

Abstract
Proteins and their domains evolve by a set of events commonly including the duplication and divergence of small motifs. The presence of short repetitive regions in domains has generally constituted a difficult case for structural domain classifications and their hierarchies. We developed the Evolutionary Classification Of protein Domains (ECOD) in part to implement a new schema for the classification of these types of proteins. Here we document the ways in which ECOD classifies proteins with small internal repeats, widespread functional motifs, and assemblies of small domain‐like fragments in its evolutionary schema. We illustrate the ways in which the structural genomics project impacted the classification and characterization of new structural domains and sequence families over the decade.
Keywords: structural bioinformatics, protein classification, protein motifs, internal, repeats, structural genomics
Introduction
The divergence of proteins from a common ancestor, and the evolutionary pathways involved in this process, can be instructive for understanding and rationalizing protein function. Duplication, deterioration, fusion, and mutation events can alter both the structure and function of homologs proteins; sometimes to the extent that their ancestry can be difficult to discern by sequence similarity or they may adopt fold changes.1, 2, 3 Protein domains represent distinct folding units that can evolve and function independently.4 Domains may evolve at dissimilar rates through point mutations, insertions, and deletions; and can be shuffled, lost, and/or duplicated. In particular, the duplication of short repetitive motifs within a domain, when modified by insertion or deletion of secondary structure elements, can result in fold change or deterioration.5 It has been previously hypothesized that the extant set of protein domains results from a series of these evolutionary events operating on a smaller, peptide‐like antecedents,3 and that remnants of this repertoire are still detectable.6
Structural classifications of protein domains have been used to study more distant ancestries in protein relationships; both due to the difficulty in detecting sequence similarity in distant homologs and because structure diverges more slowly than sequence.7, 8 Structural similarity is potentially confounded by the possibility of analogy, or convergent evolution. Constraints imposed by function can lead to motifs shared between domains with similar function but no other discernible similarity. Also, homologs relationships between topologically distinct proteins, or fold‐changes between groups of distant homologs, complicate the determination of ancestry based solely on structure. Finally, these multiple components of domain definitions: functional, structural, and homology‐based, do not necessarily lead to a single universal definition, and different priorities assigned to these components by different classifications can lead to different views of the protein universe.9
We developed the Evolutionary Classification of protein Domains (ECOD) in part to tackle these issues. ECOD is distinct from other structural domain classification in two primary factors: (1) It allows topological and structural fold dissimilarity between homologs (such as β‐propellers) by placing topology level below homology level and (2) it recognizes more distant homologs relationships than other classifications.10 The top hierarchical level of ECOD is the architecture, which classifies domains by secondary structure type and arrangement (e.g., mainly α, mainly β). There are 20 major architectures in ECOD. Below architectures, we define the X‐group, or clusters of “possible homologs.” These are sets of domains that exhibit some level of similarity (usually distant structural similarity), but where definitive evidence for homology is currently lacking. Below the X‐group, homologs groups (H‐groups) cluster those protein domains with ample evidence for homology. Homologs groups may be further split into topology groups (or T‐groups) that have a distinct topological difference from other T‐groups in the H‐group. Finally, F‐groups define sequence families with close relationships in a manner analogs to other sequence classifications.11, 12 Here we review how multiple levels of duplication in evolution at different repeat lengths, both above and below the domain level, are represented in our classification of proteins by evolutionary descent, ECOD, and how these cases can help detection of evolutionarily related domains.
Results and Discussion
Subdomains and motifs: evolutionary signals below the domain level
Domains are conserved evolutionary units that can be shuffled, duplicated, and lost.1 However, domains themselves are comprised of both conserved and diversified regions. Regions of a domain can be under differential selective pressure depending on the degree to which they contribute to function, stability, and/or folding. Accordingly, considerable sequence and structural diversity can be observed between homologs domains. Domains may arise with independent structural subunits (or subdomains) contributing to a common function (e.g., where that function occurs in a cleft10). Such subdomains have been noted to link diverse folds,6 suggesting they represent remnants of an ancient pool of functional peptide modules.3 Homologs domains can share a conserved functional motif (e.g., His‐Me endonucleases), while having considerable structural diversity. However, these motifs are not necessarily evidence of homology as they can arise by convergence (e.g., psi‐loop motifs). Finally, determination of new structures (e.g., ANTAR domains) can lead to reclassification of domains not previously recognized in earlier structures.10
The His‐Me finger homing endonucleases are a structurally diverse homologs group of proteins present across all domains of life. They are characterized by a distinct zinc‐binding site and corresponding DNA‐binding modes.13 This conserved functional motif is responsible for DNA‐binding, and occurs in the context of multiple other functions. ECOD clusters the His‐Me endonucleases into a single homologs group, incorporating the deteriorated MH1, the recombinase endonuclease VIII [Fig. 1(A)], the HNH CRISPR‐associated CAS9 endonucleases [Fig. 1(B)], and the nonspecific endonuclease family [Fig. 1(C)]. Although these domains differ notably in length and secondary structure content, they retain a conserved uncommon functional motif. This example is characteristic of the types of homologs relationships ECOD aims to systematically relate.
Figure 1.
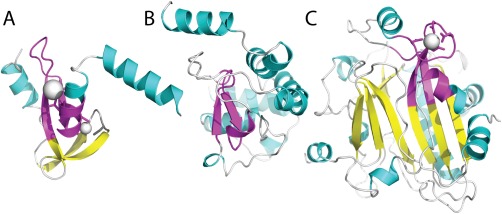
Diverse architectures surrounding the His‐Me finger endonuclease motif. Domains in the His‐Me finger endonucleases H‐group contain a conserved functional supersecondary structure motif β‐hairpin followed by a short α‐helix (colored magenta). The motif tends to include an N‐terminal α‐helix on the alternate side of the functional α‐helix and is embedded in various different architectures, including (A) a Zn‐finger in restriction endonxuclease HPY99I (e3fc3A3), (B) an α‐helical fold in CRISPR ‐associated Cas9 endonuclease (e4un3B4), and (C) an α + β 3‐layer sandwich in ectonucleotide pyrophosphatase/phosphodiesterase‐1 (e4b56A2).
Several noted motifs such as P‐loop,14, 15 helix‐turn‐helix,16, 17 and Asp box3, 18 retain similar local structures that are dictated by conserved sequence. These localized motifs represent unusual structural features often maintained within protein folds by functional constraints. Accordingly, such features can provide evidence for common evolutionary origins of protein structures,19 even between families displaying different fold architectures and topologies.1, 20 ECOD considers such published motifs as evidence for homologs classification. For example, the P‐loop domains‐related H‐group combines two P‐loop motif folds of different topologies that have a suggested evolutionary relationship.21 One of these T‐groups, the P‐loop containing nucleoside triphosphate hydrolases, contains mixed or parallel β‐sheets of differing sizes with α‐helices packed on either side [Fig. 2(A)]; whereas the other, PEP carboxykinase‐like, includes mixed β‐sheets that fold into a barrel‐like structure with helices packed on one side [Fig. 2(B)].
Figure 2.
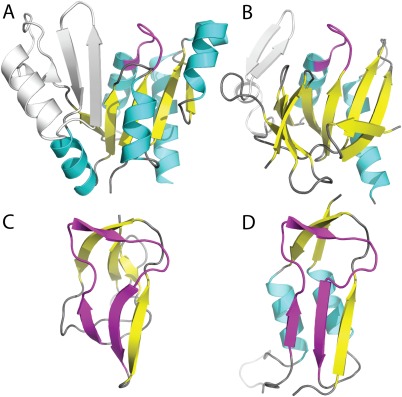
Motifs within divergent folds. The P‐loop domains‐related H‐group includes two T‐groups colored according to secondary structural element, with the P‐loop in magenta. α‐helices are in cyan and β‐sheets in yellow, with any remaining regions in gray. (A) The P‐loop containing nucleoside triphosphate hydrolases exemplified by Guanylate Kinase (e4qrhA1) has a main α/β/α core topology with five parallel β‐strands, while the (B) PEP carboxykinase‐like group exemplified by HPr kinase/phosphatase has a β‐strand core that wraps into an open barrel with α‐helices flanking one side. The β‐hammerhead motif (magenta) unifies the α/β‐hammerhead/Barrel‐sandwich hybrid H‐group including (C) the all‐β single‐hybrid motif (e1dczA1), and (D) the α/β‐hammerhead (e1brwA2).
