

Abstract
Congenital anomalies of the kidney and urinary tract (CAKUT) are common birth defects and the leading cause of end-stage renal disease in children. There is a wide spectrum of renal abnormalities, from mild hydronephrosis to more severe cases, such as bilateral renal dysplasia. The etiology of the majority of cases of CAKUT remains unknown, but there is increasing evidence that genomic imbalance contributes to the pathogenesis of CAKUT. Advances in human and mouse genetics have contributed to increased understanding of the pathophysiology of CAKUT. Mutations in genes involved in both transcription factors and signal transduction pathways involved in renal development are associated with CAKUT. Large cohort studies suggest that copy number variants, genomic, or de novo mutations may explain up to one-third of all cases of CAKUT. One of the major challenges to the use of genetic information in the clinical setting remains the lack of strict genotype–phenotype correlation. However, identifying genetic causes of CAKUT may lead to improved diagnosis of extrarenal complications. With the advent of decreasing costs for whole genome and exome sequencing, future studies focused on genotype–phenotype correlations, gene modifiers, and animal models of gene mutations will be needed to translate genetic advances into improved clinical care.
Keywords: CAKUT, congenital anomalies of kidney, renal development, genetics
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) are present in 3 to 7 out of 1,000 births, accounting for 20 to 30% of all anomalies detected in the prenatal period.1 There is a spectrum of severity of CAKUT, spanning from mild hydronephrosis to unilateral renal agenesis to dysplasia (Table 1). The most severe cases of CAKUT (bilateral aplasia, hypoplasia, dysplasia, obstructive uropathy, and reflux nephropathy) are the leading cause of pediatric end-stage renal disease (ESRD) in children, accounting for almost one-third of all cases of pediatric ESRD (North American Pediatric Renal Trial and Collaborative studies [NAPRTCS] 2011). There are few long-term studies examining the life course effects of CAKUT on the adult, but a recent study demonstrated the concerning finding that there is a higher risk of ESRD in adulthood than previously recognized.2
Table 1. Spectrum of CAKUT: Multiple manifestations of defective renal/urinary tract development.
| Defects in ureteric bud outgrowth/branching |
| • Vesicoureteral reflux (VUR) |
| • Ureterovesical junction (UVJ) obstruction |
| • Ureteropelvic junction (UPJ) obstruction |
| • Duplicated collecting system |
| • Renal agenesis |
| • Hydronephrosis |
| • Ectopic kidney |
| Defects in nephron induction/differentiation |
| • Agenesis/dysplasia/hypoplasia |
| • Multicystic dysplastic kidney (MCDK) |
| Defects in fusion |
| • Horseshoe kidney |
| Defects in bladder/urethral development |
| • Posterior urethral valves (PUVs) |
The etiology of the majority of CAKUT cases remains unknown. While environmental exposures may contribute to some cases of CAKUT,3 the preponderance of evidence suggests a strong genetic component to the pathogenesis of these congenital abnormalities. Familial aggregation studies indicate that 10 to 50% of children with CAKUT will report a family history of kidney abnormalities or urinary tract disease.4 5 Screening can demonstrate renal abnormalities in one out of four asymptomatic first-degree relatives of children with CAKUT.5 This strong familial heritability has led some to recommend screening young (age <3 years old) siblings of patients with vesicoureteral reflux (VUR).6 In addition to familial CAKUT, more than 500 genetic syndromes are associated with renal or urologic anomalies.4 Genetic testing of cohorts of children with nonfamilial, isolated CAKUT reveals genetic mutations in up to 10 to 17%.7 8 Many of these mutations occur de novo, and occur in genes that are also associated with syndromic CAKUT.8 9 In sum, genetic factors contribute strongly to the pathophysiology of CAKUT and these genetic mutations can be present in CAKUT patients without other evident abnormalities. Thus, even isolated kidney malformations should alert the clinician of a possible genomic imbalance.
In the era of prenatal ultrasounds, the majority of patients with CAKUT are now diagnosed prior to birth. This affords the opportunity to intervene when possible in cases of posterior urethral valves (PUVs), VUR, or obstruction. CAKUT is defined by the radiologic appearance of the kidneys, and includes a diverse set of phenotypes as indicated in Table 1. There are few clinical criteria available to predict risk of progression to ESKD in CAKUT. Before birth, amniotic fluid concentrations of sodium, β2 microglobulin, or other peptides may be helpful to determine the severity of disease in patients with PUV.10 11 After birth, small kidney size by ultrasound or serum creatinine > 1 mg/dL at 12 months of age in patients with PUV does predict a poor prognosis.2 12 13 Little is known about genetic factors' influence on either likelihood of renal failure or rate of progression in childhood. In adults, several genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) associated with progression of chronic kidney disease.14 15 16 17 18 However, GWAS typically require large numbers and there are limited data on the effect of SNPs on progression of childhood CKD.19 20 There is an ongoing, multicenter collaborative study, the Chronic Kidney Disease in Children Study (CKiD), a prospective cohort study of 586 children aged 1 to 16 years with chronic kidney disease, which may provide new data on this in the coming years.21 22
CAKUT Results from Disruption of Distinct Phases of Kidney and Urinary Tract Development
Disruption of kidney and urinary tract development at different stages leads to the spectrum of abnormalities observed in CAKUT. The kidney develops from the intermediate mesoderm, and renal development starts at the third week of gestation.23 There are three primitive kidneys, the pronephros, mesonephros, and metanephros. The pronephros are rudimentary tubules which form in the third week, and involute by the fourth week. The mesonephros develops in the fourth week, and is composed of well-developed nephrons with vascularized glomeruli, which drain into the mesonephric duct. The metanephros forms during the fifth week of gestation and develops into the permanent kidney. The deepest nephrons mature between the 6th and 10th week of gestation, with urine production beginning at 9 weeks. Nephrogenesis continues until the 36th week of gestation.
The first step needed to form the adult kidney is outgrowth of the ureteric bud (caudal portion of the mesonephric duct) (Fig. 1). Renal agenesis results if the ureteric bud fails to form.24 If two ureteric buds arise, a duplicated collecting system or duplicated kidney may form.24 Both are often associated with urinary tract obstruction or VUR.25 26
Fig. 1.
