

Abstract
Small molecules are powerful tools for investigating protein function and can serve as leads for new therapeutics. Most human proteins, however, lack small-molecule ligands, and entire protein classes are considered “undruggable” 1,2. Fragment-based ligand discovery (FBLD) can identify small-molecule probes for proteins that have proven difficult to target using high-throughput screening of complex compound libraries 1,3. Although reversibly binding ligands are commonly pursued, covalent fragments provide an alternative route to small-molecule probes 4–10, including those that can access regions of proteins that are difficult to access through binding affinity alone 5,10,11. In this manuscript, we report a quantitative analysis of cysteine-reactive small-molecule fragments screened against thousands of proteins. Covalent ligands were identified for >700 cysteines found in both druggable proteins and proteins deficient in chemical probes, including transcription factors, adaptor/scaffolding proteins, and uncharacterized proteins. Among the atypical ligand-protein interactions discovered were compounds that react preferentially with pro- (inactive) caspases. We used these ligands to distinguish extrinsic apoptosis pathways in human cell lines versus primary human T-cells, showing that the former is largely mediated by caspase-8 while the latter depends on both caspase-8 and −10. Fragment-based covalent ligand discovery provides a greatly expanded portrait of the ligandable proteome and furnishes compounds that can illuminate protein functions in native biological systems.
A major constraint of FBLD methods is their reliance on assaying purified proteins ex vivo. This requirement has restricted FBLD to proteins that can be produced in large quantities, and it is not clear how many human proteins can be targeted by small-molecule fragments or whether these interactions can be optimized to furnish chemical probes for studying protein function in complex biological systems. Here, we aimed to address these questions on a global scale by performing a quantitative analysis of the interactions between fragment electrophiles and thousands of cysteine residues in native human proteomes and cells.
We adapted a chemical proteomic method for quantifying cysteine reactivity – termed isoTOP-ABPP (isotopic Tandem Orthogonal Proteolysis-Activity-Based Protein Profiling 12,13) – to perform covalent FBLD in native biological systems. Lysate or intact cells are pre-treated with DMSO or an electrophilic small-molecule fragment and then exposed to a broad-spectrum cysteine-reactive probe iodoacetamide (IA)-alkyne 1 (Fig. 1a). Proteins harboring IA-alkyne-labeled cysteine residues from DMSO- and fragment-treated samples are then conjugated by copper-mediated azide-alkyne cycloaddition chemistry 14 to isotopically differentiated azide-biotin tags (heavy and light, respectively), combined, enriched by streptavidin, and proteolytically digested on-bead to yield isotopic peptide pairs that are analyzed by LC-MS. Quantification of MS1 chromatographic peak ratios for peptide pairs identifies fragment-competed Cys residues as those displaying high competition ratios, or R values, in DMSO/fragment comparisons.
Figure 1. Proteome-wide screening of covalent fragments.

a, General protocol for competitive isoTOP-ABPP. Competition ratios, or R values, are measured by dividing the MS1 ion peaks for IA-alkyne (1)-labeled peptides in DMSO-treated (heavy, or blue) versus fragment-treated (light, or red) samples. b, General structure of electrophilic fragment library, where the reactive (electrophilic) and binding groups are colored green and black, respectively. c, Competitive isoTOP-ABPP analysis of the MDA-MB-231 cell proteome pre-treated with the electrophilic 3,5-di(trifluoromethyl)aniline chloroacetamide 3 and acrylamide 14 fragments, along with the non-electrophilic acetamide analogue 17 (500 µM each). Proteomic reactivity values, or liganded cysteine rates, for fragments were calculated as the percentage of total cysteines with R values ≥ 4 in DMSO/fragment (heavy/light) comparisons. d, Representative MS1 peptide ion chromatograms from competitive isoTOP-ABPP experiments marking liganded cysteines selectively targeted by one of three fragments 3, 4, and 23.
We constructed a fragment library predominantly containing chloroacetamide or acrylamide electrophiles (Fig. 1b and Extended Data Fig. 1), which are well-characterized cysteine-reactive groups 10,15–17,18. These electrophiles were appended to structurally diverse small-molecule recognition (or binding) elements to create library members with an average molecular weight of 284 Da. Since our goal was to probe the ligandability of cysteines in the human proteome, we screened the electrophile library at a higher concentration (500 µM) than typically used in FBLD experiments 3. A subset of the fragment library was initially assayed by competitive profiling in a human MDA-MB-231 breast cancer cell line proteome using IA-rhodamine probe 16, which permitted SDS-PAGE detection of cysteine reactivity events. This experiment identified several proteins that showed reductions in IA-rhodamine labeling in the presence of one or more fragments (Extended Data Fig. 2a).
We then used competitive isoTOP-ABPP to globally map human proteins and the cysteine residues within these proteins that are targeted by fragment electrophiles. Each fragment was tested against two human cancer cell proteomes (MDA-MB-231 and Ramos cells), and most fragments were screened in duplicate against at least one of these proteomes. On average, 927 cysteines were quantified per data set, and we required that individual cysteines were quantified in at least three data sets. Based on these criteria, ~6150 cysteines from ~2900 proteins were quantified in aggregate across all data sets with an average quantification frequency of 22 datasets/cysteine (Extended Data Fig. 2b). Fragment-competed cysteine residues, or “liganded” cysteines, were defined as those showing ≥ 75% reductions in IA-alkyne labeling (R values ≥ 4 for DMSO/fragment). To minimize the potential for false-positives, only cysteines that showed R values ≥ 4 in two or more data sets and met additional criteria for data quality control (see Supplementary Methods) were considered as targets of the fragment electrophiles. The proteomic reactivity values, or liganded cysteine rates, of individual fragments were then calculated as the percentage of liganded/total quantified cysteines in isoTOP-ABPP experiments performed on that fragment.
Most fragment electrophiles showed a tempered reactivity across the human proteome, with a median liganded cysteine rate of 3.8% for the library (Extended Data Fig. 2c). Substantial differences in reactivity were observed, with individual electrophiles showing liganded cysteine rates of <0.1% and others displaying rates >15% (Extended Data Fig. 2c). A subset of fragments was also screened at lower concentrations (25–50 µM), which confirmed that their proteomic reactivities were concentration-dependent (Extended Data Fig. 2d). The relative reactivity of fragment electrophiles was similar in MDA-MB-231 and Ramos cell proteomes (Extended Data Fig. 2e), indicating that this parameter is an intrinsic property of the compounds. Fragments also showed consistent reactivity profiles when assayed in biological replicate experiments (Extended Data Fig. 2f). We found that the proteomic reactivity of fragment electrophiles was only marginally correlated with their glutathione adduction potential, which is a commonly used surrogate assay for measurements of proteinaceous cysteine reactivity 19 (Extended Data Fig. 2g). We attribute these differences to the impact of the recognition element of fragment electrophiles on their interactions and reactivity with proteins.
A comparison of fragments 3, 14, 17, and 23–26 provided insights into the relative proteomic reactivity of different electrophilic groups coupled to a common recognition element (3,5-bis(trifluoromethyl)phenyl group). Chloroacetamide 3 exhibited greater reactivity than acrylamide 14 (Fig. 1c), with cyanoacrylamide 23, but not more sterically congested electrophiles (24–26) exhibiting similar reactivity to 14 (Extended Data Fig. 2h). Importantly, the non-electrophilic acetamide control fragment 17 showed negligible activity in competitive isoTOP-ABPP experiments (Fig. 1c), indicating that the vast majority of detected fragment-cysteine interactions reflected covalent reactions versus non-covalent binding events. Also in support of this conclusion, “clickable” alkyne analogues of 3 and 14 (compounds 19 and 18, respectively) exhibited different concentration-dependent proteome labeling profiles (19 > 18; Extended Data Fig. 2i) that mirrored the respective liganded cysteine rates of 3 and 14 determined by isoTOP-ABPP (3 > 14; Fig. 1c). Despite the greater overall proteomic reactivity of 3 relative to 14 and 23, we found clear examples of cysteines that were preferentially liganded by the latter fragments (Fig. 1d and Supplementary Table 1).
Across all isoTOP-ABPP data sets combined, 758 liganded cysteines were identified on 637 distinct proteins, which corresponded to ~12 and 22% of the total quantified cysteines and proteins, respectively (Fig. 2a and Supplementary Table 1). Only a modest fraction of the proteins harboring liganded cysteines were found in the DrugBank database (15%; Fig. 2b), indicating the fragment electrophiles targeted many proteins that lack small-molecule probes. Among protein targets with known covalent ligands, the fragment electrophiles frequently targeted the same cysteine residues as these known ligands (Extended Data Table 1a). For one of these targets – the protein kinase BTK – we confirmed that interaction with the drug ibrutinib could be detected by isoTOP-ABPP, which also identified a known ibrutinib off-target – MAP2K720 – in Ramos cell lysates (Extended Data Fig. 3a).
Figure 2. Analysis of cysteines and proteins liganded by fragment electrophiles.

a, Fraction of total quantified cysteines and proteins that were liganded by fragment electrophiles in competitive isoTOP-ABPP experiments. b, Fraction of liganded proteins found in DrugBank. c, Functional classes of DrugBank and non-DrugBank proteins containing liganded cysteines. d, Comparison of the ligandability of cysteines as a function of their intrinsic reactivity with the IA-alkyne probe. Cysteine reactivity values (left y-axis) were taken from reference12, where lower ratios correspond to higher cysteine reactivity. A moving average with a step-size of 50 is shown in blue for the percentage of liganded cysteines within each reactivity bin (percent values shown on right y-axis).
DrugBank proteins with liganded cysteines mostly originated from classes that are regarded as “druggable”, including enzymes, channels, and transporters (Fig. 2c). Non-DrugBank proteins with liganded cysteines, on the other hand, showed a broader class distribution that included proteins, such as transcription factors and adaptor/scaffolding proteins, that are considered challenging to target with small-molecule ligands (Fig. 2c). We previously found that active-site and redox-active cysteines show, in general, greater intrinsic reactivity (as measured with the IA-alkyne probe) compared to other cysteines 12. While this heightened reactivity appears to be a contributory factor to the ligandability of cysteines, as reflected in the high proportion of hyperreactive (and active-site and redox-active) cysteines discovered as targets of fragment electrophiles, liganded cysteines were also well-represented across a broad range of intrinsic reactivities and included many non-active residues (Fig. 2d, Extended Data Fig 3b, c and Supplementary Discussion). Finally, most proteins were found to harbor a single liganded cysteine among the several cysteines that were, on average, quantified per protein by isoTOP-ABPP (Extended Data Fig. 3d, e and Supplementary Discussion).
Liganded cysteines, including those found in active and non-active sites of proteins, displayed strikingly distinct SARs with the fragment electrophile library (Fig. 3a, Extended Data Fig. 3g–l, Supplementary Table 1 and Supplementary Discussion). We also found that, for the majority of liganded cysteines (> 60%), electrophile (IA-alkyne or fragment) reactivity was blocked by heat denaturation of the proteome, while only a modest fraction of unliganded cysteines (~20%) showed decreased IA-alkyne labeling following heat denaturation (Extended Data Fig. 3m, n and Supplementary Discussion). These results indicate that the ligand-cysteine interactions are, in general, specific in that they depend on both the binding groups of ligands and structured sites in proteins (see Supplementary Discussion).
