

Abstract
The recent Ebola virus outbreak in western Africa highlights the need for novel therapeutics that target Ebola virus and other filoviruses. Filoviruses require processing by host cell-derived cysteine cathepsins for productive infection. Here we report the generation of a focused library of cysteine cathepsin inhibitors and subsequent screening to identify compounds with potent activity against viral entry and replication. Our top compounds show highly potent and broad-spectrum activity against cysteine cathepsins and were able to effectively block entry of Ebola and Marburg viruses. These agents are promising leads for development as antifilovirus therapeutics.
Keywords: filovirus, cathepsin inhibitor, Ebola virus, inhibition of host cell entry
Graphical abstract
Members of the family Filoviridae of enveloped negative-strand RNA viruses (filoviruses), including Ebola virus (EBOV) and Marburg virus (MARV), are associated with sporadic, but highly lethal, outbreaks of viral disease. The recent, devastating Ebola virus epidemic in western Africa has highlighted the need for vaccines and therapeutics to prevent and treat filovirus infections. Although cocktails of monoclonal antibodies specific for the viral surface glycoprotein (GP) and small molecule inhibitors of the viral polymerase have shown promise,1 targeting host factors required for viral infection is a promising alternative strategy.2 These therapeutics are more likely to provide broad protection against multiple viral strains and may be less likely to engender resistance through viral mutation.
The complex cell entry mechanism of filoviruses relies heavily on host proteins, providing the opportunity to develop antivirals that target these host factors. Following attachment at the cell surface, the filamentous virions (infectious particles) of filoviruses are internalized into the endocytic pathway and delivered to late endo/lysosomal compartments. Here, lysosomal cysteine proteases (“cysteine cathepsins”), including cathepsin B (CatB) and cathepsin L (CatL), cleave the GP to remove variable, highly glycosylated sequences,3,4 unmasking the binding site for the essential filovirus receptor, Niemann-Pick C1 (NPC1).5–7 The interaction between cleaved GP and NPC1 is absolutely required to activate downstream steps in the entry process, culminating in GP-catalyzed fusion between viral and cellular membranes and escape of the viral nucleocapsid payload into the cytoplasm.5 The current understanding of filovirus entry thus provides a strong rationale to develop cysteine cathepsin inhibitors as antifilovirus drugs.
Previous work with available class-specific inhibitors has demonstrated that inhibiting cysteine cathepsins is an effective and specific antiviral strategy, at least in tissue culture.2,3 Although the protective efficacy of such compounds in mouse models of filovirus challenge is unknown, studies with CatB- and CatL-knockout mice suggest that inhibitors highly selective for specific cysteine cathepsins (e.g., CatS vs CatB+CatL) are unlikely to provide therapeutic benefit, due to the redundant use of different cysteine cathepsins by filoviruses.4,8
E-64, an irreversible epoxide inhibitor of cysteine cathepsins,9 represents an ideal starting point for the development of antifilovirus drugs targeting these enzymes. First, it is well tolerated in vivo; E-64 analogues have been evaluated in human clinical trials as therapeutics against muscular dystrophy with overall low toxicity and few reported side effects,10,11 and analogues of E-64 have been shown to block tumor growth and invasiveness in mouse models of cancer.12 Second, it is highly specific for cysteine cathepsins with reactivity toward only a small number of other CA clan cysteine proteases (e.g., calpains). Third, it affords broad inhibition of all of the cysteine cathepsins. However, the poor membrane permeability of E-64 and analogues limits their antiviral potency as they must be added to cells at high concentrations to sufficiently permeate lysosomes and block CatB and CatL activities. Esterification of the free carboxylic acid in E-64 alleviates this problem in tissue culture, but these esters are not suitable for in vivo administration, because they are rapidly and efficiently hydrolyzed by esterases abundant in plasma.10
We sought to develop epoxide-based inhibitors of filovirus infection that are cell permeable, highly potent, and suitable for in vivo administration. Accordingly, we synthesized a library of epoxysuccinate-based cysteine cathepsin inhibitors and screened them using a filovirus entry assay. We chose to generate a library in which some of the inhibitors contained a basic substituent as these compounds can become protonated in the acidic lysosome compartment, resulting in increased retention. Although this is a liability for lead compounds that would need to be chronically administered, it is not likely to result in significant toxicity when used acutely to treat Ebola virus disease. Epoxysuccinate carboxylic acids are highly potent cysteine cathepsin inhibitors; however, they exhibit poor cell permeability and are unlikely to be highly potent inhibitors of viral entry. Therefore, we focused our design efforts on esterase-resistant epoxysuccinate inhibitors that retain high levels of cell permeability and are expected to have increased plasma stability in vivo.
