

Abstract
The human KCNE gene family comprises five genes encoding single transmembrane-spanning ion channel regulatory subunits. The primary function of KCNE genes appears to be regulation of voltage-gated potassium (Kv) channels, and the best-understood KCNE complexes are with the KCNQ1 Kv α subunit. Here, we review the often opposite effects of KCNE1 and KCNE3 on Kv channel biology, with an emphasis on regulation of KCNQ1. Slow-activating IKs channel complexes formed by KCNQ1 and KCNE1 are essential for human ventricular myocyte repolarization, while constitutively active KCNQ1-KCNE3 channels are important in the intestine. Inherited sequence variants in human KCNE1 and KCNE3 cause cardiac arrhythmias but by different mechanisms, and each is important for hearing in unique ways. Because of their contrasting effects on KCNQ1 function, KCNE1 and KCNE3 have proved an invaluable tool in the mechanistic understanding of how channel gating can be manipulated, and each may also provide a window into novel insights and new therapeutic opportunities in K+ channel pharmacology. Finally, findings from studies of Kcne1−/− and Kcne3−/− mouse lines serve to illustrate the complexity of KCNE biology and KCNE-linked disease states.
Keywords: auditory, cardiac arrhythmia, intestine, inherited deafness, Long QT syndrome, potassium channel, voltage-gated
Graphical abstract
Introduction
Bioelectricity - the electrical activity generated by living organisms - is observable across all six kingdoms of life, and essential in higher animals for processes including fertilization, neuronal firing, and muscular contraction. Electrical signals are conducted primarily by ion channels, which have evolved the capability to selectively and rapidly conduct charged, aqueous ions across the hydrophobic barrier established by the plasma membrane.
Within the ion channels, the S4 superfamily contains those that respond to changes in membrane potential via a voltage sensor that includes the S4 domain. The S4 is so named because it is the fourth transmembrane (TM) segment in the 6-TM module that constitutes the pore-forming (α) subunit of eukaryotic voltage-gated potassium (Kv) channels and some prokaryotic voltage-gated sodium (Nav) and calcium (Cav) channels1. While each of these voltage-gated channel classes achieve their respective ion selectivity by distinct mechanisms, the atomic structure and signature sequence for K+ selectivity is highly conserved between primitive tetrameric bacterial K+ channels lacking a voltage sensor2, tetrameric mammalian Kv channels carrying the S43, and even dimeric “two-pore” K2P channels such as human K2P14. Mammalian Nav and Cav channel α subunits consist of four repeats of the 6-TM module whereas mammalian Kv channels form as a tetramer of four non-covalently bound 6-TM α subunits (Figure 1A).
Figure 1. The KCNE gene family.

A. Left, Topology diagram of a 6TM spanning Kv α subunit with voltage sensing domain (VSD) and pore domain highlighted. Right, 1TM topology of a KCNE subunit. Ext, extracellular; Int, intracellular.
B. 4:2 α:β stoichiometry of a KCNQ1-KCNE1 complex.
C. Sequence alignment of the KCNE gene family. Transmembrane (TM) domain boxed; highly conserved residues/features indicated with gray shading.
In Kvs, Navs and Cavs, the voltage sensor is attached to the channel “gate” and when the voltage sensor senses a change in membrane potential and moves in response to this, the movement is communicated to the gate to open or close the pore depending on the channel type; all three channel types are opened by membrane depolarization1. Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels are activated (opened) by membrane hyperpolarization; these channels pass both Na+ and K+ and are referred to as pacemaker channels because they trigger subsequent action potentials in response to undershoot from the previous action potential, and are essential for controlling the heart beat and some rhythmic neuronal firing5.
Of all the known Kv channel α subunits, only one, KCNQ1, has thus far been discovered to have the ability to ostensibly lose its voltage dependence. KCNQ1 co-assembles with members of the KCNE family of single transmembrane segment K+ ion channel ancillary or β subunits (Figure 1A–C) to enable it to juggle such contrasting roles as cardiac myocyte repolarization, and epithelial cell ion homeostasis/transporter regulation6. KCNQ1 exists as a tetramer of α subunits in complexes with two KCNE1 subunits at the plasma membrane7–9, although stoichiometric variability (between 4:2 and 4:4, α:β) has also been proposed 10,11 (Figure 1B). The stoichiometry of other Kv-KCNE complexes other than KCNQ1-KCNE1 has not been reported. Channels formed by KCNQ1 and KCNE1 - originally named slowly activating K+ current (IsK)12 and also referred to as Minimal K+ channel (MinK) 13 - open very slowly, and less easily at negative voltages than homomeric KCNQ1 channels14,15 (Figure 2). In sharp contrast, KCNE3 (originally named MinK-related peptide 2, MiRP2) 16 has even more striking effects, locking open the KCNQ1 S4 to almost entirely remove the voltage dependence of KCNQ1 activation, converting it to a K+ leak channel17,18. This review covers the mechanisms and physiological roles of KCNE1 and KCNE3 regulation of KCNQ1 and other Kv channels. A recent companion review was focused on KCNE219.
Figure 2. Functional effects of KCNE1 and KCNE3 on KCNQ1.

Example traces recorded by two-electrode voltage clamp from oocytes injected with cRNA encoding KCNQ1 alone or with KCNE1 or KCNE3. Voltage protocol shown at top. Scale bars: vertical, 2 μA; horizontal, 1s. Dotted line indicates zero current level. Traces are from 3 and 47.
KCNE1 and KCNE3 in the context of the KCNE gene family
The first-recognized member of the KCNE family, KCNE1 was discovered by expression cloning via injection of rat kidney mRNA into Xenopus laevis oocytes. One fraction of RNA generated a slowly activating, K+-selective and voltage-dependent current measurable by two-electrode voltage-clamp (TEVC) electrophysiology12. This finding initially caused confusion because the sequenced KCNE1 protein was much smaller than, and bore no sequence similarity to, the known Kv α subunits20,21. The current generated by KCNE1 strongly resembled the mammalian cardiac Kv current known as IKs (slow-activating K+ current). Eight years after KCNE1 was cloned, KCNQ1 (then named KvLQT1, also referred to as Kv7.1) was positionally cloned via its linkage to inherited Long QT syndrome (LQTS), a defect in ventricular repolarization that can result from mutations in KCNQ122. KCNQ1 was found, like KCNE1, to be expressed in human heart. Furthermore, Xenopus laevis KCNQ1 was found to be endogenously expressed in Xenopus oocytes. Co-expression of KCNQ1 and KCNE1 recapitulated the characteristics of IKs in cells that, unlike Xenopus oocytes, do not express sufficient endogenous KCNQ1 to yield currents upon heterologous expression of KCNQ1 alone, i.e., Chinese Hamster ovary (CHO) cells. KCNE1 was thus recast as a regulatory, or β, subunit of KCNQ114,15, and was swiftly discovered, like KCNQ1, to associate with LQTS23,24.
KCNE1 was a white elephant for a decade, until the discovery of its relatives using Basic Local Alignment Search Tool (BLAST) screening of online libraries of expressed sequence tags (ESTs)16. Although the five KCNE genes share relatively little similarity with one another, they each exhibit 1-TM topology with an extracellular N-terminal domain and intracellular C-terminal domain. In addition, each possesses a consensus protein kinase C (PKC) phosphorylation site in the membrane-proximal portion of the cytoplasmic C-terminal region19. BLAST searching with features of KCNE1 known to be important for its function circumvented the difficulties presented by relatively low family-wide sequence identity, and uncovered KCNE2, 3 and 4 – which we initially termed MinK-related peptides (MiRPs) 1, 2 and 316. A fifth member, KCNE1L or KCNE5, was also discovered25.
KCNE1 and KCNE3 regulation of KCNQ1
KCNQ1 is a highly versatile Kv α subunit, which is regulated by a number of different types of protein to produce functionally distinct channel complexes6. All five of the KCNE proteins regulate KCNQ1 with distinct functional outcomes, and the effects of KCNE1 and KCNE3 are particularly striking. When heterologously co-expressed with KCNQ1 in mammalian cell lines such as Chinese Hamster ovary (CHO) or human embryonic kidney (HEK) cells, KCNE1 right-shifts the voltage dependence of KCNQ1 activation (opening), slows its activation 5–10 fold14,15, increases its unitary conductance 4-fold (as measured by noise variance analysis)26, slows its deactivation (closing) and eliminates its inactivation27. In sum, this increases KCNQ1 peak currents at positive voltages several-fold (Figure 2).
In contrast, KCNE3 holds open KCNQ1 such that the complex is constitutively active, no longer relying on membrane depolarization to open17. Instead, current magnitude (and direction) through KCNQ1-KCNE3 is dictated almost exclusively by driving force (Figure 2). Direct structural studies of the KCNE proteins in isolation (not complexed with α subunits) revealed that the TM segment of KCNE1 is α-helical, and that approximately half the KCNE1 extracellular domain adopts an α helical conformation28–31. Despite relatively low sequence similarity with KCNE1, the secondary structure distributions of the KCNE3 TM and extracellular domains are quite similar to those of KCNE1 32. A plethora of structure-function studies have been directed toward understanding the molecular bases for these differences, with The McDonald lab discovered a single residue in the TM region (T58 in KCNE1; V72 in KCNE3) that when switched between KCNE1 and KCNE3 could swap their activation properties. Thus, V72T-KCNE3 slows KCNQ1 activation almost as much as KCNE1 does, and eliminates KCNQ1-KCNE3 constitutive activation33. Conversely, T58V KCNE1 introduces half as much constitutive current as observed for KCNQ1-KCNE3, and increases the KCNQ1-KCNE1 activation rate 3-fold. Furthermore, concomitant introduction of two KCNE3 residues, to yield F57T, T58V-KCNE1, yielded KCNQ1-KCNE1 channels that were only twofold slower activating than KCNQ1-KCNE3 and almost matched its fractional constitutive current; these channels could be closed by holding for 3 s at −120 mV instead of the normal −80 mV33.
We subsequently found, using tryptophan tolerance scanning of KCNQ1 S6 followed by mutagenesis of the KCNE1 and KCNE3 residues identified by the McDonald lab, that KCNE1 T58 and L59 interact with KCNQ1 F339 and F340, whereas KCNE3 V72 interacts with KCNQ1 S33834; of note, T58 and L59 mutants were among the first described in KCNE1-linked Jervell and Lange-Nielsen syndrome (JLNS), a cardioauditory syndrome comprising LQTS and sensorineural deafness13 (see below). It should be noted that the McDonald lab study utilized the CHO cell expression system33 whereas our study involved Xenopus oocytes. An interesting phenomenon of KCNQ1-KCNE3 channels is that they exhibit an almost perfectly linear current-voltage relationship when expressed in Xenopus laevis oocytes, while in the typical mammalian heterologous expression systems they exhibit some constitutive activation but a much less linear IV curve. The basis for this difference, be it an artifact of the different techniques used to record whole-cell currents in oocytes (two-electrode voltage clamp, TEVC) versus mammalian cells (patch clamp), or a difference in co-factor expression between the systems, has not to the author’s knowledge been reported. While one may assume the mammalian cells are a truer representation of what might occur in vivo in mammalian tissues, the oocyte system more dramatically highlights the capabilities of KCNE3.
