscope slides and stained with toluidine blue. In every case, L5 of the cerebral cortex was identified by examining semithin sections under the light microscope. The pia matter and deep white matter were used as landmarks for identifying cortical layers. Ultrathin sections (70 nm in thickness) containing L5 were further cut on the ultramicrotome (Leica EM UC7) and collected on Formvar-coated single-slot copper grids, stained with uranyl acetate and lead citrate. Sections were examined in a Philips CM120 Electron Microscope at 80 kV. Images were acquired using a high-resolution 2000 × 2000 CCD camera (Gatan). In general, EM images were taken at 5800× for synaptic density analysis or at 9700–13,500× for synaptic structure analysis and counting. L5 pyramidal cell somata were imaged at 1850× for synaptic density analysis.
The quantification of the density of different types of synapses was done as follows. Presynaptic terminals of asymmetrical synapses contain clear presynaptic vesicles with a synaptic cleft and prominent thickening postsynaptic density (Peters et al., 1991); they are considered to use glutamate as a neurotransmitter and usually form asymmetrical synapses with dendritic spines and a small portion of dendritic shafts. Synapses with these features were identified and counted in EM images (the unit area for each image is ∼60 μm2); the total number of asymmetrical synapses in this unit area was counted based on a method described in detail previously (Glynn et al., 2011). For each genotype, 96–100 images were analyzed. The density of asymmetrical synapses was calculated as the number of asymmetrical synapses per square micrometer.
Perisomatic synapses on L5 pyramidal cells were identified as the symmetrical type (described in detail in previous studies; Peters et al., 1991). They are inhibitory in function using GABA as neurotransmitter. We first identified pyramidal cells by examining their general morphology at 1850×, usually by examining the main portion of the somata (which have one round- or oval-shaped nucleus with a prominent nucleolus). We next examined the symmetrical synapses on the somata. The total number of symmetrical synapses on the somata in one thin section were counted. The perimeter of the soma was measured in NIH Image J using the “Measure” function in Analyze. Thus, the density of the synapses was calculated as the total number of synapses counted/perimeter of the soma (micrometers) or number of synapses per micrometer. For each genotype, a total of 35–36 images were analyzed.
To quantify the synaptic distribution, images used for synaptic density quantification were selected. For each genotype, 21–22 EM images taken from L5 of the motor cortex were selected for counting. In each EM image (area is ∼60 μm2), identifiable asymmetrical synapses were counted, and three types of synaptic contacts were classified based on their postsynaptic targets: (1) dendritic spine; (2) dendritic shaft; and (3) perforated synapse. Thus, for each image, the number of synaptic contacts on each of the three targets was counted.
Morphology studies (Golgi staining).
Tissue blocks were Golgi stained (Rapid Golgi variant; Morest, 1981; Valverde, 1993). Briefly, tissue blocks were placed in an ultraclean staining jar containing Rapid Golgi solution composed of 0.25% osmium tetroxide and 2.4% potassium dichromate. The jar was kept in the dark for 6 d while gently shaken. Tissue blocks were then thoroughly rinsed in a freshly prepared solution of 0.75% silver nitrate, blotted, placed in another staining jar containing silver nitrate solution, and kept in the dark while gently shaken for 38–42 h. The blocks were then dehydrated in a series of graded alcohol and embedded in increasingly concentrated solutions of nitrocellulose. The nitrocellulose was then hardened by exposure to chloroform. These hardened nitrocellulose blocks of tissue were then affixed to a sectioning block and cut (at 120 μm in thickness) on an American Optical sliding microtome (Global Medical Instrumentation). The sections were cleared in α-terpineol, thoroughly rinsed in xylenes, mounted on slides, and coverslipped using Permount (Thermo Fisher Scientific). All slides were coded so that data could be collected in a blinded manner.
The Sholl analysis was used to quantify the amount and distribution of dendritic branches at increasing distances from the soma (Sholl, 1953). The Golgi-impregnated neurons were visualized using a Zeiss bright-field research microscope (Carl Zeiss). Long-working distance neofluor oil-immersion objectives (40, 63, and 100×) were used during different phases of the analysis. Camera lucida drawings were prepared of randomly selected L5 neurons from each brain. A camera lucida (two-dimensional) tracing is made from the three-dimensional microscopic image. A template composed of a series of concentric circles is placed over the camera lucida drawing with the soma in the center. The number of intersections of the dendrites with each of the concentric shells is quantified. In these studies, the analysis was made of the basilar tree of the L5 pyramids of the cortex.
Statistics.
In general, to examine significance across multiple comparisons and depending on the normality and the equal variance test of the distributions, we performed a one-way ANOVA (Kruskal–Wallis test), followed by Dunn's post hoc test when the distributions did not pass the normality test or the equal variance test. In case of ammonia analyses, we used the Student–Newman–Keuls all pairwise post hoc test. In cases of normal distributions and when the equal variance test was passed, we performed one-way ANOVA, followed by Bonferroni's post hoc test. Statistical calculations were performed using SigmaStat version 3.5. (Systat Software). Error bars in all cases represent SEM.
To analyze the excitability, means were compared nonparametrically using the mixed-effects linear regression model with bootstrapping, that is, the SEMs were estimated empirically based on 500 resamplings of the data. Genotype, current, and genotype × current were modeled as fixed effects, whereas the animal was modeled as a random effect in the above model, thus taking into account that observations in the same animal were not independent. Post hoc adjustments were performed for six pairwise comparisons at a given current using the Holm criterion. Errors bars represent SEM.
For Sholl analysis, we used repeated-measures ANOVA, followed by Bonferroni's multiple comparisons test. Error bars represent SEM.
Results
Intrinsic excitability is decreased in Arg1-deficient mice and is rescued with neonatal gene therapy
To determine whether Arg1 deficiency alters the intrinsic excitability of motor cortical neurons, we performed whole-cell recordings from L5 motor cortical pyramidal neurons (a major output layer) in slices of 13- to 16-d-old wild-type (WT), heterozygous (HET; single-copy loss of Arg1), KO (double-copy loss), and adeno-associated virus (AAV)-based Arg1 gene therapy (Treated KO) mice (Fig. 1B). We focused on this developmental age because this is a stage when the animals began to show motor difficulties. Unlike untreated KO mice, which die at 13–16 d of age, mice treated with AAV-based Arg1 gene therapy (Fig. 1C) survive to adulthood and have controlled plasma arginine (Fig. 1D) and normal plasma ammonia (Fig. 1E) levels. Like arginine, taurine levels are markedly elevated in untreated KO mice, whereas plasma levels in Treated KO mice are not significantly different from the WT (Fig. 1F).