The β‐hammerhead motif, first noted in the biotin carboxyl carrier protein (BCCP) subunit of acetyl‐coenzyme A carboxylase,22 forms a unique structure that resembles a hammer from two antiparallel β‐strands and an elongated connecting loop. Despite notable sequence and structural conservation of the β‐hammerhead,22, 23 the motif occurs in the context of different protein architectures: including barrel‐sandwich hybrids of the all‐β class and α/β‐hammerheads of the α and β class. For similar localized motifs found in different architectures, evolutionary scenarios for their distribution among folds have been postulated, including both convergent and divergent mechanisms of fold evolution.3, 18
On the basis of significant local structural and sequence similarity contained within the defined β‐hammerhead motif, ECOD combines the globally distinct architectures into a single α/β‐Hammerhead/Barrel‐sandwich hybrid H‐group. Evolutionary scenarios for the emergence of distinct modern day all‐β hammerhead‐containing folds (Fig. 3) include duplications and fusions recurring throughout protein fold evolution.5, 24 The single‐hybrid T‐group exemplified by the BCCP subunit contains two hammerhead motifs in symmetry‐related halves of the molecule [Fig. 3(A)]. Notable sequence similarity exists between the halves, suggesting the present‐day domain evolved from a simple domain half that previously functioned as a dimer.23 The duplicated‐hybrid motif [Fig. 3(B)] likely arose from a duplication and fusion of the single‐hybrid motif, while indels and hammerhead motif deterioration define the Ribosomal L27 protein T‐group [Fig. 3(C)].
Figure 3.
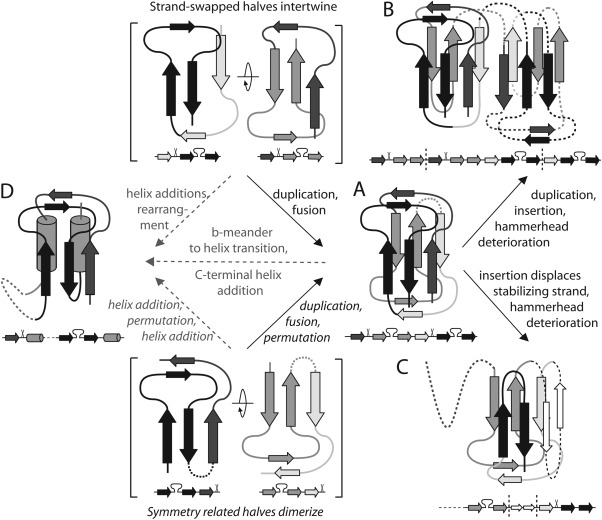
Potential β‐hammerhead motif evolution. Two alternate ancient three‐stranded units could dimerize to form a stable structure (in brackets). Evolutionary scenarios including fusion of the ancient duplicated units lead to (A) present day single‐hybrid motif. Insertion of a duplicated single‐hybrid motif combined with hammerhead deterioration lead to (B) duplicated‐hybrid motifs. Indels and hammerhead deterioration of single‐hybrid motif lead to (C) Ribosomal L27 protein. Several alternate pathways could lead to various (D) α/β‐hammerhead folds from all‐β hammerheads (gray dotted arrows).
While the evolution of the all‐β group hammerhead‐containing domains (Duplicated hybrid motif, Single hybrid motif, and Ribosomal L27 protein) involve typical fold changes1, 5 that are supported by structure and sequence similarity evidence, their evolutionary relationship to the α/β‐hammerhead‐containing domains remains less clear. The α/β‐hammerhead T‐groups (CO dehydrogenase molybdoprotein N‐domain‐like, Molybdopterin synthase subunit MoaE, Pyrimidine nucleoside phosphorylase C‐terminal domain, Nicotinate/Quinolinate PRTase N‐terminal domain‐like, and Ribosomal protein L10e) could have arisen from the addition of α‐helices to the simple domain half. Alternately, the α/β‐hammerhead folds could transition from the all‐β hammerhead folds by adding an α‐helix and converting a hammerhead β‐meander to an α‐helix. Similar β‐meander‐to‐α‐helix transitions occur in homologous proteins (i.e., Lactate dehydrogenase/NADH peroxidase and d‐Ala‐d‐Ala ligase/synapsin1). Various indels and fusions to additional domains distinguish the α‐helix‐containing T‐groups.
Psi‐loop motif: diverse evolutionary relationships
The psi‐loop structural motif consists of two antiparallel β‐strands separated by an intervening β‐strand making hydrogen bonds with both.25 The psi‐loop “+2” crossover connection occurs relatively infrequently in protein structures, making the motif an unusual structural feature that helps define homology. For example, the conserved core fold of zincin metalloproteases includes an α‐helix, psi‐loop, α‐helix (α‐psi‐α), with the two α‐helices flanking the psi‐loop on one side. The psi‐loop and C‐terminal helix, which includes the zinc‐binding motif HExxH, form the metalloprotease active site. The zincin H‐group includes numerous different families (currently 44) with diverse elaborations marked by different SSEs elaborating the α‐psi‐α core. For example, the peptidase M56 family represented by uncharacterized protein MJ1213 (e4qhfA1) includes the zincin α‐psi‐α core modified by two additional C‐terminal a‐helices [Fig. 4(B), core SSEs denoted below the structure]. In the reprolysin 5 family tumor necrosis factor‐α converting enzyme ADAM17 (e2i47A1), the β‐sheet formed by the psi‐loop motif becomes extended by an N‐terminal βαβα that adopts a Rossmann‐like crossover connection [Fig. 4(C)]. The peptidase M27 family represented by neurotoxin A (e3bonA1) contains additional SSE elaborations, with an N‐terminal β‐meander extending the psi‐loop sheet as well as multiple α‐helices and β‐strands adding to the C‐terminus [Fig. 4(D)]. Such cases of homologs with alternately elaborated structures provide a challenge for fold classification into ECOD protein architectures. The psi‐α‐psi core and additional helices in MJ1213 adopt an α + β two layers architecture, while the elaborated ADAM17 resembles an α/β three‐layered sandwich and the elaborated neurotoxin A represents an α + β complex topology. Not knowing which fold most signifies the ancestor zincin, we placed their X‐group into the ‘mixed α + β and α/β architecture’.
Figure 4.