Figure 3. Analysis of fragment-cysteine interactions.

a, Heatmap showing R values for representative cysteines and fragments organized by proteomic reactivity values (high to low, left to right) and percentage of fragment hits for individual cysteines (high to low, top to bottom). R values ≥ 4 designate fragment hits (colored medium and dark blue). White color – not detected (ND). b, Representative example of reactive docking predictions shown for XPO1 (PDB ID: 3GB8). All accessible cysteines were identified and reactive docking was conducted with all fragments from the library within a 25 Å docking cube centered on each accessible cysteine (cube shown in green for liganded Cys in XPO1; see Supplementary Information for more details). Legend presents categories of XPO1 cysteines based on combined docking and isoTOP-ABPP results. c, Success rate of reactive docking predictions for liganded cysteines identified by isoTOP-ABPP for 29 representative proteins.
We next asked whether docking could predict sites of fragment electrophile reactivity. Covalent docking programs have recently been introduced to discover ligands that target prespecified cysteines in proteins 21; here, however, we aimed to computationally assess the relative ligandability of all cysteines within a protein and match these outputs to the data acquired by isoTOP-ABPP (see Supplementary Discussion). The ranking of our computational predictions matched the experimental data for the majority of proteins investigated (i.e., cases where the top predicted ligandable cysteine matched the liganded cysteine determined by isoTOP-ABPP) (Fig. 3b, c and Extended Data Table 2). We also found that cysteines predicted to be ligandable were much more likely to have been detected by isoTOP-ABPP and exhibit heat-sensitive IA-alkyne reactivity (Extended Data Fig. 3o, p and Extended Data Table 2). These results indicate that reactive docking can provide a good overall prediction of the ligandability of cysteines.
To determine the functional impact of ligand-cysteine interactions mapped by isoTOP-ABPP, we initially selected two enzymes – the protein methyltransferase PRMT1 and the MAP3 kinase MLTK (or ZAK) – that possessed liganded cysteines with previously demonstrated activities 12,13. Our findings confirmed that the fragment electrophiles targeting PRMT1 and MLTK inhibited these enzymes (Extended Data Fig. 4a–d (PRMT1), Extended Data Fig. 4e–i (MLTK) and Supplementary Discussion). We next evaluated proteins that possess previously uncharacterized liganded cysteines, including the nucleotide biosynthetic enzyme IMPDH2 and p53-induced phosphatase TIGAR. In both cases, we found that ligand-cysteine interactions affected specific functions of these proteins – regulatory nucleotide binding and catalytic activity, respectively (Extended Data Fig. 5a–g (IMPDH2), Extended Data Fig. 5h–n (TIGAR) and Supplementary Discussion).
Competitive isoTOP-ABPP experiments identified distinct subsets of ligands that targeted a conserved cysteine in isocitrate dehydrogenases 1 (IDH1) and 2 (IDH2) (C269 and C308, respectively; Supplementary Table 1). IDH1 and IDH2 are mutated in a number of human cancers to produce enzyme variants with a neomorphic catalytic activity that converts isocitrate to the oncometabolite 2-hydroxyglutarate (2-HG)22. Increases in 2-HG may then inhibit α-ketoglutarate-dependent dioxygenases that function as tumor suppressors, in particular, by methylating DNA and proteins 22. Reversible inhibitors selective for mutant forms of IDH1 and IDH2 have been developed and are under clinical investigation for cancer22. The liganded cysteine is an active site-proximal residue that is 13 Å from the NADP+ molecule in a crystal structure of IDH1 (Extended Data Fig. 6a). We confirmed that fragment ligands inhibited the activity of wild type (WT), but not a C269S mutant of IDH1, and also blocked the R132H oncogenic mutant of IDH1 both in vitro and in cells (Extended Data Fig. 6b–k and Supplementary Discussion).
Encouraged by the cellular activity displayed by IDH1 ligands, we sought to more generally assess the capacity of fragment electrophiles to modify cysteines in situ. Cells were treated with ~20 representative members of the fragment library (50–200 µM) for 2 h in situ and then harvested, lysed, and analyzed by isoTOP-ABPP. The tested fragments showed a broad range of in situ reactivities that generally matched their respective reactivities in vitro, although some exceptional cases with greater or lesser reactivity in situ were noted (Extended Data Fig. 6l and Supplementary Table 1). These differences could reflect the impact of transport and/or metabolic pathways on the cellular concentrations of fragment electrophiles. A substantial fraction (64%) of the liganded cysteines identified in cell lysates were also sensitive to the same electrophilic fragments in cells (Extended Data Fig. 6m). Four fragment-cysteine interactions were observed in situ, but not in lysates, including C182 of p53 (TP53), a redox-regulated residue at the dimerization interface of the DNA binding domain23 (Extended Data Fig. 6n). These liganded cysteines may require an intact cellular environment to preserve their interactions with fragment electrophiles.
Several fragments targeted the catalytic cysteine nucleophile C360 of the protease caspase-8 (CASP8) in isoTOP-ABPP experiments performed in vitro and in situ (Extended Data Figure 7a and Extended Data Table 1). Curiously, however, these fragments exhibited marginal to no inhibition of active CASP8 using either substrate or activity-based probe (Rho-DEVD-AOMK probe) assays (Extended Data Fig. 7b, c). This initially puzzling outcome was explained when we discovered that the electrophilic fragments selectively labeled the inactive zymogen (pro-), but not active form of CASP8 (Fig. 4a, b, Extended Data Fig. 7b–l and Supplementary Discussion). We synthesized a clickable analogue of the most potent pro-CASP8 ligand 7 (61; Fig. 4a) and found that this probe (25 µM) strongly labeled pro-CASP8, but not a C360S–pro-CASP8 mutant (Fig. 4b and Extended Data Fig. 7i), and directly modified C360 of CASP8 in Jurkat cell lysates (Extended Data Table 1b). 7 (50 µM) blocked labeling of pro-CASP8 by 61, but did not inhibit labeling of active CASP8 or other caspases by the Rho-DEVD-AOMK probe24 (Fig. 4b and Extended Data Fig. 7j, l). Conversely, the general caspase inhibitor Ac-DEVD-CHO (20 µM) blocked Rho-DEVD-AOMK labeling of active CASP8 and other caspases, but not 61 labeling of pro-CASP8 (Fig. 6b and Extended Data Fig. 7k, l). Similar results were obtained in substrate assays, where DEVD-CHO, but not 7, blocked CASP8 and CASP3 activity (Extended Data Fig. 7b).
Figure 4. Electrophile compounds that target pro-CASP8 and pro-CASP10.

a, Compound 7 blocked 16 labeling of recombinant, purified pro-CASP8 (bearing a C409S mutation to eliminate IA-rhodamine labeling at this site; added to Ramos cell lysate at 1 µM). Note that a C360S/C409S–mutant of pro-CASP8 did not label with 16. b, 7 blocked probe labeling of pro-, but not active CASP8. Recombinant pro- and active- CASP8 (1 µM) were treated with 7 (50 µM) or Ac-DEVD-CHO (20 µM), for 1 h followed by click probe 61 (25 µM) for pro-CASP8 and the Rho-DEVD-AOMK probe (2 µM) for active-CASP8. c, Heatmap showing R values for caspases measured by quantitative proteomics in Jurkat cells treated with 7, 63R, or 62 followed by probe 61 (10 µM, 1 h). d, Comparison of effects of 7 and 63R on FasL-induced apoptosis in Jurkat cells or anti-CD3, anti-CD28-activated primary human T cells. For d, data represent mean values ± SEM for at least three independent experiments, and results are representative of multiple experiments performed with T cells from different human subjects. Statistical significance was calculated with unpaired students t-tests comparing DMSO- to fragment-treated samples; ****, p < 0.0001 and comparing Jurkat to T cells ####, p < 0.0001.
We next confirmed that 7, but not a structurally related inactive probe (62; Extended Data Fig. 7f, g, k, and m and Supplementary Discussion) blocked Fas ligand (FasL), but not staurosporine (STS) induced apoptosis in Jurkat cells (Extended Data Fig. 7n–p). Chemical proteomic experiments revealed that 7 fully inhibited CASP8, as well as the related initiator caspase CASP10 (but not other caspases, including CASP2, 3, 6, and 9) in Jurkat cells (Fig. 4c, Extended Data Fig. 8a and Supplementary Table 1). We confirmed that 7 blocked labeling of pro-CASP10 by 61 with an apparent IC50 value of 4.5 µM (Extended Data Fig. 8b–d), but did not inhibit active CASP10 as measured by labeling with the Rho-DEVD-AOMK probe (Extended Data Fig. 7l) or a substrate assay (Extended Data Fig. 8e).
The respective functions of CASP8 and CASP10 in extrinsic apoptosis and other cellular processes remain poorly understood25,26, in large part due to a lack of selective, non-peptidic, and cell-active inhibitors for these enzymes and the absence of animal models for CASP10 (which is not expressed in rodents). We therefore sought to address this challenge by improving the potency and selectivity of 7. Conversion of the 4-piperidino moiety to a 3-piperidino group and addition of a p-morpholino substituent to the benzoyl ring of 7 furnished compound 63 that was separated by chiral chromatography into its two purified enantiomers, 63-R (Fig. 4c) and 63-S (see Supplementary Methods), the former of which showed substantially improved activity against CASP8 (apparent IC50 value of 0.7 µM (95% CI, 0.5 – 0.8); Extended Data Fig. 8f–h) and negligible cross-reactivity with CASP10 (IC50 value > 100 µM; Extended Data Fig. 8c, d, f). 63-S was much less active against CASP8 (apparent IC50 value of 15 µM; Extended Data Fig. 8g, h) and also inactive against CASP10 (Extended Data Fig. 4c). With dual CASP8/10 (7) and CASP8-selective (63-R) ligands in hand, we next set out to investigate the biological functions of these proteases.
We evaluated the effects of our caspase ligands in human T cells, where both CASP8 and CASP10 are highly expressed (Extended Data Fig. 8i) and their respective roles much debated 25,26, as well as in Jurkat cells, which are a commonly studied immortalized human T cell line. We found that 63-R fully blocked FasL-induced apoptosis in Jurkat cells and did so with greater potency than 7 (Fig. 4d and Extended Data Fig. 8j) or 63-S (Extended Data Fig. 8k). Similar results were obtained in HeLa cells, which express CASP8, but not CASP1026 (Extended Data Fig. 8l). In contrast to these cell line results, FasL-induced apoptosis in primary human T cells showed substantial resistance to 63-R at all tested concentrations and instead was completely inhibited by the dual CASP8/10 ligand 7 (Fig. 4d). We confirmed by chemical proteomics with probe 61 that 7 blocked both CASP8 and CASP10, while 63-R inhibited CASP8, but not CASP10, in primary human T cells (Supplementary Table 1) and Jurkat cells (Fig. 4c and Supplementary Table 1). Consistent with these cell death results, 7, but not 63-R, prevented proteolytic processing of CASP3 and CASP10 in primary human T cells (Extended Data. 8m). Interestingly, the processing of both CASP8 and the initiator caspase substrate RIP kinase were also preferentially inhibited by 7 versus 63-R (Extended Data. 8m, indicating that CASP10 may also contribute to these proteolytic events in T cells, as has been suggested by biochemical studies27. These data, taken together, support substantive functions for both CASP8 and CASP10 in primary human T cells and are consistent with genetic findings indicating that deleterious mutations in either CASP8 or CASP10 can lead to autoimmune syndromes in humans28.