Other KCNE1 and KCNE3 residues are also important for maintaining their tight control of KCNQ1 activation, as uncovered in oocyte studies using TEVC. In KCNE1, the C-terminal domain appears to exert a strong influence on KCNQ1 activation, regulating channel assembly, destabilizing the KCNQ1 open state, altering deactivation rate and modulating rate-dependent facilitation of KCNQ1 activity35. The intracellular end of the KCNE1 transmembrane region is proposed to sit on the S5 end of the KCNQ1 S4-S5 linker (a portion of the channel that links the gate to the voltage sensor)30, and protein-protein interactions were detected between cysteines substituted for wild-type resides H363 (in the S6-proximal region of the KCNQ1 C-terminal domain) and H73, S74 and D76 in the KCNE1 C-terminal domain, and between residues H369 with D7636 (Figure 3A). These interactions probably contribute strongly to slowing KCNQ1 activation and, notably, human KCNE1 S74L and D76N mutations are among the first-described, most-studied, and most pathogenic KCNE1 variants14. The membrane-proximal portion of the extracellular domain of KCNE1 (residues G40 in the activated state and K41 in the resting state) comes close to KCNQ1 S1 (I145, at the extracellular end), as evidenced from substituted cysteine disulfide trapping. Similarly, a cysteine substituted at KCNE1 L42 can be linked to V324C in KCNQ1 S6, with the result of eliminating deactivation. These interactions themselves might not necessarily be highly influential in the gating of wild-type KCNQ1-KCNE1 complexes but they suggest how KCNE1 orientation might change with respect to KCNQ1 during gating37,38 (Figure 3A).
Figure 3. Mechanisms of KCNE1 and KCNE3 control of KCNQ1 gating and conductance.
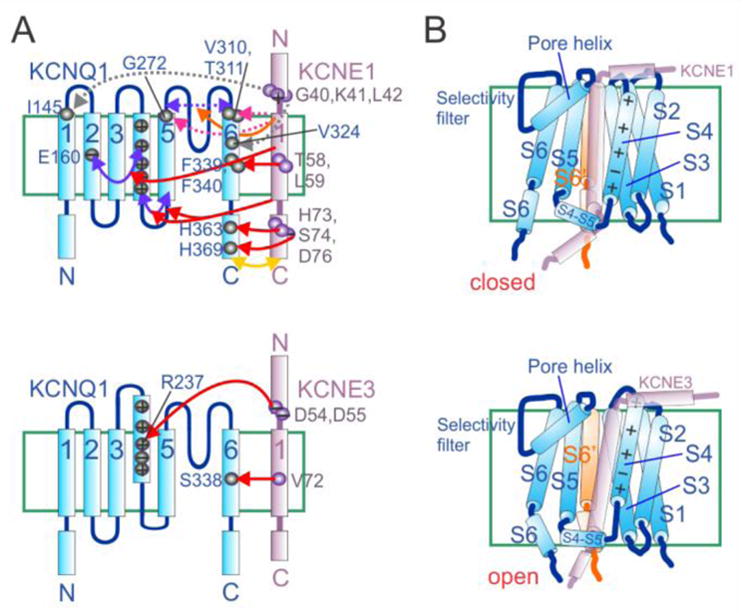
A. Topology diagrams of KCNQ1 with KCNE1 (upper) and KCNE3 (lower) showing residues (numbered where specific residues have been identified as particularly important) and domains identified as being influential in control of KCNQ1 gating by the KCNEs. Arrows: red, KCNE1 or 3 control of KCNQ1 activation; yellow, KCNE1 control of deactivation; orange, KCNE1 control of inactivation; pink, KCNE1 control of conductance; purple, inter-domain interactions within KCNQ1 that are affected by KCNE1; dashed purple, KCNQ1 outer vestibule/selectivity filter flexibility; dashed gray, physical proximity and/or interaction without major functional effects. “−” = acidic residue, “+” = basic residue. Citations appear in main text.
B. Cartoon of possible orientations with respect to KCNQ1 (blue), based on evidence summarized in panel A, of KCNE1 (upper) and KCNE3 (lower) at resting membrane potential. S6′ = the S6 of an adjoining KCNQ1 α subunit. KCNEs are depicted as semi-transparent to avoid concealing KCNQ1 features.
In contrast, in KCNE3, the transmembrane domain appears to override the influence of its C-terminal domain39. In addition, The KCNE3 N-terminal region possesses two acidic residues crucial for the robust constitutive current exhibited by KCNQ1-KCNE3 channels when expressed in oocytes. Using double-mutant thermodynamic cycle analysis, we discovered energetic coupling of KCNE3 residues D54 and D55 specifically to residue R237 in the voltage-sensing S4 helix of KCNQ140. We had previously found that the relative charge paucity of KCNQ1 S4 (+3, the lowest of all known eukaryotic voltage-dependent ion channels) was instrumental in the functional flexibility of KCNQ1 in terms of regulation by KCNE subunits18. Neutralization of R231 within KCNQ1 S4, by replacement with alanine, converts KCNQ1 to a K+ leak channel in the absence of KCNEs, and the same is achieved in related α subunit KCNQ4 by a triple mutation that mimics the charge balance of R231A-KCNQ118,41. Homomeric R237A-KCNQ1 channels exhibit slightly right-shifted activation voltage dependence compared to homomeric KCNQ1, but the former accumulate in the open state with repetitive membrane depolarizations, deactivating very slowly upon repolarization18. In KCNQ1-KCNE3 complexes, KCNQ1-R237 appears to engage in electrostatic interactions with KCNE3 D54 and D55 to help stabilize the KCNQ1 S4, and therefore also the pore, in the activated state, even at negative voltages (deactivation can be seen to occur over several seconds when hyperpolarizing to −120 mV) (Figure 3A). Interestingly, KCNE3 D54 partially protects KCNQ1-KCNE3 channels from the inhibitory effects of loss of plasma membrane phospholipid head groups. Normally, these negatively charged phosphocholines stabilize the activated conformation of S4, facilitating voltage-dependent activation. Removal of these head groups via hydrolysis by, for example, sphingomyelinase C, inhibits activity of channels including Kv2.142 and KCNQ1, but KCNE3 (and not KCNE1) helps maintain KCNQ1 activity in the face of sphingomyelinase C because KCNE3-D54 mimics the S4-stabilizing action of membrane phosphocholines40.
As striking as the constitutive activation of KCNQ1 endowed by KCNE3, is the laboriously slow activation endowed by KCNE1. As covered above, the KCNE1 C-terminal domain and to some extent the transmembrane domain may be important in slowing KCNQ1 activation, but is this achieved by slowing the voltage sensor, the pore opening, or both? There are at least two camps in terms of this aspect of the mechanistic underpinnings of slow KCNQ1-KCNE1 activation. The Goldstein and Bezanilla labs approached the problem using cut-open oocyte recording, a technique that permits much better voltage clamp and improved time resolution compared to conventional TEVC, facilitating measurement of gating currents – the current generated from movement of the charged voltage sensor itself. The cut-open oocyte recordings were combined with site-directed fluorimetry to also monitor voltage sensor movement by a complementary approach. They discovered that unlike homomeric KCNQ1, during KCNQ1-KCNE1 activation S4 moves too slowly to permit resolution of the gating current; by fluorimetry S4 was found to move 30-fold slower than that of the oft-studied Shaker Kv channel43.
Nakajo and Kubo found, using methanethiosulfonate (MTS) modification of cysteine-substituted KCNQ1 residues, that accessibility of A226C-KCNQ1, a substituted residue near the extracellular side of KCNQ1 S4, was 13-fold slower when KCNE1 was co-expressed compared to KCNQ1 alone. In contrast, when KCNE3 was co-expressed, KCNQ1 A226C accessibility was voltage-independent. This is consistent with the slow S4 movement model for KCNQ1-KCNE1 activation, although it is also consistent with S4 equilibrium rather than speed of movement being modulated by KCNE144. Subsequent work by Nakajo and Kubo also suggested KCNE1 modulates interactions between S4 and S5 to affect activation gating, which could affect S4 movement speed and/or coupling of S4 to the channel gate45. The Seebohm lab found that KCNE1 interacts with both S4 and S5/S6 to form what is probably a stable pre-open state that may slow activation46. Utilizing voltage clamp fluorimetry of Xenopus oocyte-expressed channels, the Kass lab formulated a dual-effect model for KCNE1 regulation of KCNQ1, in which KCNQ1 can open after activation of just one S4, while KCNQ1-KCNE1 needs all four S4s to shift before the pore opens. This model incorporates both slow S4 movement, and the need for all four S4s to open, as the basis of slow KCNQ1-KCNE1 activation47,48. The Fedida lab found using single channel analysis that KCNQ1-KCNE1 flits between a variety of subconductance states, behavior that may or may not also exist in faster channels but could be too rapid to resolve in these cases, and may strongly influence opening and closing rates of KCNQ1-KCNE149.
The jury is clearly still out over the specifics, but in general terms it appears KCNE1 interaction with S4 retards KCNQ1-KCNE1 activation, directly and/or or via altered interaction with S4 and S5/S6. Discrepancies between different studies could arise from different preparations, protocols and interpretations. Interaction of KCNE1 with KCNQ1 S4 may also generate the right-shift in voltage dependence of activation induced by KCNE1, possibly by altering the manner in which S4 senses the stabilizing acidic residues in other, nearby KCNQ1 TM domains46,50,51.
Low mobility of the KCNE1-bound KCNQ1 S4 was also recently suggested to underlie the slow deactivation conferred by KCNE152. In this comprehensive combination of experimental work, and modeling using elastic network analysis, loss of KCNQ1 C-type (slow) inactivation, another change conferred by KCNE1 co-assembly, was assessed in the context of acidic residues that influence slow inactivation of KcsA (the Steptomyces lividans K+ channel used by the McKinnon lab to produce the first high-resolution K+ channel structure2). It was suggested that KCNE1 limits positional fluctuations in the KCNQ1 outer vestibule and selectivity filter that drive slow inactivation, thereby eliminating or at least dampening the inactivation process. Finally, the increased conduction of KCNQ1 upon KCNE1 co-assembly was attributed in this study to a shift in KCNQ1 residues G272, V310 and T311 such that they move in concert with an entire “dynamic domain” in the pore. This is proposed to increase the scale of pore movement and therefore increase conductance, and is consistent with the results of previous mutagenesis studies52. The contrasting mechanisms by which KCNE1 and KCNE3 exert their effects on KCNQ1 gating and conductance, and possible locations of KCNE1 and KCNE3 within complexes with KCNQ1, are summarized in Figure 3A and B.
In addition, KCNE1 modulates KCNQ1 internalization from the plasma membrane. While KCNQ1 channels can be internalized by Nedd4/Nedd4-like- and Rab5-dependent processes with or without KCNE1 co-assembly53,54, KCNE1 mediates an alternative process, clathrin-mediated endocytosis of KCNQ155. This dynamin-dependent process is enhanced by PKC phosphorylation of KCNE1 at residue S10356, explaining previous findings that S102 phosphorylation diminished currents generated by KCNE1 (in complex with endogenous KCNQ1) in Xenopus oocytes57,58.
KCNE1 and KCNE3 also exhibit contrasting effects on the functional outcomes of KCNQ1 regulation by other factors. Serine 27 in the KCNQ1 N-terminal region can be phosphorylated by protein kinase A (PKA), a physiologically important mechanism for β-adrenergic stimulation of cardiac IKs. Although S27 can be phosphorylated regardless of the co-expressed KCNE isoform, the functional effect also requires KCNE1 (or KCNE2) and the cytoplasmic protein yotiao in the complex, despite the PKA site being on KCNQ1 itself. KCNQ1-KCNE3 complexes do not show the same upregulation in response to PKA phosphorylation of KCNQ1 S2759. Of note, KCNE1 increases the sensitivity of KCNQ1 channels to phosphatidylinositol 4,5-bisphosphate (PIP2), a plasma membrane lipid that acts as a co-factor for a variety of ion channels and augments IKs. It is not yet known whether KCNE3 has similar effects, but all members of the KCNE family possess similar residues to those important for modifying the manner in which PIP2 interacts with KCNQ160.