As a starting point for library design, we chose the previously reported epoxysuccinate inhibitor AMS36, which is a potent inhibitor of multiple cathepsins but suffers from poor solubility and difficult formulation.13 Therefore, to generate a small focused library of inhibitors, we used the main core structure of AMS36 but coupled a set of 24 drug-like amines to the Phe(Me)-epoxysuccinate (Figure 1A). This set of amines contains basic scaffolds, because basic compounds are known to be lysosomotropic and thus have substantially enhanced cellular potency against lysosomal proteases.14 We used both the S,S- and R,R-epoxysuccinate to evaluate the effect of the epoxysuccinate stereochemistry on cathepsin inhibition, given that this stereochemistry has been shown to influence inhibitor selectivity and potency.13 To initiate library synthesis we coupled Fmoc-Phe(Me)-OH to Boc-hydrazine, after which we removed the Fmoc group using diethylamine in DMF (Figure 1A). The resulting amine was reacted with either S,S- or R,R-epoxysuccinate monoethyl ester.15 After removal of the Boc protecting group, the hydrazides were converted to the acyl azides under the influence of tBuONO and HCl, which were subsequently reacted with the 24 amines from the library. This yielded 44 inhibitors in total.
Figure 1.

(A) Synthesis of the library of cathepsin inhibitors. (B) Heatmap of cathepsin inhibition. RAW cells were incubated with 10 nM of inhibitor for 1 h, followed by 1 h of incubation with BMV109 (1 μM). Cells were lysed, proteins separated by SDS-PAGE, and the gels scanned for fluorescence; signals were quantified with ImageJ, and the heatmap was generated using Excel.
We initially tested the potency of the compounds against native cathepsins from relevant cellular sources. To do this, we performed competition labeling studies wherein each compound was added to intact RAW macrophages and then residual cathepsin activity measured using the activity-based cathepsin probe BMV109.16 We initially screened the full library of 44 compounds containing both the R,R and S,S stereochemistries and found that the compounds containing the R,R stereochemistry were overall more potent (Figure 1B) than the S,S compounds (Supporting Information Figure 1). We also found that the compounds containing a basic amine were substantially more potent than compounds that did not contain a protonatable amine, consistent with the lysosomotropy of the basic compounds.14
Importantly, we identified a number of promising lead compounds that were able to completely inactivate multiple cathepsins in intact cells at low nanomolar concentrations. However, all of the compounds in the library contained the ethyl ester, which is labile in vivo.13 To evaluate the effect of conversion of the ethyl ester to the free carboxylic acid in serum, we synthesized the carboxylic acid derivative of several of the most potent hits, including one basic amine containing compound (R11) and one that did not contain a basic amine (R7). We found that conversion to the free acid resulted in a reduction in potency of several orders of magnitude for the basic amine containing inhibitor R11 in intact cells and a much smaller drop in the potency for the R7 scaffold (Figure 2). Furthermore, similar competition experiments performed in cell lysates confirmed that both free acid compounds were potent inhibitors of the cathepsins and suggest that the main reason for the loss of activity in cells was due to poor uptake by the negatively charged free acid. These results are again consistent with the notion that the basic amine containing compounds show enhanced potency due to cellular uptake and, furthermore, that conversion of the ethyl ester to the free carboxylic acid prevents target inhibition in the lysosome. Similar potencies of R11Et and R11H are observed in less-phagocytic U2OS cells (Supporting Information Figure 3), suggesting the observed potencies in RAW cells are not due to high phagocytic activity of these cells.
Figure 2.

Effect of ethyl ester hydrolysis on potency of compounds R11 (A) and R7 (B). RAW cells or RAW cell lysates were incubated with the indicated concentration of inhibitor. Probe BMV109 was used, as indicated in Figure 1, to label remaining cathepsin activity; disappearance of fluorescent signal corresponds to inhibition of cathepsin.