By combining chemical proteomics with FBLD, we discovered that the human proteome contains many ligandable cysteines. Many of these cysteines were found in proteins not previously known to interact with small molecules, revealing that covalent chemistry can be used to expand the druggable content of the human proteome. Our results for pro-CASP8 and others for G12C–K-Ras10,11 indicate that it is possible to improve the potency and selectivity of covalent fragment hits for protein targets, though the optimization of covalent ligands for cysteines that reside in shallow pockets may prove more challenging. Some covalent ligands may target cysteines at non-functional sites on proteins, and, in these cases, there is potential to convert the ligands into functional probes using emergent platforms for directing liganded proteins to degradation pathways in the cells29,30 . We envision that this platform could be used to discover covalent ligands that target other nucleophilic amino acids in proteins, thereby increasing the impact covalent chemistry will have on proteome-wide ligand and drug discovery.
Online Methods
Preparation of human cancer cell line proteomes
All cell lines were obtained from ATCC, were used with a low passage number and were grown at 37 °C with 5% CO2. MDA-MB-231 cells, HeLa and HEK-293T cells were grown in DMEM supplemented with 10% fetal bovine serum, penicillin, streptomycin and glutamine. Jurkat, Ramos and MUM2C cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum, penicillin and streptomycin. For in vitro labeling, cells were grown to 100% confluence for MDA-MB-231 cells or until cell density reached 1.5 million cells/mL for Ramos and Jurkat cells. Cells were washed with cold PBS, scraped with cold PBS and cell pellets were isolated by centrifugation (1,400 g , 3 min, 4 °C), and stored at −80 °C until use. Cell pellets were lysed by sonication and fractionated (100,000 g, 45 min) to yield soluble and membrane fractions, which were then adjusted to a final protein concentration of 1.5 mg/mL for proteomics experiments and 1 mg/mL for gel-based ABPP experiments. The soluble lysate was prepared fresh from frozen pellets directly before each experiment. Protein concentration was determined using the Bio-Rad DC™ protein assay kit.
Proteomic sample preparation
IsoTOP-ABPP, SILAC and REDIME samples were prepared and analyzed as has been reported previously12,31–33 . For details see Supplementary Information.
In vitro covalent fragment treatment
All compounds were made up as 50 mM stock solutions in DMSO and were used at a final concentration of 500 µM. Due to its low solubility in aqueous medium, fragment 4 was screened at a final concentration of 250 µM. Soluble lysates were adjusted to 1.5 mg/mL and, for each profiling sample, 0.5 mL of lysate was treated with 5 µL of the 50 mM compound stock solution or 5 µL of DMSO.
In situ covalent fragment treatment
For in situ labeling, MDA-MB-231 cells were grown to 95% confluence and Ramos cells were grown to 1 million cells/mL. The media in all samples was replaced with fresh media, containing 200 µM of the indicated fragments and the cells were incubated at 37 °C for 2 h, washed with cold PBS, scraped into cold PBS and harvested by centrifugation (see preparation of human cancer cell proteomes). Fragments 2, 3, 8, 9, 10, 12, 13, 14, 21, 27, 28, 29, 31, 33, 38, 45, 51 and 56 were screened at 200 µM in situ. Fragments 4 and 11 were screened at 100 µM in situ. Fragments 2, 3, 8, and 20 were tested at 50 µM in situ.
Heat inactivation
For heat inactivation experiments, 500 µL of MDA-MB-231 soluble proteome was denatured (95 °C, 10 min) and allowed to cool to ambient temperature. The denatured sample and corresponding non-denatured, native proteome (500 µL) were then each labeled with 100 µM iodoacetamide alkyne (IA-alkyne, 5 µL of 10 mM stock in DMSO) and evaluated by isoTOP-ABPP.
R value calculation and processing
The ratios of heavy/light for each unique peptide (DMSO/compound treated; isoTOP-ABPP ratios, R values) were quantified with in-house CIMAGE software 12,34, using default parameters (3 ms1s per peak and signal to noise threshold 2.5). Site-specific engagement of electrophilic fragments was assessed by blockade of IA-alkyne probe labeling. For peptides that showed a ≥ 95% reduction in MS1 peak area from the fragment treated proteome (light TEV tag) when compared to the DMSO treated proteome (heavy TEV tag), a maximal ratio of 20 was assigned. Ratios for unique peptide entries are calculated for each experiment; overlapping peptides with the same modified cysteine (e.g. different charge states, MudPIT chromatographic steps or tryptic termini) are grouped together and the median ratio is reported as the final ratio (R). The peptide ratios reported by CIMAGE were further filtered to ensure the removal or correction of low quality ratios in each individual dataset. The quality filters applied were the following: removal of half tryptic peptides; for ratios with high standard deviations from the median (90% of the median or above) the lowest ratio was taken instead of the median; removal of peptides with R=20 and only a single ms2 event triggered during the elution of the parent ion; manual annotation of all the peptides with ratios of 20, removing any peptides with low-quality elution profiles that remained after the previous curation steps. Proteome reactivity values for individual fragments were computed as the percentage of the total quantified cysteine-containing peptides with R values ≥4 (defined as liganded cysteines) for each replicate experiment and the final proteome reactivity value was calculated as the mean for all replicate experiments for each fragment from both MDA-MB-231 and Ramos cellular proteomes. See Supplementary Information for additional details.
Functional annotation of liganded cysteines
Custom python scripts were used to compile functional annotations available in the UniProtKB/Swiss-Prot Protein Knowledge35 database (release-2012_11). Relevant Uniprot entries were mined for available functional annotations at the residue level, specifically for annotations regarding enzyme catalytic residues (active sites), disulfides (redox active and structural) and metal binding sites. Liganded proteins were queried against the Drugbank database (Version 4.2)36 and fractionated into DrugBank and non-Drugbank proteins. Functional keywords assigned at the protein level were collected from the Uniprot database and the Drugbank and non-drugbank categories were further classified into protein functional classes. Previously collected cysteine reactivity data12 was re-processed using ProLuCID as detailed above (Peptide and protein identification). Cysteines found in both the reactivity and ligandability datasets were sorted based on their reactivity values (lower ratio indicates higher reactivity). The moving average of the percentage of total liganded cysteines within each reactivity bin (step-size 50) was taken. Custom python scripts were developed to collect relevant NMR and X-ray structures from the RCSB Protein Data Bank (PDB)37. For proteins without available PDB structures, sequence alignments, performed with BLAST38 to proteins deposited in the PDB, were used to identify structural homologues. For annotation of active-site and non-active cysteines, enzymes with structures in the PDB were manually inspected to evaluate the location of the cysteine. Cysteines were considered to reside in enzyme active sites if they were within 10 Å of active-site ligand or residue(s). Cysteines outside of the 10 Å range were deemed non-active-site residues. Histograms of fragment hit-rates across high-coverage, ligandable cysteines, active-site and non-active site cysteines were calculated from the subset of ligandable cysteines quantified in 10 or more separate experiments. The fragment hit rate is reported as the percentage of the total quantification events with R ≥ 4. For analyses of trends within the whole data, including histograms and heatmaps, a cell-line merged dataset was used where data from the MDA-MB-231 experiments was taken first and the Ramos data was used if there was no data from MDA-MB-231 experiments for a particular fragment and cysteine. Heatmaps were generated in R (version 3.1.3) using the heatmap.2 algorithm. Protein structures were rendered using Pymol39.
Reactive cysteine docking
In silico fragment library containing all chloroacetamide and acrylamide fragments from Extended Data Fig. 1 was prepared using Open Babel library40 with custom Python scripts. Fragments were modeled in their reactive form (i.e., with explicit chloroacetamide and acrylamide warheads). 3D coordinates were generated from SMILES strings, calculating their protonation state at pH 7.4, and then minimizing them using MMF94s forcefield (50K iterations steepest descent; 90K conjugate gradient); for chiral molecules with undefined configuration, all enantiomers were generated, resulting in 53 total fragments. For each protein, the UniProtKB ID was used to filter the PDB37. Structures determined by X-ray crystallography were selected, privileging higher sequence coverage and structure resolution (See Extended Data Table 2 for selected PDB IDs). When no human structures were available, the closest homologous organism available was selected (e.g. PRMT1: R. norvegicus). Protein structures were prepared following the standard AutoDock protocol. Waters, salts, and crystallographic additives were removed; AutoDockTools41 was used to add hydrogens, calculate Gasteiger-Marsili charges and generate PDBQT files. MSMS reduced surface method42 was used to identify accessible cysteines. The protein volume was scanned using a probe radius of 1.5 Å; residues were considered accessible if they had at least one atom in contact with either external surfaces or internal cavities. The fragment library was docked independently on each accessible cysteine using AutoDock 4.241. A grid box of 24.4×24.4×24.4 Å was centered on the geometric center of the residue; thiol hydrogen was removed from the side-chain, which was modeled as flexible during the docking; the rest of the structure was kept rigid. A custom 13-7 interaction potential was defined between the nucleophile sulfur and the reactive carbon in the ligands. The equilibrium distance (req) was set to the length of the C-S covalent bond (1.8 Å); the potential well depth (ɛeq) varied between 1.0 and 0.175 to model to the reactivity of the different ligands. For each fragment, potential well depth was determined by dividing its proteomic reactivity percentage by 20, and the value for iodoacetamide was approximated as the maximum (2.5) for reference. The potential was implemented by modifying the force field table of AutoDock. Fragments were docked with no constraints, generating 100 poses using the default GA settings. For each fragment, the best docking score pose was analyzed: if the distance between the nucleophilic sulfur and the reactive carbon was ≤2.0 Å, the cysteine was considered covalently modified. If a residue was alkylated by at least one ligand, it was considered labeled. The docking score (i.e., negative binding energy) was calculated based on the estimated interaction energy of each fragment in its docked pose. The docking score of the best alkylating fragment defined the labeling score. The residue with the best labeling score was considered the most probable to be labeled.
CASP3 and CASP8 in vitro activity assays
Caspase 3 and 8 assays were conducted with CASP8 activity assay kit (BioVision, K112-100) and Caspase 3 activity assay kit (Invitrogen, EnzChek® Caspase-3 Assay Kit), following the manufacturer’s instructions. For further details see Supplementary Information.