KCNE1 and KCNE3 regulation of other Kv α subunits
Although KCNE1 and KCNE3 regulation of other Kv channels is not generally as well understood as their effects on KCNQ1, it is interesting to note that their regulation of other α subunits also typically results in disparate outcomes (Table 1). The first non-KCNQ1 α subunit found to be regulated by KCNE1 was the human ether-a-go-go related gene product (hERG, also termed Kv11.1 or KCNH2), the channel primarily responsible for driving human ventricular repolarization, especially at resting heart rates. KCNE1 increases hERG current density by a still unresolved mechanism in heterologous expression studies61, and probably regulates hERG in the heart62–64. In contrast, KCNE3 strongly inhibits hERG, again by an unresolved mechanism17, although the same KCNE3 substitution (R83H) that disrupts ability to constitutively open KCNQ1 channels and augment Kv3.4 currents65 also blunts the ability of KCNE3 to inhibit hERG66. Interestingly, KCNE1 and KCNE3 are reported to form tripartite complexes with Kv12.2 (also termed ELK2 or KCNH3), inhibiting its surface expression and activation in Xenopus oocytes; this may also occur in mouse brain67.
Table 1. Summary of main effects of KCNE1 and KCNE3 on Kv α subunits.
See main text for expanded description of effects.
| Kv α subunit | Effects of KCNE1 | Effects of KCNE3 |
|---|---|---|
| KCNQ1 | Slows activation, strongly increases peak current in oocytes, CHO and HEK cells, and cardiac myocytes.12,13 | Instantaneous activation, small increase in peak current in oocytes and CHO cells.17,33 |
| KCNQ4 | Mild upregulation in Xenopus oocytes.67 | Inhibits in Xenopus oocytes.17,78 |
| KCNQ5 | Slows activation, increases peak current by decreasing inward rectification in HEK, oocytes. 77 | Inhibits in HEK cells and in Xenopus oocytes. 77 |
| hERG | Doubles current density in CHO cells.61 | Strongly inhibits in Xenopus oocytes.8 |
| Kv1.1 | No effect in CHO cells.69,70 | ? |
| Kv1.1-Kv1.4 | Increases current density 2-fold in CHO cells.69,70 | ? |
| Kv1.4 | Traps in ER/Golgi in CHO cells. 69,70 | ? |
| Kv2.1 | Slows activation/deactivation in CHO cells.57 | Slowed activation/deactivation in CHO cells.76 |
| Kv3.1 | Strongly slows activation/deactivation, accelerates inactivation in CHO cells.71 | Weakly slows activation/deactivation, accelerates inactivation in CHO cells.71 |
| Kv3.1-Kv3.2 | Strongly slows activation/deactivation, accelerates inactivation in CHO cells.71 | Weakly slows activation/deactivation, accelerates inactivation in CHO cells.71 |
| Kv3.1-Kv3.4 | Attenuates inactivation by “mopping” up homomeric Kv3.4 in CHO cells.69,70 | ? |
| Kv3.2 | Strongly slows activation/deactivation, accelerates inactivation in CHO cells.71 | Weakly slows activation/deactivation, accelerates inactivation in CHO cells.71 |
| Kv3.3 | Inhibits, probably by trapping in ER/Golgi in CHO cells.69,70 | ? |
| Kv3.4 | Traps in ER/Golgi in CHO cells. 69,70 | Augments activity, left-shifts voltage dependence in CHO cells.65 |
| Kv4.2 | No effect (with α,β staggered injection) in Xenopus oocytes.73 | Inhibits in CHO cells and In SGNs.72 |
| Kv4.3 | Slows gating, increases current in HEK cells.75 | Inhibits in CHO cells.74 |
| Kv4.3-KChIP2 | Accelerates activation. Mildly speeds, slows recovery of, and left-shifts steady state inactivation; all in CHO cells.76 | Accelerates activation. Strongly speeds, slows recovery of, and left-shifts steady state inactivation; all in CHO cells.76 |
| Kv12.2 | Inhibits in Xenopus oocytes.67 | Inhibits Xenopus oocytes.67 |
KCNE1 and KCNE3 both slow Kv2.1 activation and deactivation, possibly increasing the diversity of Kv2.1 currents in heart and brain64,68, and one of the rare instances in which these two KCNEs exhibit comparable effects on the activity of a specific Kv channel. In contrast, KCNE1 and KCNE3 have opposite effects on Kv3.4, a rapidly inactivating, N-type α subunit important in skeletal muscle and the CNS – albeit by different mechanisms. KCNE1 robustly inhibits Kv3.4 activity by retaining it early in the secretory pathway (endoplasmic reticulum and/or Golgi apparatus). We discovered that this provides a mechanism by which cells can ensure only mixed N-type/delayed rectifier channel complexes with intermediate inactivation kinetics, and not homomeric N-type α subunit channels, reach the plasma membrane. Both KCNE1 and KCNE2 exhibit this retention ability, and they can retain all three classic N-type, inactivation domain-containing Kv α subunits (Kv1.4, Kv3.3 and Kv3.4) until rescue by same-subfamily delayed rectifiers69,70. Conversely, KCNE3 augments activity of Kv3.4 channels, by a mechanism involving left-shifted voltage dependence of activation, increased unitary conductance, accelerated recovery from inactivation, and suppression of cumulative inactivation65. With respect to regulation of Kv3.1, Kv3.2, and Kv3.1-Kv3.2 heteromers, KCNE1 and KCNE3 both slow activation and deactivation and accelerate inactivation, but the effects of KCNE1 are of much greater magnitude71.
KCNE3 was recently found to inhibit Kv4.2, and Kv4.2-KCNE3 complexes may contribute to spike frequency and other properties in auditory neurons72 (see below), while no effects were observed for KCNE1 with Kv4.2 in Xenopus oocyte co-expression studies (albeit using a staggered, α subunit-first, cRNA injection protocol)73. Further, KCNE3 was found to inhibit Kv4.3 (fourfold reduction in current density) in CHO cells74, while in contrast, in HEK cells KCNE1 augmented Kv4.3 current density fivefold and slowed its inactivation 75. Association of the cytosolic ancillary subunit, KChIP2, altered these effects. Thus, in tripartite complexes, all KCNEs accelerated activation and inactivation of Kv4.3-KChIP2 channel complexes, although compared to complexes formed with KCNE1, Kv4.3-KChIP2-KCNE3 channels exhibited lower current density, and slightly more negative half-maximal voltage dependence of steady-state activation and inactivation76. One or more of the KCNE subunits may thus contribute to functional diversity of cardiac Ito.
Returning to the KCNQ family, in both Xenopus oocytes and HEK cells, KCNE1 reportedly slows the activation of KCNQ5, decreases its inward rectification at highly depolarized voltages, and increases its peak current; conversely, KCNE3 inhibits KCNQ577. Similarly, KCNE1 appears to mildly augment KCNQ4 current when co-expressed in Xenopus oocytes, whereas KCNE3 strongly inhibits KCNQ4 in this expression system17,78.
KCNE1, KCNE2 and KCNE3 show sex-dependent differences in expression that arise from hormonal influence and determine some sex-specific aspects of mammalian physiology. Cardiac KCNE2 expression is upregulated by estrogen via a direct genomic mechanism79. KCNE1 and KCNE3 (but not KCNE2) are expressed in rat colonic crypts and highly enriched in male versus female rodents; during the estrus cycle, colonic expression of KCNE3 (and colonic partner KCNQ1) fluctuates in a manner consistent with KCNE3 downregulation by estrogen, via PKC phosphorylation of KCNE3 serine 82. In heterologous expression experiments, estrogen inhibited KCNQ1-KCNE3 activity, but not that of KCNQ1-KCNE180–82.
In contrast, KCNE1, 3 and 5 expression is upregulated in concert with that of the orphan nuclear receptor, estrogen-related receptor γ (ERRγ), in human syncytiotrophoblasts, and is downregulated by hypoxia; KCNE4 displays the reverse pattern. KCNE1 and KCNE3 expression in trophoblast cells is downregulated by shRNA suppression of ERRγ and KCNE1 contains a putative ERRγ response element within its 5′ flanking region83. KCNE1 and KCNE2 expression was also found to be downregulated in ERRγ null mouse hearts, a similar effect was observed for KCNE2 in parietal cells, and KCNE2 (like KCNE1) was found to contain a ERRγ responsive element that binds ERRγ84.
Physiological roles for KCNE1: the heart and inner ear
The best-understood physiological roles for KCNE1 are encapsulated by JLNS, an inherited cardioauditory syndrome that results from severe disruption of KCNQ1-KCNE1 channels such as occurs with biallelic mutations in human KCNE1 or KCNQ123,85. The cardiac element of JLNS is LQTS, caused by loss of function of ventricular KCNQ1-KCNE1 channels, eroding the repolarization reserve of ventricular myocytes and delaying ventricular repolarization. Thus, the slow- and relatively positive membrane potential-activating KCNQ1-KCNE1 channel, which generates IKs, is important for repolarizing human ventricular myocytes to end their action potential in readiness for the next heartbeat24,86. IKs appears to be most important during physical exertion or other contexts when β–adrenergic stimulation increases IKs density to compensate for higher heart rates. At other times, IKr (generated by hERG, which is regulated in vivo by KCNE1 and KCNE2) is probably dominant in human ventricles87,88.
KCNE1 mutants more commonly cause the LQT5 form of LQTS, without overt auditory symptoms, and presumably though disruption of IKs complex function, although these mutants can also cause loss-of-function of hERG-KCNE1 channels and therefore potentially ventricular IKr in vivo89. To the author’s knowledge, KCNE1 sequence variation has not be linked to Brugada syndrome or to Short QT syndrome (SQTS), either of which (opposite to LQTS) can result from gain of function in ventricular Kv channels90,91. However, a KCNQ1 mutation was recently discovered that appears to shorten the QT interval by impairing association of KCNQ1 with KCNE1, leading to accelerated activation and a negative shift in the voltage dependence of activation of IKs; the mutation also decreased inactivation of homomeric KCNQ192. Human KCNE1 sequence variants in its extracellular portion are associated with the relatively rare, idiopathic form of atrial fibrillation (AF), with the underlying mechanism considered to be gain of function of atrial myocyte KCNQ1-KCNE1 channels. In addition, a more common KCNE1 polymorphism, S38G, has been linked to altered predisposition to AF and LQTS, with environmental and other genetic factors influencing this predisposition93–96. The same polymorphism influences predisposition to postoperative atrial fibrillation97, a prevalent arrhythmia following cardiothoracic surgery, and KCNE1 is downregulated in the atria of pigs following lung lobectomy98. Human KCNE1 sequence variants associated with human disease are summarized in Table 2.
Table 2. KCNE1 gene variants associated or putatively associated with human disease.