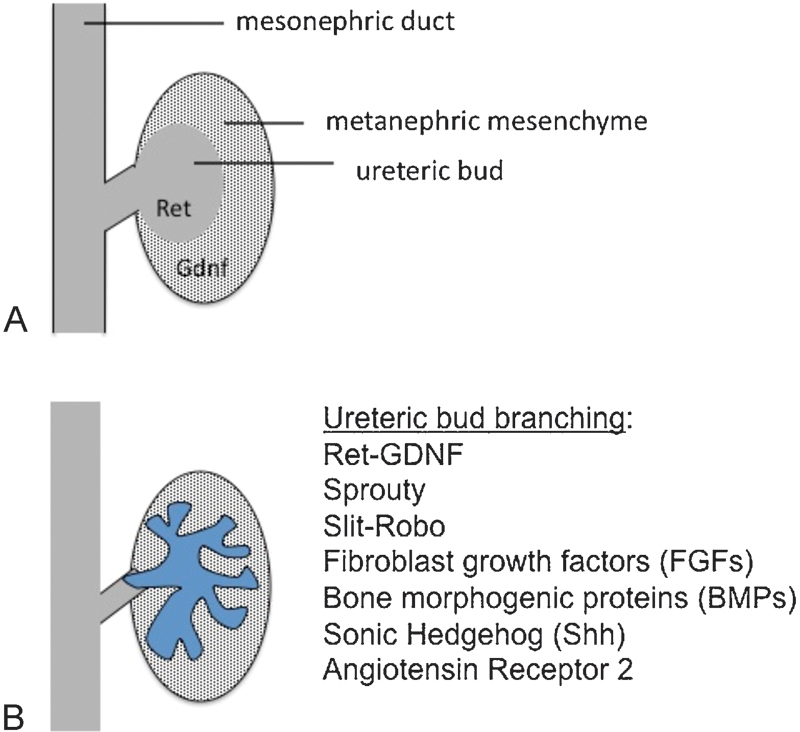
Reciprocal interactions between the metanephric mesenchyme and the ureteric bud are required for kidney development. (A) GDNF and Ret signaling are required for outgrowth of the ureteric bud into the metanephric mesenchyme. (B) Ret-GDNF, Sprouty, Slit-Robo, fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), sonic hedgehog (Shh), and angiotensin receptor 2 signaling pathways regulate ureteric bud branching. Mutations in Ret-GDNF, Robo2, and Shh, and variants in the renin–angiotensin system contribute to human CAKUT.
Next, the ureteric bud invades the surrounding metanephric mesenchyme, and undergoes branching.27 This branching is required to establish the radial organization of the nephrons and also determines nephron number. Defects in branching also contribute to hypoplasia. The tips of the ureteric bud induce the surrounding mesenchyme to undergo a transition to epithelial cells (mesenchymal–epithelial transition). Reciprocal interactions between the metanephric mesenchyme and ureteric bud are required to maintain cell survival and for further nephrogenesis, and disruption of these pathways results in renal dysplasia, renal hypoplasia, and renal agenesis.28 Later defects in nephron development and differentiation can result in a multicystic dysplastic kidney.3
The ureters form from the mesonephric duct at 5 weeks of gestation and become occluded and recanalized between 5 and 9 weeks.29 Abnormalities of ureteral development may result in ureteropelvic junction (UPJ) or ureterovesical junction (UVJ) obstruction. The bladder develops at the same time as the kidney from the urogenital sinus and fuse with the developing ureters. A perpendicular, rather than angled, insertion of the ureter into the bladder musculature predisposes children to VUR.30 31
The developing kidney is initially located in the pelvic area and migrates to its permanent location in the lumbar area at the eighth week of gestation. An ectopic (e.g., pelvic) kidney may result from ectopic ureteric budding, failure of vascularization, or abnormal migration of the kidneys during development.32 Mispatterning of the ureteric tree or defects in the kidney capsule formation may lead to fusion anomalies (e.g., horseshoe kidney).33
The bladder musculature continues to mature until 12 weeks of gestation. The male urethra develops between 9 and 14 weeks and derives from the urogenital sinus (prostatic and membrane urethra) and the urethral plate (bulbar and pendulous urethra).34 Several theories have been proposed to explain the origins of PUVs, including abnormal integration of the developing ejaculatory ducts (derived from the mesonephric/Wolffian ducts) into the developing urethra, leading to a membrane formation.34 Filling and emptying of the bladder up until the third trimester lead to remodeling of the bladder wall.35 Obstruction by PUVs impairs this process, contributing to long-term bladder dysfunction.35
Gene Mutations in Renal Developmental Signaling Pathways Result in CAKUT
Our understanding of the precise role of genes in regulating the various stages renal development has been advanced significantly over the past few decades by two factors. First, sequencing of the human genome and genetic mapping technologies facilitated the identification of multiple gene mutations associated with CAKUT. Second is the newfound ability to manipulate gene expression in animal models. The use of knockout and conditional mutants has enabled investigators to dissect out the role of signaling pathways in kidney development, often in cell- and time-specific fashion.
Clearly, the first step in kidney development is the specification of the renal progenitor populations. Genetic studies using animal models have identified key transcription factors including Six2, Pax2, Sall1, and Wt1 that are expressed in nephron progenitors, and many of these are also common mutations identified in human CAKUT.24 36 Animal models have also identified major signaling pathways that regulate later stages of kidney development, including ureteric bud branching and nephron induction.23 27 These include signaling by glial-derived neurotrophic factor GDNF/Ret, Wnt, fibroblast growth factor (FGFs), members of the transforming growth factor β superfamily (e.g., bone morphogenetic proteins), and Notch and Sonic hedgehog pathways.23 While detailed discussion of the cellular and molecular biology of kidney development is beyond the scope of this review, we will highlight a few gene mutations in both transcription factors and signaling pathways identified in human CAKUT (Table 2, Figs. 1 and 2).
Table 2. Selected gene mutations associated with CAKUT syndromes.
| Syndrome | Gene | Inheritance | Renal Phenotypes |
|---|---|---|---|
| Alagille |
JAG1
(Notch signaling pathway) |
AD | Agenesis, hypoplasia, cystic dysplasia, UPJ obstruction, VUR |
| Aniridia-Wilms tumor (WAGR) | WT1 | AD | Tumor, nephromegaly |
| ATR-X | ATRX | X-linked | Hypoplasia, agenesis, VUR, hydronephrosis |
| Branchio-oto-renal | EYA1, SIX1 | AD | Hypoplasia, VUR |
| Caudal regression | VANGL1 | AD, AR | Agenesis, ectopic kidney, hypoplasia, hydronephrosis, VUR |
| CHARGE | CHD7 | AD | Agenesis, hypodysplasia, hydronephrosis, hydroureter |
| Cornelia de Lange | NIPBL | AD | Hypoplasia, VUR, pelvic dilatation, ectopia |
| Fraser | FRAS1, FREM2 | AR | Renal agenesis, cystic dysplasia, hypoplasia, hydronephrosis, bladder agenesis |
| Hypoparathyroidism–deafness–renal | GATA3 | AD | Agenesis, hypodysplasia, VUR |
| Kallmann | KAL1, KAL2 | X-linked or AD | Unilateral renal agenesis, VUR, cystic dysplasia |
| Maturity-onset diabetes of the young type V | HNF1β | AD | Cystic dysplasia, hypoplasia, glomerulocystic, agenesis, oligomeganephronia |
| Meckel–Gruber | MKS1–4 | AR | Cystic dysplasia, hypoplasia |
| Noonan | NS1 | AD | Cystic dysplasia, duplication, hydronephrosis |
| Okihiro (acro-renal-ocular) | SALL4 | AD | Pelvic or horseshoe kidney, hypoplasia, VUR |
| Pallister–Hall |
GLI3
(member of sonic hedgehog signaling pathway) |
AD | Agenesis, hydronephrosis, hydroureter, renal ectopia, horseshoe kidney |
| Renal coloboma | PAX2 | AD | Hypoplasia, VUR, UPJ obstruction |
| Renal-hepatic-pancreatic dysplasia | EVC, EVC2 | AR | Cystic dysplasia |
| Rubinstein–Taybi |
CBP
EP300 |
AD | Unilateral agenesis, cystic hypodysplasia, urethral defects |
| Roberts | ESCO2 | AR | Agenesis, hypodysplasia, hydronephrosis |
| Silverman (dyssegmental dwarfism) | HSPG2 | Uncertain | Hydronephrosis |
| Simpson–Golabi–Behmel | GPC-3 | X-linked | Medullary cystic dysplasia |
| Smith–Lemli–Opitz | DHCR7 | AR | UPJ obstruction, hydronephrosis, VUR, cystic dysplasia |
| Townes–Brocks | SALL1 | AD | Agenesis, hypoplasia, PUV, VUR, meatal stenosis |
| Zellweger | PEX | AR | Cystic dysplasia, hydronephrosis |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive.
