

Abstract
We have previously reported that obesity attenuates pulmonary inflammation in both patients with acute respiratory distress syndrome (ARDS) and in mouse models of the disease. We hypothesized that obesity-associated hyperleptinemia, and not body mass per se, drives attenuation of the pulmonary inflammatory response and that this effect could also impair the host response to pneumonia. We examined the correlation between circulating leptin levels and risk, severity, and outcome of pneumonia in 2 patient cohorts (NHANES III and ARDSNet-ALVEOLI) and in mouse models of diet-induced obesity and lean hyperleptinemia. Plasma leptin levels in ambulatory subjects (NHANES) correlated positively with annual risk of respiratory infection independent of BMI. In patients with severe pneumonia resulting in ARDS (ARDSNet-ALVEOLI), plasma leptin levels were found to correlate positively with subsequent mortality. In obese mice with pneumonia, plasma leptin levels were associated with pneumonia severity, and in obese mice with sterile lung injury, leptin levels were inversely related to bronchoalveolar lavage neutrophilia, as well as to plasma IL-6 and G-CSF levels. These results were recapitulated in lean mice with experimentally induced hyperleptinemia. Our findings suggest that the association between obesity and elevated risk of pulmonary infection may be driven by hyperleptinemia.
Circulating leptin levels correlate with the risk of respiratory infection independent of BMI in patients and mice.
Introduction
Pulmonary infection is the ninth leading cause of death (1), and accounts for greater than $40 billion/year in direct and associated costs in the US alone (2). Despite initial gains in the late 19th through mid-20th centuries, recent efforts to further reduce the morbidity and mortality associated with bacterial and viral infection have been largely ineffective (3). Recent epidemics, such as the pandemic (pH1N1) influenza outbreaks, have highlighted the continued threat of emerging pathogens for which we have few if any effective treatments.
Epidemiological studies suggest that, in addition to pathogen prevalence and virulence, host factors play a critical role in determining both susceptibility to and outcome from pulmonary infections (4–6). Yet, our understanding of these factors remains limited. In this era of increased antibiotic resistance and novel pathogens, there is a pressing need to identify therapeutic approaches that may restore or enhance host defenses in at-risk populations. Several well-described immunocompromised states (e.g., chemotherapy-induced neutropenia, AIDS), the presence of underlying lung disease, smoking, alcoholism, and the extremes of age (4, 7–9) are associated with elevated risk for pneumonia. Some recent studies have demonstrated associations between obesity and risk for both bacterial and viral pneumonias, as well as increased disease severity and mortality (6, 10, 11). However, others have suggested a protective effect of increasing BMI on mortality in this setting (12, 13). Reasons for this inconsistency remain unclear. Studies in murine models of obesity have shown failure to contain both influenza (14, 15) and bacterial (16, 17) pneumonias with consequent lung injury and death. Subsequent work examining influenza has focused on the role of altered adaptive immune response in obesity (18–21). Our previous studies have revealed obesity-associated defects in the immune response to lung injury in patients with ARDS (22), and we have replicated these findings in obese mouse models of lung injury, in which both the inflammatory cytokine response and development of airspace neutrophilia are attenuated (23). Many of the effects of obesity have previously been ascribed to the development of a baseline proinflammatory state, yet levels of circulating inflammatory cytokines are only mildly elevated (24) compared with the levels typically thought to induce immune paralysis states such as seen with sepsis (25). The dysregulation of adipokines in obesity, however, can be quite severe. The adipokine leptin, for which leukocytes express receptors, is found in extremely high levels in obesity (10- to 50-fold increase compared with lean individuals; refs. 26 and 27). Interestingly, several host factors that have also been inconsistently associated with pneumonia risk, including pregnancy (6), diabetes (28), and chronic renal failure (29), are also associated with hyperleptinemia (30–38). The inconsistency of these associations may reflect the variability of leptin levels in these conditions as studied, raising the possibility that chronic hyperleptinemia may impair immune function.
We hypothesized that obesity-associated hyperleptinemia, and not body mass per se, is linked to an impaired response to pulmonary pathogens and an increased susceptibility to infections. In this study, we present evidence that elevated circulating leptin levels are correlated with an increased risk and severity of respiratory infection, regardless of BMI in both humans and mouse models.
Results
Hyperleptinemia is independently associated with increased risk of respiratory infection in the general population.
The National Health and Nutrition Examination Survey III (NHANES III) database was used to test the association between host factors and annual risk of pulmonary infection. In univariate logistic regression on subjects with complete data (n = 30,818), age (P < 0.0001), female gender (P < 0.0001), race (P = 0.0003), and smoking status (P = 0.0003) were found to correlate significantly with the annual risk of infection (Table 1), concordant with previous studies (4, 39). Similar examination of metabolic variables showed associations between pulmonary infection and BMI (P < 0.0001), glycosylated hemoglobin (HbA1c, P < 0.0001), and renal function (creatinine clearance, P < 0.0001) (Table 1), but neither diabetic status per se nor lipid profile (triglycerides, total cholesterol, LDL, or HDL) were found to be associated.
Table 1. The annual risk for respiratory infection is significantly correlated with age, sex (male), race, BMI, smoking status, glycosylated hemoglobin, and renal function. There were 30,818 subjects from NHANES III. P ≤ 0.05 is considered significant.

Further analysis of a smaller subset of this cohort in which plasma leptin levels were measured (n = 6,252) revealed a significant association between leptin and respiratory infection (odds ratio [OR] = 1.109 per ng/ml; 95% CI 1.003, 1.034; P = 0.019) while controlling for age, sex, BMI, social status, smoking, diabetes, and renal function (Table 2). Several metabolic variables were positively associated with leptin levels in this cohort, including BMI (P < 0.0001), HbA1c (P < 0.0001), and renal function (P < 0.0001), as has been reported in previous studies. Interestingly, these same metabolic variables were associated with respiratory infection in the larger cohort but not in the smaller cohort, which controlled for leptin. These same associations have previously been shown to be variable in other small cohorts (11, 28, 29). Together, these findings suggest the possibility that the effects of these variables on respiratory infection are mediated by leptin.
Table 2. The annual risk for respiratory infection is significantly correlated with serum leptin level, sex (male,) and smoking status. There were 6,252 subjects from NHANES III. P ≤ 0.05 is considered significant.

We also examined the incidence of urinary tract infections in the cohort in which leptin levels were measured. Although the risk for urinary tract infection was associated with serum leptin levels in univariate analysis (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/jci.insight.82101DS1), it was not in multivariate analysis, suggesting that high leptin levels may be associated specifically with respiratory infection but not other infections.
Hyperleptinemia is associated with increased risk of death in patients with severe pneumonia.