AF, Atrial Fibrillation; AV block, atrioventricular block; BrS, Brugada Syndrome; diLQTS, drug induced Long QT Syndrome; diTdP, drug induced torsade de pointes; fs, frame shift; JLNS, Jervell and Lange-Nielsen Syndrome; n.r., not reported; RWS, Romano-Ward Syndrome; Sources include cited papers and 162. KCNE1 gene accession number: P15382.
| Mutation | Disease | In vitro cellular effects |
|---|---|---|
| T7I | JLNS85 | n.r. |
| A8V | LQT5, AF | IKs +9 mV positive shift in V1/2 activation. No change in deactivation or activation time constants. IKr loss of function. 134,135 |
| T10M | LQT5135 | n.r. |
| W17X | LQT5135 | n.r. |
| G25V | Lone AF | IKs gain of function 95 |
| S28L | LQT5, AV block (neonatal) | n.r. 135,136 |
| R32H | LQT5 | Gating not altered135,137,138 |
| 38G versus 38S | AF, AF in elderly patients, postoperative AF, LQT5 | IKs loss of function, reduced membrane KV7.1 (KvLQT1) channel93,97,139–144 |
| 38G versus 38S | Ménière’s disease (Japanese cohort; not recapitulated in study of Caucasian population)122,123 | n.r |
| V47F | LQT5 | hERG Gain of Function145 |
| L51H | LQT5 | Does not process properly, not found in membrane145 |
| G52R | LQT5 | IKs reduced 50%146 |
| G55S | LQT5135 | n.r. |
| T58P | LQT5135 | n.r. |
| L59P | LQT5135 | n.r. |
| G60D | Lone AF | IKs gain of function; faster deactivation95 |
| R67C | LQT5135 | n.r. |
| R67H | LQT5135 | n.r. |
| K70M | LQT5135 | n.r. |
| K70N | LQT5147 | n.r. |
| S74L D76N |
LQT5 (JLNS+RW)24 diLQTS148 |
D76N: IKs - smaller unitary currents and open probabilities; dominant negative26 IKr – loss of function145 S74L: IKs - loss of function149 |
| N75-fs+34X | LQT5135 | n.r. |
| Y81C | LQT5? | IKs loss of function, Positive shift for voltage of activation150 |
| E83K | LQT5135 | n.r. |
| D85N | LQT5? DiLQTS DiTdP |
No effect on IKs V1/2 of activation, or deactivation properties 148,151–159 |
| W87R | LQT5 | IKs loss of function, altered gating145 |
| R98W | LQT5? | Disrupts IKs trafficking, right-shifts voltage dependence of activation160 |
| V109I | LQT5 | IKs loss of function (36%)161 |
| Q117X | LQT5135 | n.r. |
| T125M | LQT5135 | n.r. |
| P127T | LQT5, RWS137 | n.r. |
While KCNQ1-KCNE1 channels activate much faster at body temperature than the classic slow activation they exhibit at room temperature99 - which makes the effects of KCNE1 particularly apparent - KCNE1 induces other changes in KCNQ1 important for their function together in the heart. As explained above, KCNE1 (together with a raft of cytosolic regulatory proteins) facilitates PKA upregulation of IKs in response to β adrenergic stimulation100–105. KCNE1 also appears to prime KCNQ1 to be ready to respond to the need for elevated heart rate by inducing a reserve of near-open closed states prepared for rapid activation when needed87, such that IKs complexes can be thought of as the ventricles’ ‘minutemen’. As explained earlier, KCNE1 mediates PKC-stimulated endocytosis of KCNQ1-KCNE1 channels, providing an alternate pathway for downregulation of IKs (and also perhaps a therapeutic window for counteracting QT prolongation, if this process can be disrupted)56. Finally, elimination of KCNQ1 slow inactivation by KCNE1 may also better enable IKs to sustain rapid heart rates without current diminishment.
The non-inactivating status endowed by KCNE1 may also facilitate the established function of KCNQ1-KCNE1 in the inner ear. Although KCNQ1-KCNE1 channels activate voltage-dependently, they are capable of sustained activity at the membrane potentials typical for the apical membrane of strial marginal cells and dark cells of the inner ear (0 to +10 mV). KCNQ1-KCNE1 provides a conduit for K+ secretion, thus recycling K+ in the endolymph of the inner ear. In the human ear, loss of this recycling is sufficient to cause bilateral sensorineural deafness; Kcnq1 deletion in mice revealed that this is associated with morphological abnormalities because of reduction in the volume of endolymph106. KCNQ1-KCNE1 channels are, in contrast, not prominent in adult musine ventricular function. Neonatal musine neonatal myocytes exhibit IKs and expression of both KCNQ1 and KCNE1 proteins56,89,107. Adult mouse ventricles express an adrenergic-sensitive steady-state current that is disrupted by Kcnq1 deletion, does not involve KCNE1108, and could potentially be generated by KCNQ1-KCNE2 complexes. Kcne1 deletion reportedly causes atrial fibrillation in mice, and KCNE1 can be detected in adult mouse atria and conduction system109, but it is not known which α subunit(s) KCNE1 regulates in these tissues in the mouse – Kv2.1 being one possibility.
Controversies and discrepancies (I). KCNE1 in the brain and kidneys
Typically not considered a neuronal complex, it was recently proposed that KCNQ1-KCNE1 channels may operate in some neurons, and that this could introduce a neural component to cardiac arrhythmias. Thus, mutations in KCNE1 or KCNQ1 might cause seizures that in turn trigger cardiac arrhythmias (perhaps exacerbated by the channels also being dysfunctional in the heart and therefore increasing arrhythmia susceptibility in the face of a trigger)110. Interestingly, Kcnq1−/− mouse hearts exhibit impaired repolarization in vivo but not when isolated, suggesting Kcnq1-linked LQTS in mice arises from an extracardiac source106. The controversy continues, centering on whether KCNE1 and KCNQ1 are actually expressed in neurons in mice and people.
Kcne1−/− mice exhibit altered salt and glucose homeostasis, and reduced NaCl consumption - the latter suggestive of a role for KCNE1 in mouse kidneys or salivary glands111. KCNQ1 and KCNE1 are expressed in mouse kidneys but they do not necessarily form a channel there, and the pharmacology and gating kinetics of K+ currents in proximal convoluted tubule epithelial cells –where KCNE1 is expressed – are not consistent with KCNQ1-KCNE1 properties. Counterintuitively, the KCNQ1-preferential antagonist, chromanol 293B, had similar functional effects in the proximal tubule to those of Kcne1 deletion, but this may reflect nonspecific action of chromanol 293B at the dosage used (100 μM). The cell-specific expression, biological functions and α subunit partners of KCNE1 in the kidneys remain a matter of debate112–116. KCNE1 was also previously suggested to form channels with KCNQ1 in the airway epithelium, but it now appears much more likely that KCNQ1-KCNE3 complexes are expressed there instead (see below)117.
Physiological roles for KCNE3: the gut, airway and auditory neurons
KCNE3 is best known for its regulation of KCNQ1 in the intestine. KCNQ1-KCNE3 are located on the basolateral membrane of colonic crypt cells. Their constitutive activation and lack of inactivation enable them to provide a K+ recycling conduit that facilitates electrogenic intestinal Cl− secretion. All available evidence for the importance of this process is from the mouse, and specifically the Kcne3−/− mouse line, and there are currently no reported human disease associations linking KCNE3 (or KCNQ1) mutations to altered intestinal Cl− secretion17,118. As mentioned above, estrogen downregulates intestinal KCNE3 expression, causing the water retention that occurs in female mice during proestrus. As observed for the intestine, Kcne3 deletion in mice also disrupts tracheal Cl− secretion, and basolateral KCNQ1-KCNE3 complexes are thought to be required for cAMP-stimulated Cl− secretion in the airway epithelium. KCNQ1-KCNE3 channels may also facilitate Na+ reabsorption in the airways of mice117. Again, data for the potential role of KCNQ1-KCNE3 in human airway epithelia are currently lacking.
In a different study of Kcne3−/− mice, a role for KCNE3 in auditory neurons was uncovered. KCNE3 was found to be expressed in spiral ganglion neurons (SGNs), with Kcne3−/− SGNs utilized as a convincing negative control for expression. Kcne3 deletion age-dependently altered the firing properties and membrane potential variability of SGNs, and resulted in an increase in the transient outward current in these cells. Kv4.2, and not Kv4.3, was found to be expressed in both apical and basal SGNs, where it co-localized with KCNE3. Accordingly, KCNE3 was found to diminish Kv4.2 activity when the two were co-expressed in CHO cells72. Therefore, KCNE3 may act as a dampener for Kv4.2 activity in SGNs, permitting dynamic regulation of the transient K+ current in these cells without the cell being required to remove Kv4.2 from the membrane, which might be too slow for the cells’ requirements or too energetically costly. The functional effects in SGNs of Kcne3 deletion were only observed during onset of hearing, during the first 12 days. At day 56, only two effects could be observed in SGNs – slightly enhanced afterhyperpolarizations of basal but not apical SGNs, and reduced interspike interval. Thus, KCNE3 might be more important during development of hearing than after auditory maturity, or else compensatory remodeling of other channels might lessen the deleterious consequences of Kcne3 deletion over time72.
Controversies and discrepancies (II). KCNE3 in the brain, muscle and auditory system
We previously found expression of KCNE3 transcript by Northern blot in all regions tested of human brain, and by RT-PCR from 12 DIV cultured rat primary hippocampal neurons isolated from embryonic stage 18 Sprague Dawley rats68. In addition, we detected KCNE3 protein by western blot in crude membrane preparations isolated from adult Sprague Dawley rat whole brains. We detected KCNE3-Kv2.1 and KCNE3-Kv3.1 complexes in these preparations, using co-immunoprecipitations. Our studies showed that endogenous KCNE3 co-localized with endogenous Kv2.1 but not Kv3.1 in cultured hippocampal neurons, while siRNA knockdown of endogenous KCNE3 in the PC12 cell line upregulated endogenous PC12 cell delayed rectifier K+ current. Heterologous co-expression studies in CHO cells were consistent with the functional effects we had observed by knocking down endogenous KCNE3 in PC12 cells, i.e., KCNE3 slowed the activation of and downregulated Kv2.1 current (and downregulated overall activity but reduced inactivation of Kv3.1b)68. As mentioned above, KCNE3 was detected by others in auditory neurons, with the added advantage of having a Kcne3−/− negative control72. However, in contrast to our findings, Preston et al reported a failure to detect KCNE3 in mouse brain118.
Similarly, Preston and colleagues reported a lack of KCNE3 expression in mouse skeletal muscle, and a lack of effects of Kcne3 deletion on the skeletal muscle-related parameters they tested in mice: muscle morphology, spontaneous paralysis, myotonia, “movement”, and rotarod performance118. Our studies of KCNE3 in skeletal muscle were driven by the findings of colleagues that the human KCNE3 R83H mutation is associated with periodic paralysis – a skeletal muscle disorder in which sufferers experience bouts of debilitating paralysis. We detected KCNE3 transcript by northern blot in human skeletal muscle, and found KCNE3 protein in rat sartorius muscle by western blot and in the mouse C2C12 myoblast cell line by western blot and immunofluorescence65. We determined that KCNE3 co-assembles with and functionally regulates Kv3.4, an N-type Kv channel α subunit known to be expressed in skeletal muscle, and that the R83H mutation in KCNE3, found to associate in individuals with periodic paralysis within two families, impaired function of Kv3.4-KCNE3 channels. Specifically, the mutant disrupted the ability of KCNE3 to left-shift the voltage dependence of Kv3.4 activation, leading to depolarization of resting membrane potential in skeletal myocytes65. We subsequently found that the R83H mutation also impairs the ability of KCNE3 to regulate other Kv channels66. Further, we found that the histidine introduced in this substitution makes Kv3.4-KCNE3 channels susceptible to block by protons, and could therefore make carriers particularly prone to impaired channel function during rest after exercise, when muscle pH drops drastically. Additionally, we showed that the neighboring residue, S82, is a serine phosphorylation site and that PKC phosphorylation of this residue is required for the left-shift in voltage dependence of Kv3.4 activation endowed by KCNE3119. However, the disease association has been highly controversial, with others finding the R83H sequence variant in unaffected individuals, raising the possibility that it is a benign polymorphism120. Another possibility is that carriers need another genetic ‘hit’ or specific environmental conditions to provoke paralysis121.