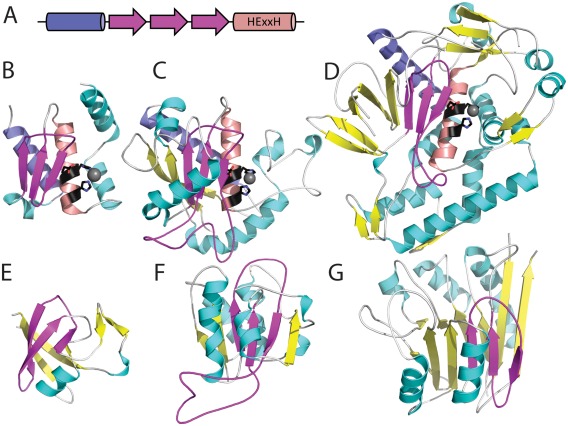
Divergent and convergent evolution of psi‐loop motifs. The psi‐loop motif (magenta) helps define the active site (HExxH in black stick) of zincin homologs with diverse folds. (A) The common zincin core includes an N‐terminal helix (slate), followed by the psi‐loop (magenta), and the HExxH‐containing active site helix (salmon). This core is decorated by various different SSEs (helix in cyan and strand in yellow) with (B) a peptidase M56 family structure (e4qhfA1) decorated by a single C‐terminal helix, (C) a reprolysin 5 family structure (e2i47A1) decorated by multiple α‐helices as well as an elongation of the psi‐loop sheet with parallel β‐strands, and (D) a peptidase M27 structure (e3bonA1) decorated by multiple α‐helices and β‐strands, elongating the psi‐loop sheet with an anti‐parallel β‐meander. The psi‐loop motif also occurs as part of the evolutionary core of unrelated folds, such as (E) double‐psi β‐barrels (e4avrA1), (F) Rossmann‐like structures of bacterial fluorinating enzyme N‐terminal domains (e1rqrA2), and (G) four‐layered metallo‐dependent phosphatase sandwiches (e2zo0B1).
Proteins with completely different architectures and evolutionary histories also contain psi‐loops. The double‐psi barrels shared by several protein superfamilies26 include two ββαβ psi‐loop units that interleave symmetrically to form a β‐barrel with α‐helix crossovers on either side (classified in ECOD as RIFT‐related27). This symmetry marks the RIFT‐related H‐group, which likely stemmed from an ancient duplication of the ββαβ unit marked by a Gly‐Asp sequence motif.28 Thus, the psi‐loops in double‐psi barrels arose from a different evolutionary history than those in the zincins, suggesting that the presence of the psi‐loop motif alone cannot justify homology. To characterize the evolutionary history of the psi‐loop motif (strand order 132), we evaluated the extent of its presence in ECOD domains. Searches of the ECOD domain database with the motif suggested that although it can unify numerous homologs with diverse folds, as in the case of zincins, it also defines part of the conserved core of numerous nonhomologs domains (e.g., psi‐loops are found in a majority of families in 16 different X‐groups). The psi‐loop motif belongs to architectures as diverse as double‐psi β‐barrels [Fig. 4(D)], bacterial fluorinating enzyme α/β three‐layered sandwiches [Fig. 4(F)], and metallo‐dependent phosphatase α + β four‐layered sandwiches [Fig. 4(G)].
The presence of identified psi‐loops in the ECOD database also suggests that evolutionarily conserved core folds can acquire (or lose) the motif through indels or other alterations of SSEs. An additional 56 H/T‐groups contain psi‐loops that belong to only a fraction of the family members. The fractional presence of the motif suggests that it can be formed by noncore elements or lost through deterioration of the motif. For example, the C‐terminal TIM β/α barrel domain from the GxGYxYP_C family (e3sggA4) of glycoside hydrolase/deacetylases includes an alteration of SSEs to the core that results in a psi‐loop replacing three of the β/α units of the typical 8‐strand TIM barrel. A related polysaccharide deacetylase homolog (e4m1bA1) from the same ECOD H‐group replaces the β/α units with a simple loop. Acquisition of a peripheral psi‐loop can also occur through insertion. The concanavalin A‐like structure of the glycoside hydrolase family 16 enzyme β‐agarase A (e1o4yA1) includes an edge β‐strand of the psi‐loop contributed by an elongated loop connecting the first two strands of the fold. A close homolog from the same glycoside hydrolase family 16 family, endo‐xyloglucanase (e2uwaA1), exhibits a shortened loop without the β‐strand. Finally, one of the zincin‐like structures from family DUF2342 (e3cmnA1) has experienced deterioration of the distinctive psi‐loop that marks the active site. Although this structure maintains the active site motif HExxH in the zincin helix, a short helix has replaced the third strand of the neighboring psi‐loop.
Repeats and evolution
Extension and reuse of small supersecondary structure element (SSE) subunits is a recurrent feature in protein evolution.29, 30 These subunits can duplicate to form interleaved, globular domains such as the cradle‐loop barrels31 or α‐β plaits.30 Alternately, they can form higher order tandem repeats in protein (TRP) structures. TRP structures can adopt closed (i.e., where both termini of the TRP are near in space) repeats such as the β‐propellers or β‐trefoils, or open repeats (i.e., where both termini are distant in space) such as the ARM or ankyrin repeats. TRPs tend to form integrated assemblies in structures, and their individual repeats are unlikely to form stable monomers alone. As such, TRP groups do not conform to the conventional concept of domains and their classification requires special considerations. In ECOD we define the largest possible number of contiguous repeats as the “domain” for open TRP folds and we base our TRP classification on SSE type and assembly into tertiary structures.29 Alternative classifications for repeat proteins exist based on length and interaction type.32 Additionally, we distinguish among domains with TRPs, globular proteins with visible internal repeats, and obligate multimers composed of repeated subunits.
The widespread nature of folds with TRPs suggests that the repetition might confer advantages for protein evolution.29, 30 Repeat expansion/contraction occurs frequently, and open TRPs can theoretically expand to have large repeat numbers, having no steric impediment for subunit additions. Upon expansion of a repeat, sequence similarities of repeating subunits can erode quickly leading to rapid evolution of the duplicated subunits. Such mechanisms of diversification allow selective adaptation to cellular functions. At the same time, this diversity presents a considerable challenge for sequence and structural similarity detection methods.33 TRPs, especially those that occur in coiled‐coils, pose difficult challenges for automated classification schemes. We exclude the coiled‐coil and collagen repeats from our current classification, as their low‐sequence complexity makes evolutionary classification by traditional methods exceedingly difficult.
Domains with small internal repeats in ECOD are classified into architectures primarily by secondary structure type. The open (or solenoid) repeat domains, which can be easily extended by axial duplication, are separated into primarily α (α superhelices) and primarily β (β duplicates and obligate multimers) categories. Ten distinct X‐groups are identified as α superhelices. α superhelical domains are concentrated (90.4% of 40% redundant domains) in the “Repetitive α hairpins” X‐group, with the remaining distributed among 20 X‐groups. These other X‐groups generally contain domains with pairs of α‐helices oriented along a screw axis, although they are not necessarily observed to form a superhelix in a biological context. The “Repetitive α hairpins” X‐group contains many armadillo repeats (ARM), HEAT, and Tetratricopeptide repeats (TPR) collected into a single homologs group [Fig. 5(A)].34 The ankyrin [Fig. 5(B)] and α/α toroid [Fig. 5(C)] H‐groups are siblings to the ARM/HEAT/TPR H‐group. The α/α toroids are closed, rather than open, α‐helical hairpin repeats that include the pectate lyase domains and the farnesyltransferase β subunits.
Figure 5.
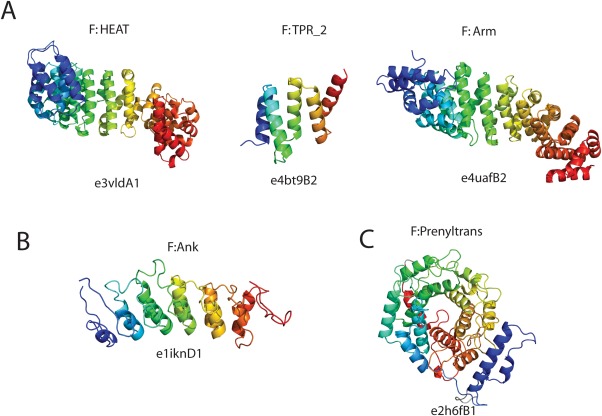
α‐hairpin repeat domains in ECOD. (A) Alpha internal repeat domains in ECOD from the HEAT, TPR, and Armadillo repeats are classified as homologs. (B) The ankyrin domains are classified as possibly homologs to ARM/HEAT/TPR domains. (C) Alpha/alpha toroids, such as farnesyltransferase, are alpha‐hairpin repeats that are closed, rather than open, and not as easily expanded by axial duplication.