We found that Arg1 KO and, unexpectedly, HET neurons fired fewer APs than WT neurons to equivalent current step injections, with the phenotype more pronounced in KO neurons. Interestingly, recordings from neurons of KO animals treated with AAV-based Arg1 (Treated KO) dramatically increased the excitability of KO neurons to WT levels, such that gene therapy was able to recover the excitability completely to that of the WT (Fig. 2A,C). Arg1 KO neurons had slower rise times compared with HET and Treated KO mice and were broader at half-width as a result of slowed repolarization rates compared with those recorded WT, HET, and Treated KO neurons (Fig. 2B,D). Although the input resistance was decreased in HET and KO compared with WT (Fig. 2D), the resting membrane potential (RMP) was more depolarized in the Treated KO compared with the rest of the groups (Fig. 2D); this change cannot explain the rescue of the excitability because neurons were all held at −70 mV before current injection.
Figure 2.
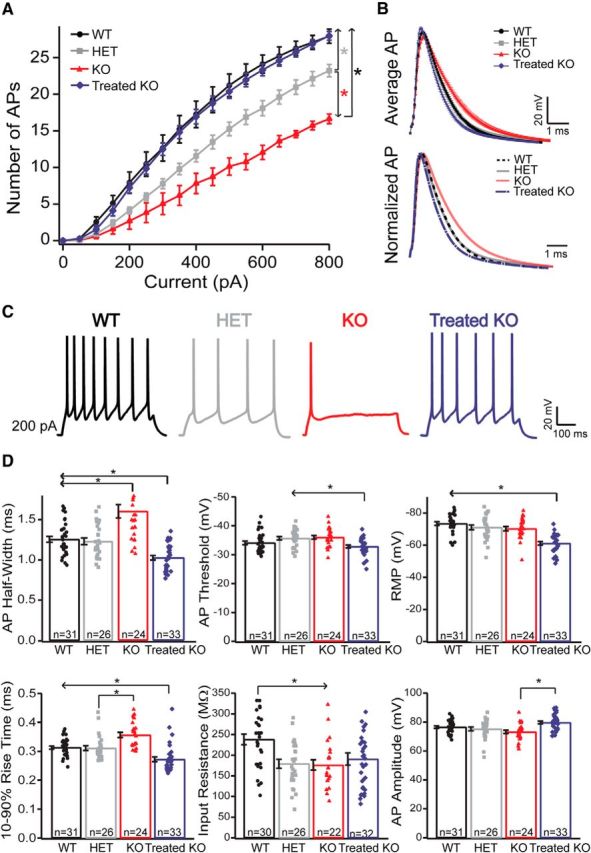
Intrinsic excitability in Arg1-deficient mice is altered. This alteration was recovered with neonatal gene therapy. A, Intensity–frequency plot in response to somatic current injection (16 steps of 50 pA, 500 ms duration) in WT, HET, KO, and Treated KO neurons. Note the decreased firing rate in KO neurons (red line) versus HET (gray line) and versus WT controls (black line). There is an intermediate intrinsic excitability phenotype found in HET and a total recovery of the excitability in Treated KO neurons (WT, n = 28 neurons; HET, n = 23 neurons; KO, n = 22 neurons; Treated KO, n = 32 neurons; means were compared nonparametrically using the mixed-effects linear regression model with bootstrapping). Post hoc adjustments were performed for six pairwise comparisons at a given current using the Holm criterion. Overall p values (Holm adjusted) are by pair comparisons: WT-KO and Treated KO-KO, *p < 0.001; for KO-HET, *p < 0.001; HET-WT and HET-Treated KO, *p ≤ 0.001; for WT-Treated KO, p = 0.764. Error bars are ± bootstrapped SEMs). B, Top, AP average (1 ms) from WT (black trace), HET (gray trace), KO (red trace), and Treated KO (blue trace) neurons. (Bottom) Normalized average of AP traces from the four genotypes. Note that the rising phase of the AP is slower in KO mice (dotted red trace) compared with Treated KO (blue trace) and HET (gray trace), and the half-width is broader compared with the WT control (dashed black trace), the HET, and the Treated KO. There is a recovery in AP shape with the gene therapy (Treated KO). C, Example of voltage traces elicited by current injection (200 pA) in an L5 pyramidal neuron from WT (black), HET (gray), KO (red), and Treated KO (blue) mice in acute slice preparations. Note the differences in excitability for each genotype. D, Bar graphs with scatter plots representing some intrinsic properties of neurons for each genotype. Note that there is a significant recovery in the Treated KO neurons when compared with KO in most of the intrinsic properties analyzed. Although the input resistance was decreased in HET and KO compared with WT [WT, 238.09 ± 12.9 MΩ, n = 30 neurons; HET, 179.18 ± 10.9 MΩ, n = 26 neurons; KO, 176.2 ± 12.37 MΩ, n = 22 neurons; Treated KO, 190.7 ± 14.7 MΩ, n = 32 neurons; one-way ANOVA, p = 0.003; Bonferroni's post hoc test (all pairwise), p < 0.05; Fig. 1D), the RMP was higher in the Treated KO compared with the rest of the groups [WT, −73.42 ± 1.11 mV, n = 31 neurons; HET, −71.06 ± 1.47 mV, n = 26 neurons; KO, −70.24 ± 1.35 mV, n = 24 neurons; Treated KO, −61.14 ± 1.1 mV, n = 33 neurons; one-way ANOVA, p ≤ 0.001; Bonferroni's post hoc test (all pairwise), p < 0.05]. In all traces, error bars indicate ± SEM.