Primary human T cell isolation and stimulation
All studies using samples from human volunteers follow protocols approved by the TSRI institutional review board. Blood from healthy donors were obtained after informed consent and peripheral blood mononuclear cells (PBMCs) were purified over Ficoll-Hypaque gradients (Sigma Aldrich). T cells were purified via negative selection with magnetic beads (EasySep, STEMCELL). The purified T cells were washed with sterile PBS and resuspended in RPMI-1640 supplemented with penicillin, streptomycin and glutamine (2 million cells/mL) and 200,000 cells/well were seeded on non-tissue culture treated, 96-well transparent plates that had been coated with anti-CD3 (1:200, BioXcell) and anti-CD28 (1:500, Biolegend) in PBS (100 µL/well). The T cells were removed from stimulus after 3 days and cultured in complete RPMI-1640 supplemented with IL2 (10 µg/mL, eBioscience) for 3–4 additional days.
Apoptosis assays in primary human T cells with CASP8 inhibitors
Primary human T cells were stimulated for 3 days with anti-CD3 and anti-CD28, and the cells were then washed and cultured in complete RPMI with IL2 (10 µg/mL) for 4 additional days. For western blot analysis, 10 mL of stimulated primary human T cells (1.5 million cells/mL) in RPMI with IL2 were then treated with the indicated compounds for 1h prior to addition of FasL (1 µL of 100 µg/µL stock solution of MegaFasLigand™ in water, final concentration =10 ng/mL, Adipogen). After 3 hours, cells were harvested by centrifugation, washed in PBS and lysed in cell lysis buffer (BioVision, 1067-100) with 1 × cOmplete protease inhibitor (Roche) and 40 µg of each sample were separated by SDS-Page on 14% polyacrylamide gels. The gels were transferred to nitrocellulose membranes and were immunoblotted overnight with the indicated antibodies. For measurements of cell viability, in triplicate for each condition, 150,000 cells (100 µL of 1.5 million cells/mL ) were plated in 96-Well Optical-Bottom Plates. FasL was used at the same concentration indicated above with a 30 minute pre-incubation with compounds at the indicated concentrations, followed by 4 hours with FasL or DMSO. 20 × compound stock solutions were made in RPMI immediately before use. Cell viability was measured with CellTiter-Glo® Luminescent Cell Viability Assay (Promega) and was read on a Biotech Synergy 4 plate reader.
Western blotting
For apoptosis studies, cell pellets were resuspended in cell lysis buffer from (BioVision ,1067-100) with 1 × cOmplete protease inhibitor (Roche) and allowed to incubate on ice for 30 min prior to centrifugation (10 min, 16,000 g). For all other studies, cell pellets were resuspended in PBS and lysed with sonication prior to centrifugation (10 min, 16,000 g). The proteins were then resolved by SDS-PAGE and transferred to nitrocellulose membranes, blocked with 5% milk in TBST and probed with the indicated antibodies. The primary antibodies and the dilutions used are as follows: anti-parp (Cell Signaling, 9532, 1:1000), anti-casp3 (Cell Signaling, 9662, 1:1000), anti-casp8 (cleaved form, Cell Signaling, 9746, 1:1000), anti-casp8 (pro-form, Cell Signaling, 4970, 1:1000), anti-IDH1 (Cell Signaling, 1:500, 3997s), anti-actin (Cell Signaling, 3700, 1:3000), anti-gapdh (Santa Cruz, sc-32233, 1:2000) anti-flag (Sigma Aldrich, F1804, 1:3000), anti-casp10 (MBL, M059-3, 1:1000), anti-ripk (Cell Signaling, 3493S, 1:1000) . Blots were incubated with primary antibodies overnight at 4 °C with rocking and were then washed (3 × 5 min, TBST) and incubated with secondary antibodies (LICOR, IRDye® 800CW or IRDye® 800LT, 1:10,000 ) for 1 h at ambient temperature. Blots were further washed (3 × 5 min, TBST) and visualized on a LICOR Odyssey Scanner. The percentage cleavage was determined by quantifying the integrated optical intensity of the bands (n = 3 for STS and n = 2 for FasL), using ImageJ software43. For CASP8, the 43 and 41 kDa bands were quantified together. For CASP3, the 17 kDa subunit band was quantified.
Statistical analysis
Data are shown as mean ± SEM. P values were calculated using unpaired, two-tailed Student’s t-test. P values of <0.05 were considered significant.
Extended Data
Extended Data Figure 1.

Composition of fragment electrophile library and structures of additional tool compounds, click probes, and fragments.
Extended Data Figure 2. Analysis of proteomic reactivities of fragment electrophiles in human cell lysates.
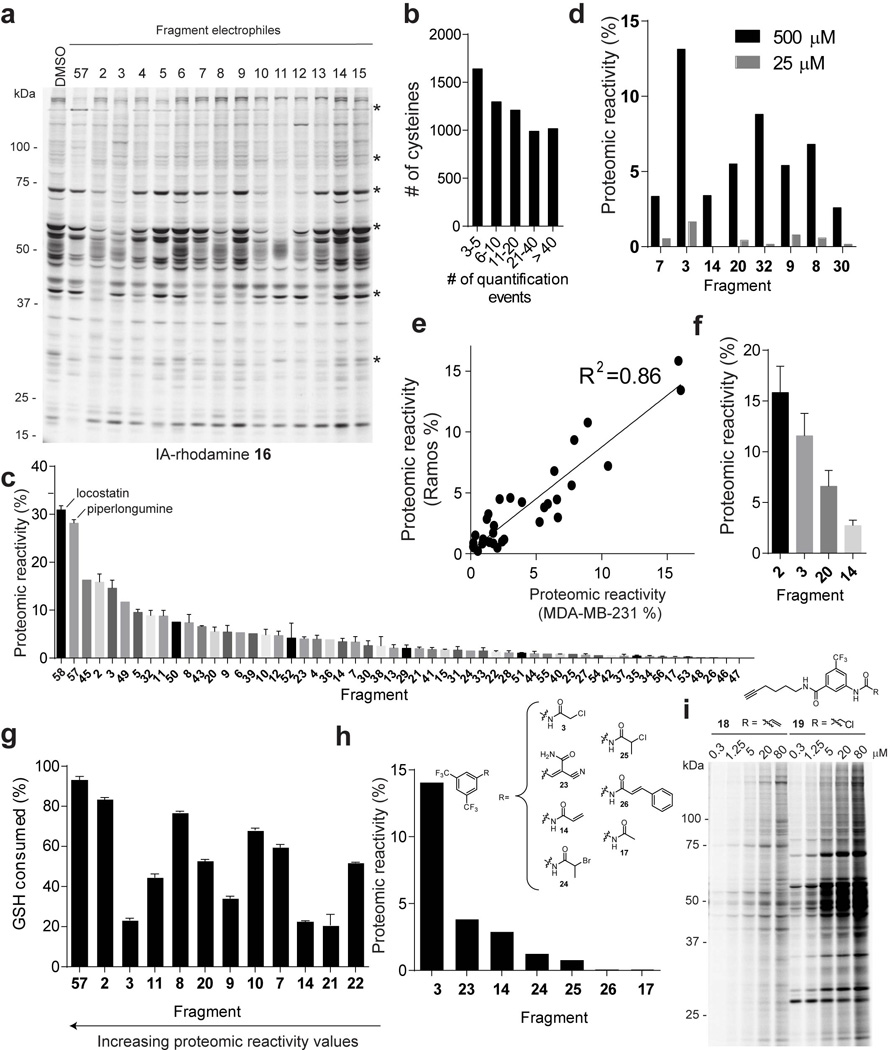
a, Initial analysis of the proteomic reactivity of fragments using an IA-rhodamine probe 16. Soluble proteome from Ramos cells was treated with the indicated fragments (500 µM each) for 1 h, followed by labeling with IA-rhodamine (1 µM, 1 h) and analysis by SDS-PAGE and in-gel fluorescence scanning. Several proteins were identified that show impaired reactivity with IA-rhodamine in the presence of one or more fragments (asterisks). Fluorescent gel shown in grayscale. b, Frequency of quantification of all cysteines across the complete set of competitive isoTOP-ABPP experiments performed with fragment electrophiles. Note that cysteines were required to have been quantified in at least three isoTOP-ABPP data sets for interpretation. c, Rank order of proteomic reactivity values (or liganded cysteine rates) of fragments calculated as the percentage of all quantified cysteines with R values ≥ 4 for each fragment. The majority of fragments were evaluated in 2–4 replicate experiments in MDA-MB-231 and/or Ramos cell lysates, and their proteomic reactivity values are reported as mean ± SEM values for the replicates. d, Comparison of the proteomic reactivities of representative fragments screened at 500 versus 25 µM in cell lysates. e, Comparison of proteomic reactivity values for fragments tested in both Ramos and MDA-MB-231 lysates. f, Proteomic reactivity values of representative fragments. g, Relative GSH reactivity for representative fragment electrophiles. Consumption of GSH (125 µM) was measured using Ellman’s reagent (5 mM) after 1 h incubation with the indicated fragments (500 µM). h, Proteomic reactivity values for fragments electrophiles (500 µM) possessing different electrophilic groups attached to a common binding element. i, Concentration-dependent labeling of MDA-MB-231 soluble proteomes with acrylamide 18 and chloroacetamide 19 click probes detected by CuACC with a rhodamine-azide tag and analysis by SDS-PAGE and in-gel fluorescence scanning. For panels f and g, data represent mean values ± SEM for at least three independent experiments.
Extended Data Figure 3. Analysis of cysteines liganded by fragment electrophiles in competitive isoTOP-ABPP experiments.