The tetratricopeptide repeat (TPR) family is a diverse group of protein domains composed of 34‐residue repeats that form an antiparallel α‐hairpin.35 As with other repeat families (such as the leucine‐rich repeats), their expandable and nonglobular structure leads to their use as scaffolds, and their hypermutability36 leads to their use as molecular recognition domains.37 Pfam‐A clans link sets of sequence families believed to originate from a single evolutionary origin,11 analogs to ECOD H‐groups. We compared the distribution of ECOD domains and F‐groups mapped to Pfam‐A families belong to the TPR clan. Pfam‐A v27.0 contains 117 distinct families in the TPR clan, 79 of which contain at least one structurally characterized protein domain classified by ECOD. TPR domains in ECOD are largely gathered in this “ARM/HEAT/TPR” H‐group. 84.1% of ECOD F40 domains mapped to Pfam‐A families belonging to the TPR clan are clustered in the ECOD “Repetitive α hairpins” H‐group. Some TPR domains could not be definitively linked by homology to the primary TPR group. Domains belonging to F‐groups linked by the Pfam‐A TPR clan appear in distinct H‐groups for the Nup133 C‐terminal domain, alkylsulfatase SdsA1 linker domains, and proteasome/cyclosome (PC) repeats. A TPR clan relationship for the GUN4‐like family was spurious and removed in a subsequent version of Pfam. We separate the β‐solenoids by handedness, for the right‐handed β‐helices and left‐handed β‐helices. The leucine‐rich repeats (LRR), predominantly β, are classified under the right‐handed β helices. The β‐hairpin stacks, which include choline‐binding protein, are placed in a single X‐group.
β‐propellers are distinct in that their topology is both closed (i.e., their N‐ and C‐terminal ends are close in space) and consists of varying numbers of repeats.38 Other closed solenoid repeats, such as the α/α toroids are observed, but not in the same diversity of distinct numbers of repeats. Recently, homologs relationships between the β‐propellers and the β‐prism II domains and IRE1‐LD domains were suggested on the basis of a combination of profile‐profile similarity searches and structural analysis.39 The IRE1‐LD domains were classified as a topological group within the β‐propellers, and the β‐prism II domains became a sibling H‐group to the β‐propellers within the “β‐propeller‐like” X‐group. The diversity of topologies observed within β‐propellers is demonstrative of the purpose of the topological group (T‐group) in ECOD. This includes groups for propellers with differing numbers of repeats, as well as domains with deteriorated blades, or blades that have been substituted for other motifs (Fig. 6). “Closed” domains with varying numbers of internal repeats serve as a bridge in our understanding between open repeats, such as the ARM and TPR open repeats, which are easily expandable and observable in many differing numbers of repeats, and the transition to closed globular domains where internal repeats are observable (e.g., TIM barrels, Rossmann folds, the cradle‐loop barrels), but the multiple‐repeat globular unit is conserved.
Figure 6.
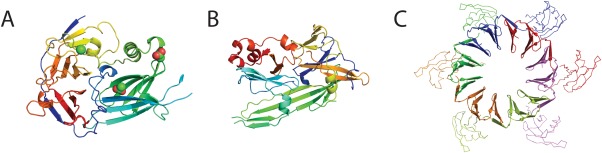
Diverse topologies of the β‐propellers. Examples of non‐canonical topologies in the β‐propeller homologs group. (A) One blade of the deteriorated propeller domain of adsorption PRD1 P2 (e1n7vA1) has been replaced with a novel β domain with complex topology (e1n7vA2). (B) The luminal domain of endoribonuclease IRE1 (e2be1A1) is topologically dissimilar to other β‐propellers, but strong sequence homology to propeller repeats is detected between IRE1 and other canonical propellers. (C) PH1500 is 12‐bladed propeller composed of a hexamer of double propeller repeats, both an obligate multimer and composed of internal repeats (PDB: 2m3x).
Promiscuity of domain arrangements
When proteins are composed of multiple domains the N‐ to C‐terminal ordering of these domains is known as domain arrangement or domain architecture. We refer to it as domain arrangement to disambiguate this concept from ECOD domain architectures (i.e., SSE content and orientation). In addition to containing small internal repeats, domains can contain other inserted domains, which complicates detection and consideration of domain arrangements. In addition to the accumulation of internal repeats or subdomain‐based evolution, proteins can evolve by recombination, duplication, or terminal deletion.5, 40 Synteny is evidence of homology between multidomain proteins in different organisms. The events which lead to domain shuffling, such as terminal deletion, gene fusion, and fission, have been studied extensively on sequence databases and across whole genomes.40, 41, 42 Many studies have examined the frequency of domain shuffling both across domains of life, fully sequenced genomes, and in the host‐pathogen context of comparative genomics.43 The biological mechanisms that lead to small repeat deletion/extensions are distinct from those that lead to larger scale domain shuffling.32 ECOD both attempts to split all domain duplications it finds, and uses domain arrangement as a component of the automatic assignment process, so an analysis of its constituent domain arrangements is necessary to understand how ECOD was created and is maintained.
Domain classifications that use three‐dimensional structure for domain partitioning can provide better and more consistent domain boundary definitions. In cases of internal repetition or whole‐domain duplication, precise boundaries can be difficult to delineate across groups of proteins. In addition, domain discontinuity introduced by insertion can complicate detection of that domain arrangement. However, structural classifications are biased towards proteins that are more easily structurally characterized and/or the subject of investigator interest. Sixty‐eight percentage of ECOD domains are single domain by structure, and 8% are matched to a reference UniProt domain where more than 30 residues are uncovered by the experimental construct. Discerning the domain arrangement of multidomain structures can aid in the determination of new groups. Detection of the same domain in multiple independent contexts reinforces its definition as an independent evolutionary unit.
Differences in synteny between orthologs proteins can be instructive towards analyzing their phylogeny. Similarly, we can observe alterations of domain arrangement within orthologs proteins as a set of intraprotein (rather than interprotein) events of the same type. Previous studies have examined the domain arrangements in single organisms41 and multiple fully sequenced organisms from the archaea, eubacteria and prokaryotes44 on the sequence level. The promiscuity of domains in the context of the repertoire of structurally characterized proteins can be instructive towards events such as insertion, domain deterioration, and multimerization that are not necessarily visible at the level of sequence events. Consideration of structural domains has the advantage of more clearly delineated domain boundaries and more distant homology detection methods for uncovered or unclassified regions. However, the structurally characterized sequence space has different biases than the sequence space of complete genomes and cannot be directly compared.
We considered unique ECOD domain arrangements within both families (F‐groups) and homologs groups (H‐groups). ECOD contained 3203 homologs groups and 12,357 families, classifying 314,989 protein chains from 110,085 PDB depositions. Filtered for 95% nonredundancy, 25,870 protein chains remain. Among these nonredundant protein chains, 8,284 were multidomain by ECOD, with 5104 and 2696 unique domain arrangements considering families and homologs groups as distinct, respectively. The most structurally promiscuous H‐groups are the Ig domains with 192 distinct arrangements, followed by the helix‐turn‐helix (HTH), P‐loop domains, and Rossmann folds. The most structurally promiscuous families are the N‐terminal HTH domain of the tetracycline repressors (68 distinct arrangements), followed by the α‐amylase periplasmic binding proteins, a novel family of EGF repeat, and the pyridine nucleotide‐disulfide oxidoreductase Rossmann domains. We see that in H‐groups, most promiscuous domains combine with domains from the same H‐group but different families (e.g., Ig repeats), but in the most promiscuous F‐groups, domains combine mostly with nonhomologs domains.