Neurons from treated mice had a slightly more depolarized AP threshold than neurons from HET or KO animals, which again does not explain the treatment-induced changes in excitability. We also measured the afterhyperpolarization in the four groups 25 ms after AP onset. There were no significant differences between groups in this parameter [WT, 18.7 ± 0.4 mV, n = 31 neurons; HET, 17.5 ± 0.5 mV, n = 26 neurons; KO, 17.3 ± 0.6 mV, n = 24 neurons; Treated KO, 18.8 ± 0.5 mV, n = 33 neurons; one-way ANOVA; Bonferroni's post hoc test (all pairwise), p > 0.05]. In general, except for the RMP, all other intrinsic properties and parameters were recovered in the Treated KO compared with the untreated KO neurons (Fig. 2D). These data indicate that Arg1 deficiency significantly decreases the intrinsic excitability of motor cortex L5 pyramidal neurons in a dosage-dependent manner and that AAV-based gene therapy is able to recover the intrinsic excitability and the majority of intrinsic properties when compared with untreated KO neurons.
Circuit connectivity is affected in KO neurons and gene therapy is able to recover some parameters
To examine whether synaptic connectivity and excitatory/inhibitory balance is altered in Arg1-deficient mice, we performed whole-cell patch-clamp recordings from L5 pyramidal neurons from motor cortex. We examined both miniature glutamatergic and GABAergic transmission. To investigate excitatory synaptic transmission, we recorded mEPSCs at −70 mV in the presence of tetrodotoxin (TTX; 1 μm) and the GABAA antagonist picrotoxin (50 μm). There were no changes in the distribution of amplitudes of mEPSCs in general between groups, and the cumulative probability distribution of mEPSC amplitudes was not significantly different (Fig. 3A). However, the frequency of mEPSCs was decreased in KO neurons compared with WT. There was no significant difference observed between WT and Treated KO and between WT and HET groups (Fig. 3B).
Figure 3.

Circuit connectivity is affected in KO neurons. Gene therapy is able to recover some parameters. mEPSCs: Decreased mEPSC frequency in L5 KO pyramidal neurons and recovery in the Treated KO. A, Histogram and cumulative distributions of mEPSC amplitudes from the four genotypes of the L5 pyramidal neurons (WT, n = 34 neurons; HET, n = 31 neurons; KO, n = 21 neurons; Treated KO, n = 41 neurons; one-way ANOVA Kruskal–Wallis test, p = 0.131). B, Bar graph with scatter plots representing the mean frequency in the four genotypes. The mEPSC frequency is reduced in KO neurons and is recovered in Treated KO neurons [WT, 12.75 ± 0.81 Hz, n = 34 neurons; HET, 14.51 ± 0.8 Hz, n = 31 neurons; KO, 9.25 ± 0.7 Hz, n = 21 neurons; Treated KO, 10.84 ± 0.53 Hz, n = 41 neurons; one-way ANOVA, p ≤ 0.001; Bonferroni's post hoc test (all pairwise), p < 0.05], whereas no difference was observed between WT-Treated KO and WT-HET. Error bars indicate ± SEM. mIPSCs: Decreased mIPSCs amplitude, but not frequency, in L5 KO pyramidal neurons compared with WT. Recovery using the gene therapy (Treated-KO). C, Histogram and cumulative distributions of mIPSC amplitudes from the four groups of the L5 pyramidal neurons. (Inset, mIPSC average in the four genotypes.) There is a significant reduction in amplitude in HET and KO when compared with WT (D). Bar graph with scatter plot representing the mean amplitudes in the four genotypes. Note that the amplitude is significantly decreased in HET and KO compared with WT. There is recovery in the amplitude when using the gene therapy (WT, −45.09 ± 3.32 pA, n = 32 neurons; HET, −34.79 ± 3.01 pA, n = 31 neurons; KO, −35.58 ± 3.28 pA, n = 30 neurons; Treated KO, −43.25 ± 3.90 pA, n = 37 neurons; one-way ANOVA Kruskal-Wallis test, p = 0.031; Dunn's post hoc test vs WT, p < 0.05). Error bars indicate ± SEM.
To investigate the spontaneous GABAergic transmission, we recorded mIPSCs in the presence of TTX (1 μm) and the AMPA and NMDA blockers CNQX (10 μm) and AP-5 (50 μm), respectively. mIPSC amplitude was significantly decreased with single- and double-copy loss of Arg1 compared with WT neurons. (Fig. 3C, inset in C, D). A partial recovery in amplitude was observed in Treated KO neurons (Fig. 3C, inset in C, D). There was no difference in the frequency of mIPSCs between groups. These results suggest that treatment differentially modulates different components of excitatory and inhibitory transmission.
Electron microscopy demonstrates a decreased density in asymmetrical and perisomatic synapses in HET and homozygous KO neurons and a significant recovery in Treated KO neurons
To detect whether the synapse number and the morphology of those synapses are affected in Arg1 deficiency, we performed EM analyses. We found that, in WT mice, a large number of asymmetrical synapses (putative excitatory synapses) in L5 of the cerebral cortex were formed with dendritic spines (Fig. 4Aa); in addition, a small portion of these synapses exhibited perforations in postsynaptic densities, a feature of excitatory synapses expressing AMPA receptors (Fig. 4Ab). Quantitative analysis from the WT mice demonstrated that the mean asymmetrical synaptic density in L5 was the highest in this group (Fig. 4B, black bar). Homozygous Arg1 KO mice showed a dramatically decreased number of asymmetrical synapses (Fig. 4B, red bar). In HET mice, the mean synaptic density was between the WT and KO (Fig. 4B, gray bar), again demonstrating an intermediate phenotype related to copy number of Arg1.
Figure 4.