Another controversy surrounds the possible association of KCNE3 (and KCNE1) with Ménière’s disease – an inner ear disorder that manifests as tinnitus, periodic hearing loss, and spontaneous episodes of vertigo. Single nucleotide polymorphisms (SNPs) in KCNE1 (112G/A, coding for the well-studied S38G polymorphism) and KCNE3 (198T/C, a synonymous switch within the codon for F66) were found in one study to be enriched in Japanese Ménière’s cohorts compared to unaffected individuals122. However, in a study of Caucasians, KCNE1 and KCNE3 SNPs were not found to associate with Ménière’s123. Deep resequencing of KCNE3 in Caucasian people with chronic tinnitus failed to either support or disprove a link, with the authors concluding that more power was required124. While both genes are expressed in the auditory system (KCNE3 has been detected in auditory neurons, and all five KCNEs were detected in outer hair cells78), and KCNE1 is tightly associated with JLNS23,85, establishing a firm link between human KCNE3 sequence variation and auditory dysfunction will require further population or linkage studies.
Finally, not so much a controversy or necessarily a discrepancy, KCNE3 has been detected in human heart104 (although the relative expression levels of KCNEs varies depending on the study76,125 and may be affected by the sex of the donors, which was not always reported) and mutations in human KCNE3 that disrupt its ability to inhibit Kv4.3 channels in human ventricle are thought to cause some cases of Brugada syndrome74,126. Human KCNE3 mutations that cause gain-of-function of KCNQ1-KCNE3 channels are associated with atrial fibrillation, also suggested to arise from altered atrial myocyte channel activity127. Human KCNE3 sequence variants associated with human disease are summarized in Table 3.
Table 3. KCNE3 gene variants associated or putatively associated with human disease.
AF, Atrial Fibrillation; BrS, Brugada Syndrome; fs, frame shift; LQTS, Long QT syndrome; n.r., not reported; Sources include cited papers and 162. KCNE3 Gene accession number: Q9Y6H6.
| Mutation | Disease | In vitro cellular effects |
|---|---|---|
| T4A | LQTS? BrS |
No effect on IQ1-E3
163 Ito gain of function126 |
| V17M | AF | KV4.3 and Kv11.1 (hERG) gain of function 127 |
| R53H | AF | IQ1-E3 gain of function164 |
| F66F (198T/C synonymous switch) | Ménière’s disease (Japanese cohort; not recapitulated in study of Caucasian population) 122,123 | n.r. |
| R99H | BrS LQTS? |
KV4.3, Ito gain of function74 IQ1-E3 Loss of function163 |
| R83H | Periodic paralysis (but also detected in asymptomatic individuals in a later study)65,120,121 | Kv3.4 loss of function and increased sensitivity to proton block65 |
KCNE3 transcript was also detected in horse heart43. However, Kcne3 was not detected in either the atria or ventricles of adult mice, by two different labs using different Kcne3−/− mouse strains as negative controls118,128, and Kcne3 deletion did not alter Kv currents in isolated ventricular myocytes128. Yet, Kcne3 deletion predisposed to ventricular arrhythmogenesis in mice, lengthening the QT interval in older adults and predisposing to sustained ventricular tachycardias following ischemia/reperfusion128. The underlying mechanism is unprecedented, and involves hyperaldosteronism in Kcne3−/− mice associated with autoimmune attack on the adrenal glands. Kcne3 is not expressed in mouse adrenals, but its deletion causes cytokines to be released that attract activated lymphocytes specifically to the adrenal glands. Administration of spironolactone, an aldosterone receptor antagonist, to Kcne3−/− mice ameliorated their QT prolongation and predisposition to ventricular tachycardia128. Two questions arise: first, what causes the adrenals in Kcne3−/− mice to release signals to attract activated B cells? Second, can this mechanism also be a component of human KCNE3-linked arrhythmogenesis? Resolution of these condundra will require further investigation.
Conclusions
In many ways, KCNE1 and KCNE3 represent the yin and yang of Kv channel regulation because of the often opposite yet complementary outcomes of their co-assembly with Kv α subunits. As more and more functional data emerge from studies of the KCNE family in expression systems and from mouse and human genetics studies, it becomes apparent that the KCNEs constitute a molecular toolkit that diversifies Kv channel functionality but also facilitates dynamic alteration of channel properties in response to other signaling elements – including membrane lipids, hormones, kinases and even larger membrane proteins such as solute transporters129. Their human disease associations will likely not be as common as those of the Kv α subunits for a variety of reasons – although in LQTS, base-for-base they match up rather well to α subunit genes in terms of population-wide contribution to pathogenicity130. Nevertheless, the phenotypes of Kcne null mice are fascinating and can inform us of their physiological roles and the consequences of their ablation (typically an altogether different lesion than a point mutation). Because KCNEs can alter Kv channel responses to native signals and also to pharmacological agents (in the context of both therapeutic targeting and side effects)11,16,130–133, it behooves us to consider their presence in Kv and other channel complexes in different tissues, cell types and life stages, as they may yet provide a crucial ally in tuning drug specificity and efficacy.
Highlights.
KCNE1 and KCNE3 regulate K+ channel gating, voltage dependence and trafficking
KCNE1 and 3 often have opposite effects on channel function, especially so for KCNQ1
KCNE1-KCNQ1 channels are best known for their role in cardiomyocyte repolarization
KCNE3-KCNQ1 channels are important in various secretory epithelia
Human KCNE1 and KCNE3 sequence variants cause various cardiac arrhythmias
Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series--a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM089820 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. G.W.A. is grateful for financial support from the National Institutes of Health (HL079275, DK41544 and GM115189).
Abbreviations
- AV block
atrioventricular block
- BLAST
basic local alignment search tool
- cAMP
cyclic adenosine monophosphate
- Cav channel
voltage-gated calcium channel
- CHO
Chinese Hamster ovary
- di
drug-induced
- ERRγ
estrogen-related receptor γ
- EST
expressed sequence tag
- fs
frameshift
- HCN
hyperpolarization-activated, cyclic nucleotide-gated
- HEK
human embryonic kidney
- IKs/IsK
slowly activating K+ current
- JLNS
Jervell and Lange-Nielsen syndrome
- KChIP
K+ channel interacting protein
- Kv channel
voltage-gated potassium channel
- LQTS
Long QT syndrome
- MiRP
MinK-related peptide
- MTS
methanethiosulfonate
- Nav channel
voltage-gated sodium channel
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKA
protein kinase A
- PKC
protein kinase C
- QTc
QT interval corrected for heart rate
- SGN
spiral ganglion neuron
- SNP
single nucleotide polymorphism
- SQTS
Short QT syndrome
- TdP
torsade de pointes
- TEVC
two-electrode voltage clamp
- TM
transmembrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67(6):915–28. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 3.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309(5736):897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 4.Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science. 2012;335(6067):432–6. doi: 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- 5.Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66(3):470–94. doi: 10.1007/s00018-008-8525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbott GW. Biology of the KCNQ1 potassium channel. New Journal of Science. 2014;2014 [Google Scholar]
- 7.Chen H, Kim LA, Rajan S, Xu S, Goldstein SA. Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron. 2003;40(1):15–23. doi: 10.1016/s0896-6273(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 8.Plant LD, Xiong D, Dai H, Goldstein SA. Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1323548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang KW, Goldstein SA. Subunit composition of minK potassium channels. Neuron. 1995;14(6):1303–9. doi: 10.1016/0896-6273(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Xia J, Kass RS. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J Biol Chem. 1998;273(51):34069–74. doi: 10.1074/jbc.273.51.34069. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Lin Z, Mattmann ME, Zou B, Terrenoire C, Zhang H, Wu M, McManus OB, Kass RS, Lindsley CW, Hopkins CR, Li M. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc Natl Acad Sci U S A. 2013;110(21):8732–7. doi: 10.1073/pnas.1300684110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takumi T, Ohkubo H, Nakanishi S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science. 1988;242(4881):1042–5. doi: 10.1126/science.3194754. [DOI] [PubMed] [Google Scholar]
- 13.Kaczmarek LK. Voltage-dependent potassium channels: minK and Shaker families. New Biol. 1991;3(4):315–23. [PubMed] [Google Scholar]
- 14.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 15.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–3. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 16.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97(2):175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch TJ. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403(6766):196–9. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- 18.Panaghie G, Abbott GW. The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J Gen Physiol. 2007;129(2):121–33. doi: 10.1085/jgp.200609612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abbott GW. The KCNE2 K(+) channel regulatory subunit: Ubiquitous influence, complex pathobiology. Gene. 2015;569(2):162–72. doi: 10.1016/j.gene.2015.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papazian DM, Schwarz TL, Tempel BL, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237(4816):749–53. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- 21.Tempel BL, Jan YN, Jan LY. Cloning of a probable potassium channel gene from mouse brain. Nature. 1988;332(6167):837–9. doi: 10.1038/332837a0. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 23.Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, Aslaksen B, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet. 1997;6(12):2179–85. doi: 10.1093/hmg/6.12.2179. [DOI] [PubMed] [Google Scholar]
- 24.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17(3):338–40. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 25.Piccini M, Vitelli F, Seri M, Galietta LJ, Moran O, Bulfone A, Banfi S, Pober B, Renieri A. KCNE1-like gene is deleted in AMME contiguous gene syndrome: identification and characterization of the human and mouse homologs. Genomics. 1999;60(3):251–7. doi: 10.1006/geno.1999.5904. [DOI] [PubMed] [Google Scholar]