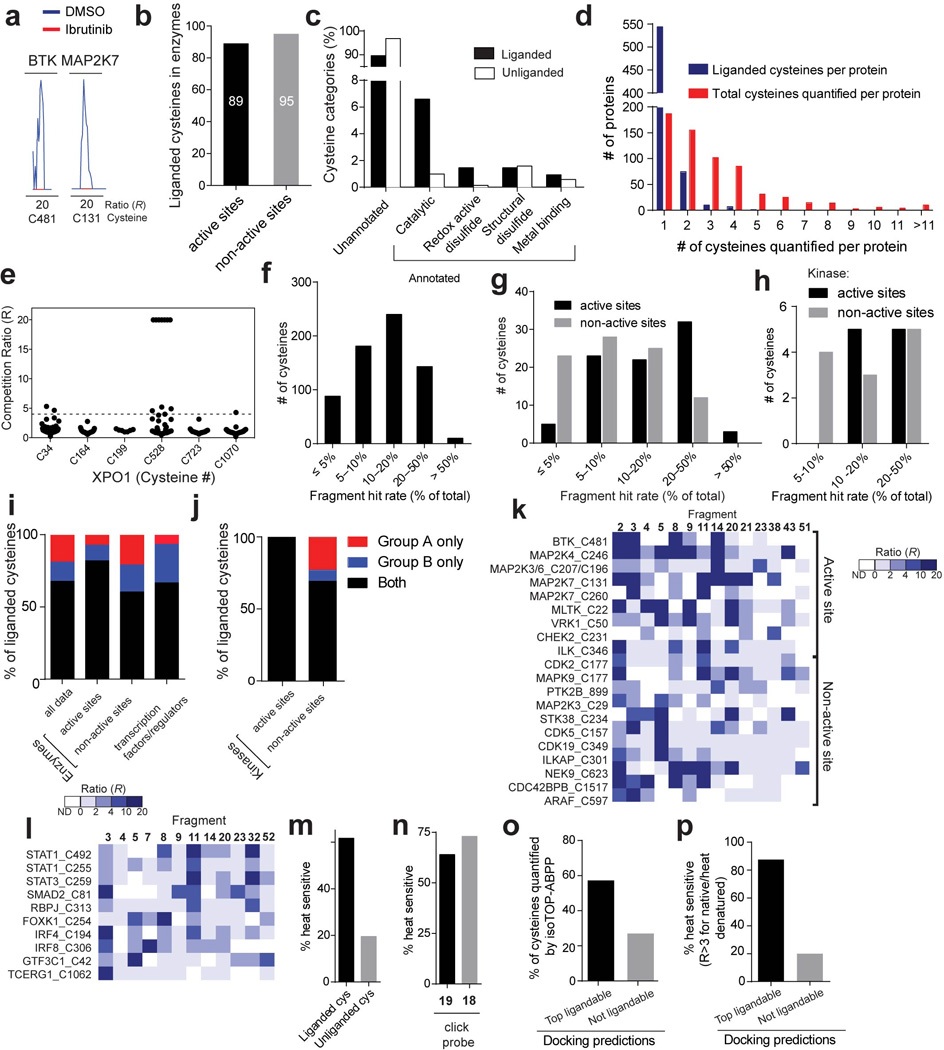
a, Representative MS1 ion chromatograms for peptides containing C481 of BTK and C131 of MAP2K7, two cysteines known to be targeted by the anti-cancer drug ibrutinib. Ramos cells were treated with ibrutinib (1 µM, 1 h, red trace) or DMSO (blue trace) and evaluated by isoTOP-ABPP. b, Total number of liganded cysteines found in the active sites and non-active sites of enzymes for which X-ray and/or NMR structures have been reported (or reported for a close homologue of the enzyme). c, Functional categorization of liganded and unliganded cysteines based on residue annotations from the Uniprot database. d, Number of liganded and quantified cysteines per protein measured by isoTOP-ABPP. Respective average values of one and three for liganded and quantified cysteines per protein were measured by isoTOP-ABPP. e R values for six cysteines in XPO1 quantified by isoTOP-ABPP, identifying C528 as the most liganded cysteine in this protein. Each point represents a distinct fragment-cysteine interaction quantified by isoTOP-ABPP. f-h Histograms depicting the percentage of fragments that are hits (R ≥ 4) for all 768 liganded cysteines (f), for liganded cysteines found in enzymes for which X-ray and/or NMR structures have been reported (or reported for a close homologue of the enzyme) (g), or for active- and non-active site cysteines in kinases (h). i, Percentage of liganded cysteines targeted only by group A (red) or B (blue) fragments or both group A and B fragments (black). Shown for all liganded cysteines, liganded cysteines in enzyme active and non-active sites, and liganded cysteines in transcription factors/ regulators. For g, i, active-site cysteines were defined as those that reside < 10 Å from established active-site residues and/or bound substrates/inhibitors in enzyme structures. j, The percentage of liganded cysteines in kinases that were targeted by only group A, only group B, or both group A and B compounds. k, Heatmap showing representative fragment interactions for liganded cysteines found in the active sites and non-active sites of kinases. l, Heatmap showing representative fragment interactions for liganded cysteines found in transcription factors/regulators. m, The fraction of liganded (62%; 341 of 553 quantified cysteines) and unliganded (20%; 561 of 2870 quantified cysteines) cysteines that are sensitive to heat denaturation measured by IA-alkyne labeling (R > 3 native/heat denatured). n, Percentage of proteins identified by isoTOP-ABPP as liganded by fragments 3 and 14 and enriched by their corresponding click probes 19 and 18 that are sensitive to heat denaturation (64% (85 of 133 quantified protein targets) and 73% (19 of 26 quantified protein targets), respectively). Protein enrichment by 18 and 19 was measured by whole protein capture of isotopically-SILAC labeled MDA-MB-231 cells using quantitative (SILAC) proteomics. o, The fraction of cysteines predicted to be ligandable or unligandable by reactive docking that were quantified in isoTOP-ABPP experiments. p, The fraction of cysteines predicted to be ligandable or unligandable by reactive docking that show heat-sensitive labeling by the IA-alkyne probe (R > 3 native/heat denatured).
Extended Data Figure 4. Confirmation and functional analysis of fragment-cysteine interactions in PRMT1 and MLTK.
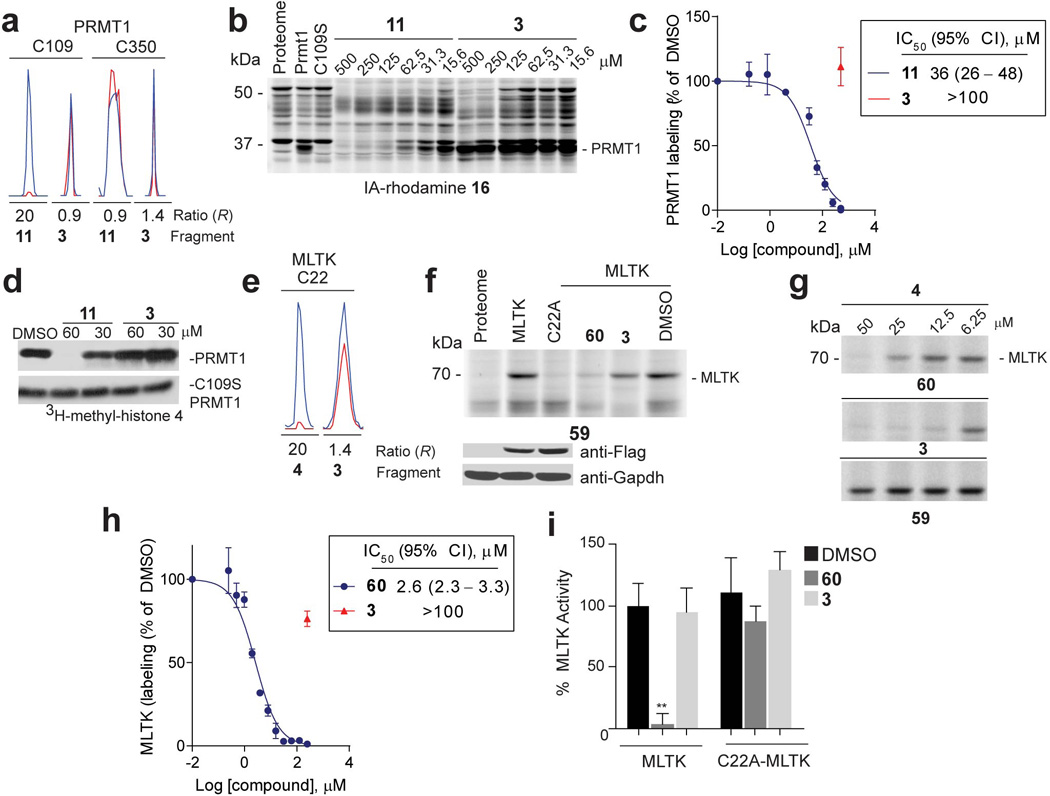
a, Representative MS1 chromatograms for the indicated Cys-containing peptides from PRMT1 quantified in competitive isoTOP-ABPP experiments of MDA-MB-231 cell lysates, showing blockade of IA-alkyne 1 labeling of C109 by fragment 11, but not control fragment 3. b, 11, but not 3 blocked IA-rhodamine (2 µM) labeling of recombinant, purified WT-PRMT1 (1 µM protein doped into HEK293T cell lysates). Note that a C109S–PRMT1 mutant did not react with IA-rhodamine. c, Apparent IC50 curve for blockade of 16 labeling of PRMT1 by 11. CI, 95% confidence intervals. d, Effect of 11 and control fragment 3 on methylation of recombinant histone 4 by recombinant PRMT1. Shown is one representative experiment of three independent experiments that yielded similar results. e, Representative MS1 ion chromatograms for the MLTK tryptic peptide containing liganded cysteine C22 quantified by isoTOP-ABPP in MDA-MB-231 lysates treated with fragment 4 or control fragment 3 (500 µM each). f, 60, but not control fragment 3 (50 µM of each fragment) blocked labeling of recombinant MLTK (or ZAK) kinase by a previously reported ibrutinib-derived activity probe 59 20 (upper panel). A C22A–MLTK mutant did not react with the ibrutinib probe. Anti-FLAG blotting confirmed similar expression of WT- and C22A–MLTK proteins, which were expressed as FLAG-fusion proteins in HEK293T cells (lower panel). g, Lysates from HEK293T cells expressing WT- or C22A–MLTK treated with the indicated fragments and then an ibrutinib-derived activity probe 5920 at 10 µM . MLTK labeling by 59 was detected by CuAAC conjugation to a rhodamine-azide tag and analysis by SDS-PAGE and in-gel fluorescence scanning. h, Apparent IC50 curve for blockade of ibrutinib probe-labeling of MLTK by 60. i, 60, but not control fragment 3 (100 µM of each fragment) inhibited the kinase activity of WT-, but not C22A–MLTK. For panels c, h and i, data represent mean values ± SEM for at least three independent experiments. Statistical significance was calculated with unpaired students t-tests comparing DMSO- to fragment-treated samples; **, p < 0.1.
Extended Data Figure 5. Confirmation and functional analysis of fragment-cysteine interactions in IMPDH2 and TIGAR.