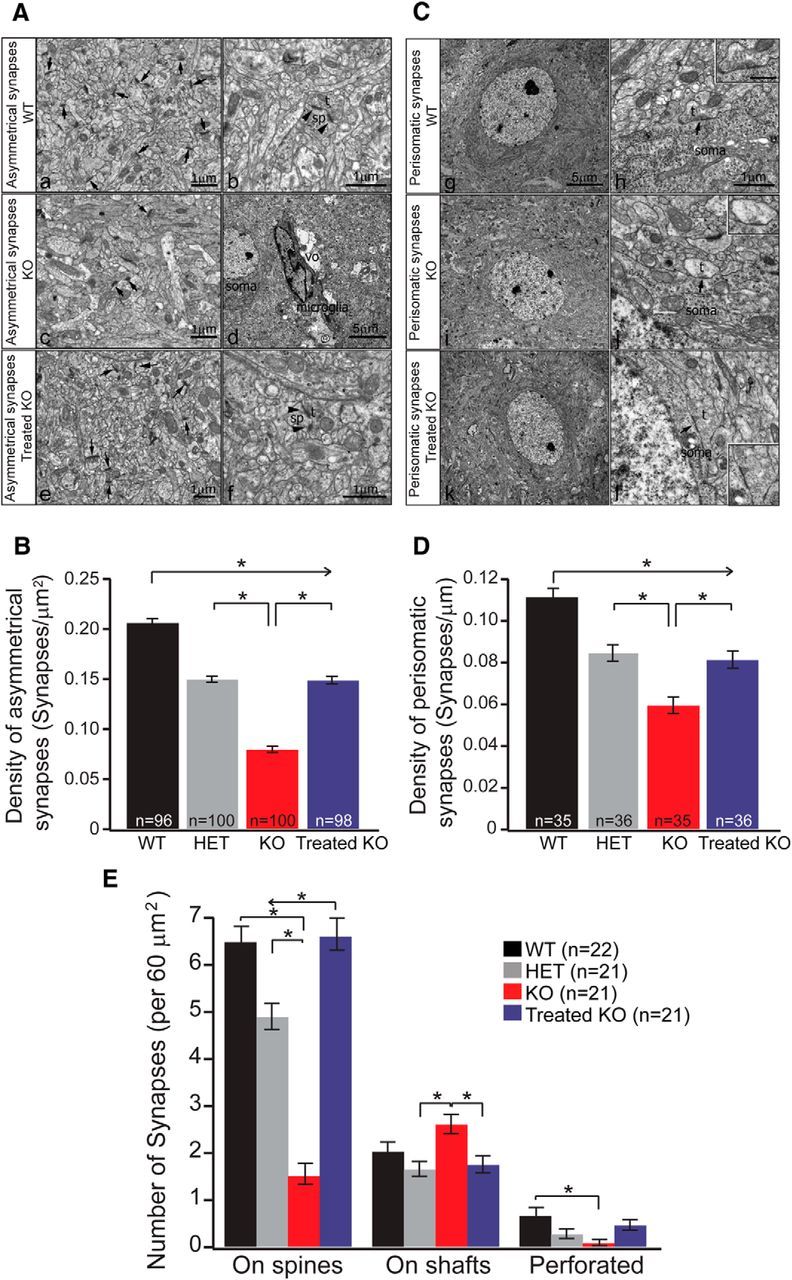
Electron microcopy studies show a decreased density in asymmetrical and perisomatic synapses in KO neurons and a significant recovery in Treated KO neurons, and a significant change in synaptic distribution of L5 neurons was found between groups. A, Comparison of subcellular distribution of asymmetrical synapses in L5 of cerebral cortex of WT, KO, and Treated KO mice. a, b, WT samples, a low-power EM image (a) showing a wide distribution of small axon terminals (t) forming asymmetrical synaptic contacts with many small dendritic spines (sp) presumably derived from pyramidal cells (arrows point to the synaptic contacts). At higher magnification as shown in b, an axon terminal (t) forms an asymmetrical synapse with a dendritic spine (sp) that exhibits a perforated PSD as noted by two arrowheads. Scale bars, 1 μm. c, d, In KO samples, as shown in a low-power image (c), the number of asymmetrical synaptic contacts on dendritic spines (arrows) is drastically reduced compared with WT samples. In d, a low-power EM image shows a microglia cell (microglia) next to a pyramidal cell (soma) and is associated with a vacuolar (vo) structure indicating signs of degeneration. Scale bars: c, 1 μm; d, 5 μm. e, f, Treated KO samples: a low-power EM image in e shows restoration in number of asymmetrical synapses on dendritic spines (arrows) when compared with KO samples. f is a high-power image showing a terminal (t) forming asymmetrical synaptic contacts with a perforated PSD on a spine (sp). Scale bars, 1 μm. B, Quantitative analysis of density of asymmetrical synapses in L5 of the mouse cerebral cortex in WT, HET, KO, and Treated KO samples. Histograms showing the distribution of the mean density from different genotypes. It is shown that the density is lowest in KO samples and highest in WT samples, HET remains in the middle between WT and KO, and the density in the Treated KO shows significant restoration when compared with KO [WT, 0.21 ± 0.0038 synapses/μm2, n = 96 images, 3 mice; HET, 0.15 ± 0.0031 synapses/μm2, n = 100 images, 3 mice; KO, 0.079 ± 0.0032 synapses/μm2, n = 100 images, 3 mice; Treated KO, 0.15 ± 0.0037 synapses/μm2; n = 98 images, 3 mice; one-way ANOVA Kruskal–Wallis test, p ≤ 0.001; Dunn's post hoc test (all pairwise), p < 0.05]. C, Change in perisomatic synaptic organization of pyramidal cells in L5 of the cerebral cortex of WT, KO, and Treated KO mice. g is a low-power EM image showing a pyramidal soma in L5; note the prominent nucleolus in a WT mouse sample. Scale bar, 5 μm (applies also to i and k). h is a high-power image showing an axon terminal (t) forming a typical symmetrical synaptic contact with the soma (arrow); the inset shows higher magnification of the same synaptic contact. Scale bar, 1 μm (applies also to j and l). i shows a low-power EM image of a pyramidal soma in L5 of a KO mouse. j shows one small axon terminal (t) lacking synaptic vesicles and forming a symmetrical synaptic contact with the soma. Note few synaptic vesicles in the terminal as shown in the inset. k shows a low-power EM image of a pyramidal soma in L5 of a Treated KO mouse. l shows a symmetrical synaptic contact (t) with the soma. The inset is the high-magnification image of the same synaptic contact. D, Quantitative analysis of the density of perisomatic synapses in L5 of the mouse cerebral cortex in WT, HET, KO, and Treated KO samples. Histograms showing the distribution of the mean density in different genotypes. It is clearly shown that the density is the lowest in KO samples and the highest in WT samples, HET remains intermediate between WT and KO, and the density in Treated KO shows significant restoration when compared with KO [WT, 0.11 ± 0.004 synapses/μm; n = 35 images, 3 mice; HET, 0.085 ± 0.004 synapses/μm, n = 36 images, 3 mice; KO, 0.059 ± 0.004 synapses/μm, n = 35 images, 3 mice; Treated KO, 0.081 ± 0.004 synapses/μm, n = 36 images, 3 mice; one-way ANOVA Kruskal–Wallis test, p ≤ 0.001; Dunn's post hoc test (all pairwise), p < 0.05]. E, Quantitative analysis of three different types of asymmetrical synaptic contacts based on their targets: (1) dendritic spine; (2) dendritic shaft; and (3) perforated synapses [WT, 6.5 ± 0.32 spine synapses/60 μm2, 2.04 ± 0.19 shaft synapses/60 μm2, 0.68 ± 0.16 perforated synapses/60 μm2, n = 22 images, 3 mice; HET, 4.9 ± 0.27 spine synapses/60 μm2, 1.66 ± 0.15 shaft synapses/60 μm2, 0.28 ± 0.1 perforated synapses/60 μm2, n = 21 images, 3 mice; KO, 1.52 ± 0.22 spine synapses/60 μm2, 2.61 ± 0.2 shaft synapses/60 μm2, 0.09 ± 0.06 perforated synapses/60 μm2, n = 21 images, 3 mice; Treated KO, 6.61 ± 0.34 spine synapses/60 μm2, 1.76 ± 0.18 shaft synapses/60 μm2, 0.47 ± 0.11 perforated synapses/60 μm2, n = 21 images, 3 mice; one-way ANOVA Kruskal–Wallis test, p ≤ 0.001 (on spine); p = 0.005 (on shafts); p = 0.008 (perforated); Dunn's post hoc test (all pairwise), p < 0.05]. All values represent mean ± SEM.