Domain insertions are more difficult to distinguish by sequenced‐based methods. Of the unique H‐group domain arrangements in ECOD, 556 contain at least one domain insertion. Furthermore, 957 of the unique F‐group domain arrangements in ECOD contain inserted domains. Domain insertions are a boundary case between evolutionary events at the domain level and below the domain level. Indeed, it can be difficult to distinguish between insertion and fusion and extension.4 Domains can evolve both by duplication and modification of internal motifs, similar to (although by different biochemical mechanisms) to how proteins can shuffle, lose, and gain domains by fusion, duplication, or deletion. At the boundary between subdomain evolution and repeat proteins exist those domains that are composed of small obligate peptide components. Like internal repeat proteins, these obligate multimeric domains are illustrative of how small subunits can be built up to form protein domains.
Obligate multimeric domains as intermediates between TRPs and globular domains
It has been hypothesized that the ancient protein world was composed of small peptide segments (“ancestral peptides” or “antecedent domain segments”) and that the subsequent protein universe can be described as the result of evolution operating on these segments.3 These segments formed both homo‐ and heteromeric complexes. The duplication of elements of small homomeric complexes lead to larger proteins, such as the aforementioned internal repeat proteins. By combinations of permutation, deletion, and subsequent additional duplication, domains evolved into interleaved and globular forms. In ECOD, domain assemblies are defined where multiple domain elements contribute to a single evolutionary unit. These encompass both obligate multimers such as the archaeal cradle loop barrels and cases of domain swapping.
ECOD defines domain assemblies as collections of domains that are either a single evolutionary unit or interacting over some large area. Assemblies are divided into a category of order‐independent domain assemblies (i.e., domain swaps or domains with oligomerization subdomains) and order‐dependent domain assemblies primarily resulting from post‐translational modification (e.g., insulin, viral polyprotein). The principle difference between the order‐independent and order‐dependent assemblies is that the sequence of the order‐dependent subunits can be reassembled in a single sequence for search purposes. The order‐independent assemblies are divided into the nonobligate and obligate multimers. Obligate multimers comprise a group of assemblies where the subunits are not observed independently and are not sufficiently compact to be considered domains. Nonobligate multimers are comprised of domain swaps and or oligomerization helices where the recognition of the interaction with nearby subunits is vital for understanding the boundary assignment, but the individual subunits can be considered domains. One hundred seventy four nonobligate assemblies are divided among 133 topological groups, 22 obligate assemblies are divided among 13 topological groups. Ninty nonredundant order‐dependent assemblies are defined across 55 topology groups. The order‐independent assemblies include dimerization domains, obligate domains in the viral capsid and tail‐spike assemblies, and observed domain swaps. Domain assemblies tend to be formed by domains evolved to function as oligomerization modules, 15% of F40 domain assemblies in ECOD are composed of such domains. These assemblies also tend to occur between capsid proteins in virus, 11% of F40 domain assemblies in ECOD are viral in origin, as compared to 4% among F40 domains in general. Finally, domain assemblies tend to be singleton and unpublished. Of 119 total X‐groups containing domain assemblies, 51 contain only a single domain assembly (and no other unassembled domains). These domains often have little evolutionary signal in terms of structural or sequence similarity, which reflects the ease with which they arise. Twenty‐seven percentage of domain assemblies in ECOD are from structural genomics centers, which is similar to the 25% fraction of ECOD domains from SG projects in general. Domain assemblies in ECOD are currently only found by manual inspection, accordingly, some domains have yet to be assembled and result in mixed F‐groups wherein domain assemblies and domains comingle.
The four‐helix bundle represents a common protein structural motif found independently and as a component of larger folding units in globular proteins. While the motif can be classified by the topology and geometry of interacting helices, the diversity of structural contexts and functions of class members hinders detection of their relatives. Thus, four‐helix bundles appear to consist of many small groups of possibly evolutionarily unrelated domains, suggesting the regular architecture of two interacting α‐helix hairpins has arisen independently multiple times. The ECOD Ferritin/Heme oxygenase H‐group provides an interesting example of four‐helix bundle evolution from an obligate oligomeric α‐helix hairpin.
The ferritin‐like fold retains a core four‐helix bundle with a left‐handed twist and a single crossover connection between the two pairs of conserved helices.45 The secondary structure composition of the crossover connection in ferritin‐like structures ranges from a simple elongated loop, as seen in rubrerythrin (e1lkoA1), to multiple helices as in phenylacetic acid degradation protein (PaaC) (e1otkA1) or both helices and strands as in ribonucleotide reductase (e1mxrA1). While ferritin (e1lb3A1) forms the least complex four‐helix bundle, additional helices can decorate this conserved core in ribonucleotide reductases and heme oxygenases. For example, ribonucleotide reductase includes an N‐terminal helical extension as well as a C‐terminal α‐hairpin that packs against the core four‐helix bundle.
Despite the presence of different crossover compositions and additional secondary structure elements decorating the core fold, the ferritin/heme oxygenase H‐group retains a similar active site position at the center of the four‐helix bundle that unites the folds. The ferritin di‐iron center closely resembles that found in the redox enzyme class that includes ribonucleotide reductase and other related enzymes, suggesting a previously described evolutionary linkage between the two families.46 While heme oxygenase forms the heme binding site towards the exterior of the four‐helix bundle using an N‐terminal α‐helix extension, its ferrous O2 points into the center of the core bundle where the active site residues are located.47 The active sites of heme oxygenase‐like enzymes such as pyrroloquinolinquinone synthase C (PqqC) (e4ny7A1) and transcriptional activator A thiaminase (TenA) (e1rtwA1) also fall in the center of the core fold.
The proposed evolution of the ECOD Ferritin/Heme oxygenase H‐group is illustrated in Figure 7. An ancient “half‐ferritin” α‐hairpin, exemplified by the structure of unknown protein ne0167 (e3k6cA1), assembles head‐to‐tail into the ferritin‐like four‐helix bundle core, which forms a higher order homododecamer similar to that seen in the dodecameric ferritin homolog Dps (e1dpsA1). Duplication and fusion of the “half‐ferritin” α‐hairpin into the four‐helix bundle requires a crossover connection that defines the fold. ECOD splits the ferritin/heme oxygenase H‐group, which adopts the crossover connection adjacent to the first two helices, from the cobalamin adenosyltransferase‐like H‐group with the crossover on the other side. Additional α‐helices can decorate the core ferritin‐like bundle leading to ribonucleotide reductase or to MiaE, which both possess similarly positioned C‐terminal α‐helices. The ribonucleotide reductase fold is further decorated in toluene monooxygenase (e1t0qA1), which contains two homologs subunits that probably arose from another duplication event. One of the toluene monooxygenase subunits exhibits loss of the di‐metallic active site as well as deteriorations of the decorating helices with respect to the active‐site containing subunit. The heme oxygenase‐like folds adapted the di‐metallic active site, possibly from a MiaE‐like fold, into that of an oxidoreductase (PqqC or heme oxygenase) or a thiaminase (TenA).
Figure 7.