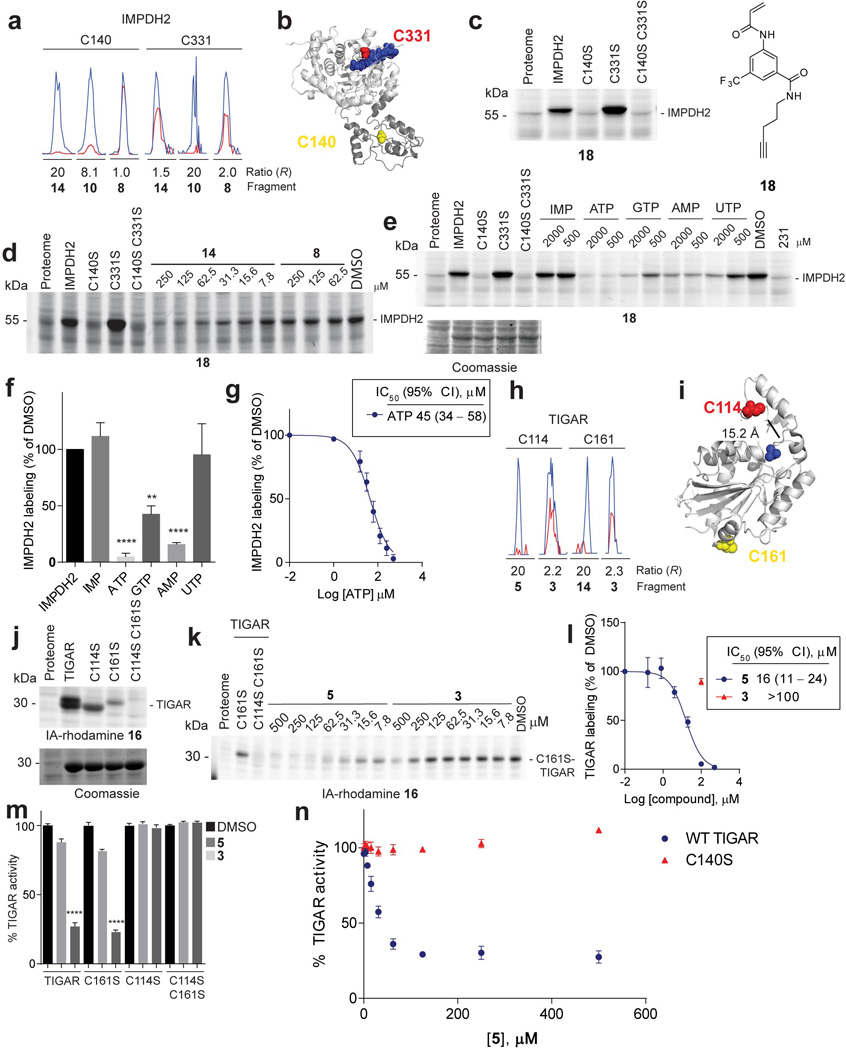
a, Representative MS1 ion chromatograms for IMPDH2 tryptic peptides containing the catalytic cysteine, C331, and Bateman domain cysteine, C140, quantified by isoTOP-ABPP in cell lysates treated with the indicated fragments (500 µM each). b, Structure of human IMPDH2 (PDB ID: 1NF7) (light grey) and its structurally unresolved Bateman domain modeled by ITASSER55 (dark grey) showing the positions of C331 (red spheres), ribavirin monophosphate and C2-mycophenolic adenine dinucleotide (blue), and C140 (yellow spheres). c, Click probe 18 (25 µM) labeled WT-IMPDH2 and C331S–IMPDH2, but not C140S–IMPDH2 (or C140S/C331S–IMPDH2). Labeling was detected by CuAAC conjugation to a rhodamine-azide reporter tag and analysis by SDS-PAGE and in-gel fluorescence scanning. Recombinant IMPDH2 WT and mutants were expressed and purified from E. coli and added to Jurkat lysates to a final concentration of 1 µM protein. d, Fragment reactivity with recombinant, purified IMPDH2 added to Jurkat lysates to a final concentration of 1 µM protein, where reactivity was detected in competition assays using the click probe 18 (25 µM). Note that 18 reacted with WT- and C331S–IMPDH2, but not C140S or C140S/C331S–IMPDH2. e, Nucleotide competition of 18 (25 µM) labeling of WT-IMPDH2 added to MDA-MB-231 lysates to a final concentration of 1 µM protein. f, Nucleotide competition profile for 18-labeling of recombinant WT-IMPDH2 (500 µM of each nucleotide). g, Apparent IC50 curve for blockade of 18 labeling of IMPDH2 by ATP. h, Representative MS1 chromatograms for TIGAR tryptic peptides containing C114 and C161 quantified by isoTOP-ABPP in cell lysates treated with the indicated fragments (500 µM each). i, Crystal structure of TIGAR (PDB ID: 3DCY) showing C114 (red spheres), C161 (yellow spheres), and inorganic phosphate (blue). j, Labeling of recombinant, purified TIGAR and mutant proteins by the IA-rhodamine (2 µM) probe. TIGAR proteins were added to MDA-MB-231 lysates, to a final concentration of 2 µM protein. k, 5, but not control fragment 3 blocked IA-rhodamine (2 µM) labeling of recombinant, purified C161S–TIGAR (2 µM protein doped into Ramos cell lysates). l, Apparent IC50 curve for blockade of IA-rhodamine labeling of C161S–TIGAR by 5. m, 5, but not control fragment 3 (100 µM of each fragment) inhibited the catalytic activity of WT-TIGAR, C161S–TIGAR, but not C114S–TIGAR or C114S/C161S–TIGAR. n, Concentration-dependent inhibition of WT-TIGAR by 5. Note that the C140S–TIGAR mutant was not inhibited by 5. Data represent mean values ± SEM for 4 replicate experiments at each concentration. For panels f, g and l-n, data represent mean values ± SEM for at least three independent experiments. Statistical significance was calculated with unpaired students t-tests comparing DMSO- to fragment-treated samples; **, p < 0.01, ****, p < 0.0001.
Extended Data Figure 6. IDH1-related and general in situ activity of fragment electrophiles.

a, X-ray crystal structure of IDH1 (PDB ID: 3MAS) showing the position of C269 and the frequently mutated residue in cancer, R132. b, Blockade of 16 labeling of WT-IDH1 by representative fragment electrophiles. Recombinant, purified WT-IDH1 was added to MDA-MB-231 lysates at a final concentration of 2 µM, treated with fragments at the indicated concentrations, followed by IA-rhodamine probe 16 (2 µM) and analysis by SDS-PAGE and in-gel fluorescence scanning. Note that a C269S mutant of IDH1 did not label with IA-rhodamine 16. c, d, Reactivity of 20 and control fragment 2 with recombinant, purified WT-IDH1 (b) or R132H–IDH1 (c) added to MDA-MB-231 lysates to a final concentration of 2 or 4 µM protein, respectively. Fragment reactivity was detected in competition assays using the IA-rhodamine probe (2 µM); note that the C269S–IDH1 mutant did not react with IA-rhodamine. e, f, Apparent IC50 curve for blockade of IA-rhodamine-labeling of IDH1 (e) and R132H–IDH1 (f) by 20. Note that the control fragment 2 showed much lower activity. g, Representative MS1 ion chromatograms for the IDH1 tryptic peptides containing liganded cysteine C269 and an unliganded cysteine C379 quantified by isoTOP-ABPP in MDA-MB-231 lysates treated with fragment 20 (25 µM). h, 20, but not 2, inhibited IDH1-catalyzed oxidation of isocitrate to α-ketoglutarate (α-KG) as measured by an increase in NADPH production (340 nm absorbance). 20 did not inhibit the C269S–IDH1 mutant. i, 20 inhibited oncometabolite 2-hydroxyglutarate (2-HG) production by R132H–IDH1. MUM2C cells stably overexpressing the oncogenic R132H–IDH1 mutant or control GFP-expressing MUM2C cells were treated with the indicated fragments (2 h, in situ). Cells were harvested, lysed and IDH1-dependent production of 2-HG from α-KG and NADPH was measured by LC-MS and from which 2-HG production of GFP-expressing MUM2C cells was subtracted (GFP-expressing MUM2C cells produced < 10% of the 2-HG generated by R132H–IDH1-expressing MUM2C cells). j, Western blot of MUM2C cells stably overexpressing GFP (mock) or R132H–IDH1 proteins. k, Representative MS1 chromatograms for the IDH1 tryptic peptides containing liganded cysteine C269 and an unliganded cysteine C379 quantified by isoTOP-ABPP in R132H–IDH-expressing MUM2C lysates treated with 20 or control fragment 2 (50 µM, 2 h, in situ). l, Proteomic reactivity values for individual fragments are comparable in vitro and in situ. One fragment (11) marked in red showed notably lower reactivity in situ versus in vitro. Reactivity values were calculated as in Fig. 1c. Dashed line mark 90% prediction intervals for the comparison of in vitro and in situ proteomic reactivity values for fragment electrophiles. Blue and red circles mark fragments that fall above (or just at) or below these prediction intervals, respectively. m, Fraction of cysteines liganded in vitro that are also liganded in situ. Shown are liganded cysteine numbers for individual fragments determined in vitro and the fraction of these cysteines that were liganded by the corresponding fragments in situn, Representative cysteines that were selectively targeted by fragments in situ, but not in vitro. For in situ-restricted fragment-cysteine interactions, a second cysteine in the parent protein was detected with an unchanging ratio (R ~ 1), thus controlling for potential fragment-induced changes in protein expression. For panels e, f, h and i, data represent mean values ± SEM for at least three independent experiments. Statistical significance was calculated with unpaired students t-tests comparing DMSO- to fragment-treated samples; ****, p < 0.0001.
Extended Data Figure 7. Fragment electrophiles that target pro-CASP8.