EM analysis of KO animals treated with AAV Arg1 gene therapy demonstrated a consistent recovery in synaptic density (Fig. 4B, blue bar). There was also a redistribution of the synapses in L5, with a majority of the asymmetrical synapses contacting dendritic spines similar to WT mice (Fig. 4Ae), unlike the untreated KO mice in which the largest number of synapses occurred on dendritic shafts. In addition, there was an increase in the proportion of perforated asymmetrical synapses compared with the KO (Fig. 4Af). Significant differences were detected between WT and all other groups and between the homozygous Arg1 KO and the other three groups (Fig. 4B).
We also measured the density of symmetrical perisomatic synapses (putative inhibitory synapses) on pyramidal cell bodies in L5 of mouse cerebral cortex. In WT, only symmetrical synapses filled with synaptic vesicles and mitochondria were identified along the somatic surfaces (Fig. 4Cg,Ch), with the highest mean synaptic density of all groups studied (Fig. 4D, black bar). A pattern similar to what we saw with the asymmetrical synapses was found for perisomatic synapses for the HET, KO, and Treated KO groups. The HET samples showed a lower density of symmetrical synapses than WT but still higher than KO mice, again showing an intermediate phenotype (Fig. 4D, gray bar). For KO mice, we observed some changes associated with perisomatic synapses: (1) the number of symmetrical synapses on the somata of the pyramidal cells was decreased (Fig. 4D, red bar); and (2) some synapses showed alteration in the terminals with shrinkage in the sizes and lack of synaptic vesicles (Fig. 4Cj, inset).
Gene therapy with AAV Arg1 partially rescued the decreased perisomatic synapse density in KO animals (Fig. 4D, blue bar).The perisomatic synapses showed features similar to the WT with clear symmetrical synaptic contacts (Fig. 4Cl, inset). Between the four groups, we found significant differences in perisomatic synapses between WT and all other groups and between KO and the other three groups (Fig. 4D).
We quantified the number of synaptic contacts with different targets (dendritic spines, dendritic shafts, and perforated synapses; Fig. 4E). We found an approximately fourfold decrease in the number of synapses on dendritic spines in the untreated KO group when compared with the WT group; a complete recovery of the spine synapse number was observed in the Treated KO group. A moderate reduction was observed in the HET. The number of shaft synapses (synapses on dendritic shafts) was not changed drastically in the four groups, but we could detect a significant difference between KO versus HET and KO versus Treated KO. Overall, the number of perforated synapses was not high in the four groups, but the number in the KO group was reduced, a sevenfold decrease when compared with the WT. There was a significant recovery in the Treated KO group. HETs remained at an intermediate level.
In summary, we found the following. (1) The density of asymmetrical synapses in L5 of the KO mouse cerebral cortex was decreased significantly compared with WT and HET mice. (2) In Treated KO mice, the density of asymmetrical synapses was higher than the KO mice, suggesting a significant recovery. (3) The density of perisomatic synapses in L5 of KO mice was reduced when compared with WT and HET mice. (4) In the treated KO mouse, the density of perisomatic synapses was partially restored. Regarding the change in synaptic contacts, we found a significant loss of excitatory synaptic contacts with dendritic spines, as well as the reduction of perforated synapses of L5 neurons in KO mice. Significantly, a complete recovery of the number of spine synapses in the Treated KO group implies that gene therapy applied to these mice is highly effective.
Development of dendritic arborization is altered in Arg1 deficiency and normalization is achieved with gene therapy
To examine whether Arg1 deficiency has an effect on dendritic complexity and in the development of dendritic arborization, we performed Sholl analysis and measured the dendritic complexity of basal dendritic arbors of Golgi-stained L5 cortical neurons in WT, HET, KO, and Treated KO neurons at 2 weeks of age. Figure 5A shows representative micrographs of the basal dendritic arbor of the different genotypes studied. The WT and Treated KO neurons had increased dendritic length and complexity compared with Arg1-deficient neurons, with homozygous KO neurons being the most affected. The greatest differences between the groups were seen more proximal in relationship to the cell body. More distally in the branching profiles, there were relatively less differences between the four groups. There was also a total rescue of the dendritic length and complexity in Treated KO neurons (blue trace). We found significant differences between the KO and all other groups and HET and all other groups (Fig. 5B).
Figure 5.