- 26.Sesti F, Goldstein SA. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J Gen Physiol. 1998;112(6):651–63. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tristani-Firouzi M, Sanguinetti MC. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol. 1998;510(Pt 1):37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercer EA, Abbott GW, Ramesh B, Haris PI, Chapman D, Srai SK. Structural characterisation of a slowly activating potassium channel (IsK) Biochem Soc Trans. 1995;23(3):478S. doi: 10.1042/bst023478s. [DOI] [PubMed] [Google Scholar]
- 29.Mercer EA, Abbott GW, Brazier SP, Ramesh B, Haris PI, Srai SK. Synthetic putative transmembrane region of minimal potassium channel protein (minK) adopts an alpha-helical conformation in phospholipid membranes. Biochem J. 1997;325(Pt 2):475–9. doi: 10.1042/bj3250475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang C, Tian C, Sonnichsen FD, Smith JA, Meiler J, George AL, Jr, Vanoye CG, Kim HJ, Sanders CR. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry. 2008;47(31):7999–8006. doi: 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coey AT, I, Sahu D, Gunasekera TS, Troxel KR, Hawn JM, Swartz MS, Wickenheiser MR, Reid RJ, Welch RC, Vanoye CG, Kang C, Sanders CR, Lorigan GA. Reconstitution of KCNE1 into lipid bilayers: comparing the structural, dynamic, and activity differences in micelle and vesicle environments. Biochemistry. 2011;50(50):10851–9. doi: 10.1021/bi2009294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang C, Vanoye CG, Welch RC, Van Horn WD, Sanders CR. Functional delivery of a membrane protein into oocyte membranes using bicelles. Biochemistry. 2010;49(4):653–5. doi: 10.1021/bi902155t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melman YF, Krumerman A, McDonald TV. A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J Biol Chem. 2002;277(28):25187–94. doi: 10.1074/jbc.M200564200. [DOI] [PubMed] [Google Scholar]
- 34.Panaghie G, Tai KK, Abbott GW. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J Physiol. 2006;570(Pt 3):455–67. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zheng R, Melman YF, McDonald TV. Functional interactions between KCNE1 C-terminus and the KCNQ1 channel. PLoS One. 2009;4(4):e5143. doi: 10.1371/journal.pone.0005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lvov A, Gage SD, Berrios VM, Kobertz WR. Identification of a protein-protein interaction between KCNE1 and the activation gate machinery of KCNQ1. J Gen Physiol. 2010;135(6):607–18. doi: 10.1085/jgp.200910386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung DY, Chan PJ, Bankston JR, Yang L, Liu G, Marx SO, Karlin A, Kass RS. Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc Natl Acad Sci U S A. 2009;106(3):743–8. doi: 10.1073/pnas.0811897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu X, Jiang M, Hsu KL, Zhang M, Tseng GN. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J Gen Physiol. 2008;131(6):589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gage SD, Kobertz WR. KCNE3 truncation mutants reveal a bipartite modulation of KCNQ1 K+ channels. J Gen Physiol. 2004;124(6):759–71. doi: 10.1085/jgp.200409114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi E, Abbott GW. A shared mechanism for lipid- and beta-subunit-coordinated stabilization of the activated K+ channel voltage sensor. FASEB J. 2010;24(5):1518–24. doi: 10.1096/fj.09-145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panaghie G, Purtell K, Tai KK, Abbott GW. Voltage-dependent C-type inactivation in a constitutively open K+ channel. Biophys J. 2008;95(6):2759–78. doi: 10.1529/biophysj.108.133678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milescu M, Bosmans F, Lee S, Alabi AA, Kim JI, Swartz KJ. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat Struct Mol Biol. 2009;16(10):1080–5. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruscic KJ, Miceli F, Villalba-Galea CA, Dai H, Mishina Y, Bezanilla F, Goldstein SA. IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci U S A. 2013;110(7):E559–66. doi: 10.1073/pnas.1222616110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakajo K, Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J Gen Physiol. 2007;130(3):269–81. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakajo K, Kubo Y. Steric hindrance between S4 and S5 of the KCNQ1/KCNE1 channel hampers pore opening. Nat Commun. 2014;5:4100. doi: 10.1038/ncomms5100. [DOI] [PubMed] [Google Scholar]
- 46.Strutz-Seebohm N, Pusch M, Wolf S, Stoll R, Tapken D, Gerwert K, Attali B, Seebohm G. Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell Physiol Biochem. 2011;27(5):443–52. doi: 10.1159/000329965. [DOI] [PubMed] [Google Scholar]
- 47.Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP, Kass RS. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci U S A. 2010;107(52):22710–5. doi: 10.1073/pnas.1016300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osteen JD, Barro-Soria R, Robey S, Sampson KJ, Kass RS, Larsson HP. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci U S A. 2012;109(18):7103–8. doi: 10.1073/pnas.1201582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Werry D, Eldstrom J, Wang Z, Fedida D. Single-channel basis for the slow activation of the repolarizing cardiac potassium current, I(Ks) Proc Natl Acad Sci U S A. 2013;110(11):E996–1005. doi: 10.1073/pnas.1214875110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu D, Delaloye K, Zaydman MA, Nekouzadeh A, Rudy Y, Cui J. State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J Gen Physiol. 2010;135(6):595–606. doi: 10.1085/jgp.201010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu D, Pan H, Delaloye K, Cui J. KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys J. 2010;99(11):3599–608. doi: 10.1016/j.bpj.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gofman Y, Shats S, Attali B, Haliloglu T, Ben-Tal N. How does KCNE1 regulate the Kv7.1 potassium channel? Model-structure, mutations, and dynamics of the Kv7.1-KCNE1 complex. Structure. 2012;20(8):1343–52. doi: 10.1016/j.str.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Seebohm G, Strutz-Seebohm N, Birkin R, Dell G, Bucci C, Spinosa MR, Baltaev R, Mack AF, Korniychuk G, Choudhury A, Marks D, Pagano RE, Attali B, Pfeufer A, Kass RS, Sanguinetti MC, Tavare JM, Lang F. Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ Res. 2007;100(5):686–92. doi: 10.1161/01.RES.0000260250.83824.8f. [DOI] [PubMed] [Google Scholar]
- 54.Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I, Abriel H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 2007;74(1):64–74. doi: 10.1016/j.cardiores.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 55.Xu X, V, Kanda A, Choi E, Panaghie G, Roepke TK, Gaeta SA, Christini DJ, Lerner DJ, Abbott GW. MinK-dependent internalization of the IKs potassium channel. Cardiovasc Res. 2009;82(3):430–8. doi: 10.1093/cvr/cvp047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanda VA, Purtell K, Abbott GW. Protein kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm. 2011;8(10):1641–7. doi: 10.1016/j.hrthm.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varnum MD, Busch AE, Bond CT, Maylie J, Adelman JP. The min K channel underlies the cardiac potassium current IKs and mediates species-specific responses to protein kinase C. Proc Natl Acad Sci U S A. 1993;90(24):11528–32. doi: 10.1073/pnas.90.24.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang ZJ, Jurkiewicz NK, Folander K, Lazarides E, Salata JJ, Swanson R. K+ currents expressed from the guinea pig cardiac IsK protein are enhanced by activators of protein kinase C. Proc Natl Acad Sci U S A. 1994;91(5):1766–70. doi: 10.1073/pnas.91.5.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurokawa J, Bankston JR, Kaihara A, Chen L, Furukawa T, Kass RS. KCNE variants reveal a critical role of the beta subunit carboxyl terminus in PKA-dependent regulation of the IKs potassium channel. Channels (Austin) 2009;3(1):16–24. doi: 10.4161/chan.3.1.7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Zaydman MA, Wu D, Shi J, Guan M, Virgin-Downey B, Cui J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci U S A. 2011;108(22):9095–100. doi: 10.1073/pnas.1100872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388(6639):289–92. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- 62.Anantharam A, Abbott GW. Does hERG coassemble with a beta subunit? Evidence for roles of MinK and MiRP1. Novartis Found Symp. 2005;266:100–12. discussion 112–7, 155–8. [PubMed] [Google Scholar]
- 63.Finley MR, Li Y, Hua F, Lillich J, Mitchell KE, Ganta S, Gilmour RF, Jr, Freeman LC. Expression and coassociation of ERG1, KCNQ1, and KCNE1 potassium channel proteins in horse heart. Am J Physiol Heart Circ Physiol. 2002;283(1):H126–38. doi: 10.1152/ajpheart.00622.2001. [DOI] [PubMed] [Google Scholar]
- 64.McCrossan ZA, Roepke TK, Lewis A, Panaghie G, Abbott GW. Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J Membr Biol. 2009;228(1):1–14. doi: 10.1007/s00232-009-9154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 2001;104(2):217–31. doi: 10.1016/s0092-8674(01)00207-0. [DOI] [PubMed] [Google Scholar]
- 66.Abbott GW, Goldstein SA. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 2002;16(3):390–400. doi: 10.1096/fj.01-0520hyp. [DOI] [PubMed] [Google Scholar]
- 67.Clancy SM, Chen B, Bertaso F, Mamet J, Jegla T. KCNE1 and KCNE3 beta-subunits regulate membrane surface expression of Kv12.2 K(+) channels in vitro and form a tripartite complex in vivo. PLoS One. 2009;4(7):e6330. doi: 10.1371/journal.pone.0006330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCrossan ZA, Lewis A, Panaghie G, Jordan PN, Christini DJ, Lerner DJ, Abbott GW. MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J Neurosci. 2003;23(22):8077–91. doi: 10.1523/JNEUROSCI.23-22-08077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanda VA, Lewis A, Xu X, Abbott GW. KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel alpha-subunit composition. Biophys J. 2011;101(6):1364–75. doi: 10.1016/j.bpj.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanda VA, Lewis A, Xu X, Abbott GW. KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys J. 2011;101(6):1354–63. doi: 10.1016/j.bpj.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lewis A, McCrossan ZA, Abbott GW. MinK, MiRP1, and MiRP2 diversify Kv3.1 and Kv3.2 potassium channel gating. J Biol Chem. 2004;279(9):7884–92. doi: 10.1074/jbc.M310501200. [DOI] [PubMed] [Google Scholar]
- 72.Wang W, Kim HJ, Lee JH, Wong V, Sihn CR, Lv P, Perez Flores MC, Mousavi-Nik A, Doyle KJ, Xu Y, Yamoah EN. Functional significance of K+ channel beta-subunit KCNE3 in auditory neurons. J Biol Chem. 2014;289(24):16802–13. doi: 10.1074/jbc.M113.545236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang M, Jiang M, Tseng GN. minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ Res. 2001;88(10):1012–9. doi: 10.1161/hh1001.090839. [DOI] [PubMed] [Google Scholar]
- 74.Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman-Bang J, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1(3):209–18. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deschenes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528(1–3):183–8. doi: 10.1016/s0014-5793(02)03296-9. [DOI] [PubMed] [Google Scholar]