Note: Not included are nephronophthisis and polycystic kidney diseases, which are addressed in a separate review.
Fig. 2.
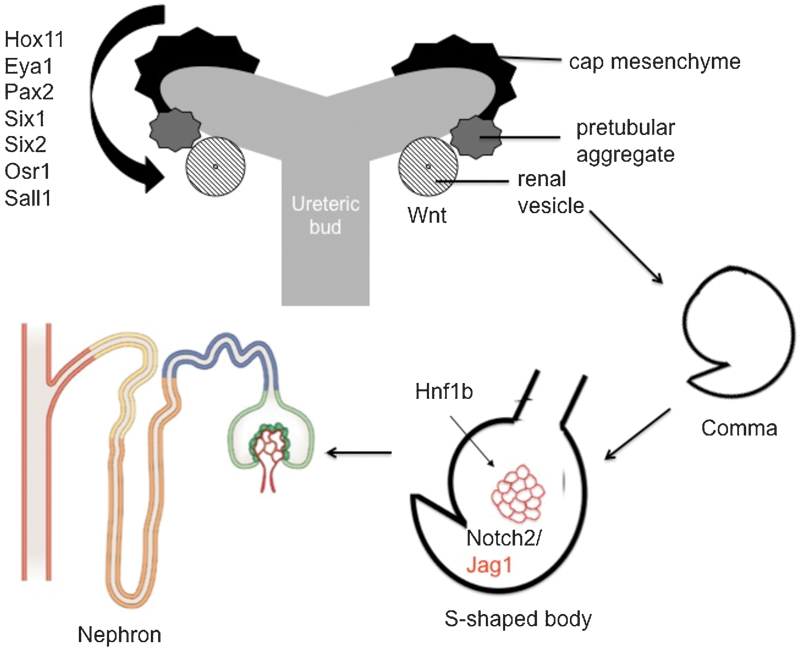
CAKUT is associated with mutations in transcriptions factors and signaling pathways that are required for nephron differentiation. Multiple transcription factors (including Hox11, Eya1, Pax2, Six1, Six2, Osr1, Sall1) regulate the balance between differentiation and maintenance of nephron progenitors. Wnt signaling is required for differentiation into the renal vesicle. HNF1B and Notch signaling contribute to specification of the proximal tubules and terminal nephron differentiation.
Distinct sets of transcription factors are expressed in the nephron, vascular, and stromal progenitors (Fig. 2). Some of these transcription factors within the nephron progenitors, such as Pax2, Hox11, and Eya1, physically associate and form DNA regulatory complexes.37 The Hox-Eya-Pax complex activates Six2 expression, which in turn activates Osr1, leading to a positive feedback loops that maintain the nephron progenitor population.23 37 38 39 40 41 42 Six1 also interacts with Eya1 and is required to maintain nephron progenitors.43 Activation of Wnt4 signaling leads to differentiation of the mesenchyme into epithelial cells.44 Interactions between transcription factors in nephron progenitors, including Six2, Osr1, and Sall1, tightly regulate the balance between renewal and proliferation versus differentiation.38 39 40 41 42 45 Either premature differentiation due to loss of these factors or failure to differentiate leads to renal hypoplasia in mice.39 45 46 47 Interestingly, epigenetic regulation by both microRNA and histone methylation contributes to maintenance of the renal progenitor population,48 49 50 although the exact role defects in epigenetic regulation may play in human CAKUT is unclear. Many of the transcription factors critical in nephron progenitors also contribute to development of other organs. For example, Six1, Six2, Eya1, and Osr1 play roles in eye and ear development, and patients with mutations in these genes present with renal, oto, and ophthalmologic manifestations.51 In particular, EYA1 and, less commonly, SIX1 mutations are associated with branchio-oto-renal (BOR) syndrome, an autosomal dominant disorder, characterized by branchial arch anomalies, hearing loss, and renal malformations. Similarly, Sall1 functions in anal, limb, and ear development, as well as kidney development. Sall1 mutations leads to Townes–Brock syndrome.52
CAKUT may also result from defects in transcription factors in other renal progenitor populations. For example, the stromal transcription factor FoxD1 regulates renal patterning, and loss of FoxD1 in mice leads to duplex kidneys.33
Many cases of CAKUT are linked to defective ureteric bud branching (Fig. 1). One of the major pathways regulating ureteric bud branching is GDNF. GDNF is a growth factor ligand that is secreted by the metanephric mesenchyme and binds the retinoic acid receptor (Ret) expressed on the tips of the ureteric buds. Binding of GDNF to Ret stimulates cell proliferation and ureteric bud branching. Defects in Ret are associated with bilateral renal aplasia.53 A recent study demonstrated rare variants or novel mutations of GDNF, Ret, or GFRα (a Ret co-receptor) in 5% of a cohort of unrelated patients affected with CAKUT.54 Ret mutations are also associated with Hirschsprung's disease, and up to 20% of Hirschsprung's patients have renal anomalies.55
Ureteric bud branching is modified by multiple factors, including FGF, BMP, Shh, Slit-Robo, and angiotensin-2 receptor signaling (Fig. 1).56 57 58 59 Mutations in Shh signaling members are causes of syndromic renal malformations (Pallister–Hall syndrome).57 Slit and Robo are neural guidance proteins that regulate ureteric bud outgrowth, branching, and the UVJ formation.60 Robo2 mutations are associated with familial VUR.60 Genetic variants in the renin angiotensin system may be linked to VUR, but this relationship has not been consistent in all populations.61 62
CAKUT can also result from mutations in genes involved in later stages of nephron differentiation. Induction by the ureteric bud results in aggregation of the mesenchyme that becomes a sphere of epithelial cells known as the renal vesicle. Wnt signaling regulates this process and genetic defects in Wnt4 lead to renal hypodysplasia (RHD).63 The vesicle then undergoes a series of morphologic changes to become a comma and then an s-shaped body. The epithelial cells in the s-shaped nephron closest to the ureteric bud form the distal tubules, the mid-portion forms the proximal tubule and loop of Henle, and those furthest from the ureteric bud form the parietal epithelium and the podocytes. The Notch signaling pathway is required for development of podocytes and proximal tubules.64 65 Notch is a cell–cell signaling pathway.66 Signal-sending cells express Notch ligands (i.e., Jag1) on their cell membrane.66 Ligand binding of Notch receptors on signal-receiving cells leads to a series of proteolytic cleavages, releasing the intracellular domain of Notch (NICD).66 This NICD translocates to the nucleus, stimulating expression of the Notch effectors, Hes and Hey, which lead to cell proliferation and differentiation.66 Gene mutations in Jag1 or the notch receptor Notch2 lead to Alagille syndrome, which is associated with a paucity of bile ducts and also renal abnormalities (Table 2).