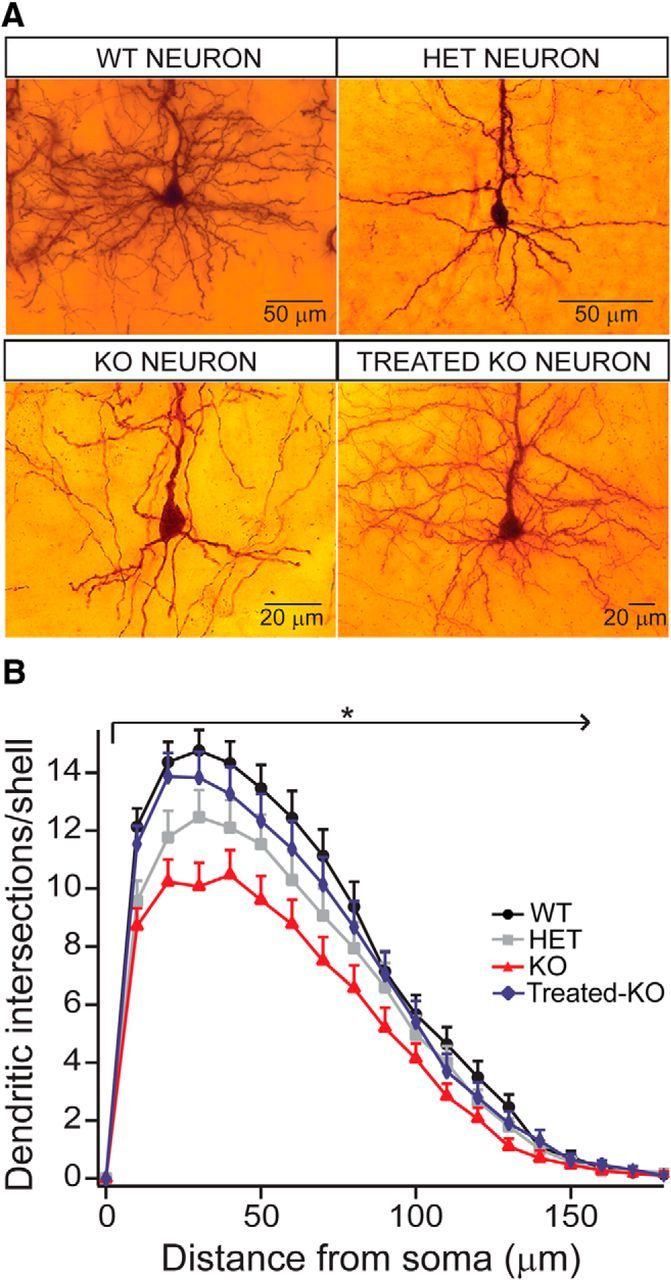
Sholl analysis demonstrates affected dendritic complexity (development) in Arg1-deficient mice. This complexity is recovered using gene therapy. A, Micrographs showing the basal dendritic arbor in the four genotypes. Note that the dendritic complexity is highest in WT, followed by Treated KO (equivalent to WT), HET, and KO, which shows the lowest complexity. B, Sholl analysis distribution diagram. The WT neurons present the most complex dendritic arbor (black trace). The dendritic arbor of KO neurons is affected to the highest degree (red trace), and the HET neurons show an intermediate phenotype between WT and KO. There is a complete rescue of the complexity in Treated KO neurons [blue trace; WT, n = 30 neurons; HET, n = 30 neurons; KO, n = 30 neurons; Treated KO, n = 30 neurons (repeated-measures ANOVA, p < 0.0001; Bonferroni's multiple comparisons test)]. All values are mean ± SEM.
In summary, these studies show that the basal dendritic length and complexity is dramatically decreased by loss of Arg1 in a graded manner and that rescue is achieved with neonatal gene therapy.
Discussion
We have demonstrated in neurons by multiple modalities that, in arginase deficiency, a gradation of abnormalities exists based on the functional copy number of arginase, with heterozygote animals showing an intermediate phenotype. We found that the intrinsic excitability is altered in proportion to the copy number loss of arginase; in addition, there is total recovery in the neurons of AAV-treated animals. Circuit connectivity is altered in KO neurons as shown by decreased frequencies of mEPSCs and the decrease in amplitude of mIPSCs, again with recovery in AAV-treated animals. Electron microscopy demonstrated decreased density in both asymmetrical and perisomatic synapses along with the number of synapses on spines and those that were perforated; with AAV-based gene therapy, there was a significant recovery of both asymmetrical and perisomatic synapses and a redistribution of the number of synapses on spines and those that were perforated. Finally, quantitative analysis of the dendritic complexity demonstrated this to be lowest in the KO and that, with AAV-based neonatal gene therapy, it was completely recovered to that of the WT. Together, these alterations may lead to functional abnormalities and developmental defects and that, with neonatal expression of arginase, these can be prevented.
L5 neurons are a major output projection from the cerebral cortex. They provide the principal descending projections to the thalamus, striatum, and brainstem, as well as the spinal cord (for review, see Shepherd, 2004). These pyramidal cells receive dense inputs from the thalamus and other cortical areas via commissural and communicating fibers (Shepherd, 2004). A reduction in the synaptic input on dendritic spines and somata in the KO indicates that information processing at the cortical level may be severely compromised. Furthermore, we found a reduction of perforated synapses of L5 neurons in KO mice, further supporting that excitatory synaptic transmission may be compromised and, in particular, that the synaptic plasticity (which is frequently associated with an increase in perforated synapses; Connor et al., 2006) is possibly disrupted.
Despite intensive study in the clinic, little is known about the mechanism of brain injury in the urea cycle disorders and in particular arginase deficiency. It has been suggested that these disorders may share mechanisms of injury, such as disrupted astrocyte function, excitotoxic energy, and energy failure (Gropman, 2012). Neuroimaging has demonstrated that there are typical anatomic patterns of vulnerability in certain disorders; however, these are not always easily understood and may be focal or diffuse or may affect gray or white matter (Gropman, 2012).
Although arginase deficiency is a distal defect, ammonia has been implicated with proximal disorders, and several hypotheses have been formulated based on biochemical and structural changes that occur (Summar and Tuchman, 2001). Ammonia is freely diffusible across the blood–brain barrier, and, although astroglial cells can provide short-term buffering, osmotically active glutamine leads to edema in addition to alteration of neurotransmitter systems and signal transduction pathways, energy metabolism, and oxidative stress (Butterworth et al., 1987; Braissant et al., 2013; Helman et al., 2014). Ammonia also compromises potassium buffering of astrocytic membranes and causes clinical neurological abnormalities by impairing cortical inhibition (Rangroo Thrane et al., 2013).