We next examined whether leptin levels might be associated with outcomes in critically ill patients with pneumonia. We determined plasma leptin levels in banked samples of critically ill patients with ARDS due to pneumonia (n = 147) who had been enrolled in the ARDSnet/ALVEOLI study of high vs. low PEEP ventilation strategy (40). High circulating leptin levels at day 1 of enrollment were associated with increased mortality (OR = 1.3142 per log leptin; 95% CI= 1.0163, 1.6996; P = 0.037) when adjusted for BMI, gender, disease severity (based on the APACHE II ICU mortality prediction score), and diabetic status. Interestingly, when analyzing the entire cohort, including patients without pneumonia (n = 385), no association was found. Furthermore, elevated leptin levels at enrollment were found to correlate with the diagnosis of pneumonia as the identified risk factor for developing ARDS in the cohort as a whole (OR = 0.616; 95% CI = 0.0875, 1.1447; P = 0.022; n = 385).
Plasma leptin levels are more strongly correlated with lung bacterial burden than body weight in a mouse model of obesity.
We next investigated whether the observed associations between hyperleptinemia and pneumonia risk and severity could be replicated in animal models of obese pneumonia. We examined Klebsiella pneumoniae (K. pneumoniae) infection in a diet-induced obesity (DIO) mouse model (23) to mimic human obesity and pneumonia. Pneumonia severity, as measured by whole lung bacterial CFU at 48 hours, was increased in the obese mice as a group (Figure 1A). The association between plasma leptin levels and lung bacterial burden (Figure 1C) appeared to be stronger than the association between weight and lung bacterial counts (Figure 1B).
Figure 1. Pneumonia severity as measured by bacterial burden correlates with plasma leptin levels in K. pneumoniae–infected lean and obese mice.

(A) Lung bacterial CFU were determined at 48 hours after K. pneumoniae infection in lean (10% fat diet) and diet-induced obese (60% fat diet) mice. In addition, (B) lung CFU was compared with mouse weight and (C) plasma leptin levels by linear regression. n = 11 (10% diet) and n = 9 (60% diet) animals. Black circles indicate lean mice; black squares indicate obese mice. Dashed lines indicate 95% CI. ***P ≤ 0.001 as determined by an unpaired Student’s t test (2-tailed).
Hyperleptinemia, and not body weight, is independently associated with impairment of pulmonary neutrophilia in response to inhaled LPS exposure in obese mice.
We have previously reported an inverse association between body weight and airspace neutrophilia following LPS-induced lung injury in mouse models of obesity (r2 = 0.32, P = 0.02) (23). To determine the relationship between obesity-associated hyperleptinemia and the neutrophil response following lung injury in this model, lavage neutrophil levels were compared with plasma leptin levels 24 hours after injury in obese and lean mice. We found a strong inverse association between bronchoalveolar lavage (BAL) neutrophil counts and plasma leptin levels (P = 0.0002) (Figure 2). Furthermore, in multivariate analysis, including weight and plasma leptin levels, leptin remained significant (P = 0.021), whereas weight did not (P = 0.796). These results suggest that the previously reported defects in the pulmonary neutrophil response to LPS challenge in obese mice (23) may be driven by obesity-associated hyperleptinemia and not obesity per se.
Figure 2. Plasma leptin levels inversely correlate with airspace neutrophilia in LPS-injured lean and obese mice.
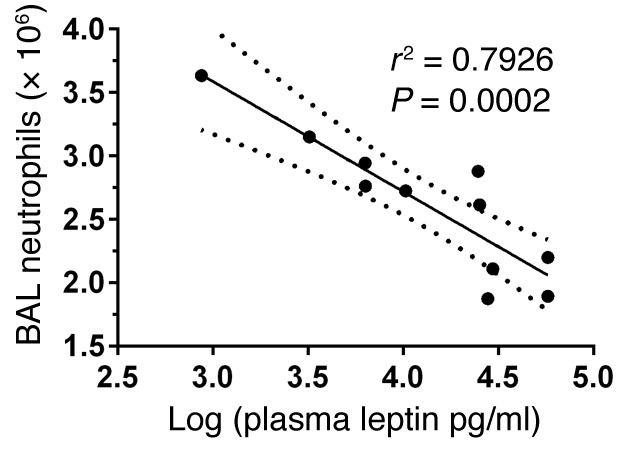
Previously, we have reported that bronchoalveolar lavage (BAL) neutrophil counts and mouse body weight are inversely associated (r2 = 0.32, P = 0.02) following LPS-induced lung injury in diet-induced obese mice (23). We now compared BAL neutrophil counts and plasma leptin levels at 24 hours after LPS-induced lung injury in diet-induced obese (60% fat diet, n = 6) and lean (10% fat diet, n = 5) mice by linear regression. Dashed lines indicate 95% CI.
Hyperleptinemia is associated with decreased plasma cytokine levels independent of body weight in both ARDS patients and obese mice following LPS-induced lung injury.
As in obese patients with ARDS (22), murine obesity is associated with an attenuated release of inflammatory cytokines following LPS-induced acute lung injury (23). To determine whether these relationships might reflect associations between hyperleptinemia and impaired cytokine response, we examined inflammatory cytokine levels in both the ALVEOLI cohort and our murine model of acute lung injury. We found that plasma IL-6 levels were inversely associated with circulating leptin levels independent of BMI (P = 0.047; n = 359) in the ALVEOLI cohort of ARDS patients, whereas IL-8 levels were not associated (P = 0.499; n = 362). In our obese mouse model of acute lung injury, an inverse association was found between plasma leptin levels and plasma IL-6 levels (P =0.01) (Figure 3A), whereas KC (a murine homologue of human IL-8) was not associated (r2 = 0.0025; P = 0.88). Given the changes observed in airspace neutrophil counts in this mouse model following injury, we also examined the neutrophilic cytokine G-CSF and found it to be inversely associated with plasma leptin levels (P = 0.04) (Figure 3B). However, no association was found between body weight and IL-6 or G-CSF in this model (Figure 3, C and D). These results suggest that obesity-associated hyperleptinemia underlies the previously established associations between obesity and impaired cytokine response following lung injury.
Figure 3. Plasma leptin levels are inversely associated with plasma neutrophilic cytokine levels following LPS-induced lung injury in lean and obese mice.

Plasma IL-6 and G-CSF levels were compared with either (A and B) plasma leptin levels or (C and D) body weight at 24 hours after LPS-induced lung injury in diet-induced obese (60% fat diet) and lean (10% fat diet) mice by linear regression. n = 6 for each group. Dashed lines indicate 95% CI.
A mouse model of isolated hyperleptinemia.