Mouse genetic studies have also provided some insight into the wide spectrum of phenotypes observed in CAKUT. The cell- and time-specific effects may contribute to the different observed phenotypes. For example, FGFs have been implicated in both ureteric bud branching and metanephric mesenchyme differentiation.56 67 68 69 70 71 Another likely possibility is the presence of modifier genes and incomplete penetrance.67 Even genetically identical mutant mice do not all have the same phenotype. Finally, as microRNAs and histone methylation regulate renal development,24 48 49 epigenetic modification induced by the environment may also affect the phenotype.
There is a diversity of the CAKUT phenotype observed with mutations in the same gene. A good example of this is mutations in hepatocyte nuclear factor 1β (HNF1B). Gene deletions or mutations in HNF1B are one of the most commonly identified mutations associated with human CAKUT.72 73 HNF1B is a transcription factor, and mouse studies of HNF1B function demonstrate a role in proximal tubular differentiation.74 75 76 Kidney-specific inactivation of Hnf1b in the mouse leads to cystic disease, and HNF1B has been shown to regulate expression of genes whose mutations are involved in cystic kidney disease (i.e., PKHD1, NPHP1, PKD2).75 77 HNF1B mutations in humans are associated with a broad spectrum of disease, including renal cysts and diabetes syndrome, maturity onset diabetes of the young type 5, and hepatic, genital, and pancreatic abnormalities with variable expression of renal and extrarenal manifestations.7 72 78 79 80 81 82 83 84 85 86
Another example of a gene mutation involved in diverse CAKUT phenotypes is PAX2. PAX2 is a critical transcription factor expressed in the nephron progenitors.37 87 However, it is also expressed in the ureteric bud, where it regulates ureteric bud branching.88 It is also expressed in the eye, ear, and central nervous system.89 90 91 Mouse studies have shown that homozygous PAX2 mutant mice develop renal and ureteral agenesis,92 whereas PAX2 hypomorphic mice exhibit mild-to-severe RHD.93 There are mutational hotspots in a seven base pair polyguanidine region of exon2 (the DNA binding region).94 PAX2 mutations are most commonly associated with renal coloboma syndrome, also known as papillorenal syndrome, an autosomal dominant disorder characterized by optic nerve malformations (optic nerve coloboma, optic nerve dysplasia) and renal defects (oligomeganephronia, hypodysplasia with or without VUR, and renal cysts).30 84 88 91 94 95 Other extrarenal manifestations which are less common include sensorineural hearing loss and brain malformations.96 Just as described with HNF1B, the same gene mutation has different manifestations, even within the same family.88 96 Mouse models indicate that the Pax2 phenotype is modified by gene–gene interactions, including with WT-1 and HNF1B.76 97 PAX2 is a target of epigenetic regulation, and PAX2 expression is modified by the prenatal environment in mice.98 99 Such environmental and gene–gene interactions likely contribute to the variable PAX2 phenotype.
Genetic Studies in Populations with CAKUT
While studies of genes in animal models have advanced our understanding of the cellular and molecular mechanisms required for kidney development, we are still far from routinely applying these to clinical management of patients. However, recent studies of genetics in CAKUT cohorts added to our understanding of the types and frequency of genetic causes of CAKUT.
Copy number variation (CNV) has been implicated in the pathogenesis of many developmental disorders, and CNV may be a frequent cause of CAKUT. Sanna-Cherchi et al examined large, rare, CNVs (size > 100 kb and frequency < 1%) in a large cohort of patients with RHD, and identified known copy-number disorders in 10.5% of RHD cases.73 In 6.1% of RHD cases, they identified novel or rare copy-number disorders, which may help identify potential candidate genes or loci involved in the disruption of kidney development leading to CAKUT.73 Larger gene-disrupting events were associated with RHD cases, and deletions at the HNF1B locus were most frequent. HNF1B is highly susceptible to CNV as it is flanked by areas of segmental duplications, which are sites for recurrent rearrangements.73 Rearrangements in chromosomal region 17q12 were the most common genomic disorder. Interestingly, neuropsychiatric disease is also an increasingly recognized complication of rearrangements in this chromosomal region.73 Moreover, the majority of the known CNV disorders detected in the RHD cohort have previous associations with developmental disease or neuropsychiatric diseases.73 These findings suggest shared pathways between renal and neural development, and implicate genetic factors in cognitive defects associated with kidney disease.
Genetic Mutations in CAKUT in US and European Cohorts
Data from the Chronic Kidney Disease in Children Cohort Study (CKiD) demonstrated that 10% of all patients and 14% of North American Caucasian patients with RHD had pathogenic mutations for either HNF1B or PAX2.7 The majority of children (particularly non-Caucasian) in the cohort did not have HNF1B or PAX2 mutations, which suggests that there are other undiscovered genes that may cause CAKUT.7 There was no significant difference in progression of renal disease between patients with mutations in HNF1B or PAX2 and those without. The ESCAPE (European multicenter Effect of Strict Blood Pressure Control and ACE Inhibition on Progression in Pediatric Patients) study screened for genetic mutations, including HNF1B (also known as TCF2), PAX2, SALL1, EYA1, and SIX1, in a cohort of 100 patients with RHD and mild-to-moderate kidney disease. Genetic mutations were found in 17% of patients, of which 15% had mutations in TCF2 or PAX2.8
Together, these studies suggest that up to 30% of patients with CAKUT may have either large copy number variant or gene mutations. Roughly 50% of copy number variants73 and gene mutations may occur de novo85 and are not inherited. This raises the questions of the potential use of genomics in the clinical setting with patients with isolated CAKUT.