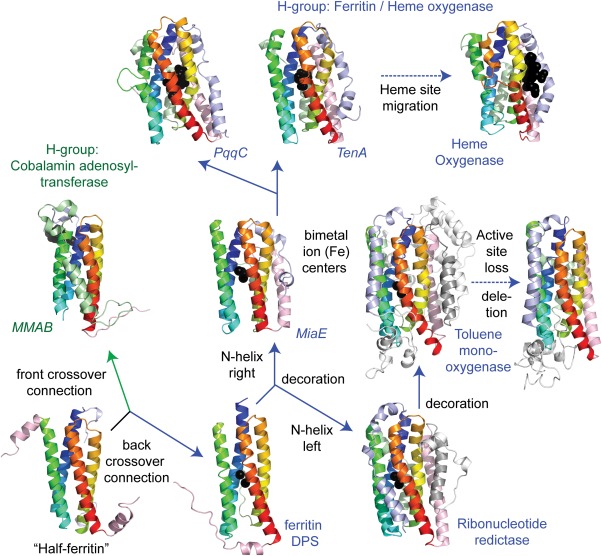
Evolution of the ferritin domains. The evolution of the core four‐helix bundle (in rainbow, from blue to red) of the ferritin/heme oxygenase H‐group from duplication and fusion of two “half ferritin” helix‐hairpins (lower left panel). The head to tail arrangement of the helix‐hairpins requires a crossover connection, which occurs between the first two helices (ferritin/heme oxygenase labeled in blue) or the last two (cobalamin adenosyltransferase labeled in green). Secondary structure decorations of the core fold (N‐terminal decoration in slate, C‐terminal decoration in salmon) as well as alternate crossover connection compositions (colored in light green) occur in several ferritin/heme oxygenase families. The positioning of the active sites marked by di‐iron centers or other ligands (black spheres) in the core of the four‐helix bundle provides the basis for uniting the different families. Duplication combined with secondary structure deletion and active site loss occurred in a subunit of toluene monooxygenase, while a migration of a heme binding site occurred in heme oxygenase.
Delineation of domain boundaries using structural classifications
The domain boundaries of a protein family can be partitioned consistently given structural evidence, considering the complex scenario of evolutionary events at the aforementioned domain and subdomain levels. In addition, the multiple concepts comprising domain definitions lead to different perspectives between different types of protein classifications. Whereas structural classifications may have clearer domain boundaries, sequence‐based classifications can access larger datasets that more comprehensively sample protein space (including entire genomes). In ECOD, we consider domains as independent evolutionary units and manually curate the domain boundary for structural representatives. ECOD F‐groups cluster domains into families by using the sequence‐based Pfam‐A family definitions as a seed. The boundaries of Pfam families were then compared with ECOD domains. Out of a 40% sequence redundant representative set, 25,989 (81.7%) domains are mapped to exactly one Pfam family; 1334 (4.2%) domains contain multiple nonoverlapping mappings to distinct Pfam families, indicating potential Pfam families to merge; 4476 (14.1%) domains have no significant Pfam hits. For the one‐to‐one mapping, the coverage of Pfam families by ECOD domain exhibits an exponential distribution and 91.2% of these domains have more than 50% coverage by a Pfam family [Fig. 8(A)]. Those ECOD domains that can be classified by Pfam are consistent with the Pfam domain definition. ECOD domains with low coverage by Pfam families can be attributed to continual internal repeats (e.g., β‐propeller, ARM repeat, β‐helix, etc.), where Pfam only defines one or several repeating units as a family and ECOD tends to cover all. Others are usually explained by the nature of slower evolution of structure.2 The sequence family may only capture the most conserved core of the actual domain especially when it is established before any structural information is available, while individual structures can diverge in sequence space and develop assorted insertions and decorations. The percentage of Pfam overlap on ECOD domain suggests the degree to which structures diverge while keeping detectable conserved sequence signal. However, there are also some Pfam families that are very short and are better described as a conserved motif.
Figure 8.
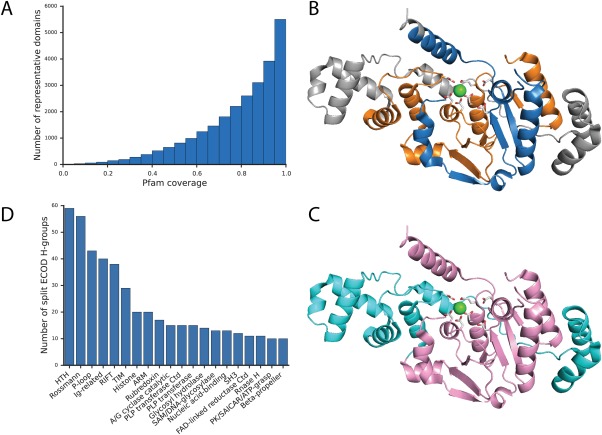
Comparison of domain definitions between ECOD and Pfam. (A) The distribution of Pfam family coverage on a nonredundant set of ECOD domains that have a one‐to‐one mapping to Pfam families. (B) Mapping of Pfam family XPG_N (PF00752, blue) and XPG_I (PF00867, orange) on RAD2 structure (PDB: 4q0w). (C) Mapping of HAD‐related domain (e4q0wA2, pink) and SAM‐like domain (e4q0wA1, cyan) from ECOD on the same structure. Side chains of catalytic residues are shown in stick, with the coordinating calcium ion in green sphere. (D) Top 20 H‐groups where split Pfam families are assigned.
Out of 1334 ECOD domains with nonoverlapping regions that map to distinct Pfam families, 426 are mapped to unique Pfam domain arrangements where the individual families contain no structurally compact domain and do not occur independently. Often the co‐occurrence of these families is noted by Pfam. For example, XPG_N (PF00752) and XPG_I (PF00867) were first discovered as two highly conserved N‐terminal and internal regions of Xeroderma Pigmentosum Complementation Group G (XPG) proteins.48 The XPG family includes various structure‐specific nucleases, such as XPG/RAD2, flap endonuclease 1 (FEN1), and exonuclease 1 (EXO1).49 Initially identified via comparison to yeast RAD2, XPG_N and XPG_I are separated by more than 600 amino acids in the alignment, but in FEN1 and EXO1 the spacer is shorter than 50 amino acids.48, 49 Later, crystal structures showed that XPG_N and XPG_I intertwine to form a compact α/β three‐layered sandwich50, 51, 52 [Fig. 8(B)], which suggests that both belong to the large HAD‐like superfamily.53 A number of protein domains in the XPG_I family incorrectly include the C‐terminal SAM‐like domain H2TH motif [Fig. 8(B,C)]. The XPG family active site is located above the β‐sheet where the N and I regions meet and is composed of carboxylate groups from both segments [Fig. 8(B)].54 These two families also co‐occur with high frequency in Pfam. Therefore, we determine that XPG‐N and XPG‐I are best represented as a single family [Fig. 8(C), pink], where the H2TH motif belongs to a separate C‐terminal domain [Fig. 8(C), cyan). A similar merge was made with respect to incorporation of PAC motif into PAS domain.55 Conversely, ECOD domains sometimes split Pfam defined domains. Often, functional sites form at the intersection of structural domains. For such cases, sequence‐based classifications tend to merge the structural domains into a single sequence domain due to similar conservation patterns that define the functional site. We found 771 Pfam families mapped to multiple ECOD domains in different H‐groups in the aforementioned 40% redundant set. The most commonly split H‐groups are HTH, Rossmann‐related, P‐loop domain‐related, and immunoglobulin‐related, which are also the most populated groups generally in ECOD [Fig. 8(D)].
The definition of domain could evolve as our understanding of proteins advances. In a structural classification database, we can identify remote homology beyond the capacity of sequence detection methods to gain a better view of protein evolution, and we keep refining it with updates of ECOD every week. The inconsistency between Pfam family and ECOD domain reflects a need to improve current definitions of protein families that are purely based on sequence information by incorporating evolutionary insights from protein structures.
Impact of structural genomics on classification
Structural genomics was a global initiative to determine structures from uncharacterized protein families in order to increase the coverage of the known protein structural space.56 The Protein Structure Initiative (PSI) funded by the NIH, began in 2000 and ended in 2015. Now that this project has concluded, we are in an ideal position to examine the increase in structural coverage. Since ECOD classifies each structure in the known protein structural space, we can locate domains from structural genomics and analyze their relationship to other domains from structures determined by traditional structural biology methods. We assessed domains from structural genomics targets in ECOD both to determine whether they initially contributed to the formation of new hierarchical groups or the extent to which hierarchical groups are entirely composed of structural genomics domains.