To determine whether the association between hyperleptinemia and impaired pulmonary immune response could be causal, we developed a model of induced hyperleptinemia in the absence of obesity and other elements of the metabolic syndrome. In this model, lean mice received daily i.p. injections of PEGylated leptin, at a dose we previously titrated not to alter weight (2 μg), or PBS control for 14 days. Although plasma leptin levels were markedly elevated in leptin-treated mice compared with controls (123.54 ± 26.69 ng/ml vs. 5.07 ± 0.44 ng/ml at 14 days; P = 0.0022) — similar to those in uninjured obese vs. lean mice (98.39 ± 13.52 ng/ml vs. 23.71 ± 5.75 ng/ml; P = 0.0066) — no differences in mouse body weights nor food consumption were observed between these 2 groups during or after 14 days of treatment (Figure 4, A–C). Furthermore, both fasting glucose levels and cholesterol levels were decreased in the hyperleptinemic mice at baseline, whereas LDL levels were unchanged compared with control mice (Figure 4, D–F), demonstrating that this model of induced hyperleptinemia does not cause glucose intolerance or hypercholesterolemia and may actually enhance metabolic control in these mice. These results are in line with previously published findings with leptin replacement therapy in hypoleptinemic lipodystrophy patients, in which it was demonstrated that administration of leptin not only leads to restoration of leptin levels, but also substantially reduces hyperglycemia and hyperlipidemia in these patients (41). To determine whether this model of lean hyperleptinemia might have a proinflammatory effect, we assessed airspace and blood leukocyte counts, as well as inflammatory cytokine levels (KC, MCP-1, MIP-2, G-CSF, TNF-α, IL-1β, and IL-6) in uninjured leptin-treated and control mice. No differences were observed in blood or BAL leukocyte counts (Supplemental Figure 1) or cytokine levels (data not shown) between conditions.
Figure 4. Mouse weight remained unchanged, and plasma leptin levels are increased in hyperleptinemic mice at baseline.

(A) Body weight and (B) food intake/mouse were monitored for 14 days in HL (hyperleptinemia) and control (PBS) mice. Differences between mouse weights and food intake over time between 2 groups were analyzed by repeated measures ANOVA. A drop in food intake after overnight fast for blood glucose measurement on day 12 is indicated with a black arrow. (C) Average daily food intake/mouse/day over 14 days was calculated in HL and control mice. (D) Fasting blood glucose levels were measured on day 12. (E) Plasma cholesterol and (F) LDL levels were measured on day 14. n = 4/5 per group. *P ≤ 0.05, **P ≤ 0.01 as determined by an unpaired Student’s t test (2-tailed).
Pneumonia severity is increased in mice with isolated hyperleptinemia.
To further determine whether the association between plasma leptin levels and pneumonia severity in the diet-induced obese mouse model represents causality and not the effects of other obesity-related factors, we next examined K. pneumoniae infection in our lean mouse model of hyperleptinemia. Whole lung bacterial CFU were significantly increased in the lungs of hyperleptinemic compared with control mice 48 hours after infection (Figure 5A), suggesting that pneumonia severity is increased in this model of lean hyperleptinemia and recapitulating our findings in obese mice. However, airspace neutrophil levels did not differ between lean hyperleptinemic and control mice at 48 hours after infection (Figure 5B). Interestingly, as leptin injections were stopped in the leptin-treated mice at the time of bacterial inoculation, plasma leptin levels were found to be similar between leptin-treated and control mice at the time of euthanasia, 48 hours after the last leptin dose was given (6.37 ± 1.89 ng/ml vs. 10.71 ± 3.12 ng/ml, P = 0.2665), suggesting that chronic hyperleptinemia may have a lasting effect on pulmonary host defense, even after circulating leptin levels fall to normal range.
Figure 5. Pneumonia severity is increased in a mouse model of isolated hyperleptinemia.
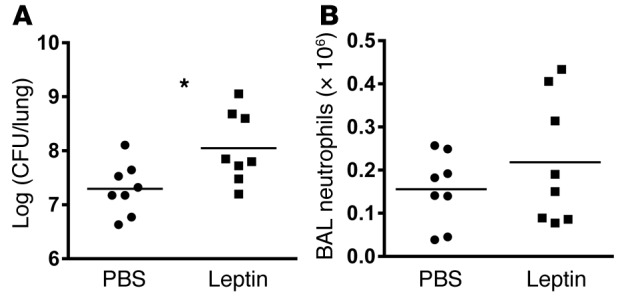
(A) Whole lung K. pneumoniae CFU and (B) BAL neutrophil levels were determined in leptin-treated (2 ug i.p. × 14 days) mice compared with i.p. PBS-treated controls at 48 hours after K. pneumoniae–induced respiratory infection. n = 8 per group. *P ≤ 0.05 as determined by an unpaired Student’s t test (2-tailed).
LPS-induced airspace neutrophilia is attenuated in isolated hyperleptinemia.
To determine whether the previously described effects of DIO on pulmonary neutrophil recruitment and cytokine response seen in our mice might be conferrable by isolated hyperleptinemia, we examined the inflammatory response to inhaled LPS in lean hyperleptinemic mice. BAL neutrophil counts were decreased in hyperleptinemic mice compared with control mice at 24 hours after lung injury (Figure 6A), and linear regression analysis showed a significant inverse correlation between BAL neutrophil counts and plasma leptin levels in these lung-injured mice (Figure 6B). In addition, BAL IL-6 levels were significantly decreased in the hyperleptinemic mice (Supplemental Figure 2). Unfortunately, plasma IL-6 and G-CSF levels were undetectable in both control and hyperleptinemic mice. These results are in line with our observations in the DIO mouse model.
Figure 6. LPS-induced lung injury is attenuated in a mouse model of isolated hyperleptinemia.
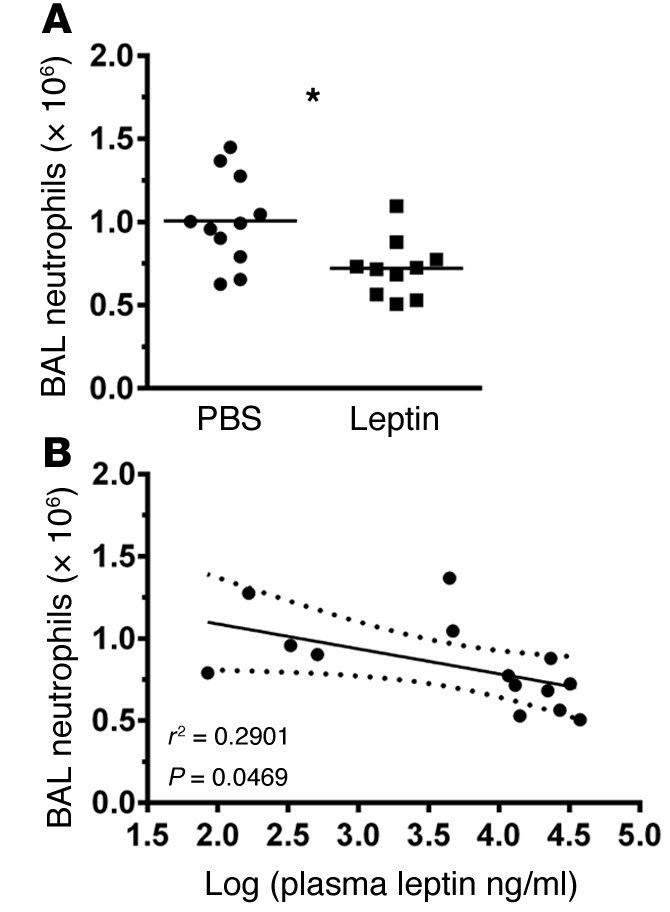
(A and B) BAL neutrophil counts (n = 11 [control] and n = 10 [HL]) were determined in i.p. leptin-treated (14 days) C57Bl/6 WT mice compared with i.p. PBS-treated controls at 24 hours after LPS-induced lung injury and compared with plasma leptin levels by linear regression (n = 6 [control] and n = 8 [HL]). Dashed lines indicate 95% CI. *P ≤ 0.05 as determined by an unpaired Student’s t test (2-tailed).