Clinical Implications of Gene Mutations
The major challenge of use of genetic evaluation in the clinical arena is the lack of strict genotype–phenotype correlation. The severity of renal disease associated with genetic mutation is extremely variable. Heidet et al examined a large cohort (377) of unrelated cases with various renal phenotypes (multicystic dysplastic kidney, renal agenesis, renal hypoplasia, cystic dysplasia) and screened for HNF1β mutation, which was found in 20% of cases.72 The gene alterations included whole gene deletion in 42 cases, small mutations (missense, nonsense, frameshift, splice site mutations) in 32 cases, and exon deletion in 1 case. Of 42 patients with heterozygous HNF1B deletion, the radiologic appearance of the kidneys varied from bilateral hypoplastic kidneys to enlarged hypoechoic kidneys.72 Furthermore, the prenatal ultrasound was often normal. The consequences of radiologic findings on renal function also varied, and some adult patients had normal glomerular filtration rate, whereas some infants had neonatal-onset renal failure.72 This illustrates the difficulty of using genetics in predicting renal outcome, even in the setting of complete gene deletion.
Another study demonstrating the lack of genotype–phenotype correlation was reported by Madariaga et al. 84 They screened for HNF1β and PAX2 genes in a cohort of 103 fetuses from 91 families with severe CAKUT that led to termination of pregnancy. They detected a mutation rate of 17% (HNF1β in 12 cases; PAX2 in 4 cases). However, in many cases, they observed marked intrafamilial phenotype variability with the same inherited mutation with respect to severity of disease. The lack of genotype–phenotype correlation and the wide variability observed even within a single family make genetic counseling difficult.100
The most obvious clinical application is to enhance screening for extrarenal manifestations. Many of the genes involved in kidney development are also critical in organogenesis of several tissues, resulting in other organ involvement. For example, HNF1B plays a crucial role in the development of pancreas, liver, gonads, gut, and thymus. Besides diabetes, other extrarenal manifestations related to HNF1B mutations include liver and pancreas abnormalities, hyperuricemia with or without gout, and genital malformations. BOR syndrome (with EYA1 or SIX1 mutations) is most commonly associated with hearing impairment, which can occur in various forms (conductive, sensorineural, mixed type) with variable progression to hearing loss. As described earlier, PAX2 is also expressed in the eye, ear, and central nervous system. Patients with PAX2 mutations and renal coloboma syndrome have extrarenal manifestations of optic nerve malformations and, less commonly, sensorineural hearing loss or brain malformations. It is important to note that many extrarenal manifestations may be subtle or subclinical and can be missed. In the ESCAPE trial, ocular abnormalities were found in five of seven patients with PAX2 mutations, and these ocular findings had not been previously detected.8 Thus, identification of PAX2 mutations would allow for thorough ophthalmologic evaluation to detect optic malformations. For patients with HNF1B mutation, diabetes screening is warranted. Regular auditory testing would be indicated for patients with EYA1 or SIX1 mutations. As described earlier, renal malformations may also be associated with neuropsychiatric disease, and CNV screen might identify the potential for complications such as developmental delay, autism, or other cognitive defects. In the follow-up phase of the CKiD study, detailed information about extrarenal manifestations is being collected and may provide more data to support mutation screening of children with CAKUT.21
On the Frontier: Translation of Improved Genomic Analysis to Improved Clinical Care
It is likely that the cost of whole genome and exome sequencing will continue to decrease over the next few decades. Analysis of such large datasets will require use of comparative genomics approaches and will enable us to identify new genes associated with CAKUT.51 But interpretation of sequencing results will also require better understanding of the mechanisms regulating phenotypic variation in CAKUT. First, detailed clinical phenotyping will be necessary to better define genotype–phenotype correlations101 . Second, we will need to identify genetic and epigenetic modifiers of phenotypes. Whole exome and genome sequencing will make it possible to investigate the role of gene–gene interactions in humans. But we will also need to understand the role of noncoding DNA in modifying phenotype. For example, epigenetic changes or genetic variation in promotor or enhancer regions can alter transcription factor binding, leading to alterations in gene expression.102
Continued advances in genetic technologies may help in these efforts. The majority of animal studies to date have relied upon whole gene deletion. With newer technologies in gene targeting and editing, such as the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas system,103 animal models may established to better understand the mechanisms behind specific genetic mutations. Genetic advances may lead to discovery of the effect of specific mutations and genetic modifiers of outcome that will facilitate a personalized medicine approach to CAKUT, and potentially even provide new targets for therapies for specific genotypes.104
References
- 1.Livera L N, Brookfield D S, Egginton J A, Hawnaur J M. Antenatal ultrasonography to detect fetal renal abnormalities: a prospective screening programme. BMJ. 1989;298(6685):1421–1423. doi: 10.1136/bmj.298.6685.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanna-Cherchi S, Ravani P, Corbani V. et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76(5):528–533. doi: 10.1038/ki.2009.220. [DOI] [PubMed] [Google Scholar]
- 3.Hains D S, Bates C M, Ingraham S, Schwaderer A L. Management and etiology of the unilateral multicystic dysplastic kidney: a review. Pediatr Nephrol. 2009;24(2):233–241. doi: 10.1007/s00467-008-0828-8. [DOI] [PubMed] [Google Scholar]
- 4.Weber S. Novel genetic aspects of congenital anomalies of kidney and urinary tract. Curr Opin Pediatr. 2012;24(2):212–218. doi: 10.1097/MOP.0b013e32834fdbd4. [DOI] [PubMed] [Google Scholar]
- 5.Bulum B, Ozçakar Z B, Ustüner E. et al. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol. 2013;28(11):2143–2147. doi: 10.1007/s00467-013-2530-8. [DOI] [PubMed] [Google Scholar]
- 6.Menezes M Puri P Familial vesicoureteral reflux—is screening beneficial? J Urol 2009182(4, Suppl):1673–1677. [DOI] [PubMed] [Google Scholar]
- 7.Thomas R, Sanna-Cherchi S, Warady B A, Furth S L, Kaskel F J, Gharavi A G. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol. 2011;26(6):897–903. doi: 10.1007/s00467-011-1826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber S, Moriniere V, Knüppel T. et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17(10):2864–2870. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- 9.Ulinski T, Lescure S, Beaufils S. et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17(2):497–503. doi: 10.1681/ASN.2005101040. [DOI] [PubMed] [Google Scholar]