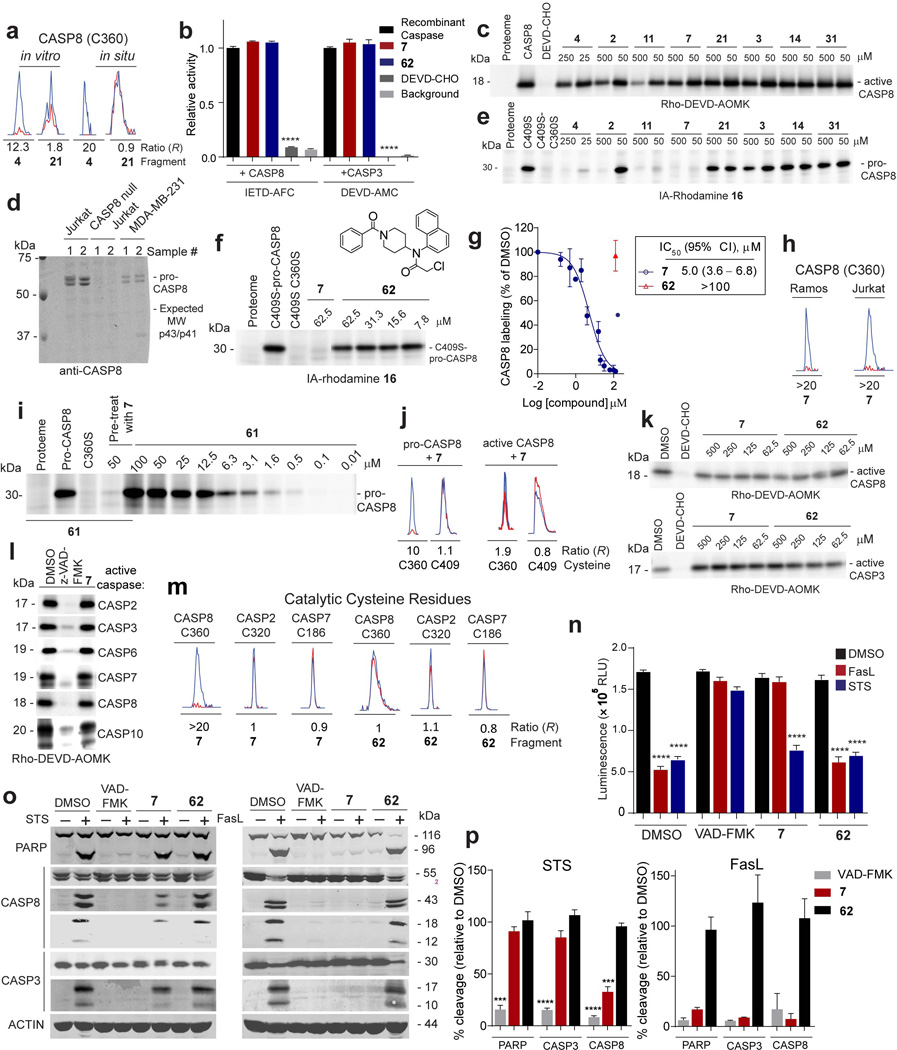
a, Representative MS1 chromatograms for CASP8 tryptic peptide containing the catalytic cysteine C360 quantified by isoTOP-ABPP in cell lysates or cells treated with fragment 4 (250 µM, in vitro; 100 µM, in situ) and control fragment 21 (500 µM, in vitro; 200 µM, in situ). b, Neither 7 nor control fragment 62 (100 µM each) inhibited recombinant, purified active CASP3 and CASP8 assayed using DEVD-AMC and IETD-AFC substrates, respectively. DEVD-CHO (20 µM) inhibited both caspases. c, Fragment reactivity with recombinant, purified active CASP8 added to cell lysates, where reactivity was detected in competition assays using the caspase activity probe Rho-DEVD-AOMK probe (2 µM, 1 h). d, Western blot of proteomes from MDA-MB-231, Jurkat, and CASP8-null Jurkat proteomes showing that CASP8 was only found in the pro-enzyme form in these cells. e, Fragment reactivity with recombinant, purified pro-CASP8 (D374A, D384A, C409S) added to cell lysates to a final concentration of 1 µM protein, where reactivity was detected in competition assays with the IA-rhodamine probe (2 µM). Note that mutation of both cysteine-360 and cysteine-409 to serine prevented labeling of pro-CASP8 by IA-rhodamine. f, inactive control fragment 62 did not compete IA-rhodamine labeling of C360 of pro-CASP8. g, Apparent IC50 curve for blockade of IA-rhodamine labeling of pro-CASP8 (C409S) by 7. h, 7 (50 µM) fully competed IA-alkyne-labeling of C360 of endogenous CASP8 in cell lysates as measured by isoTOP-ABPP. Representative MS1 chromatograms are shown for the C360-containing peptide of CASP8. i, Concentration-dependent reactivity of click probe 61, with recombinant, purified pro-CASP8 (D374A, D384A) added to cell lysates to a final concentration of 1 µM protein. Note that pre-treatment with 7 blocked 61 reactivity with pro-CASP8 and mutation of C360 to serine prevented labeling of pro-CASP8 by 61 (25 µM). ). j, 7 (30 µM) blocked IA-alkyne labeling of C360 of pro-CASP8, but not active-CASP8 as measured by isoTOP-ABPP. Recombinant pro- and active-CASP8 were added to Ramos lysates at 1 µM and then treated with 7 (30 µM) followed by isoTOP-ABPP. k, Fragments 7 and 62 did not block labeling by Rho-DEVD-AOMK (2 µM) of recombinant, purified active-CASP8 and active-CASP3 added to MDA-MB-231 cell lysates to a final concentration of 1 µM protein. l, 7 does not inhibit active caspases. Recombinant, active caspases were added to MDA-MD-231 lysate to a final concentration of 200 nM (CASP2, 3, 6, 7) or 1 µM (CASP8, 10), treated with z- VAD-FMK (25 µM) or 7 (50 µM), followed by labeling with the Rho-DEVD-AOMK probe (2 µM). m, Representative MS1 chromatograms for tryptic peptides containing the catalytic cysteines of CASP8 (C360), CASP2 (C320), and CASP7 (C186) quantified by isoTOP-ABPP in Jurkat cell lysates treated with 7 or 62 (50 µM, 1 h). n, 7, but not control fragment 62, blocked extrinsic, but not intrinsic apoptosis. Jurkat cells (1.5 million cells/mL) were incubated with 7 or 62 (30 µM) or the pan-caspase inhibitor VAD-FMK (100 µM) for 30 min prior to addition of staurosporine (2 µM) or SuperFasLigand™ (100 ng/mL). Cells were incubated for 4 h and viability was quantified with CellTiter-Glo®. RLU- relative light unit. o, For cells treated as described in n, cleavage of PARP (96 kDa), CASP8 (p43/p41, p18), and CASP3 (p17) was visualized by western blot. p, 7 protects Jurkat cells from extrinsic, but not intrinsic apoptosis. Cleavage of PARP, CASP8, and CASP3 detected by western blotting as shown in panel o was quantified for three (STS) or two (FasL) independent experiments. Cleavage products (PARP (96 kDa), CASP8 (p43/p41), CASP3 (p17)) were quantified for compound treatment and the % cleavage relative to DMSO-treated samples was calculated. For panels b, g and n, data represent mean values ± SEM for at least three independent experiments. For panel p, STS data represent mean values ± SEM for three independent experiments, and FasL data represent mean values ± SD for two independent experiments. Statistical significance was calculated with unpaired students t-tests comparing active compounds (VAD-FMK and 7) to control compound 62; **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
Extended Data Figure 8. CASP10 is involved in intrinsic apoptosis in primary human T cells.
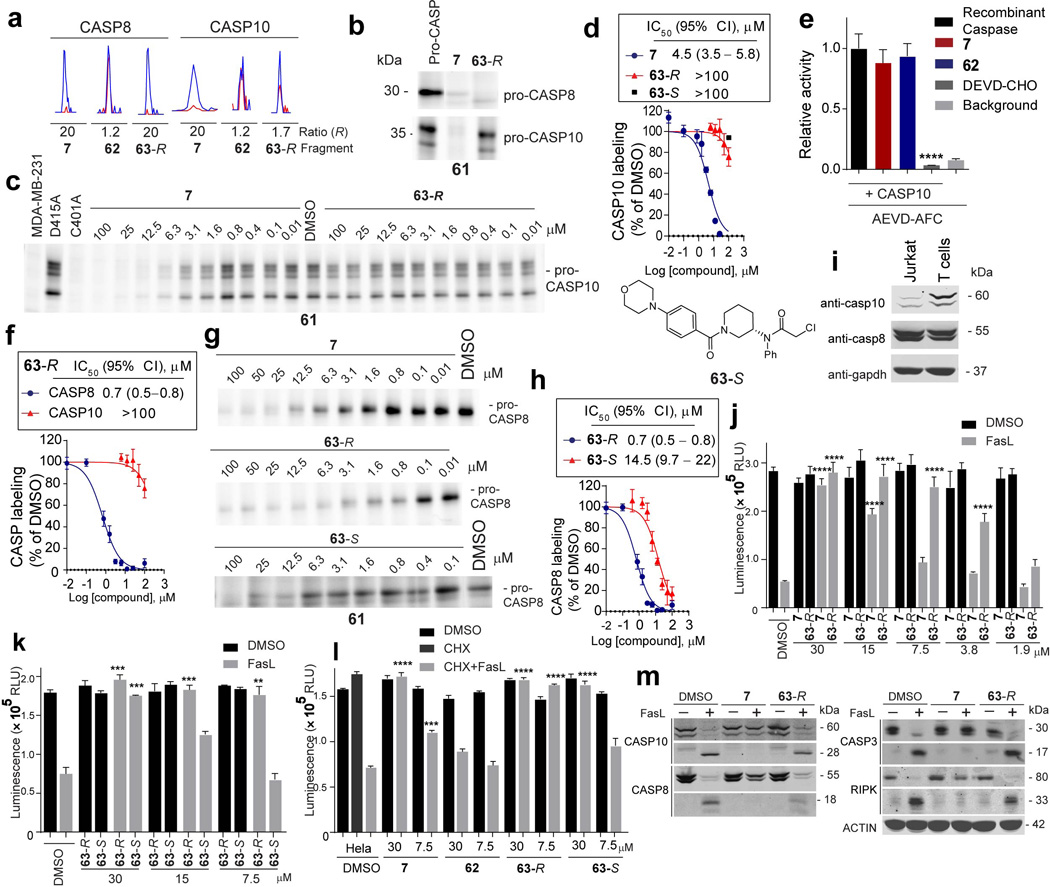
a, Representative MS1 peptide signals showing R values for caspases detected by quantitative proteomics using probe 61. ABPP-SILAC experiments. Jurkat cells (10 million cells) were treated with either DMSO (heavy cells) or the indicated compounds (light cells) for 2 h followed by probe 61 (10 µM, 1 h). b, 7 competed 61-labeling of pro-CASP8 and CASP10, whereas 63-R selectively blocked probe labeling of pro-CASP8. c, 7, but not 63-R block probe labeling of pro-CASP10. Recombinant pro-CASP10 was added to MDA-MB-231 lysates to a final concentration of 300 nM, treated with the indicated compounds, and labeled with probe 61. Mutation of the catalytic cysteine C401A fully prevented labeling by 61. d, Apparent IC50 curve for blockade of 61-labeling of pro-CASP10 by 7, 63-R or 63-Se, Neither 7 nor 63 (25 µM each) inhibited the activity of recombinant, purified active CASP10 (500 nM), which was assayed following addition of the protein to MDA-MB-231 lysate using fluorometric AEVD-AMC substrate. DEVD-CHO (20 µM) inhibited the activity of CASP10. f, Apparent IC50 curve for blockade of 61 labeling of pro-CASP8 and pro-CASP10 by 63-Rg, 63-R shows increased potency against pro-CASP8. Recombinant pro-CASP8 was added to MDA-MB-231 lysates to a final concentration of 300 nM, treated with the indicated compounds and labeled with probe 61. h, Apparent IC50 curve for blockade of 61 labeling of pro-CASP8 by 63R compared with 63- S. The structure of 63-S is shown. i, CASP10 is more highly expressed in primary human T cells compared to Jurkat cells. Western blot analysis of full-length CASP10, CASP8 and GAPDH expression levels in Jurkat and T-cell lysates (2 mg/mL). j, Jurkat cells (150,000 cells/well) were incubated with 7 or 63R at the indicated concentrations for 30 min prior to addition of staurosporine (2 µM) or SuperFasLigand™ (100 ng /mL). Cells were incubated for 4 h and viability was quantified with CellTiter-Glo®. k, Jurkat cells treated as in j, but with 63-Ror 63-Sl, HeLa cells (20,000 cells/well) were seeded and 24 h later treated with the indicated compounds for 30 minutes prior to addition of SuperFasLigand™ (100 ng /mL) and cycloheximide (CHX, 2.5 ng/mL). Cells were incubated for 6 h and viability quantified with CTG. m, For T cells treated as in Fig. 4d cleavage of CASP10 (p22), CASP8 (p18), CASP3 (p17) and RIPK (33 kDa) was visualized by western blotting. For panels d-f, h, and j-k, data represent mean values ± SEM for at least three independent experiments. Statistical significance was calculated with unpaired students t-tests comparing DMSO- to fragment-treated samples; **, p < 0.01, ****, p < 0.0001.