Neutrophil function is impaired in isolated hyperleptinemia.
As neutrophil function plays a critical role in the response to both respiratory infection and LPS-induced lung injury, we examined 2 aspects of neutrophil function in vitro using BM-derived neutrophils from uninjured lean hyperleptinemic and control mice. We observed impaired neutrophil chemotaxis toward chemokine KC (also known as CXCL1) and the chemotactic peptide N-formylmethionyl-leucyl-phenylalanine (fMLP) (Supplemental Figure 3A), as well as impaired cytokine transcriptional response to in vitro LPS stimulation (Supplemental Figure 3B). Taken together, these results suggest that hyperleptinemia may play an important role in suppressing the pulmonary inflammatory response, at least in part, through suppressive effects on neutrophil function.
Discussion
These analyses demonstrate a significant association between elevated circulating plasma leptin levels and increased risk and severity of respiratory infection in humans, independent of BMI and other established risk factors. These findings can be recapitulated in a mouse model of obesity, in which hyperleptinemia is associated with not only impaired pulmonary bacterial clearance, but also suppression of plasma inflammatory cytokines and attenuated pulmonary neutrophilia following LPS-induced inflammation. Finally, using a model of induced hyperleptinemia, these hyperleptinemia-associated effects can be replicated in the absence of obesity and other associated metabolic defects in mice.
A recent meta-analysis has confirmed the clinical association between increased BMI and risk of both bacterial and viral pneumonias in humans (11). Studies in murine models of obesity and the metabolic syndrome have also shown that the defense against both bacterial and viral respiratory infections is profoundly attenuated (42, 43). Evidence of obesity-associated impairment of the pulmonary innate immune response has recently been demonstrated in the context of acute lung injury/ARDS, as well (44). Although obesity is associated with baseline increases in the same inflammatory cytokines that drive ARDS, counterintuitively, obese patients have better survival from ARDS (45, 46). Although discrete elements of the metabolic syndrome, including both diabetes and hypercholesterolemia, may contribute to obesity’s effects on the pulmonary immune response (42, 47), human and murine studies controlling for these comorbid conditions show that much of obesity’s effects may be independent of these conditions (19, 20), leaving the underlying mechanisms of this impairment poorly defined.
One of the hallmarks of obesity and the metabolic syndrome is a state of leptin resistance with persistent, markedly increased circulating leptin levels compared with lean subjects (27). High leptin levels have also been observed in several nonobese conditions, including diabetes, pregnancy, and renal failure (28, 29), making hyperleptinemia far more prevalent than obesity. Several studies have shown that leptin-deficient mice have impaired host defenses and that restoration of leptin levels in these mice can reverse this impaired response, indicating that leptin may be an important mediator of the pulmonary immunity (42, 47). However, our data suggest that, not only leptin deficiency, but also extremely elevated leptin levels may impair the pulmonary host defense in both obesity-associated hyperleptinemia and isolated lean hyperleptinemia.
The effects of increased leptin levels on the immune response appear to be, in part, dependent on the acute or chronic nature of the exposure. In addition to states of chronic hyperleptinemia, such as obesity, leptin may also be elevated as an acute phase reactant (48). Acute in vitro exposure to leptin has been shown to enhance immune cell function, such as monocyte and macrophage activation, phagocytosis, and cytokine secretion (42, 49, 50). Additional work has shown that leptin acts as a neutrophil chemoattractant and an antiapoptotic (17, 51, 52); it also shows that not only the presence of leptin, but also intact leptin signaling, is important to maintain pulmonary host defense (17). However, in multiple studies using mouse models of obesity, and thus presumed chronic hyperleptinemia, the obese milieu has a protective and antiinflammatory effect in induced acute lung injury, leading to an attenuated pulmonary inflammatory response (23, 53). Furthermore, in studies of the association between lavage leptin levels in ARDS patients, the associations between high leptin levels and clinical outcomes appear to differ based on the context (54). In nonobese patients (BMI < 30), high leptin levels (presumably acute) are associated with poor outcomes, whereas in the obese, high levels (most likely reflecting preceding, baseline hyperleptinemia) have no such association.
The attenuated inflammatory response that we report in both human ARDS patients and murine models of lung injury with hyperleptinemia suggests that such defects may underlie the demonstrated associations between high leptin levels and risk for and severity of pulmonary infections. Such associations have not previously been described and appear to be independent of comorbid conditions such as obesity and the metabolic syndrome, given our ability to recapitulate our findings in obese mice using a lean model of hyperleptinemia.
Potential mechanisms underlying the associations between hyperleptinemia and blunted pulmonary immune response remain unclear. We have previously reported that obese ARDS patients have lower plasma levels of both IL-6 and IL-8 compared with lean patients with ARDS (22), and our current study demonstrates that these associations may reflect the presence of obesity-associated hyperleptinemia in these patients — not obesity itself or other elements of the metabolic syndrome. This suggests that the hyperleptinemic state may underlie or at least contribute to the impaired inflammatory response seen in the obese. Such an inverse association between leptin and IL-6 has been previously demonstrated in patients with sepsis (55), and we describe similar effects in our mouse models of obesity and LPS-induced lung injury, in which not only circulating leptin levels were inversely associated with plasma IL-6 levels independent of weight, but also with plasma G-CSF and airspace neutrophil levels. These results suggest for the first time to our knowledge that the impaired inflammatory response seen in obese ARDS patients and obese mouse models of lung injury may be driven, at least in part, by the effects of obesity-associated hyperleptinemia, and further, that these effects may increase the susceptibility to and severity of pulmonary infection in the general population.
As leptin has previously been shown to behave as an acute phase reactant (48), the possibility of “reverse causation” must be considered in several of the murine and clinical pneumonia studies presented, such that worsening inflammation and infection could drive plasma leptin elevation and not the other way around. In order to assess this possibility, we examined plasma leptin levels in both uninjured and K. pneumoniae–infected mice in our models and found them to be similar (5.08 ± 0.44 ng/ml vs. 6.37 ± 1.89 ng/ml, P = 0.5267), suggesting that infection does not significantly alter circulating leptin levels in this model, and arguing against reverse causation. Furthermore, in order to help discriminate between acute and chronic elevations in circulating leptin levels in our lean hyperleptinemia model, we held leptin injections after K. pneumoniae infection and observed that, despite comparable levels of plasma leptin between control and lean hyperleptinemic mice at 48 hours after infection (6.37 ± 1.89 ng/ml vs. 10.71 ± 3.12 ng/ml, P = 0.2665), the leptin-treated mice had increased lung bacterial counts and, thus, worse infection (Figure 5A), implying that it is the leptin level at the time of initial exposure to pathogen (prior to infection) that influences pneumonia severity. A similar dissection of possible reverse causation is not possible in our examinations of the ALVEOLI cohort, yet it is worth noting that all of the patients in the cohort had severe inflammation related to their precipitating cause of ARDS — most relevantly including the sepsis subset — but the relationship between mortality and leptin is only evident in the pneumonia subset. Furthermore, considering the NHANES cohort — given the relative relatively rare occurrence of respiratory infection on an annual basis — it is extremely unlikely that leptin elevation, as captured by the study, could be significantly influenced by such infections, making possibility of reverse causation quite low in these studies.