- 10.Klein J, Lacroix C, Caubet C. et al. Fetal urinary peptides to predict postnatal outcome of renal disease in fetuses with posterior urethral valves (PUV) Sci Transl Med. 2013;5(198):198ra106. doi: 10.1126/scitranslmed.3005807. [DOI] [PubMed] [Google Scholar]
- 11.Muller F, Dommergues M, Mandelbrot L, Aubry M C, Nihoul-Fekete C, Dumez Y. Fetal urinary biochemistry predicts postnatal renal function in children with bilateral obstructive uropathies. Obstet Gynecol. 1993;82(5):813–820. [PubMed] [Google Scholar]
- 12.Pulido J E, Furth S L, Zderic S A, Canning D A, Tasian G E. Renal parenchymal area and risk of ESRD in boys with posterior urethral valves. Clin J Am Soc Nephrol. 2014;9(3):499–505. doi: 10.2215/CJN.08700813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heikkilä J, Holmberg C, Kyllönen L, Rintala R, Taskinen S. Long-term risk of end stage renal disease in patients with posterior urethral valves. J Urol. 2011;186(6):2392–2396. doi: 10.1016/j.juro.2011.07.109. [DOI] [PubMed] [Google Scholar]
- 14.Böger C A, Gorski M, Li M. et al. Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD. PLoS Genet. 2011;7(9):e1002292. doi: 10.1371/journal.pgen.1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köttgen A, Glazer N L, Dehghan A. et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 2009;41(6):712–717. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Köttgen A, Pattaro C, Böger C A. et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42(5):376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C T, Garnaas M K, Tin A. et al. Genetic association for renal traits among participants of African ancestry reveals new loci for renal function. PLoS Genet. 2011;7(9):e1002264. doi: 10.1371/journal.pgen.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pattaro C, Köttgen A, Teumer A. et al. Genome-wide association and functional follow-up reveals new loci for kidney function. PLoS Genet. 2012;8(3):e1002584. doi: 10.1371/journal.pgen.1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Eerde A M, Duran K, van Riel E. et al. Genes in the ureteric budding pathway: association study on vesico-ureteral reflux patients. PLoS ONE. 2012;7(4):e31327. doi: 10.1371/journal.pone.0031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laksmi N K, Khullar M, Kaur B. et al. Association of angiotensin converting enzyme and angiotensin type 2 receptor gene polymorphisms with renal damage in posterior urethral valves. J Pediatr Urol. 2010;6(6):560–566. doi: 10.1016/j.jpurol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Wong C J, Moxey-Mims M, Jerry-Fluker J, Warady B A, Furth S L. CKiD (CKD in children) prospective cohort study: a review of current findings. Am J Kidney Dis. 2012;60(6):1002–1011. doi: 10.1053/j.ajkd.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staples A, Wong C. Risk factors for progression of chronic kidney disease. Curr Opin Pediatr. 2010;22(2):161–169. doi: 10.1097/MOP.0b013e328336ebb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reidy K J, Rosenblum N D. Cell and molecular biology of kidney development. Semin Nephrol. 2009;29(4):321–337. doi: 10.1016/j.semnephrol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dressler G R. Advances in early kidney specification, development and patterning. Development. 2009;136(23):3863–3874. doi: 10.1242/dev.034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doğan C S, Torun Bayram M. Renal outcome of children with unilateral renal agenesis. Turk J Pediatr. 2013;55(6):612–615. [PubMed] [Google Scholar]
- 26.Hunziker M, Kutasy B, D'Asta F, Puri P. Urinary tract anomalies associated with high grade primary vesicoureteral reflux. Pediatr Surg Int. 2012;28(2):201–204. doi: 10.1007/s00383-011-2986-1. [DOI] [PubMed] [Google Scholar]
- 27.Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 2010;18(5):698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis T K, Hoshi M, Jain S. To bud or not to bud: the RET perspective in CAKUT. Pediatr Nephrol. 2014;29(4):597–608. doi: 10.1007/s00467-013-2606-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnenpoll T, Kispert A. Ureter growth and differentiation. Semin Cell Dev Biol. 2014;36:21–30. doi: 10.1016/j.semcdb.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 30.Murawski I J, Myburgh D B, Favor J, Gupta I R. Vesico-ureteric reflux and urinary tract development in the Pax2 1Neu+/- mouse. Am J Physiol Renal Physiol. 2007;293(5):F1736–F1745. doi: 10.1152/ajprenal.00221.2007. [DOI] [PubMed] [Google Scholar]
- 31.Murawski I J, Gupta I R. Vesicoureteric reflux and renal malformations: a developmental problem. Clin Genet. 2006;69(2):105–117. doi: 10.1111/j.1399-0004.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 32.Ichikawa I, Kuwayama F, Pope J C IV, Stephens F D, Miyazaki Y. Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int. 2002;61(3):889–898. doi: 10.1046/j.1523-1755.2002.00188.x. [DOI] [PubMed] [Google Scholar]
- 33.Levinson R S, Batourina E, Choi C, Vorontchikhina M, Kitajewski J, Mendelsohn C L. Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development. 2005;132(3):529–539. doi: 10.1242/dev.01604. [DOI] [PubMed] [Google Scholar]
- 34.Krishnan A, de Souza A, Konijeti R, Baskin L S. The anatomy and embryology of posterior urethral valves. J Urol. 2006;175(4):1214–1220. doi: 10.1016/S0022-5347(05)00642-7. [DOI] [PubMed] [Google Scholar]
- 35.Thomas J. Etiopathogenesis and management of bladder dysfunction in patients with posterior urethral valves. Indian J Urol. 2010;26(4):480–489. doi: 10.4103/0970-1591.74434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang D Y, Dworschak G C, Kohl S. et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85(6):1429–1433. doi: 10.1038/ki.2013.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong K Q, Yallowitz A R, Sun H, Dressler G R, Wellik D M. A Hox-Eya-Pax complex regulates early kidney developmental gene expression. Mol Cell Biol. 2007;27(21):7661–7668. doi: 10.1128/MCB.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basta J M, Robbins L, Kiefer S M, Dorsett D, Rauchman M. Sall1 balances self-renewal and differentiation of renal progenitor cells. Development. 2014;141(5):1047–1058. doi: 10.1242/dev.095851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanda S, Tanigawa S, Ohmori T. et al. Sall1 maintains nephron progenitors and nascent nephrons by acting as both an activator and a repressor. J Am Soc Nephrol. 2014;25(11):2584–2595. doi: 10.1681/ASN.2013080896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Liu H, Park J S, Lan Y, Jiang R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 2014;141(7):1442–1452. doi: 10.1242/dev.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Wong E Y, Cheng C. et al. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell. 2014;31(4):434–447. doi: 10.1016/j.devcel.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou P, Chen T, Fang Y. et al. Down-regulated Six2 by knockdown of neurofibromin results in apoptosis of metanephric mesenchyme cells in vitro. Mol Cell Biochem. 2014;390(1-2):205–213. doi: 10.1007/s11010-014-1971-0. [DOI] [PubMed] [Google Scholar]