We next tested the ability of our most potent inhibitors to block the processing of EBOV or MARV GP that is required for productive infection. We tested inhibitors in an infection model in which vesicular stomatitis virus (VSV) particles bearing EBOV or MARV GP (VSV-GP) are used to infect human cells in culture.3 This allows assessment of overall capacity of the inhibitors to block GP processing in cells without the need to work with authentic filoviruses in BSL4 facilities. The nonbasic thiazole containing compound R7Et showed good potency with complete protection in the low nanomolar range (Figure 3A). As expected from our cellular studies, the basic amine containing compound R11Et was exceptionally potent and could completely block VSV infectivity in the sub-nanomolar concentration range (Figure 3B). We also tested several other top hits from the library including R4Et, R5Et, and R14Et, and all showed highly potent inhibition of VSV infectivity (Suporting Information Table 1). As expected, all of the free acid versions of the compounds were substantially less potent, with activities in the micromolar range (Figure 3A,B). Overall, we found R11Et to be the most potent inhibitor of VSV-GP infection, having an IC50 values of 30 and 50 pM for VSV-EBOV GP and VSV-MARV GP, respectively. This illustrates that lysosomal cysteine cathepsins indeed are the effectors of GP cleavage and are an important gateway for EBOV entry into host cells.
Figure 3.

Inhibition of VSV-EBOV and VSV-MARV infection by cathepsin inhibitor: (A) R7Et, R7H; (B) R11Et, R11H. (C) Inhibition of VSV-MARV infection by R11-amides. (D) Competition of R11P in RAW cells and RAW cell lysate versus BMV109 as indicated in Figure 2.
Although we identified an inhibitor of GP processing with picomolar potency in cells, we had to find a solution to the overall poor stability of the ethyl ester in vivo. To circumvent this liability, we synthesized a range of epoxysuccinate analogues, in which the ethyl ester was replaced by amide groups that were likely to have better in vivo stability properties. We determined their IC50 values in RAW cells (Supporting Information Table 2) and analyzed their ability to block VSV-GP infection (Figure 3C). From this SAR, we determined that the propylamide provided the greatest potency in the viral entry assay. Although conversion of the ethyl ester to the propylamide resulted in some loss of potency (Figure 3D), the resulting amide retained low nanomolar potency activity for both EBOV and MARV GP processing in the VSV assay. Furthermore, direct analysis of the overall serum stability of R11Et and R11P confirmed that conversion of the highly labile ethyl ester group to the amide analogue resulted in dramatically increased serum stability (Supporting Information Figure 4).
As a final and important test of our lead compounds, we evaluated potency in an infection assay using authentic EBOV and MARV. For these studies, we selected the propylamide analogues of three primary compound scaffolds, R11, R7, and R23. We selected these leads because R11 was the most potent nonracemic basic inhibitor scaffold, whereas R7 and R23 were the most potent nonbasic scaffolds that also had minimal chiral centers. We confirmed that all of the compounds in the ethyl ester and propylamide forms efficiently blocked cathepsin activity in intact cells (Supporting Information Table 3). We then measured the capacity of each to block authentic filovirus infection (Figure 4A,B; Supporting Information Figure 2A,B). Consistent with our result using the VSV assay, we found that R11Et was the most potent inhibitor of both EBOV and MARV infection, with an IC50 of 4 nM. Importantly, its more serum-stable equivalent, R11P, was highly potent as well with an IC50 of 70 nM. We also found that the R7 and R23 scaffolds were effective inhibitors with activities in the low nanomolar range for the ethyl ester analogues but showed a substantial drop in potency to the micromolar range for the corresponding propyl amides. Furthermore, authentic EBOV and MARV were inhibited with similar potencies by all compounds tested, indicating that the novel epoxysuccinates described herein confer pan-filovirus antiviral activity (Figure 4C).
Figure 4.

Inhibition of authentic Ebola virus by (A) epoxysuccinate esters or (B) epoxysuccinate propylamides. (C) Correlation of IC50 values from Ebola and Marburg virus inhibition.
Overall our results from the reported small focused library synthesis and screening yielded highly potent, broad-spectrum inhibitors of the lysosomal cysteine cathepsins. Our studies identify a novel lead scaffold that is a highly potent antifilovirus small molecule. Furthermore, we describe methods to control cellular uptake and increase in vivo stability of the epoxysuccinate inhibitors. These compounds have great promise for development into novel antifilovirus agents. We are currently evaluating these lead compounds in mouse models of EBOV and MARV challenge.