We have previously shown that neutrophils isolated from uninjured obese mice demonstrate functional defects, including impaired chemotaxis at baseline (23, 56), that likely attenuate neutrophil recruitment to the lung early in the course of infection and contribute to the failure to contain infection in these mice. In the current study, we demonstrate similar functional defects in neutrophils isolated from lean hyperleptinemic mice, indicating that such defects may contribute to the failure to contain bacterial pneumonia in hyperleptinemic mice, as well, and further suggesting that the hyperleptinemic state itself may contribute to these defects in obesity. Given the noted hyperleptinemia-associated defects in neutrophil chemotaxis, and the witnessed impairment in airspace neutrophilia seen in hyperleptinemic mice after LPS exposure, it is interesting that BAL neutrophil levels in lean hyperleptinemic mice at 48 hours after K. pneumoniae infection were found to be similar to those seen in the infected control mice. We have recently shown that a similar phenomenon occurs in diet-induced obese mice after K. pneumoniae infection, in which BAL neutrophil levels are also similar compared with lean controls (56) despite finding impaired airspace neutrophilia following LPS in the obese mice. This parity in airspace neutrophil levels may reflect a compensatory response to worsening infection in the mice with failed bacterial clearance, such that additional neutrophil recruitment signals are released and neutrophils are recruited, though less effectively, as we have previously shown to be the case in obese db/db mice (17). However, further examination of neutrophil recruitment kinetics in our model would be needed to assess this possibility.
There are potential limitations of our approach worth noting. Although standard for a large-cohort health survey study, NHANES III relied on self-reported incidents of respiratory infection, which may introduce some inaccuracy to the dataset. Despite this potential limitation, we found expected associations for previously reported risk factors of respiratory infection (e.g., smoking), suggesting that the cohort is sufficiently robust for such analyses. Both the NHANES and ALVEOLI study cohorts are historic datasets (enrolled in 1988–1994 and 1999–2002) and are thus likely to underrepresent obesity in the general population and the ICU, respectively, compared with its contemporary incidence in the US. Furthermore, in our efforts to understand hyperleptinemia’s effects in the obese state, we must recognize that obesity is a complex disease and consists of a variety of metabolic alterations in addition to hyperleptinemia. To date, 2 elements of the metabolic syndrome, hyperglycemia and dyslipidemia, have been specifically shown to impair the response to bacterial pneumonia (57, 58). In this context, it is important to note that the lean hyperleptinemic mouse model employed in these studies manifests no evidence of either hyperglycemia or dyslipidemia (Figure 4), suggesting that the influence of hyperleptinemia on bacterial burden in the lung is independent of (and likely additive to) these metabolic factors. Lastly, although the results from our mouse model of lean hyperleptinemia (a gain-of-function approach) are strongly suggestive of a causative role of leptin in our findings, further studies as yet unperformed examining the effects of leptin inhibition in hyperleptinemic conditions (loss-of-function) could further strengthen support for our hypothesis that hyperleptinemia drives immune defects predisposing to respiratory infection. In that light, the lack of such complimentary loss-of-function studies is a limitation of the current study.
In summary, the data presented provide evidence that the observed effects of obesity on respiratory infection might be attributable to increased systemic leptin levels independent of other metabolic and mechanical factors. The finding that obesity-associated hyperleptinemia, and not body mass per se, is an important factor in maintenance of pulmonary host defenses is a critical step toward understanding the complex mechanisms underlying increased susceptibility to and mortality from respiratory infections in the obese and other hyperleptinemic populations.
Methods
Human data analysis.
The NHANES III database was used to test the association of serum leptin levels to self-reported infections in a representative sample of noninstitutionalized adults. NHANES is an ongoing data collection of the US Centers for Disease Control (http://www.cdc.gov/nchs/nhanes.htm). Each sample is composed of subjects randomly selected from the US population using a stratified sampling scheme. Consenting subjects submit to an extensive interview, examination, and collection of blood.
NHANES III subjects were interviewed between 1988 and 1994. Eligible subjects included adults aged 18 years or older at the time of the interview who provided data for all the outcomes and potential confounders and had leptin measures reported. Upper respiratory infection (URI) was coded as the answer to the question, “In the past 3 weeks, have you had any respiratory infections, such as flu, pneumonia, bronchitis, or a severe cold?” Pneumonia was coded as the answer to the question, “During the past 12 months, have you had…pneumonia?” The primary outcome measure, respiratory infection, was created by combining the responses to URI and pneumonia. Subjects who reported one or more pneumonias in the last year, one or more URIs in the past 3 weeks, or both were coded as positive.
Variables with the theoretical ability to confound the relationship between leptin and infection because of their known or suspected association with leptin were selected (age, sex, race [white or nonwhite], education [number of years of school], income [above or below $20,000 per year], marital status [currently married or living as married], BMI [measured by trained research staff], smoking status [current smoker vs. former or never smoker], diabetes [“Other than during pregnancy, has a doctor ever told you that you have diabetes or sugar diabetes?”], glycated hemoglobin A1C, and estimated creatinine clearance (59).
Alveoli.
Patients with ARDS in our study participated in an randomized control trial (RCT) of lower versus higher positive-end respiratory pressure, and these patients also received 6 cc/kg tidal volume. Details of this RCT are described elsewhere (40). Briefly, patients were eligible if they required mechanical ventilation and met criteria for acute lung injury according to the American-European Consensus Conference (AECC) definition (60). Patients with body weight greater than 1 kilogram per centimeter of height were excluded. For each participant, an Acute Physiology and Chronic Health Evaluation (APACHE) score during the 24 hours following ICU admission was calculated, and the physician investigator identified the primary risk factor for the development of acute lung injury (ALI; sepsis, trauma, pneumonia, aspiration, multiple transfusions, or other) as previously described (61).
Human cytokine analysis.
In the NHANES study human serum leptin levels (after an overnight fast) were determined by radioimmunoassay with a polyclonal antibody raised in rabbits against highly purified recombinant leptin (US Department of Health and Human Services [DHHS] National Center for Health Statistics 2001). Serum IL-6 and leptin levels in samples from the ALVEOLI study were determined by ELISA (both Quantikine, R&D Systems) according to manufacturer’s protocol.
Animals.
Eight- to 12-week-old female C57Bl/6 mice (Harlan) were fed high- vs. normal-fat chow (60% vs. 10% fat; Research Diets) for 20 weeks or i.p. injected with PEGylated leptin (2 μg in 200 μl PBS) (62) or PBS control (200 μl) daily for 14 days. Mouse weights and food intake were monitored, and daily and average food intake was calculated per mouse. Animals were housed in the animal facilities at the University of Vermont, and all experimental animal procedures were approved by the University of Vermont Institutional Animal Care and Use Committee.