However, arginase-deficient patients are characterized by hyperargininemia; episodes of hyperammonemia are either infrequent or substantially fewer than those with proximal defects. In the arginase-deficient murine model, both arginine and ammonia are markedly elevated at day 14 of life (Iyer et al., 2002; Gau et al., 2009); however, HET (single-copy loss) and WT animals show no difference in ammonia or plasma arginine and yet a phenotype (cortical circuit described herein, not behavioral; Lee et al., 2013) for the heterozygote is present, suggesting that neither hyperammonemia or hyperargininemia are the mechanism for these findings.
Guanidino compounds have been suggested as a potential cause (Deignan et al., 2008). However, no confirmatory evidence exists, and some data, in both humans (Deignan et al., 2010) and mice (Lee et al., 2013), refute this hypothesis. Taurine has numerous effects on the mammalian nervous system, having been shown to be involved in many physiological functions (Foos and Wu, 2002), including as a neurotransmitter and in inhibiting Purkinje cell dendrites in the cerebellum (Okamoto et al., 1983) and pyramidal cells in the hippocampus (Taber et al., 1986). In Arg1 heterozygotes, plasma taurine is nearly 60% higher than that of WT mice, and, in the untreated KO, it is more than double that of the WT. With AAV-based gene therapy, the Treated KO has a marked reduction in plasma taurine that is only slightly above that of the WT. However, no additional investigation of its possible role has been studied, and the cause of the unique neurological findings remain unknown.
With AAV-based gene therapy beginning in the neonatal period, significant recovery was detected in the density of both asymmetrical synapses and perisomatic synapses in L5 of the Treated KO. Interestingly, both asymmetrical and perisomatic synaptic densities were restored to ∼70% compared with the WT. The plasticity of the CNS during early postnatal development can be critical in restoring the synaptic connectivity and function. Our observations with the Treated KO brains strongly indicates that the ability to recover from the deleterious effects of arginase deletion are preserved in the KO mice and that early treatment with genetic modification or other pharmacological manipulation may be critical in restoring neuronal function. At present in the United States, neonatal screening programs include measurement of arginine as a secondary target. Although the interpretive algorithms differ from state to state, either an elevated arginine or a secondary reduced arginine/ornithine ratio should lead to a presumptive diagnosis. Our previous studies with both reporter (Hu et al., 2010; Tai et al., 2015a) and therapeutic genes (Hu et al., 2011, 2014; Hu and Lipshutz, 2012; Lee et al., 2012, 2013) shows that such neonatal interventions in mice can lead to both long-term expression and alteration of phenotype. In addition, although limited in number, we have administered AAV to a neonatal nonhuman primate and demonstrated not only long-term expression of both secreted and intracellular foreign transgene-encoded proteins but also that operational immune tolerance was achieved (Tai et al., 2015b); tolerance may be necessary if a nonsense or missense mutation led to a truncated or abnormal protein that, when the correct protein is expressed postnatally, leads to a neutralizing immune response. Because arginase copy loss leads to significant changes in brain function, development, and maturation, the findings demonstrated in these studies and our other work supports potential neonatal intervention as AAV-based Arg1 gene therapy is effective in not only preventing additional deterioration in neurological function but in promoting the normalization of anatomical and physiological parameters.
In these studies, we were interested in elucidating the underlying changes in the brain that occur in arginase deficiency and hypothesized that arginase copy loss may lead to significant changes in brain function, development, and maturation. Unexpectedly, we found that heterozygotes demonstrated changes at the electrophysiological, dendritic arborization, and synaptic levels and that these changes were more severe in the homozygous KO animal. It was previously the general belief that there were no neurological differences between the HET and WT. In part, this was based on biochemical data that showed only minimal differences that were not statistically different in the plasma ammonia and arginine between these groups. However, our studies in fact demonstrate that there are both structural and functional neuronal differences. This discovery that “asymptomatic” heterozygotes of arginase deficiency demonstrate measurable differences at the neuron, synapse, and circuit level has helped to begin to elucidate the functional abnormalities in arginase deficiency even in the absence of a “clinically” definable phenotypic abnormality and provides a model to test our understanding of the underlying mechanisms and to measure the effects of novel treatments. It is possible that detailed testing of asymptomatic human carriers could show subtle changes in cognitive or motor performance. Alternatively, there may be species differences in arginine and ammonia handling that could obscure such differences between single-copy carriers and noncarriers.
It is conceivable that some of the physiological and anatomical changes we detected in KO animals could be secondary to malnutrition or general ill health. We believe this to be less likely given that HET animals show no obvious deficits and show an intermediate phenotype in many of these measures. Therefore, the physiological and anatomical deficits in the KOs are more likely to be a direct result of arginase deficiency.
Finally, the anatomical and physiological changes observed in the motor cortex are likely not limited to this cortical region but will likely be found across many regions in the cortex. Motor deficits shown by the animals may result from these widespread cortical changes in combination with other alterations in connected subcortical structures.
Footnotes
This work was supported by National Institutes of Health Grants 1R01NS071076-05A1 and 1R01NS071076-04S (G.S.L.) and 1R01MH101198-03 (P.G.). We thank the Semel Institute for Neuroscience and the Intellectual and Developmental Disabilities Research Center at University of California, Los Angeles for support; Dr. Qin Sun at the Analyte Center at the Baylor College of Medicine for performing the amino acid analysis; Dr. Pablo Garcia-Junco Clemente for assistance with the analysis and helpful discussions and advice regarding the electrophysiology studies; Sarah Zajd, Ashmini Misir, Shyam Patel, Juan Hernandez, and Alexis Pena from Neurostructural Research Labs for collecting data for the Sholl study; and Daniela Markovic from University of California, Los Angeles Biomathematics for assistance with the statistical calculations.
The authors declare no competing financial interests.