- 43.Xu P X, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130(14):3085–3094. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark K, Vainio S, Vassileva G, McMahon A P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372(6507):679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 45.Park J S, Ma W, O'Brien L L. et al. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell. 2012;23(3):637–651. doi: 10.1016/j.devcel.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi A, Valerius M T, Mugford J W. et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3(2):169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujimura S, Jiang Q, Kobayashi C, Nishinakamura R. Notch2 activation in the embryonic kidney depletes nephron progenitors. J Am Soc Nephrol. 2010;21(5):803–810. doi: 10.1681/ASN.2009040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chu J Y, Sims-Lucas S, Bushnell D S, Bodnar A J, Kreidberg J A, Ho J. Dicer function is required in the metanephric mesenchyme for early kidney development. Am J Physiol Renal Physiol. 2014;306(7):F764–F772. doi: 10.1152/ajprenal.00426.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLaughlin N, Wang F, Saifudeen Z, El-Dahr S S. In situ histone landscape of nephrogenesis. Epigenetics. 2014;9(2):222–235. doi: 10.4161/epi.26793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel S R, Ranghini E, Dressler G R. Mechanisms of gene activation and repression by Pax proteins in the developing kidney. Pediatr Nephrol. 2014;29(4):589–595. doi: 10.1007/s00467-013-2603-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brophy P D, Alasti F, Darbro B W. et al. Genome-wide copy number variation analysis of a Branchio-oto-renal syndrome cohort identifies a recombination hotspot and implicates new candidate genes. Hum Genet. 2013;132(12):1339–1350. doi: 10.1007/s00439-013-1338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kohlhase J. SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum Mutat. 2000;16(6):460–466. doi: 10.1002/1098-1004(200012)16:6<460::AID-HUMU2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 53.Skinner M A, Safford S D, Reeves J G, Jackson M E, Freemerman A J. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet. 2008;82(2):344–351. doi: 10.1016/j.ajhg.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chatterjee R, Ramos E, Hoffman M. et al. Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum Genet. 2012;131(11):1725–1738. doi: 10.1007/s00439-012-1181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pini Prato A, Rossi V, Mosconi M. et al. A prospective observational study of associated anomalies in Hirschsprung's disease. Orphanet J Rare Dis. 2013;8:184. doi: 10.1186/1750-1172-8-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sims-Lucas S, Di Giovanni V, Schaefer C, Cusack B, Eswarakumar V P, Bates C M. Ureteric morphogenesis requires Fgfr1 and Fgfr2/Frs2α signaling in the metanephric mesenchyme. J Am Soc Nephrol. 2012;23(4):607–617. doi: 10.1681/ASN.2011020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cain J E, Islam E, Haxho F. et al. GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE. 2009;4(10):e7313. doi: 10.1371/journal.pone.0007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu W, van Eerde A M, Fan X. et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet. 2007;80(4):616–632. doi: 10.1086/512735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song R, Spera M, Garrett C, El-Dahr S S, Yosypiv I V. Angiotensin II AT2 receptor regulates ureteric bud morphogenesis. Am J Physiol Renal Physiol. 2010;298(3):F807–F817. doi: 10.1152/ajprenal.00147.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertoli-Avella A M, Conte M L, Punzo F. et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J Am Soc Nephrol. 2008;19(4):825–831. doi: 10.1681/ASN.2007060692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yim H E, Jung M J, Choi B M. et al. Genetic polymorphism of the renin-angiotensin system on the development of primary vesicoureteral reflux. Am J Nephrol. 2004;24(2):178–187. doi: 10.1159/000076620. [DOI] [PubMed] [Google Scholar]
- 62.Zhou T B, Lin N, Liu Y G, Qin Y H, Shao M B, Peng D D. Association of ACE I/D gene polymorphism with vesicoureteral reflux susceptibility in children: a meta-analysis. J Renin Angiotensin Aldosterone Syst. 2012;13(2):273–281. doi: 10.1177/1470320312437892. [DOI] [PubMed] [Google Scholar]
- 63.Vivante A, Mark-Danieli M, Davidovits M. et al. Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol. 2013;24(4):550–558. doi: 10.1681/ASN.2012010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng H T, Kim M, Valerius M T. et al. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134(4):801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng H T, Miner J H, Lin M, Tansey M G, Roth K, Kopan R. Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development. 2003;130(20):5031–5042. doi: 10.1242/dev.00697. [DOI] [PubMed] [Google Scholar]
- 66.Sirin Y, Susztak K. Notch in the kidney: development and disease. J Pathol. 2012;226(2):394–403. doi: 10.1002/path.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hains D, Sims-Lucas S, Kish K, Saha M, McHugh K, Bates C M. Role of fibroblast growth factor receptor 2 in kidney mesenchyme. Pediatr Res. 2008;64(6):592–598. doi: 10.1203/PDR.0b013e318187cc12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sims-Lucas S, Cullen-McEwen L, Eswarakumar V P. et al. Deletion of Frs2alpha from the ureteric epithelium causes renal hypoplasia. Am J Physiol Renal Physiol. 2009;297(5):F1208–F1219. doi: 10.1152/ajprenal.00262.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sims-Lucas S, Cusack B, Baust J. et al. Fgfr1 and the IIIc isoform of Fgfr2 play critical roles in the metanephric mesenchyme mediating early inductive events in kidney development. Dev Dyn. 2011;240(1):240–249. doi: 10.1002/dvdy.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sims-Lucas S, Cusack B, Eswarakumar V P, Zhang J, Wang F, Bates C M. Independent roles of Fgfr2 and Frs2alpha in ureteric epithelium. Development. 2011;138(7):1275–1280. doi: 10.1242/dev.062158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walker K A, Sims-Lucas S, Di Giovanni V E. et al. Deletion of fibroblast growth factor receptor 2 from the peri-wolffian duct stroma leads to ureteric induction abnormalities and vesicoureteral reflux. PLoS ONE. 2013;8(2):e56062. doi: 10.1371/journal.pone.0056062. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 72.Heidet L, Decramer S, Pawtowski A. et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol. 2010;5(6):1079–1090. doi: 10.2215/CJN.06810909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanna-Cherchi S, Kiryluk K, Burgess K E. et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet. 2012;91(6):987–997. doi: 10.1016/j.ajhg.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferrè S, Veenstra G J, Bouwmeester R, Hoenderop J G, Bindels R J. HNF-1B specifically regulates the transcription of the γa-subunit of the Na+/K+-ATPase. Biochem Biophys Res Commun. 2011;404(1):284–290. doi: 10.1016/j.bbrc.2010.11.108. [DOI] [PubMed] [Google Scholar]