Murine exposures.
Murine K. pneumoniae (43816 serotype 2, ATCC, 2 × 103 CFU) infections were performed by oropharyngeal (o.p.) aspiration, as described (63). Lipopolysaccharide lung injury was induced by nebulized LPS (E. coli 0111:B4, Sigma-Aldrich), as described (23).
Murine lung analysis.
Airspace lavage cell counts and cytokine levels, as well as bacterial CFU where appropriate, were determined at 48 hours (K. pneumoniae) and 24 hours (LPS) after exposure (17).
Murine cytokine analysis.
Murine IL-1β, IL-6, KC, MCP-1, TNF-α, and G-CSF levels in plasma and BAL were assessed using a Bio-Plex suspension array system (Bio-Rad), as previously described (23). Plasma and BAL leptin concentrations were measured by ELISA (mouse leptin Quantikine, R&D Systems) (at 12 hours following the last leptin injection for baseline levels) according to manufacturer’s protocol.
Murine blood glucose and cholesterol determination.
Fasting blood glucose levels were determined after an overnight fast by a glucometer (Nipro Diagnostics). In addition, plasma LDL and cholesterol levels were assayed using an Advia Chemistry System (Siemens) (64).
Preparation of morphologically mature murine BM neutrophils.
Mature BM neutrophils were isolated using a 3-step Percoll gradient as previously described (65).
Neutrophil chemotaxis.
Chemotaxis of BM-derived neutrophils was assayed in response to KC (R&D Systems) and fMLP (Sigma-Aldrich) using a 48-well modified Boyden chamber with 5-μm pore polycarbonate membranes (both NeuroProbe Inc.), as previously described (17, 66).
Cytokine transcription.
BM-derived neutrophils were stimulated with LPS (100 ng/ml) for 4 hours at 37ºC, and IL-1β, KC, TNF-α, and MIP-2 expression levels were determined by quantitative PCR using SYBR universal PCR master mix and the ABI PRISM 7700 sequence detection system (Thermo Fisher Scientific) and analyzed as described in online supplement. Expression levels were analyzed using the cycle threshold (ΔΔCt) method and normalized to peptidylprolyl isomerase A (pPia) expression. A taqman AOD primer was used for pPia (Invitrogen). LPS-stimulated neutrophil cytokine expression levels were reported as the fold change over unstimulated neutrophil expression levels. The following primer sequences were used for IL-1β: GCCCATCCTCTGTGACTCAT and AGGCCACAGGTATTTTGTCG; for KC: GCTGGGATTCACCTCAAGAA and TGGGGACACCTTTTAGCATC; for TNF-α: GAACTGGCAGAAGAGGCACT and AGGGTCTGGGCCATAGAACT; and for MIP-2: AGTGAACTGCGCTGTCAATG and TTCAGGGTCAAGGCAAACTT.
Statistics.
Analyses of the data derived from the NHANES III database were performed by univariate linear regression in order to confirm that each of the potential confounders was associated with the independent variable (serum leptin level). Thereafter, univariate logistic regression analysis was used to assess the relationships between the potential confounders and respiratory infection. Those that were associated with both the predictor and the outcome with P < 0.1 were retained in an adjusted multivariate logistic model. The adjusted OR, 95% CI, and P value was calculated for each retained variable, and a P value less than 0.05 was accepted on the OR for leptin as evidence of statistical significance. NHANES analyses were performed with SAS software.
When analyzing ARDSNet data, leptin data were natural log–transformed due to nonnormal distribution. Univariate analysis were performed with linear or logistic regression for continuous and categorical variables, respectively. Multivariable linear and logistic regressions with robust standard errors were used to evaluate the associations between leptin and our dependent variable of interest (IL-6) with adjustment for confounders. In our multivariable models, BMI and APACHE score were fit as a linear continuous variable. Inflammatory biomarker levels were natural log–transformed due to nonnormality. Sex and comorbid diabetes were dichotomous, and risk factor for ALI was fit as an indicator variable. All analyses were performed with Stata 9.0 or greater. Statistical significance was defined as a 2-sided P value less than or equal to 0.05.
Murine data were represented as mean, and analysis of differences between experimental groups was performed by Student’s t test. Differences between mouse weights and food intake over time between 2 groups were analyzed by repeated measures ANOVA. Correlations between murine plasma leptin levels or weight and lung CFU levels, BAL neutrophil counts, and plasma cytokine levels were analyzed by linear regression. All analyses were performed using Prism 6 software (GraphPad). Results with P less than or equal to 0.05 were considered statistically significant.
Study approval.
The study protocol for the ARDSNet/ALVEOLI study was previously approved by IRBs of the University of Vermont and was conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent prior to their participation in the study. All animal experiments were performed in accordance with and approved by the University of Vermont Institutional Animal Care and Use Committee.
Author contributions
NDJU, RDS, BL, EFMW, and BTS conceived and designed the research. NDJU, JHJV, EB, JB, SV, and CMH performed the experiments. NDJU, RDS, EB, CMH, and BL analyzed the data. NDJU, RDS, LZ, JT, MJW, BL, MEP, PEP, AED, EFMW, and BTS interpreted the experiment results. NDJU and BTS prepared the figures and drafted the manuscript. NDJU, RDS LZ, JT, MJW, BL, MEP, PEP, AED, EFMW, and BTS edited and revised the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
This work was supported, in part, by grants from the Weijerhorst Foundation (N.D.J. Ubags, E.F.M. Wouters), an ERS advanced grant (no. 340941) (J. Tavernier), and K23 HL105654 (R.D. Stapleton), R01 AI103003 (M.J. Wargo), R01 HL107291 (M.E. Poynter, B.T. Suratt), P20 GM103496 (M.E. Poynter, B.T. Suratt), and R01 HL084200 (B.T. Suratt) from the NIH. Samples from the ALVEOLI trial were provided by the NHLBI Acute Respiratory Distress Syndrome Network.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(8):e82101. doi:10.1172/jci.insight.82101.
Contributor Information
Elianne Burg, Email: elianne.burg@gmail.com.
Jenna Bement, Email: jennalynn1980@gmail.com.
Sebastian Ventrone, Email: Sebastian.ventrone@uvm.edu.
Lennart Zabeau, Email: lennart.zabeau@vib-ugent.be.
Jan Tavernier, Email: jan.tavernier@vib-ugent.be.
Anne E. Dixon, Email: Anne.Dixon@uvm.edu.
Benjamin Littenberg, Email: Benjamin.Littenberg@uvm.edu.