METHODS
General Synthetic Materials and Methods
All solvents were purchased from Fisher Scientific (HPLC grade). All reagents were purchased from commercial sources and were used without further purifications. All water-sensitive reactions were performed in anhydrous solvents under positive pressure of argon. Reactions were analyzed by LC-MS (C18) using an API 150EX single-quadrupole mass spectrometer (Applied Biosystems). Reverse-phase HPLC was conducted with an AKTA explorer 100 (Amersham Pharmacia Biotech) using C18 columns; buffers: (A) 5% acetonitrile in H2O + 0.1% TFA and (B) acetonitrile + 0.1% TFA. NMR spectra were recorded on a Varian 400 MHz (400/100) or a Varian 500 MHz (500/125) equipped with a pulsed field gradient accessory. Chemical shifts are given in parts per million (δ) relative to tetramethylsilane as an internal standard. Coupling constants are given in hertz.
Fmoc-Phe(Me)-NHNHBoc
Fmoc-Phe(Me)-OH (500 mg, 1.25 mmol, 1 equiv) was dissolved in DMF. HBTU (520 mg, 1.37 mmol, 1.1 equiv), DiPEA (475 μL, 2.88 mmol, 2.3 equiv), and Boc-hydrazine (173 mg, 1.31 mmol, 1.05 equiv) were added, and the mixture was stirred for 1 h. Ethyl acetate was added and the mixture washed with 1 M HCl, saturated aqueous NaHCO3, and brine. After drying over MgSO4, column chromatography (hexane → 1:1 hex/EA) yielded the title compound (620 mg, 1.2 mmol, 96%).1H NMR (300 MHz, CDCl3) δ 8.23 (s, 1H), 7.73 (d, J = 7.5 Hz, 2H), 7.47 (dd, J = 12.4, 4.9 Hz, 2H), 7.36 (q, J = 7.5 Hz, 2H), 7.32–7.20 (m, 2H), 7.05 (t, J = 6.0 Hz, 4H), 6.61 (s, 1H), 5.57 (d, J = 7.4 Hz, 1H), 4.52 (s, 1H), 4.43–4.16 (m, 2H), 4.10 (t, J = 7.0 Hz, 1H), 3.06 (dt, J = 39.5, 11.0 Hz, 2H), 2.26 (s, 3H), 1.44 (s, 9H). 13C NMR (75 MHz) δ 170.80, 170.76, 155.11, 143.60, 141.19, 136.57, 132.88, 129.34, 129.15, 127.66, 127.01, 125.04, 119.90, 81.87, 67.18, 46.95, 28.07, 21.02. LCMS tR 8.83 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 516.4 (M + H+).
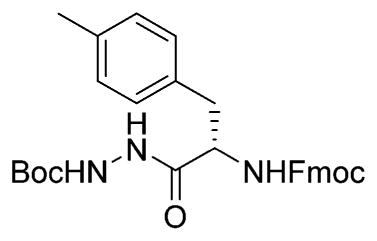
H-Phe(Me)-NHNHBoc
Fmoc-Phe(Me)-NHNHBoc (620 mg, 1.2 mmol, 1 equiv) was dissolved in THF. Ethanethiol (1 mL, 13.5 mmol, 11 equiv) and DBU (20 μL, 135 μmol, 0.1 equiv) were added, and the mixture was stirred for 5 h. After concentration, column chromatography (1:1 Hex/Ea → EA → 5% MeOH/EA) yielded the title compound (350 mg, 1.19 mmol, 99%).1H NMR (400 MHz, CDCl3) δ 7.10 (s, 4H), 3.66 (dd, J = 9.5, 4.1 Hz, 1H), 3.20 (dd, J = 13.7, 4.1 Hz, 1H), 2.68 (dd, J = 13.4, 9.6 Hz, 1H), 2.30 (s, 3H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 173.57, 155.30, 136.10, 134.15, 129.14, 128.98, 81.21, 55.55, 40.24, 27.93, 20.80. LCMS tR 4.2 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 293.8 (M + H+).