- 75.Heliot C, Desgrange A, Buisson I. et al. HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Development. 2013;140(4):873–885. doi: 10.1242/dev.086538. [DOI] [PubMed] [Google Scholar]
- 76.Paces-Fessy M, Fabre M, Lesaulnier C, Cereghini S. Hnf1b and Pax2 cooperate to control different pathways in kidney and ureter morphogenesis. Hum Mol Genet. 2012;21(14):3143–3155. doi: 10.1093/hmg/dds141. [DOI] [PubMed] [Google Scholar]
- 77.Gresh L, Fischer E, Reimann A. et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23(7):1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Adalat S, Woolf A S, Johnstone K A. et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20(5):1123–1131. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Faguer S, Chassaing N, Bandin F. et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014;86(5):1007–1015. doi: 10.1038/ki.2014.202. [DOI] [PubMed] [Google Scholar]
- 80.Faguer S, Decramer S, Chassaing N. et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011;80(7):768–776. doi: 10.1038/ki.2011.225. [DOI] [PubMed] [Google Scholar]
- 81.Hasui M, Kaneko K, Tsuji S. et al. Different phenotypes of HNF1ß deletion mutants in familial multicystic dysplastic kidneys. Clin Nephrol. 2013;79(6):484–487. doi: 10.5414/cn107136. [DOI] [PubMed] [Google Scholar]
- 82.Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide A L, Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum Mol Genet. 2006;15(15):2363–2375. doi: 10.1093/hmg/ddl161. [DOI] [PubMed] [Google Scholar]
- 83.Loirat C, Bellanné-Chantelot C, Husson I, Deschênes G, Guigonis V, Chabane N. Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol Dial Transplant. 2010;25(10):3430–3433. doi: 10.1093/ndt/gfq380. [DOI] [PubMed] [Google Scholar]
- 84.Madariaga L, Morinière V, Jeanpierre C. et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol. 2013;8(7):1179–1187. doi: 10.2215/CJN.10221012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nakayama M, Nozu K, Goto Y. et al. HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2010;25(6):1073–1079. doi: 10.1007/s00467-010-1454-9. [DOI] [PubMed] [Google Scholar]
- 86.Raile K, Klopocki E, Wessel T. et al. HNF1B abnormality (mature-onset diabetes of the young 5) in children and adolescents: high prevalence in autoantibody-negative type 1 diabetes with kidney defects. Diabetes Care. 2008;31(11):e83. doi: 10.2337/dc08-0920. [DOI] [PubMed] [Google Scholar]
- 87.Buisson I, le Bouffant R, Futel M, Riou J F, Umbhauer M. Pax8 and Pax2 are specifically required at different steps of Xenopus pronephros development. Dev Biol. 2015;397(2):175–190. doi: 10.1016/j.ydbio.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 88.Porteous S, Torban E, Cho N P. et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of Pax2(1Neu) +/- mutant mice. Hum Mol Genet. 2000;9(1):1–11. doi: 10.1093/hmg/9.1.1. [DOI] [PubMed] [Google Scholar]
- 89.Bouchard M, de Caprona D, Busslinger M, Xu P, Fritzsch B. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev Biol. 2010;10:89. doi: 10.1186/1471-213X-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Freter S, Muta Y, O'Neill P, Vassilev V S, Kuraku S, Ladher R K. Pax2 modulates proliferation during specification of the otic and epibranchial placodes. Dev Dyn. 2012;241(11):1716–1728. doi: 10.1002/dvdy.23856. [DOI] [PubMed] [Google Scholar]
- 91.Higashide T, Wada T, Sakurai M, Yokoyama H, Sugiyama K. Macular abnormalities and optic disk anomaly associated with a new PAX2 missense mutation. Am J Ophthalmol. 2005;139(1):203–205. doi: 10.1016/j.ajo.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 92.Dziarmaga A, Clark P, Stayner C. et al. Ureteric bud apoptosis and renal hypoplasia in transgenic PAX2-Bax fetal mice mimics the renal-coloboma syndrome. J Am Soc Nephrol. 2003;14(11):2767–2774. doi: 10.1097/01.asn.0000094082.11026.ee. [DOI] [PubMed] [Google Scholar]
- 93.Favor J, Sandulache R, Neuhäuser-Klaus A. et al. The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc Natl Acad Sci U S A. 1996;93(24):13870–13875. doi: 10.1073/pnas.93.24.13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amiel J, Audollent S, Joly D. et al. PAX2 mutations in renal-coloboma syndrome: mutational hotspot and germline mosaicism. Eur J Hum Genet. 2000;8(11):820–826. doi: 10.1038/sj.ejhg.5200539. [DOI] [PubMed] [Google Scholar]
- 95.Otteson D C, Shelden E, Jones J M, Kameoka J, Hitchcock P F. Pax2 expression and retinal morphogenesis in the normal and Krd mouse. Dev Biol. 1998;193(2):209–224. doi: 10.1006/dbio.1997.8794. [DOI] [PubMed] [Google Scholar]
- 96.Schimmenti L A, Cunliffe H E, McNoe L A. et al. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am J Hum Genet. 1997;60(4):869–878. [PMC free article] [PubMed] [Google Scholar]
- 97.Discenza M T, He S, Lee T H. et al. WT1 is a modifier of the Pax2 mutant phenotype: cooperation and interaction between WT1 and Pax2. Oncogene. 2003;22(50):8145–8155. doi: 10.1038/sj.onc.1206997. [DOI] [PubMed] [Google Scholar]
- 98.Patel S R, Bhumbra S S, Paknikar R S, Dressler G R. Epigenetic mechanisms of Groucho/Grg/TLE mediated transcriptional repression. Mol Cell. 2012;45(2):185–195. doi: 10.1016/j.molcel.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yan L, Yao X, Bachvarov D, Saifudeen Z, El-Dahr S S. Genome-wide analysis of gestational gene-environment interactions in the developing kidney. Physiol Genomics. 2014;46(17):655–670. doi: 10.1152/physiolgenomics.00035.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.El Andalousi J, Murawski I J, Capolicchio J P, El-Sherbiny M, Jednak R, Gupta I R. A single-center cohort of Canadian children with VUR reveals renal phenotypes important for genetic studies. Pediatr Nephrol. 2013;28(9):1813–1819. doi: 10.1007/s00467-013-2440-9. [DOI] [PubMed] [Google Scholar]
- 101.Darlow J M, Dobson M G, Darlay R. et al. A new genome scan for primary nonsyndromic vesicoureteric reflux emphasizes high genetic heterogeneity and shows linkage and association with various genes already implicated in urinary tract development. Mol Genet Genomic Med. 2014;2(1):7–29. doi: 10.1002/mgg3.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ko Y A, Mohtat D, Suzuki M. et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14(10):R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wijshake T, Baker D J, van de Sluis B. Endonucleases: new tools to edit the mouse genome. Biochim Biophys Acta. 2014;1842(10):1942–1950. doi: 10.1016/j.bbadis.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 104.Clark P, Dziarmaga A, Eccles M, Goodyer P. Rescue of defective branching nephrogenesis in renal-coloboma syndrome by the caspase inhibitor, Z-VAD-fmk. J Am Soc Nephrol. 2004;15(2):299–305. doi: 10.1097/01.asn.0000111248.23454.19. [DOI] [PubMed] [Google Scholar]