References
- 1.Vol. 64, No. 2. Hyattsville, Maryland, USA: National Center for Health Statistics; 2016. Xu JQ, Murphy SL, Kochanek KD, Bastian BA. Deaths: final data for 2013. In: National Vital Statistics Reports. [Google Scholar]
- 2.Broulette J, Yu H, Pyenson B, Iwasaki K, Sato R. The incidence rate and economic burden of community-acquired pneumonia in a working-age population. Am Health Drug Benefits. 2013;6(8):494–503. [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong GL, Conn LA, Pinner RW. Trends in infectious disease mortality in the United States during the 20th century. JAMA. 1999;281(1):61–66. doi: 10.1001/jama.281.1.61. [DOI] [PubMed] [Google Scholar]
- 4.Almirall J, et al. New evidence of risk factors for community-acquired pneumonia: a population-based study. Eur Respir J. 2008;31(6):1274–1284. doi: 10.1183/09031936.00095807. [DOI] [PubMed] [Google Scholar]
- 5.Fine MJ, et al. Prognosis and outcomes of patients with community-acquired pneumonia. JAMA. 1996;275(2):134–141. [PubMed] [Google Scholar]
- 6.Mertz D, et al. Populations at risk for severe or complicated influenza illness: systematic review and meta-analysis. BMJ. 2013;347:e82101. doi: 10.1136/bmj.f5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almirall J, Gonzalez CA, Balanzo X, Bolibar I. Proportion of community-acquired pneumonia cases attributable to tobacco smoking. Chest. 1999;116(2):375–379. doi: 10.1378/chest.116.2.375. [DOI] [PubMed] [Google Scholar]
- 8.Baik I, Curhan GC, Rimm EB, Bendich A, Willett WC, Fawzi WW. A prospective study of age and lifestyle factors in relation to community-acquired pneumonia in US men and women. Arch Intern Med. 2000;160(20):3082–3088. doi: 10.1001/archinte.160.20.3082. [DOI] [PubMed] [Google Scholar]
- 9.Samokhvalov AV, Irving HM, Rehm J. Alcohol consumption as a risk factor for pneumonia: a systematic review and meta-analysis. Epidemiol Infect. 2010;138(12):1789–1795. doi: 10.1017/S0950268810000774. [DOI] [PubMed] [Google Scholar]
- 10.Fezeu L, et al. Obesity is associated with higher risk of intensive care unit admission and death in influenza A (H1N1) patients: a systematic review and meta-analysis. Obes Rev. 2011;12(8):653–659. doi: 10.1111/j.1467-789X.2011.00864.x. [DOI] [PubMed] [Google Scholar]
- 11.Phung DT, Wang Z, Rutherford S, Huang C, Chu C. Body mass index and risk of pneumonia: a systematic review and meta-analysis. Obes Rev. 2013;14(10):839–857. doi: 10.1111/obr.12055. [DOI] [PubMed] [Google Scholar]
- 12.Corrales-Medina VF, Valayam J, Serpa JA, Rueda AM, Musher DM. The obesity paradox in community-acquired bacterial pneumonia. Int J Infect Dis. 2011;15(1):e54–e57. doi: 10.1016/j.ijid.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Singanayagam A, Singanayagam A, Chalmers JD. Obesity is associated with improved survival in community-acquired pneumonia. Eur Respir J. 2013;42(1):180–187. doi: 10.1183/09031936.00115312. [DOI] [PubMed] [Google Scholar]
- 14.Easterbrook JD, et al. Obese mice have increased morbidity and mortality compared to non-obese mice during infection with the 2009 pandemic H1N1 influenza virus. Influenza Other Respir Viruses. 2011;5(6):418–425. doi: 10.1111/j.1750-2659.2011.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith AG, Sheridan PA, Harp JB, Beck MA. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J Nutr. 2007;137(5):1236–1243. doi: 10.1093/jn/137.5.1236. [DOI] [PubMed] [Google Scholar]
- 16.Conge GA, Gouache P, Joyeux Y, Goichot J, Fournier JM. Influence of different types of experimental obesity on resistance of the mouse to infection by Salmonella typhimurium and Klebsiella pneumoniae. Ann Nutr Metab. 1988;32(3):113–120. doi: 10.1159/000177423. [DOI] [PubMed] [Google Scholar]
- 17.Ubags ND, et al. The role of leptin in the development of pulmonary neutrophilia in infection and acute lung injury. Crit Care Med. 2014;42(2):e143–1e51. doi: 10.1097/CCM.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlsson EA, Sheridan PA, Beck MA. Diet-induced obesity in mice reduces the maintenance of influenza-specific CD8+ memory T cells. J Nutr. 2010;140(9):1691–1697. doi: 10.3945/jn.110.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson EA, Sheridan PA, Beck MA. Diet-induced obesity impairs the T cell memory response to influenza virus infection. J Immunol. 2010;184(6):3127–3133. doi: 10.4049/jimmunol.0903220. [DOI] [PubMed] [Google Scholar]
- 20.Paich HA, et al. Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obesity (Silver Spring) 2013;21(11):2377–2386. doi: 10.1002/oby.20383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith AG, Sheridan PA, Tseng RJ, Sheridan JF, Beck MA. Selective impairment in dendritic cell function and altered antigen-specific CD8+ T-cell responses in diet-induced obese mice infected with influenza virus. Immunology. 2009;126(2):268–279. doi: 10.1111/j.1365-2567.2008.02895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stapleton RD, Dixon AE, Parsons PE, Ware LB, Suratt BT, Network NARDS. The association between BMI and plasma cytokine levels in patients with acute lung injury. Chest. 2010;138(3):568–577. doi: 10.1378/chest.10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kordonowy LL, et al. Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol. 2012;47(1):120–127. doi: 10.1165/rcmb.2011-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract. 2005;69(1):29–35. doi: 10.1016/j.diabres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Reddy RC, Chen GH, Tekchandani PK, Standiford TJ. Sepsis-induced immunosuppression: from bad to worse. Immunol Res. 2001;24(3):273–287. doi: 10.1385/IR:24:3:273. [DOI] [PubMed] [Google Scholar]
- 26.Sideleva O, et al. Obesity and asthma: an inflammatory disease of adipose tissue not the airway. Am J Respir Crit Care Med. 2012;186(7):598–605. doi: 10.1164/rccm.201203-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinha MK, et al. Evidence of free and bound leptin in human circulation. J Clin Invest. 1996;98(6):1277–1282. doi: 10.1172/JCI118913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valdez R, Narayan KM, Geiss LS, Engelgau MM. Impact of diabetes mellitus on mortality associated with pneumonia and influenza among non-Hispanic black and white US adults. Am J Public Health. 1999;89(11):1715–1721. doi: 10.2105/AJPH.89.11.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6):1883–1887. doi: 10.1378/chest.120.6.1883. [DOI] [PubMed] [Google Scholar]
- 30.Abate N, Chandalia M, Snell PG, Grundy SM. Adipose tissue metabolites and insulin resistance in nondiabetic Asian Indian men. J Clin Endocrinol Metab. 2004;89(6):2750–2755. doi: 10.1210/jc.2003-031843. [DOI] [PubMed] [Google Scholar]
- 31.Castellano Filho DS, et al. Body weight gain and serum leptin levels of non-overweight and overweight/obese pregnant women. Med Sci Monit. 2013;19:1043–1049. doi: 10.12659/MSM.884027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang KC, et al. Plasma leptin is associated with insulin resistance independent of age, body mass index, fat mass, lipids, and pubertal development in nondiabetic adolescents. Int J Obes (Lond) 2004;28(4):470–475. doi: 10.1038/sj.ijo.0802531. [DOI] [PubMed] [Google Scholar]