- 76.Radicke S, Cotella D, Graf EM, Banse U, Jost N, Varro A, Tseng GN, Ravens U, Wettwer E. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc Res. 2006;71(4):695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 77.Roura-Ferrer M, Etxebarria A, Sole L, Oliveras A, Comes N, Villarroel A, Felipe A. Functional implications of KCNE subunit expression for the Kv7.5 (KCNQ5) channel. Cell Physiol Biochem. 2009;24(5–6):325–34. doi: 10.1159/000257425. [DOI] [PubMed] [Google Scholar]
- 78.Strutz-Seebohm N, Seebohm G, Fedorenko O, Baltaev R, Engel J, Knirsch M, Lang F. Functional coassembly of KCNQ4 with KCNE-beta- subunits in Xenopus oocytes. Cell Physiol Biochem. 2006;18(1–3):57–66. doi: 10.1159/000095158. [DOI] [PubMed] [Google Scholar]
- 79.Kundu P, Ciobotaru A, Foroughi S, Toro L, Stefani E, Eghbali M. Hormonal regulation of cardiac KCNE2 gene expression. Mol Cell Endocrinol. 2008;292(1–2):50–62. doi: 10.1016/j.mce.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alzamora R, O’Mahony F, Bustos V, Rapetti-Mauss R, Urbach V, Cid LP, Sepulveda FV, Harvey BJ. Sexual dimorphism and oestrogen regulation of KCNE3 expression modulates the functional properties of KCNQ1 K(+) channels. J Physiol. 2011;589(Pt 21):5091–107. doi: 10.1113/jphysiol.2011.215772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.O’Mahony F, Thomas W, Harvey BJ. Novel female sex-dependent actions of oestrogen in the intestine. J Physiol. 2009;587(Pt 21):5039–44. doi: 10.1113/jphysiol.2009.177972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rapetti-Mauss R, O’Mahony F, Sepulveda FV, Urbach V, Harvey BJ. Oestrogen promotes KCNQ1 potassium channel endocytosis and postendocytic trafficking in colonic epithelium. J Physiol. 2013;591(Pt 11):2813–31. doi: 10.1113/jphysiol.2013.251678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Luo Y, Kumar P, Mendelson CR. Estrogen-related receptor gamma (ERRgamma) regulates oxygen-dependent expression of voltage-gated potassium (K+) channels and tissue kallikrein during human trophoblast differentiation. Mol Endocrinol. 2013;27(6):940–52. doi: 10.1210/me.2013-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alaynick WA, Way JM, Wilson SA, Benson WG, Pei L, Downes M, Yu R, Jonker JW, Holt JA, Rajpal DK, Li H, Stuart J, McPherson R, Remlinger KS, Chang CY, McDonnell DP, Evans RM, Billin AN. ERRgamma regulates cardiac, gastric, and renal potassium homeostasis. Mol Endocrinol. 2010;24(2):299–309. doi: 10.1210/me.2009-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H. KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17(3):267–8. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- 86.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336(22):1562–7. doi: 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- 87.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112(10):1384–91. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suto F, Zhu W, Chan A, Gross GJ. IKr and IKs remodeling differentially affects QT interval prolongation and dynamic adaptation to heart rate acceleration in bradycardic rabbits. Am J Physiol Heart Circ Physiol. 2007;292(4):H1782–8. doi: 10.1152/ajpheart.00932.2006. [DOI] [PubMed] [Google Scholar]
- 89.Ohyama H, Kajita H, Omori K, Takumi T, Hiramoto N, Iwasaka T, Matsuda H. Inhibition of cardiac delayed rectifier K+ currents by an antisense oligodeoxynucleotide against IsK (minK) and over-expression of IsK mutant D77N in neonatal mouse hearts. Pflugers Arch. 2001;442(3):329–35. doi: 10.1007/s004240100547. [DOI] [PubMed] [Google Scholar]
- 90.Brugada R, Campuzano O, Brugada P, Brugada J, Hong K. Brugada Syndrome. In: Pagon RA, et al., editors. Gene Reviews. Seattle (WA): 1993. [Google Scholar]
- 91.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30–5. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 92.Moreno C, Oliveras A, de la Cruz A, Bartolucci C, Munoz C, Salar E, Gimeno JR, Severi S, Comes N, Felipe A, Gonzalez T, Lambiase P, Valenzuela C. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc Res. 2015 doi: 10.1093/cvr/cvv196. [DOI] [PubMed] [Google Scholar]
- 93.Ehrlich JR, Zicha S, Coutu P, Hebert TE, Nattel S. Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc Res. 2005;67(3):520–8. doi: 10.1016/j.cardiores.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 94.Mao T, Miao HJ, Xu GJ, Gan TY, Zhou XH, Zhang J, Li FP, Tang BP. Association of single nucleotide polymorphism of KCNE1 and KCNE4 gene with atrial fibrillation in Xinjiang Uygur and Han population. Zhonghua Xin Xue Guan Bing Za Zhi. 2013;41(11):916–21. [PubMed] [Google Scholar]
- 95.Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunso S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012;13(1):24. doi: 10.1186/1471-2350-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yao J, Ma Y, Xie X, Liu F, Chen B, An Y. Association of rs1805127 polymorphism of KCNE1 gene with atrial fibrillation in Uigur population of Xinjiang. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2011;28(4):436–40. doi: 10.3760/cma.j.issn.1003-9406.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 97.Voudris KV, Apostolakis S, Karyofillis P, Doukas K, Zaravinos A, Androutsopoulos VP, Michalis A, Voudris V, Spandidos DA. Genetic diversity of the KCNE1 gene and susceptibility to postoperative atrial fibrillation. Am Heart J. 2014;167(2):274–280. e1. doi: 10.1016/j.ahj.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 98.Heerdt PM, Kant R, Hu Z, Kanda VA, Christini DJ, Malhotra JK, Abbott GW. Transcriptomic analysis reveals atrial KCNE1 down-regulation following lung lobectomy. J Mol Cell Cardiol. 2012;53(3):350–3. doi: 10.1016/j.yjmcc.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dong MQ, Lau CP, Gao Z, Tseng GN, Li GR. Characterization of recombinant human cardiac KCNQ1/KCNE1 channels (I (Ks)) stably expressed in HEK 293 cells. J Membr Biol. 2006;210(3):183–92. doi: 10.1007/s00232-006-0006-5. [DOI] [PubMed] [Google Scholar]
- 100.Kurokawa J, Motoike HK, Rao J, Kass RS. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci U S A. 2004;101(46):16374–8. doi: 10.1073/pnas.0405583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li Y, Chen L, Kass RS, Dessauer CW. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem. 2012;287(35):29815–24. doi: 10.1074/jbc.M112.380568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–9. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 103.Nicolas CS, Park KH, El Harchi A, Camonis J, Kass RS, Escande D, Merot J, Loussouarn G, Le Bouffant F, Baro I. IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc Res. 2008;79(3):427–35. doi: 10.1093/cvr/cvn085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96(5):e25–34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- 105.Terrenoire C, Houslay MD, Baillie GS, Kass RS. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem. 2009;284(14):9140–6. doi: 10.1074/jbc.M805366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci U S A. 2001;98(5):2526–31. doi: 10.1073/pnas.041398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knollmann BC, Casimiro MC, Katchman AN, Sirenko SG, Schober T, Rong Q, Pfeifer K, Ebert SN. Isoproterenol exacerbates a long QT phenotype in Kcnq1-deficient neonatal mice: possible roles for human-like Kcnq1 isoform 1 and slow delayed rectifier K+ current. J Pharmacol Exp Ther. 2004;310(1):311–8. doi: 10.1124/jpet.103.063743. [DOI] [PubMed] [Google Scholar]
- 108.Knollmann BC, Sirenko S, Rong Q, Katchman AN, Casimiro M, Pfeifer K, Ebert SN. Kcnq1 contributes to an adrenergic-sensitive steady-state K+ current in mouse heart. Biochem Biophys Res Commun. 2007;360(1):212–8. doi: 10.1016/j.bbrc.2007.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Temple J, Frias P, Rottman J, Yang T, Wu Y, Verheijck EE, Zhang W, Siprachanh C, Kanki H, Atkinson JB, King P, Anderson ME, Kupershmidt S, Roden DM. Atrial fibrillation in KCNE1-null mice. Circ Res. 2005;97(1):62–9. doi: 10.1161/01.RES.0000173047.42236.88. [DOI] [PubMed] [Google Scholar]
- 110.Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1(2):2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Puchalski RB, Kelly E, Bachmanov AA, Brazier SP, Kuang J, Arrighi I, Barhanin J, Tordoff MG. NaCl consumption is attenuated in female KCNE1 null mutant mice. Physiol Behav. 2001;74(3):267–76. doi: 10.1016/s0031-9384(01)00572-8. [DOI] [PubMed] [Google Scholar]
- 112.Barriere H, Rubera I, Belfodil R, Tauc M, Tonnerieux N, Poujeol C, Barhanin J, Poujeol P. Swelling-activated chloride and potassium conductance in primary cultures of mouse proximal tubules. Implication of KCNE1 protein. J Membr Biol. 2003;193(3):153–70. doi: 10.1007/s00232-003-2014-z. [DOI] [PubMed] [Google Scholar]
- 113.Millar ID, Hartley JA, Haigh C, Grace AA, White SJ, Kibble JD, Robson L. Volume regulation is defective in renal proximal tubule cells isolated from KCNE1 knockout mice. Exp Physiol. 2004;89(2):173–80. doi: 10.1113/expphysiol.2003.026674. [DOI] [PubMed] [Google Scholar]
- 114.Neal AM, Taylor HC, Millar ID, Kibble JD, White SJ, Robson L. Renal defects in KCNE1 knockout mice are mimicked by chromanol 293B in vivo: identification of a KCNE1-regulated K+ conductance in the proximal tubule. J Physiol. 2011;589(Pt 14):3595–609. doi: 10.1113/jphysiol.2011.209155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J, Volkl H, Warth R. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol. 2001;12(10):2003–11. doi: 10.1681/ASN.V12102003. [DOI] [PubMed] [Google Scholar]
- 116.Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci U S A. 2005;102(49):17864–9. doi: 10.1073/pnas.0505860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grahammer F, Warth R, Barhanin J, Bleich M, Hug MJ. The small conductance K+ channel, KCNQ1: expression, function, and subunit composition in murine trachea. J Biol Chem. 2001;276(45):42268–75. doi: 10.1074/jbc.M105014200. [DOI] [PubMed] [Google Scholar]
- 118.Preston P, Wartosch L, Gunzel D, Fromm M, Kongsuphol P, Ousingsawat J, Kunzelmann K, Barhanin J, Warth R, Jentsch TJ. Disruption of the K+ channel beta-subunit KCNE3 reveals an important role in intestinal and tracheal Cl- transport. J Biol Chem. 2010;285(10):7165–75. doi: 10.1074/jbc.M109.047829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Abbott GW, Butler MH, Goldstein SA. Phosphorylation and protonation of neighboring MiRP2 sites: function and pathophysiology of MiRP2-Kv3.4 potassium channels in periodic paralysis. FASEB J. 2006;20(2):293–301. doi: 10.1096/fj.05-5070com. [DOI] [PubMed] [Google Scholar]
- 120.Sternberg D, Tabti N, Fournier E, Hainque B, Fontaine B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology. 2003;61(6):857–9. doi: 10.1212/01.wnl.0000082392.66713.e3. [DOI] [PubMed] [Google Scholar]
- 121.Dias Da Silva MR, Cerutti JM, Arnaldi LA, Maciel RM. A mutation in the KCNE3 potassium channel gene is associated with susceptibility to thyrotoxic hypokalemic periodic paralysis. J Clin Endocrinol Metab. 2002;87(11):4881–4. doi: 10.1210/jc.2002-020698. [DOI] [PubMed] [Google Scholar]