References
- Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. J Inherit Metab Dis. 2013;36:595–612. doi: 10.1007/s10545-012-9546-2. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Horwich A. The metabolic and molecular bases of inherited disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Ed 8. New York: McGraw-Hill; 2001. pp. 1909–1963. [Google Scholar]
- Butterworth RF, Giguère JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol. 1987;6:1–12. doi: 10.1007/BF02833598. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor S, Williams PT, Armstrong B, Petit TL, Ivanco TL, Weeks AC. Long-term potentiation is associated with changes in synaptic ultrastructure in the rat neocortex. Synapse. 2006;59:378–382. doi: 10.1002/syn.20248. [DOI] [PubMed] [Google Scholar]
- Deignan JL, Marescau B, Livesay JC, Iyer RK, De Deyn PP, Cederbaum SD, Grody WW. Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol Genet Metab. 2008;93:172–178. doi: 10.1016/j.ymgme.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Deignan JL, De Deyn PP, Cederbaum SD, Fuchshuber A, Roth B, Gsell W, Marescau B. Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of patients with argininemia. Mol Genet Metab. 2010;100(Suppl 1):S31–S36. doi: 10.1016/j.ymgme.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res. 2002;27:21–26. doi: 10.1023/A:1014890219513. [DOI] [PubMed] [Google Scholar]
- Gau CL, Rosenblatt RA, Cerullo V, Lay FD, Dow AC, Livesay J, Brunetti-Pierri N, Lee B, Cederbaum SD, Grody WW, Lipshutz GS. Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector. Mol Ther. 2009;17:1155–1163. doi: 10.1038/mt.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, Elmer BM, Garay PA, Liu XB, Needleman LA, El-Sabeawy F, McAllister AK. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14:442–451. doi: 10.1038/nn.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropman AL. Patterns of brain injury in inborn errors of metabolism. Semin Pediatr Neurol. 2012;19:203–210. doi: 10.1016/j.spen.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman G, Pacheco-Colón I, Gropman AL. The urea cycle disorders. Semin Neurol. 2014;34:341–349. doi: 10.1055/s-0034-1386771. [DOI] [PubMed] [Google Scholar]
- Hu C, Lipshutz GS. AAV-based neonatal gene therapy for hemophilia A: long-term correction and avoidance of immune responses in mice. Gene Ther. 2012;19:1166–1176. doi: 10.1038/gt.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Busuttil RW, Lipshutz GS. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J Gene Med. 2010;12:766–778. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Cela RG, Suzuki M, Lee B, Lipshutz GS. Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci U S A. 2011;108:2082–2087. doi: 10.1073/pnas.1015571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Kasten J, Park H, Bhargava R, Tai DS, Grody WW, Nguyen QG, Hauschka SD, Cederbaum SD, Lipshutz GS. Myocyte-mediated arginase expression controls hyperargininemia but not hyperammonemia in arginase-deficient mice. Mol Ther. 2014;22:1792–1802. doi: 10.1038/mt.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O'Brien WE, Yu H, Grody WW, Cederbaum SD. Mouse model for human arginase deficiency. Mol Cell Biol. 2002;22:4491–4498. doi: 10.1128/MCB.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer R, Jenkinson CP, Vockley JG, Kern RM, Grody WW, Cederbaum S. The human arginases and arginase deficiency. J Inherit Metab Dis. 1998;21(Suppl 1):86–100. doi: 10.1023/a:1005313809037. [DOI] [PubMed] [Google Scholar]
- Lee EK, Hu C, Bhargava R, Rozengurt N, Stout D, Grody WW, Cederbaum SD, Lipshutz GS. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol Ther. 2012;20:1844–1851. doi: 10.1038/mt.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Hu C, Bhargava R, Ponnusamy R, Park H, Novicoff S, Rozengurt N, Marescau B, De Deyn P, Stout D, Schlichting L, Grody WW, Cederbaum SD, Lipshutz GS. AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther. 2013;20:785–796. doi: 10.1038/gt.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Schumann CM. Optimization of electron microscopy for human brains with long-term fixation and fixed-frozen sections. Acta Neuropathol Commun. 2014;2:42. doi: 10.1186/2051-5960-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morest DK. The Golgi methods. In: Heym C, Fresmann WG, editors. Neuroanatomical research. Heidelberg, Germany: Springer; 1981. pp. 124–138. [Google Scholar]
- Okamoto K, Kimura H, Sakai Y. Evidence for taurine as an inhibitory neurotransmitter in cerebellar stellate interneurons: selective antagonism by TAG (6-aminomethyl-3-methyl-4H,1,2,4-benzothiadiazine-1,1-dioxide) Brain Res. 1983;265:163–168. doi: 10.1016/0006-8993(83)91350-1. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD. The fine structure of the nervous system. New York: Oxford UP; 1991. [Google Scholar]
- Prasad AN, Breen JC, Ampola MG, Rosman NP. Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J Child Neurol. 1997;12:301–309. doi: 10.1177/088307389701200502. [DOI] [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med. 2013;19:1643–1648. doi: 10.1038/nm.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. Synaptic organization of the brain. Ed 5. Oxford UP; 2004. [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Summar M, Tuchman M. Proceedings of a consensus conference for the management of patients with urea cycle disorders. J Pediatr. 2001;138:S6–S10. doi: 10.1067/mpd.2001.111831. [DOI] [PubMed] [Google Scholar]
- Taber KH, Lin CT, Liu JW, Thalmann RH, Wu JY. Taurine in hippocampus: localization and postsynaptic action. Brain Res. 1986;386:113–121. doi: 10.1016/0006-8993(86)90147-2. [DOI] [PubMed] [Google Scholar]
- Tai DS, Hu C, Kim EH, Lipshutz GS. Augmentation of transgene-encoded protein after neonatal injection of adeno-associated virus improves hepatic copy number without immune responses. Pediatr Res. 2015a;78:239–246. doi: 10.1038/pr.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai DS, Hu C, Lee CC, Martinez M, Cantero G, Kim EH, Tarantal AF, Lipshutz GS. Development of operational immunologic tolerance with neonatal gene transfer in nonhuman primates: preliminary studies. Gene Ther. 2015b;22:923–930. doi: 10.1038/gt.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde F. The rapid Golgi technique for staining CNS neurons: light microscopy. In: Wouterlood FG, editor. Neuroscience protocols. Amsterdam: Elsevier; 1993. pp. 1–9. [Google Scholar]