The PSI was conducted in three phases between 2000 and 2015; by the conclusion of the project over 13,500 structures were deposited in the PDB. We partitioned these structures into 41,245 domains in ECOD. Then we analyzed the novelty of the distribution of these domains in ECOD, both over time and within the hierarchy. Principally we wanted to know, which SG domains were the initial members of homologs groups and which groups were solely populated by SG domains. Groups were determined to be formed by SG targets if the earliest deposition date in a group belonged to an SG domain. SG targets began to be deposited in increasing amounts around 2004, increasing through 2007, then tapering off by 2012. These depositions occurred in the background of a consistent exponential increase of domains from structures determined by traditional methods [Fig. 9(A)]. We analyzed the contents of the entire PDB so these domain trends encompass sequence‐redundant depositions. Nonetheless, these trends are recapitulated by the count of ECOD groups initially formed by SG targets, leading to 2,668 new sequence families, 709 new homologous groups, and 394 new X‐groups [Fig. 9(B)]. Interestingly, we find that although the target selection pipelines of SG projects were efficient at targeting new groups, this discovery occurred in the background of fairly steady rate of characterization of new sequence families [Fig. 9(C)] and the determination of structures fairly evolutionarily distant from existing groups [Fig. 9(D)].
Figure 9.
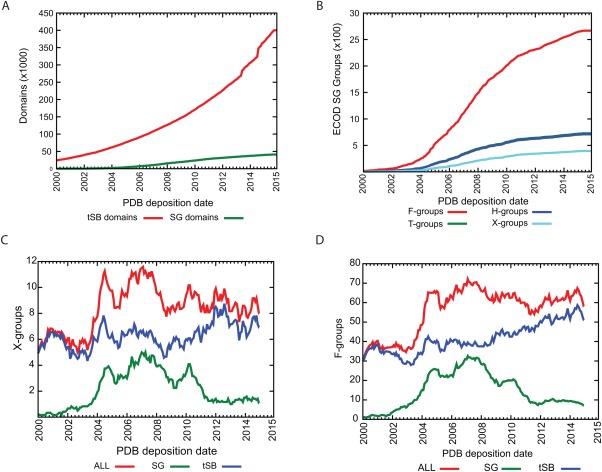
Cumulative total over time of structural genomics targets in ECOD. Distribution of domains from structural genomics targets over time (A) by domains and (B) by hierarchical groups. SG domains were considered to form a new group if they were the earliest deposited domain in that group. Moving averages (1 year) were calculated for structural genomics domains that (C) formed new X‐groups and (D) newly characterized sequence families.
The development of high‐throughput methods for genome sequencing and determination of structures has led to the so‐called “deluge of data.” Increasingly, it is unlikely to find that any single structure (or small set of structures) or genome necessarily is mapped to a single descriptive publication. Only 31% of SG domains have a primary citation recorded in their deposition that resolves to an existing publication (compared to 86% overall). However, of those SG domains with no primary citation, 83% and 95% are siblings to sequence family (F‐group) or homologs group (H‐group) members with a primary citation, respectively. As structural genomics comes to a close, it is unclear whether uncited depositions will become prevalent again in the future, but this illustrates that classifications such as ECOD might be useful for finding additional data on such structures.
Methods
ECOD versions and representative sets
All analyses were conducted using ECOD v105, which is available from the ECOD website at http://prodata.swmed.edu/ecod/. ECOD v105 contains 452288 domains from 110,085 structures. A 40% redundant set (i.e., the F40 representative set) was constructed by filtering those domains with greater than 40% sequence identity within each F‐group by BLASTCLUST, then selecting a representative from the resulting clusters, yielding 31,799 F40 domains. Structural genomics annotations and deposition dates for structures were determined from PDB metadata in PDBml files distributed by the wwPDB. The derivation of ECOD has been discussed in detail elsewhere.27
Detection of psi‐loop motif by PROSMOS
Psi‐loop motifs were detected in F40 ECOD v105 domains by Protein Structure Motif Search (ProSMoS).57 Two search matrices were used, where the cross‐over connection precedes or follows the anti‐parallel β‐sheet. β‐strands were allowed to vary between 5 and 100 residues. 3,219 F40 domains were detected where the psi‐loop preceded the antiparallel strands, 2,158 were detected where the psi‐loop followed.
Pfam analysis sources and methods
HMMER 3.1b258 was used to assign Pfam families (version 28) to the ECOD nonredundant set described above with a E‐value cutoff of 1e‐3. Pfam assignments were made sequentially based on E‐value, alignment overlap of 20 residues or less was allowed between subsequent assignments. The number of Pfam assignment on each ECOD representative domain was counted and coverage was calculated per residue. Pfam families that were assigned to ECOD domains from different H‐groups in the set were also analyzed.
Acknowledgments
The authors thank Dr. Hua Cheng for critical reading of the manuscript.
References
- 1. Grishin NV (2001) Fold change in evolution of protein structures. J Struct Biol 134:167–185. [DOI] [PubMed] [Google Scholar]
- 2. Grishin NV (2001) KH domain: one motif, two folds. Nucleic Acids Res 29:638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lupas AN, Ponting CP, Russell RB (2001) On the evolution of protein folds: are similar motifs in different protein folds the result of convergence, insertion, or relics of an ancient peptide world? J Struct Biol 134:191–203. [DOI] [PubMed] [Google Scholar]
- 4. Majumdar I, Kinch LN, Grishin NV (2009) A database of domain definitions for proteins with complex interdomain geometry. PLoS One 4:e5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kinch LN, Grishin NV (2002) Evolution of protein structures and functions. Curr Opin Struct Biol 12:400–408. [DOI] [PubMed] [Google Scholar]
- 6. Alva V, Remmert M, Biegert A, Lupas AN, Soding J (2010) A galaxy of folds. Protein Sci 19:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reeves GA, Dallman TJ, Redfern OC, Akpor A, Orengo CA (2006) Structural diversity of domain superfamilies in the CATH database. J Mol Biol 360:725–741. [DOI] [PubMed] [Google Scholar]
- 8. Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG (2008) Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 36:D419–D425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hadley C, Jones DT (1999) A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Structure 7:1099–1112. [DOI] [PubMed] [Google Scholar]
- 10. Cheng H, Liao Y, Schaeffer RD, Grishin NV (2015) Manual classification strategies in the ECOD database. Proteins 83:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marchler‐Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grishin NV (2001) Mh1 domain of Smad is a degraded homing endonuclease. J Mol Biol 307:31–37. [DOI] [PubMed] [Google Scholar]
- 14. Via A, Ferre F, Brannetti B, Valencia A, Helmer‐Citterich M (2000) Three‐dimensional view of the surface motif associated with the P‐loop structure: cis and trans cases of convergent evolution. J Mol Biol 303:455–465. [DOI] [PubMed] [Google Scholar]
- 15. Leipe DD, Koonin EV, Aravind L (2003) Evolution and classification of P‐loop kinases and related proteins. J Mol Biol 333:781–815. [DOI] [PubMed] [Google Scholar]
- 16. Rosinski JA, Atchley WR (1999) Molecular evolution of helix‐turn‐helix proteins. J Mol E 49:301–309. [DOI] [PubMed] [Google Scholar]
- 17. Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM (2005) The many faces of the helix‐turn‐helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29:231–262. [DOI] [PubMed] [Google Scholar]
- 18. Copley RR, Russell RB, Ponting CP (2001) Sialidase‐like Asp‐boxes: sequence‐similar structures within different protein folds. Protein Sci 10:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murzin AG (1998) How far divergent evolution goes in proteins. Curr Opin Struct Biol 8:380–387. [DOI] [PubMed] [Google Scholar]