- 122.Doi K, Sato T, Kuramasu T, Hibino H, Kitahara T, Horii A, Matsushiro N, Fuse Y, Kubo T. Meniere’s disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J Otorhinolaryngol Relat Spec. 2005;67(5):289–93. doi: 10.1159/000089410. [DOI] [PubMed] [Google Scholar]
- 123.Campbell CA, Della Santina CC, Meyer NC, Smith NB, Myrie OA, Stone EM, Fukushima K, Califano J, Carey JP, Hansen MR, Gantz BJ, Minor LB, Smith RJ. Polymorphisms in KCNE1 or KCNE3 are not associated with Meniere disease in the Caucasian population. Am J Med Genet A. 2010;152A(1):67–74. doi: 10.1002/ajmg.a.33114. [DOI] [PubMed] [Google Scholar]
- 124.Sand PG, Langguth B, Kleinjung T. Deep resequencing of the voltage-gated potassium channel subunit KCNE3 gene in chronic tinnitus. Behav Brain Funct. 2011;7:39. doi: 10.1186/1744-9081-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res. 2005;67(3):529–38. doi: 10.1016/j.cardiores.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 126.Nakajima T, Wu J, Kaneko Y, Ashihara T, Ohno S, Irie T, Ding WG, Matsuura H, Kurabayashi M, Horie M. KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ J. 2012;76(12):2763–72. doi: 10.1253/circj.cj-12-0551. [DOI] [PubMed] [Google Scholar]
- 127.Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol Biochem. 2008;21(1–3):47–54. doi: 10.1159/000113746. [DOI] [PubMed] [Google Scholar]
- 128.Hu Z, Crump SM, Anand M, Kant R, Levi R, Abbott GW. Kcne3 deletion initiates extracardiac arrhythmogenesis in mice. FASEB J. 2014;28(2):935–45. doi: 10.1096/fj.13-241828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Abbott GW, Tai KK, Neverisky DL, Hansler A, Hu Z, Roepke TK, Lerner DJ, Chen Q, Liu L, Zupan B, Toth M, Haynes R, Huang X, Demirbas D, Buccafusca R, Gross SS, Kanda VA, Berry GT. KCNQ1, KCNE2, and Na+-Coupled Solute Transporters Form Reciprocally Regulating Complexes That Affect Neuronal Excitability. Sci Signal. 2014;7(315):ra22. doi: 10.1126/scisignal.2005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abbott GW. KCNE genetics and pharmacogenomics in cardiac arrhythmias: much ado about nothing? Expert Rev Clin Pharmacol. 2013;6(1):49–60. doi: 10.1586/ecp.12.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, Priori SG, Roden DM, George AL, Jr, Goldstein SA. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A. 2000;97(19):10613–8. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bett GC, Morales MJ, Beahm DL, Duffey ME, Rasmusson RL. Ancillary subunits and stimulation frequency determine the potency of chromanol 293B block of the KCNQ1 potassium channel. J Physiol. 2006;576(Pt 3):755–67. doi: 10.1113/jphysiol.2006.116012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lerche C, Seebohm G, Wagner CI, Scherer CR, Dehmelt L, Abitbol I, Gerlach U, Brendel J, Attali B, Busch AE. Molecular impact of MinK on the enantiospecific block of I(Ks) by chromanols. Br J Pharmacol. 2000;131(8):1503–6. doi: 10.1038/sj.bjp.0703734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ohno S, Zankov DP, Yoshida H, Tsuji K, Makiyama T, Itoh H, Akao M, Hancox JC, Kita T, Horie M. N- and C-terminal KCNE1 mutations cause distinct phenotypes of long QT syndrome. Heart Rhythm. 2007;4(3):332–40. doi: 10.1016/j.hrthm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 135.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297–303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shim SH, Ito M, Maher T, Milunsky A. Gene sequencing in neonates and infants with the long QT syndrome. Genet Test. 2005;9(4):281–4. doi: 10.1089/gte.2005.9.281. [DOI] [PubMed] [Google Scholar]
- 137.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 138.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109(15):1834–41. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 139.Fatini C, Sticchi E, Genuardi M, Sofi F, Gensini F, Gori AM, Lenti M, Michelucci A, Abbate R, Gensini GF. Analysis of minK and eNOS genes as candidate loci for predisposition to non-valvular atrial fibrillation. Eur Heart J. 2006;27(14):1712–8. doi: 10.1093/eurheartj/ehl087. [DOI] [PubMed] [Google Scholar]
- 140.Xu LX, Yang WY, Zhang HQ, Tao ZH, Duan CC. Study on the correlation between CETP TaqIB, KCNE1 S38G and eNOS T-786C gene polymorphisms for predisposition and non-valvular atrial fibrillation. Zhonghua Liu Xing Bing Xue Za Zhi. 2008;29(5):486–92. [PubMed] [Google Scholar]
- 141.Prystupa A, Dzida G, Myslinski W, Malaj G, Lorenc T. MinK gene polymorphism in the pathogenesis of lone atrial fibrillation. Kardiol Pol. 2006;64(11):1205–11. discussion 1212–3. [PubMed] [Google Scholar]
- 142.Husser D, Stridh M, Sornmo L, Roden DM, Darbar D, Bollmann A. A genotype-dependent intermediate ECG phenotype in patients with persistent lone atrial fibrillation genotype ECG-phenotype correlation in atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2(1):24–8. doi: 10.1161/CIRCEP.108.799098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li L, Shen C, Yao Z, Liang J, Huang C. Genetic Variants of Potassium Voltage-Gated Channel Genes (KCNQ1, KCNH2, and KCNE1) Affected the Risk of Atrial Fibrillation in Elderly Patients. Genet Test Mol Biomarkers. 2015;19(7):359–65. doi: 10.1089/gtmb.2014.0307. [DOI] [PubMed] [Google Scholar]
- 144.Han HG, Wang HS, Yin Z, Jiang H, Fang M, Han J. KCNE1 112G>a polymorphism and atrial fibrillation risk: a meta-analysis. Genet Mol Res. 2014;13(4):8367–77. doi: 10.4238/2014.October.20.12. [DOI] [PubMed] [Google Scholar]
- 145.Bianchi L, Shen Z, Dennis AT, Priori SG, Napolitano C, Ronchetti E, Bryskin R, Schwartz PJ, Brown AM. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum Mol Genet. 1999;8(8):1499–507. doi: 10.1093/hmg/8.8.1499. [DOI] [PubMed] [Google Scholar]
- 146.Ma L, Lin C, Teng S, Chai Y, Bahring R, Vardanyan V, Li L, Pongs O, Hui R. Characterization of a novel Long QT syndrome mutation G52R-KCNE1 in a Chinese family. Cardiovasc Res. 2003;59(3):612–9. doi: 10.1016/s0008-6363(03)00510-8. [DOI] [PubMed] [Google Scholar]
- 147.Lai LP, Su YN, Hsieh FJ, Chiang FT, Juang JM, Liu YB, Ho YL, Chen WJ, Yeh SJ, Wang CC, Ko YL, Wu TJ, Ueng KC, Lei MH, Tsao HM, Chen SA, Lin TK, Wu MH, Lo HM, Huang SK, Lin JL. Denaturing high-performance liquid chromatography screening of the long QT syndrome-related cardiac sodium and potassium channel genes and identification of novel mutations and single nucleotide polymorphisms. J Hum Genet. 2005;50(9):490–6. doi: 10.1007/s10038-005-0283-3. [DOI] [PubMed] [Google Scholar]
- 148.Weeke P, Mosley JD, Hanna D, Delaney JT, Shaffer C, Wells QS, Van Driest S, Karnes JH, Ingram C, Guo Y, Shyr Y, Norris K, Kannankeril PJ, Ramirez AH, Smith JD, Mardis ER, Nickerson D, George AL, Jr, Roden DM. Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug-induced long QT interval syndrome. J Am Coll Cardiol. 2014;63(14):1430–7. doi: 10.1016/j.jacc.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97(2):142–6. doi: 10.1161/01.cir.97.2.142. [DOI] [PubMed] [Google Scholar]
- 150.Wu DM, Lai LP, Zhang M, Wang HL, Jiang M, Liu XS, Tseng GN. Characterization of an LQT5-related mutation in KCNE1, Y81C: implications for a role of KCNE1 cytoplasmic domain in IKs channel function. Heart Rhythm. 2006;3(9):1031–40. doi: 10.1016/j.hrthm.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 151.Nielsen NH, Winkel BG, Kanters JK, Schmitt N, Hofman-Bang J, Jensen HS, Bentzen BH, Sigurd B, Larsen LA, Andersen PS, Haunso S, Kjeldsen K, Grunnet M, Christiansen M, Olesen SP. Mutations in the Kv1.5 channel gene KCNA5 in cardiac arrest patients. Biochem Biophys Res Commun. 2007;354(3):776–82. doi: 10.1016/j.bbrc.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 152.Zeng ZY, Pu JL, Tan C, Teng SY, Chen JH, Su SY, Zhou XY, Zhang S, Li YS, Wang FZ, Gu DF. The association of single nucleotide polymorphism of slow delayed rectifier K+ channel genes with atrial fibrillation in Han nationality Chinese. Zhonghua Xin Xue Guan Bing Za Zhi. 2005;33(11):987–91. [PubMed] [Google Scholar]
- 153.Zeng Z, Tan C, Teng S, Chen J, Su S, Zhou X, Wang F, Zhang S, Gu D, Makielski JC, Pu J. The single nucleotide polymorphisms of I(Ks) potassium channel genes and their association with atrial fibrillation in a Chinese population. Cardiology. 2007;108(2):97–103. doi: 10.1159/000095943. [DOI] [PubMed] [Google Scholar]
- 154.Porthan K, Marjamaa A, Viitasalo M, Vaananen H, Jula A, Toivonen L, Nieminen MS, Newton-Cheh C, Salomaa V, Kontula K, Oikarinen L. Relationship of common candidate gene variants to electrocardiographic T-wave peak to T-wave end interval and T-wave morphology parameters. Heart Rhythm. 2010;7(7):898–903. doi: 10.1016/j.hrthm.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, Schulze-Bahr E, Haverkamp W, Breithardt G, Cohen N, Aerssens J. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med (Berl) 2004;82(3):182–8. doi: 10.1007/s00109-003-0522-z. [DOI] [PubMed] [Google Scholar]
- 156.Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, Yamamoto S, Ozawa T, Ding WG, Toyoda F, Kawamura M, Akao M, Matsuura H, Kimura T, Kita T, Horie M. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J Am Coll Cardiol. 2009;54(9):812–9. doi: 10.1016/j.jacc.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 157.Lin L, Horigome H, Nishigami N, Ohno S, Horie M, Sumazaki R. Drug-induced QT-interval prolongation and recurrent torsade de pointes in a child with heterotaxy syndrome and KCNE1 D85N polymorphism. J Electrocardiol. 2012;45(6):770–3. doi: 10.1016/j.jelectrocard.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 158.Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, Meitinger T, Peters A, Wichmann HE, Ingram C, Bradford Y, Carter S, Norris K, Ritchie MD, George AL, Jr, Roden DM. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet. 2012;5(1):91–9. doi: 10.1161/CIRCGENETICS.111.960930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13(11):1213–22. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- 160.Harmer SC, Wilson AJ, Aldridge R, Tinker A. Mechanisms of disease pathogenesis in long QT syndrome type 5. Am J Physiol Cell Physiol. 2010;298(2):C263–73. doi: 10.1152/ajpcell.00308.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schulze-Bahr E, Schwarz M, Hauenschild S, Wedekind H, Funke H, Haverkamp W, Breithardt G, Pongs O, Isbrandt D. A novel long-QT 5 gene mutation in the C-terminus (V109I) is associated with a mild phenotype. J Mol Med (Berl) 2001;79(9):504–9. doi: 10.1007/s001090100249. [DOI] [PubMed] [Google Scholar]
- 162.Crump SM, Abbott GW. Arrhythmogenic KCNE gene variants: current knowledge and future challenges. Front Genet. 2014;5:3. doi: 10.3389/fgene.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ohno S, Toyoda F, Zankov DP, Yoshida H, Makiyama T, Tsuji K, Honda T, Obayashi K, Ueyama H, Shimizu W, Miyamoto Y, Kamakura S, Matsuura H, Kita T, Horie M. Novel KCNE3 mutation reduces repolarizing potassium current and associated with long QT syndrome. Hum Mutat. 2009;30(4):557–63. doi: 10.1002/humu.20834. [DOI] [PubMed] [Google Scholar]
- 164.Zhang DF, Liang B, Lin J, Liu B, Zhou QS, Yang YQ. KCNE3 R53H substitution in familial atrial fibrillation. Chin Med J (Engl) 2005;118(20):1735–8. [PubMed] [Google Scholar]