- 33.Menon V, et al. Factors associated with serum leptin in patients with chronic kidney disease. Clin Nephrol. 2004;61(3):163–169. doi: 10.5414/CNP61163. [DOI] [PubMed] [Google Scholar]
- 34.Molvarec A, et al. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod Biol Endocrinol. 2011;9:e82101. doi: 10.1186/1477-7827-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nordfors L, Lonnqvist F, Heimburger O, Danielsson A, Schalling M, Stenvinkel P. Low leptin gene expression and hyperleptinemia in chronic renal failure. Kidney Int. 1998;54(4):1267–1275. doi: 10.1046/j.1523-1755.1998.00088.x. [DOI] [PubMed] [Google Scholar]
- 36.Schubring C, et al. Longitudinal analysis of maternal serum leptin levels during pregnancy, at birth and up to six weeks after birth: relation to body mass index, skinfolds, sex steroids and umbilical cord blood leptin levels. Horm Res. 1998;50(5):276–283. doi: 10.1159/000023290. [DOI] [PubMed] [Google Scholar]
- 37.Silha JV, Krsek M, Skrha JV, Sucharda P, Nyomba BL, Murphy LJ. Plasma resistin, adiponectin and leptin levels in lean and obese subjects: correlations with insulin resistance. Eur J Endocrinol. 2003;149(4):331–335. doi: 10.1530/eje.0.1490331. [DOI] [PubMed] [Google Scholar]
- 38.Beberashvili I, et al. Longitudinal study of leptin levels in chronic hemodialysis patients. Nutr J. 2011;10:e82101. doi: 10.1186/1475-2891-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almirall J, Bolibar I, Balanzo X, Gonzalez CA. Risk factors for community-acquired pneumonia in adults: a population-based case-control study. Eur Respir J. 1999;13(2):349–355. doi: 10.1183/09031936.99.13234999. [DOI] [PubMed] [Google Scholar]
- 40.Brower RG, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351(4):327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 41.Oral EA, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 42.Mancuso P, Gottschalk A, Phare SM, Peters-Golden M, Lukacs NW, Huffnagle GB. Leptin-deficient mice exhibit impaired host defense in Gram-negative pneumonia. J Immunol. 2002;168(8):4018–4024. doi: 10.4049/jimmunol.168.8.4018. [DOI] [PubMed] [Google Scholar]
- 43.Milner JJ, Sheridan PA, Karlsson EA, Schultz-Cherry S, Shi Q, Beck MA. Diet-induced obese mice exhibit altered heterologous immunity during a secondary 2009 pandemic H1N1 infection. J Immunol. 2013;191(5):2474–2485. doi: 10.4049/jimmunol.1202429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stapleton RD, Suratt BT. Obesity and nutrition in acute respiratory distress syndrome. Clin Chest Med. 2014;35(4):655–671. doi: 10.1016/j.ccm.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong MN, Bajwa EK, Thompson BT, Christiani DC. Body mass index is associated with the development of acute respiratory distress syndrome. Thorax. 2010;65(1):44–50. doi: 10.1136/thx.2009.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martino JL, et al. Extreme obesity and outcomes in critically ill patients. Chest. 2011;140(5):1198–1206. doi: 10.1378/chest.10-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu A, Aronoff DM, Phipps J, Goel D, Mancuso P. Leptin improves pulmonary bacterial clearance and survival in ob/ob mice during pneumococcal pneumonia. Clin Exp Immunol. 2007;150(2):332–339. doi: 10.1111/j.1365-2249.2007.03491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.La Cava A, Matarese G. The weight of leptin in immunity. Immunology. 2004;4(5):371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 49.Dixit VD, Mielenz M, Taub DD, Parvizi N. Leptin induces growth hormone secretion from peripheral blood mononuclear cells via a protein kinase C- and nitric oxide-dependent mechanism. Endocrinology. 2003;144(12):5595–5603. doi: 10.1210/en.2003-0600. [DOI] [PubMed] [Google Scholar]
- 50.Zarkesh-Esfahani H, et al. High-dose leptin activates human leukocytes via receptor expression on monocytes. J Immunol. 2001;167(8):4593–4599. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 51.Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res. 2003;37(8):809–814. doi: 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 52.Bruno A, Conus S, Schmid I, Simon HU. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol. 2005;174(12):8090–8096. doi: 10.4049/jimmunol.174.12.8090. [DOI] [PubMed] [Google Scholar]
- 53.Bellmeyer A, Martino JM, Chandel NS, Scott Budinger GR, Dean DA, Mutlu GM. Leptin resistance protects mice from hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2007;175(6):587–594. doi: 10.1164/rccm.200603-312OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain M, et al. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-gamma. Am J Respir Crit Care Med. 2011;183(11):1490–1498. doi: 10.1164/rccm.201009-1409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torpy DJ, Bornstein SR, Chrousos GP. Leptin and interleukin-6 in sepsis. Horm Metab Res. 1998;30(12):726–729. doi: 10.1055/s-2007-978967. [DOI] [PubMed] [Google Scholar]
- 56.Ubags ND, et al. A comparative study of lung host defense in murine obesity models: insights into neutrophil function. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2016-0042OC. [published online ahead of print April 29, 2016]. doi: 10.1165/rcmb.2016-0042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hunt WR, et al. Hyperglycemia impedes lung bacterial clearance in a murine model of cystic fibrosis-related diabetes. Am J Physiol Lung Cell Mol Physiol. 2014;306(1):L43–L49. doi: 10.1152/ajplung.00224.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madenspacher JH, et al. Dyslipidemia induces opposing effects on intrapulmonary and extrapulmonary host defense through divergent TLR response phenotypes. J Immunol. 2010;185(3):1660–1669. doi: 10.4049/jimmunol.0903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 60. Bernard, G.R., et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med.1994;149(3 pt 1):818–824. [DOI] [PubMed] [Google Scholar]
- 61.Eisner MD, et al. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164(2):231–236. doi: 10.1164/ajrccm.164.2.2011093. [DOI] [PubMed] [Google Scholar]
- 62.Vernooy JH, et al. Leptin modulates innate and adaptive immune cell recruitment after cigarette smoke exposure in mice. J Immunol. 2010;184(12):7169–7177. doi: 10.4049/jimmunol.0900963. [DOI] [PubMed] [Google Scholar]
- 63.Wargo MJ, et al. Hemolytic phospholipase C inhibition protects lung function during Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2011;184(3):345–354. doi: 10.1164/rccm.201103-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palvinskaya T, et al. Effects of acute and chronic low density lipoprotein exposure on neutrophil function. Pulm Pharmacol Ther. 2013;26(4):405–411. doi: 10.1016/j.pupt.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol. 2001;281(4):L913–L921. doi: 10.1152/ajplung.2001.281.4.L913. [DOI] [PubMed] [Google Scholar]
- 66.Petty JM, et al. Pulmonary stromal-derived factor-1 expression and effect on neutrophil recruitment during acute lung injury. J Immunol. 2007;178(12):8148–8157. doi: 10.4049/jimmunol.178.12.8148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.