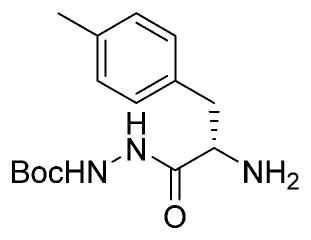
Ethyl-(S,S)-epoxysuccinate-Phe(Me)-NHNHBoc
H-Phe-(Me)-NHNHBoc (350 mg, 1.19 mmol) was dissolved in DCM. Ethyl (2S,3S)-(p-nitrophenyl)-oxirane-2,3-dicarboxylate (503 mg, 1.8 mmol, 1.5 equiv)15 and DiPEA (432 μL, 2.62 mmol, 2.2 equiv) were added, and the mixture stirred overnight. The mixture was washed with 0.1 M HCl, saturated aqueous NaHCO3 (four times), and brine. Column chromatography (hexanes →60% EA/Hex) yielded the title compound (390 mg, 896 μmol, 75%).1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 7.13 (dd, J = 12.5, 6.0 Hz, 1H), 7.12–7.03 (m, 4H), 4.83 (s, 1H), 4.29–4.15 (m, 2H), 3.58 (d, J = 1.9 Hz, 1H), 3.22 (dd, J = 13.9, 5.0 Hz, 1H), 2.99–2.89 (m, 2H), 2.31 (s, 3H), 1.46 (s,9H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.33, 166.53, 166.49, 155.38, 136.53, 132.78, 129.16, 129.03, 81.69, 62.04, 53.52, 52.44, 51.97, 28.01, 20.88, 13.85. LCMS tR 7.1 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 436.3 (M + H+).
Ethyl-(S,S)-epoxysuccinate-Phe(Me)-NHNH2 TFA Salt
Ethyl-(S,S)-epoxysuccinate-Phe(Me)-NHNHBoc (390 mg, 896 μmol) was dissolved in 1:1 DCM/TFA and stirred for 30 min before being coevaporated with toluene (three times). The crude product was used without further purification. LCMS tR 5.05 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 336.1 (M + H+).
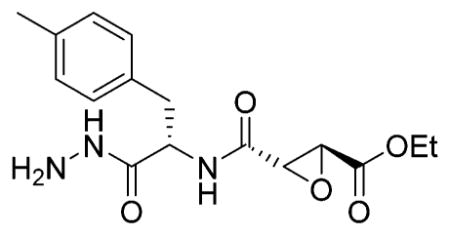
Ethyl-(R,R)-epoxysuccinate-Phe(Me)-NHNHBoc
H-Phe( Me)-NHNHBoc (94 mg, 320 μmol, 1 equiv) was dissolved in DMF. Ethyl (2R,3R)-oxirane-2,3-dicarboxylate (62 mg, 385 μmol, 1.2 equiv), HOBt hydrate (60 mg, 385 μmol, 60 mg), and EDC HCl (74 mg, 385 μmol, 1.2 equiv) were added, and the mixture was stirred for 2 h. The mixture was washed with 0.1 M HCl, saturated aqueous NaHCO3 (four times), and brine. Column chromatography (hexanes →60% EA/Hex) yielded the title compound (103 mg, 237 μmol, 74%).1H NMR (400 MHz, CDCl3) δ 8.72 (s, 1H), 7.11–7.02 (m, 5H), 6.91 (d, J = 2.4 Hz, 1H), 4.77 (d, J = 6.4 Hz, 1H), 4.25–4.15 (m, 2H), 3.59 (d, J = 1.8 Hz, 1H), 3.53 (s, 1H), 3.14 (dd, J = 13.9, 5.9 Hz, 1H), 2.94 (dd, J = 14.0, 8.4 Hz, 1H), 2.28 (s, 3H), 1.43 (s, 9H), 1.26 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.34, 166.67, 166.52, 155.17, 136.53, 132.61, 129.24, 129.05, 81.72, 62.10, 53.49, 52.40, 37.01, 28.02, 20.95, 13.88. LCMS tR 7.15 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 436.3 (M + H+).
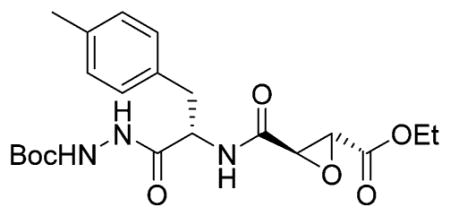
Ethyl-(R,R)-epoxysuccinate-Phe(Me)-NHNH2 TFA Salt
Ethyl-(R,R)-epoxysuccinate-Phe(Me)-NHNHBoc was dissolved in 1:1 DCM/TFA and stirred for 30 min before being coevaporated with toluene (three times). The crude product was used without further purification. LCMS tR 5.3 (linear gradient 10–90% TFA in ACN, 13.5 min). ESI-MS (m/z) 336.4 (M + H+).