- 20. Andreeva A, Murzin AG (2006) Evolution of protein fold in the presence of functional constraints. Curr Opin Struct Biol 16:399–408. [DOI] [PubMed] [Google Scholar]
- 21. Russell RB, Marquez JA, Hengstenberg W, Scheffzek K (2002) Evolutionary relationship between the bacterial HPr kinase and the ubiquitous PEP‐carboxykinase: expanding the P‐loop nucleotidyl transferase superfamily. FEBS Lett 517:1–6. [DOI] [PubMed] [Google Scholar]
- 22. Athappilly FK, Hendrickson WA (1995) Structure of the biotinyl domain of acetyl‐coenzyme A carboxylase determined by MAD phasing. Structure 3:1407–1419. [DOI] [PubMed] [Google Scholar]
- 23. Anantharaman V, Koonin EV, Aravind L (2001) Regulatory potential, phyletic distribution and evolution of ancient, intracellular small‐molecule‐binding domains. J Mol Biol 307:1271–1292. [DOI] [PubMed] [Google Scholar]
- 24. McLachlan AD (1972) Repeating sequences and gene duplication in proteins. J Mol Biol 64:417–437. [DOI] [PubMed] [Google Scholar]
- 25. Hutchinson EG, Thornton JM (1996) PROMOTIF–a program to identify and analyze structural motifs in proteins. Protein Sci 5:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castillo RM, Mizuguchi K, Dhanaraj V, Albert A, Blundell TL, Murzin AG (1999) A six‐stranded double‐psi beta barrel is shared by several protein superfamilies. Structure 7:227–236. [DOI] [PubMed] [Google Scholar]
- 27. Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH, Grishin NV (2014) ECOD: an evolutionary classification of protein domains. PLoS Comput Biol 10:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alva V, Koretke KK, Coles M, Lupas AN (2008) Cradle‐loop barrels and the concept of metafolds in protein classification by natural descent. Curr Opin Struct Biol 18:358–365. [DOI] [PubMed] [Google Scholar]
- 29. Andrade MA, Perez‐Iratxeta C, Ponting CP (2001) Protein repeats: structures, functions, and evolution. J Struct Biol 134:117–131. [DOI] [PubMed] [Google Scholar]
- 30. Soding J, Lupas AN (2003) More than the sum of their parts: on the evolution of proteins from peptides. Bioessays 25:837–846. [DOI] [PubMed] [Google Scholar]
- 31. Coles M, Djuranovic S, Soding J, Frickey T, Koretke K, Truffault V, Martin J, Lupas AN (2005) AbrB‐like transcription factors assume a swapped hairpin fold that is evolutionarily related to double‐psi beta barrels. Structure 13:919–928. [DOI] [PubMed] [Google Scholar]
- 32. Kajava AV (2012) Tandem repeats in proteins: from sequence to structure. J Struct Biol 179:279–288. [DOI] [PubMed] [Google Scholar]
- 33. Pellegrini M (2015) Tandem repeats in proteins: Prediction algorithms and biological role. Front Bioeng Biotechnol 3:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrade MA, Petosa C, O'Donoghue SI, Muller CW, Bork P (2001) Comparison of ARM and HEAT protein repeats. J Mol Biol 309:1–18. [DOI] [PubMed] [Google Scholar]
- 35. Lamb JR, Tugendreich S, Hieter P (1995) Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci 20:257–259. [DOI] [PubMed] [Google Scholar]
- 36. Ellegren H (2000) Microsatellite mutations in the germline: implications for evolutionary inference. Trends Genet 16:551–558. [DOI] [PubMed] [Google Scholar]
- 37. Smith DF (2004) Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones 9:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaudhuri I, Soding J, Lupas AN (2008) Evolution of the beta‐propeller fold. Proteins 71:795–803. [DOI] [PubMed] [Google Scholar]
- 39. Kopec KO, Lupas AN (2013) Beta‐propeller blades as ancestral peptides in protein evolution. PLoS One 8:e77074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chothia C, Gough J (2009) Genomic and structural aspects of protein evolution. Biochem J 419:15–28. [DOI] [PubMed] [Google Scholar]
- 41. Teichmann SA, Park J, Chothia C (1998) Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc Natl Acad Sci U S A 95:14658–14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore AD, Grath S, Schuler A, Huylmans AK, Bornberg‐Bauer E (2013) Quantification and functional analysis of modular protein evolution in a dense phylogenetic tree. Biochim Biophys Acta 1834:898–907. [DOI] [PubMed] [Google Scholar]
- 43. Barrera A, Alastruey‐Izquierdo A, Martin MJ, Cuesta I, Vizcaino JA (2014) Analysis of the protein domain and domain architecture content in fungi and its application in the search of new antifungal targets. PLoS Comput Biol 10:e1003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Apic G, Gough J, Teichmann SA (2001) Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J Mol Biol 310:311–325. [DOI] [PubMed] [Google Scholar]
- 45. Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540. [DOI] [PubMed] [Google Scholar]
- 46. Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203. [DOI] [PubMed] [Google Scholar]
- 47. Matsui T, Iwasaki M, Sugiyama R, Unno M, Ikeda‐Saito M (2010) Dioxygen activation for the self‐degradation of heme: reaction mechanism and regulation of heme oxygenase. Inorg Chem 49:3602–3609. [DOI] [PubMed] [Google Scholar]
- 48. Scherly D, Nouspikel T, Corlet J, Ucla C, Bairoch A, Clarkson SG (1993) Complementation of the DNA repair defect in xeroderma pigmentosum group G cells by a human cDNA related to yeast RAD2. Nature 363:182–185. [DOI] [PubMed] [Google Scholar]
- 49. Harrington JJ, Lieber MR (1994) Functional domains within FEN‐1 and RAD2 define a family of structure‐specific endonucleases: implications for nucleotide excision repair. Genes Dev 8:1344–1355. [DOI] [PubMed] [Google Scholar]
- 50. Hwang KY, Baek K, Kim HY, Cho Y (1998) The crystal structure of flap endonuclease‐1 from Methanococcus jannaschii . Nat Struct Biol 5:707–713. [DOI] [PubMed] [Google Scholar]
- 51. Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA (2011) Human flap endonuclease structures, DNA double‐base flipping, and a unified understanding of the FEN1 superfamily. Cell 145:198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mietus M, Nowak E, Jaciuk M, Kustosz P, Studnicka J, Nowotny M (2014) Crystal structure of the catalytic core of Rad2: insights into the mechanism of substrate binding. Nucleic Acids Res 42:10762–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burroughs AM, Allen KN, Dunaway‐Mariano D, Aravind L (2006) Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol 361:1003–1034. [DOI] [PubMed] [Google Scholar]
- 54. Tomlinson CG, Atack JM, Chapados B, Tainer JA, Grasby JA (2010) Substrate recognition and catalysis by flap endonucleases and related enzymes. Biochem Soc Trans 38:433–437. [DOI] [PubMed] [Google Scholar]
- 55. Hefti MH, Francoijs KJ, de Vries SC, Dixon R, Vervoort J (2004) The PAS fold. A redefinition of the PAS domain based upon structural prediction. Eur J Biochem 271:1198–1208. [DOI] [PubMed] [Google Scholar]
- 56. Chandonia JM, Brenner SE (2006) The impact of structural genomics: expectations and outcomes. Science 311:347–351. [DOI] [PubMed] [Google Scholar]
- 57. Shi S, Zhong Y, Majumdar I, Sri Krishna S, Grishin NV (2007) Searching for three‐dimensional secondary structural patterns in proteins with ProSMoS. Bioinformatics 23:1331–1338. [DOI] [PubMed] [Google Scholar]
- 58. Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR (2015) HMMER web server: 2015 update. Nucleic Acids Res 43:W30–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]