General Protocol for Azide Couplings
The appropriate hydrazide was dissolved in 1:1 DMF/DCM (v/v) and cooled to −30 °C. tBuONO (1.1 equiv) and HCl (4 M solution in 1,4-dioxane, 2.8 equiv) were added, and the mixture was stirred for 3 h at −30 °C, after which TLC analysis (10% MeOH/DCM, v/v) showed complete consumption of the starting material. The appropriate amine (1.2 equiv) was added to the reaction mixture as a solution in DMF with 1.1 equiv of DiPEA. A further 3.9 equiv of DiPEA was added to the reaction mixture, and this mixture was allowed to warm to room temperature slowly overnight. The mixture was diluted with EA and washed with H2O (three times). The organic layer was dried over MgSO4 and purified by flash column chromatograpy or HPLC.
General Protocol Ethyl Ester Hydrolysis
The appropriate ethyl ester (50 mM in DMSO, 1 equiv) was treated with 1.1 equiv of NaOH (2 M in water) until LCMS analysis revealed complete hydrolysis. The product was used without further purification.
General Protocol for Generation of Epoxysuccinamides from Epoxysuccinate Carboxylic Acids
The appropriate carboxylic acid (1 equiv) was dissolved in DMF. HBTU (1.1 equiv), DiPEA (3 or 4 equiv), and appropriate amine or amine HCl salt were added (1.1 equiv), and the mixture was stirred for 1 h. HPLC purification yielded the title compound after lyopilization.
Competition Assay in Living RAW Cells and RAW Cell Lysate
RAW cells were grown in DMEM (Gibco) supplemented with 10% fetal calf serum (Gibco), 100 units/mL penicillin, and 100 mg/mL streptomycin (Gibco) in a 5% CO2 humidified incubator at 37 °C. One hundred times inhibitor DMSO solution (1 μL) was added to 100 μL of medium (see above) containing 100K cells and incubated for 1 h at 37 °C. BMV109 (1 μM end concentration) was added and incubated for 1 h at 37 °C. Cells were spun down, washed with PBS (one time), and incubated for 10 min in 20 μL of cell extraction buffer (100 mM Tris, pH 7.4, 2 mM Na3VO4, 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM EGTA, 0.1% SDS, 1 mM NaF, 0.5% deoxycholate, 20 mM Na4P2O7) for 10 min. Seven microliters of Laemmli’s sample buffer was added, and the mixture was boiled for 5 min and run on 15% SDS-PAGE gel. In-gel detection of fluorescently labeled proteins was performed directly in the wet gel slabs on the Typhoon Variable Mode Imager (Amersham Biosciences) using Cy5 settings (λex 650 nm, λem 670 nm).
Competition Assay in RAW Cell Lysate
Cell lysate was prepared by harvesting RAW cells, which were washed twice with PBS, and permeated in lysis buffer (50 mM sodium citrate, pH 5.5, 0.5% CHAPS, 0.1% Triton X-100, 5 mM DTT) for 15 min. After 15 min of spinning down at 4 °C, the protein concentration in the supernatant was determined by the BCA assay. Forty times inhibitor stock was added to 20 μg of protein, incubated for 1 h, labeled with 1 μM BMV109 for 1 h, and run as above.
Viral Infectivity Measurements
Recombinant VSVs bearing EBOV or MARV GP and expressing enhanced green fluorescent protein (eGFP) were previously generated from cDNAs.6,17 Inhibitor dose titrations were performed as follows. Briefly, monolayers of U2OS human osteosarcoma cells in 96-well plates were pre-incubated with indicated concentrations of inhibitor or DMSO vehicle for 1 h at 37 °C and then exposed to pretitrated amounts of virus (5 μL into 50 μL of medium; ≈1 infectious unit per cell). At 12–14 h postinfection, infected (eGFP-positive) cells were enumerated by fluorescence microscopy and automated image analysis (CellProfiler18). Dose curves were normalized to vehicle controls and fit to a four-parameter logistic equation using Prism (Graphpad Software, La Jolla, CA, USA) to estimate IC50 values.
Inhibitor-treated U2OS cells were exposed to EBOV (Zaire 1995)19 or MARV (Ci67)20 (≈3 plaque-forming units per cell) for 1 h. Viral inoculum was then removed, and fresh culture medium containing inhibitor was added. At 48 h postinfection, cells were fixed with formalin and blocked with 1% bovine serum albumin. EBOV-infected, MARV-infected, and uninfected control cells were immunostained with GP-specific monoclonal antibodies, counterstained with Hoechst stain (Invitrogen), washed with PBS, and stored at 4 °C, as described.6 Infected cells were quantitated by fluorescence microscopy and automated image analysis, as described previously.6 Dose curves were normalized and analyzed as described above.
Supplementary Material
Acknowledgments
We thank S. R. Lynch at the Stanford University Department of Chemistry NMR facility for assistance. This work was supported by National Institutes of Health Grants R01 EB005011 (to M.B.) and R01 AI088027 (to K.C.) and the Defense Threat Reduction Agency funding CB3873 (J.M.D.). W.A.v.d.L. was supported by a Rubicon fellowship from The Netherlands Organization for Scientific Research (NWO).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.5b00130.
Experimental details, spectroscopic data for all new compounds, and additional images and tables (PDF) Analytical data for the compounds (PDF)
References
- 1.Li H, Ying T, Yu F, Lu L, Jiang S. Microbes Infect. 2015;17(2):109. doi: 10.1016/j.micinf.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Nyakatura EK, Frei JC, Lai JR. ACS Infect Dis. 2015;1(1):42. doi: 10.1021/id500025n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Science. 2005;308(5728):1643. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misasi J, Chandran K, Yang J-Y, Considine B, Filone CM, Côté M, Sullivan N, Fabozzi G, Hensley L, Cunningham J. J Virol. 2012;86(6):3284. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR, Cin PD, Dye JM, Whelan SP, Ch K, Ran Brummelkamp TR, Chandran K, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Nature. 2011;477(7364):340. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. EMBO J. 2012;31(8):1947. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Nature. 2011;477(7364):344. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marzi A, Reinheckel T, Feldmann H. PLoS Neglected Trop Dis. 2012;6(12):e1923. doi: 10.1371/journal.pntd.0001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanada K, Tamai M, Yamagishi M, Ohmura S, Sawada J, Tanaka I. Agric Biol Chem. 1978;42(3):523. [Google Scholar]
- 10.Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K. J Pharmacobio-Dyn. 1986;9:672. doi: 10.1248/bpb1978.9.672. [DOI] [PubMed] [Google Scholar]
- 11.Satoyoshi E. Intern Med. 1992;31(7):841. doi: 10.2169/internalmedicine.31.841. [DOI] [PubMed] [Google Scholar]
- 12.Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cancer Cell. 2004;5(5):443. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 13.Sadaghiani AM, Verhelst SHL, Gocheva V, Hill K, Majerova E, Stinson S, Joyce JA, Bogyo M. Chem Biol. 2007;14(5):499. doi: 10.1016/j.chembiol.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Black WC, Percival MD. ChemBioChem. 2006;7(10):1525. doi: 10.1002/cbic.200600149. [DOI] [PubMed] [Google Scholar]
- 15.Chehade KAH, Baruch A, Verhelst SHL, Bogyo M. Synthesis (Stuttgart) 2005;2:240. [Google Scholar]
- 16.Verdoes M, Oresic Bender K, Segal E, Van Der Linden WA, Syed S, Withana NP, Sanman LE, Bogyo M. J Am Chem Soc. 2013;135(39):14726. doi: 10.1021/ja4056068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong aC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. J Virol. 2010;84(1):163. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin Da, Chang JH, Lindquist Ra, Moffat J, Golland P, Sabatini DM. Genome Biol. 2006;7(10):R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connolly BM, Steele KE, Davis KJ, Geisbert TW, Kell WM, Jaax NK, Jahrling PB. J Infect Dis. 1999;179(Suppl 1):S203. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 20.Swenson DL, Wang D, Luo M, Warfield KL, Woraratanadharm J, Holman DH, Dong JY, Pratt WD. Clin Vaccine Immunol. 2008;15(3):460. doi: 10.1128/CVI.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.