Extended Data Table 1.
Ligandability of representative cysteines and proteins. a. Representative cysteines with known covalent ligands targeted by fragment electrophiles in isoTOP-ABPP experiments. b. Site of fragment labeling for recombinant proteins. Underlines mark the fragment-modified cysteines.
| 1a. | ||||||
|---|---|---|---|---|---|---|
| Protein | Liganded cysteine |
Fragment(s) | Other cysteines quantified by isoTOP- ABPP |
Previous covalent inhibitor(s) |
Cysteine location |
Reference |
| BTK | C481 | 2, 3,14, 31 | C145, C337 | Ibrutinib | Active site | 44 |
| TGM2 | C277 | 12,14, 32 | C10,C27,C230,C269, C290, C336, C370, C524, C545, C620 |
18d | Active Site | 45 |
| MAP2K7 | C131 |
2,3,11,14,20,21 38 |
C260, C280 | Ibrutinib | Active Site | 20 |
| XPO1 | C528 |
2,3,5,14,24,31 43,56 |
C34, C119, C164, C199, C327, C498, C723, C1070 |
KPT-330 | Non-active site |
46 |
| CASP5 | C315 | 3,50 | - | Z-WEHD- CHO/FMK |
Active Site | 47 |
| CASP8 | C360 | 2,4,11 | C236, C409 | Z-VAD-FMK, CV8/9-AOMK |
Active Site | 48,49 |
| ERCC3 | C342 | 2, 3, 5, 8, 14, 21 | - | Triptolide | Active Site | 50 |
|
PARK7 (Toxoplasma DJ-1) |
C106 |
2,9,8,11,13,43 45, 50, 52 |
, C46, C53 | WRR-086 | Active Site | 51 |
| GST01 | C32 |
2–13,16,18–22,33 27–30, 32–34, 36, 39 43, 49, 50, 52, 54 55 |
C90, C192, C237 | KT53 | Active Site | 52 |
| ALDH2 | C319 |
3,8–10,12,27,28 32, 39, 40, 43, 49 50 |
C66, C179, C386, C472 | Disulfiram | Active Site | 53 |
| CTSZ | C92 | 4,11,20,28,32 | C89, C126, C132, C154, C170, C173, C179, C214 |
Cy5DCG04 | Active Site | 54 |
| 1b. | ||||||
|---|---|---|---|---|---|---|
| Protein | Cysteine | Fragment # | Peptide | M+H calculated (m/z) |
M+H observed (m/z) |
Charge |
| IMPDH2 | C140 | 14 | R.HGFCGIPITDTGR.M | 715.86 | 715.86 | 2 |
| TIGAR | C114 | 5 | R.EECPVFTPPGGETLDQVK.M | 1123.97 | 1123.97 | 2 |
| IDH1 | C269 | 20 | K.SEGGFIWACK.N | 702.84 | 702.84 | 2 |
| CASP8 | C360 | 7 | K.VFFIQACQGDNYQK.G | 660.98 1195.58 (light) |
660.98 1195.58 (light) |
3 |
| CASP8* | C360 | 61-TEV-Tag | K.VFFIQACQGDNYQK.G | and 1198.11 (heavy) |
and 1198.59 (heavy) |
2 |
measured for endogenous protein by isoTOP-ABPP using probe 61.
Extended Data Table 2.
Reactive docking results for liganded cysteines. Shows the most ligandable cysteine predicted by reactive docking. Match indicates whether the top cysteine by docking matches that identified by isoTOP-ABPP. Heat sensitive column indicates whether the top cysteine identified by covalent docking is sensitive to heat denaturation. ND- not detected.
| Protein | PDB ID: | Most ligandable cysteine by docking. |
Cysteine location | Most ligandable cysteine by isoTOP-ABPP |
Match | Heat Sensitive |
|---|---|---|---|---|---|---|
| Aldh2 | 1O05 | C319 | Active site | C319 | Yes | Yes |
| BTK | 1K2P | C481 | Active site | C481 | Yes | ND |
| CASP8 | 1QTN | C360 | Active Site | C360 | Yes | Yes |
| CCNB1 | 2JGZ | C238 | Non-active site | C238 | Yes | Yes |
| CDKN3 | 1FQ1 | C39 | Non-active site | C39 | Yes | Yes |
| CLIC4 | 2AEH | C35 | Non-active site | C35 | Yes | Yes |
| DTYMK | 1E2G | C163 | Non-active site | C163 | Yes | No |
| IDH1 | 3MAP | C269 | Non-active site | C269 | Yes | Yes |
| IMPDH2 | 1NF7 | C331 | Active site | C331, C140 | Yes | Yes |
| GLRX5 | 2WUL | C67 | Active site | C67 | Yes | No |
| GST01 | 1EEM | C32 | Active site | C32 | Yes | Yes |
| NME3 | 1ZS6 | C158 | Non-active site | C158 | Yes | Yes |
| PKM | 4JPG | C423 | Non-active site | C423 | Yes | Yes |
| SRC | 2SRC | C277 | Active Site | C277 | Yes | ND |
| TIGAR | 3DCY | C114 | Non-active site | C114, C161 | Yes | Yes |
| TXNDC | 1WOU | C43 | Active site | C43 | Yes | Yes |
| UGDH | 3ITK | C276 | Active site | C276 | Yes | Yes |
| UPP1 | 3EUF | C162 | Non-active site | C162 | Yes | Yes |
| XP01 | 3GB8 | C528 | Non-active site | C528 | Yes | Yes |
| CDK5 | 1UNG | C157 | Non-active site | C269 | Second | Yes |
| EDC3 | 3D3K | C311 | Non-active site | C137, C413, C499 | Second | ND |
| NR2F2 | 3CJW | C213 | Non-active site | C326, C213(in situ) | Second | ND |
| PDCD6IP | 2R02 | C231 | Non-active site | C90 | Second | ND |
| PRMT1 | 10RI | C285 | Active site | C109 | Second | Yes |
| UBE2S | 1ZDN | C118 | Non-active site | C95 | Second | ND |
| FNBP1 | 2EFL | C145 | Non-active site | C70 | No | ND |
| HAT1 | 2P0W | C120 | Non-active site | C101 | No | Yes |
| MAPK9 | 3NPC | C163 | Active site | C177 | No | ND |
| STAT1 | 1YVL | C543 | Non-active site | C492, C255 | No | ND |
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (CA087660 (B.F.C.), GM090294 (B.F.C.), K.M.B (GM108208), GM069832 (S.F., A.J.O.)). We thank J. Cisar, K. Mowen, C. Wang, M. Suciu, M. Dix, G. Simon, M. Carrillo, and J. Hulce for experimental assistance, M. Lenardo, L. Zheng and R. Siegel for helpful suggestions, the Marletta and Vogt labs at TSRI for sharing instrumentation, and ITASSER for the structural modeling of IMPDH2.
Footnotes
Contributions
B.F.C and K.M.B conceived of the project. K.M.B performed mass spectrometry experiments and data analysis. B.E.C. wrote software, compiled and analyzed mass spectrometry data. S.F. wrote software and conducted reactive docking. K.M.B cloned, overexpressed and purified proteins, conducted inhibition studies in vitro and in situ. S.C. cloned and purified IDH1. K.M.B, K.M.L, B.D.H and B.R.L synthesized compounds. G.G. expressed and purified recombinant caspases and TEV protease. D.W.W. provided assistance with the caspase studies. K.M.B, B.E.C. and B.F.C designed experiments and analyzed data. K.M.B, B.E.C. and B.F.C wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
See Supporting Information for a detailed Methods section.
References
- 1.Edfeldt FN, Folmer RH, Breeze AL. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug discovery today. 2011;16:284–287. doi: 10.1016/j.drudis.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. The druggable genome. Nature reviews. Drug discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Scott DE, Coyne AG, Hudson SA, Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry. 2012;51:4990–5003. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- 4.Erlanson DA, et al. Site-directed ligand discovery. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardoso R, et al. Identification of Cys255 in HIF-1alpha as a novel site for development of covalent inhibitors of HIF-1alpha/ARNT PasB domain protein-protein interaction. Protein science : a publication of the Protein Society. 2012;21:1885–1896. doi: 10.1002/pro.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nonoo RH, Armstrong A, Mann DJ. Kinetic template-guided tethering of fragments. ChemMedChem. 2012;7:2082–2086. doi: 10.1002/cmdc.201200404. [DOI] [PubMed] [Google Scholar]
- 7.Kathman SG, Xu Z, Statsyuk AV. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. Journal of medicinal chemistry. 2014;57:4969–4974. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jost C, Nitsche C, Scholz T, Roux L, Klein CD. Promiscuity and selectivity in covalent enzyme inhibition: a systematic study of electrophilic fragments. Journal of medicinal chemistry. 2014;57:7590–7599. doi: 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- 9.Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. Journal of the American Chemical Society. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patricelli MP, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 12.Weerapana E, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nature methods. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rostovtsev VV, Green JG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry & biology. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim SM, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angewandte Chemie. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffstrom BG, et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wissner A, et al. Synthesis structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2) Journal of medicinal chemistry. 2003;46:49–63. doi: 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]
- 20.Lanning BR, et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.London N, et al. Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Biol. 2014;10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLaBarre B, Hurov J, Cianchetta G, Murray S, Dang L. Action at a distance: allostery and the development of drugs to target cancer cell metabolism. Chemistry & biology. 2014;21:1143–1161. doi: 10.1016/j.chembiol.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Held JM, et al. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Molecular & cellular proteomics : MCP. 2010;9:1400–1410. doi: 10.1074/mcp.M900643-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vickers CJ, Gonzalez-Paez GE, Wolan DW. Selective detection and inhibition of active caspase-3 in cells with optimized peptides. Journal of the American Chemical Society. 2013;135:12869–12876. doi: 10.1021/ja406399r. [DOI] [PubMed] [Google Scholar]
- 25.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nature reviews. Immunology. 2007;7:532–542. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- 26.Lafont E, et al. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PloS one. 2010;5:e13638. doi: 10.1371/journal.pone.0013638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wachmann K, et al. Activation and specificity of human caspase-10. Biochemistry. 2010;49:8307–8315. doi: 10.1021/bi100968m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annual review of immunology. 2006;24:321–352. doi: 10.1146/annurev.immunol.24.021605.090513. [DOI] [PubMed] [Google Scholar]
- 29.Winter GE, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015 doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & biology. 2015 doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for online-only Methods
- 31.Weerapana E, Speers AE, Cravatt BF. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)--a general method for mapping sites of probe modification in proteomes. Nat Protoc. 2007;2:1414–1425. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- 32.Inloes JM, et al. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:14924–14929. doi: 10.1073/pnas.1413706111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adibekian A, et al. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nature Chemical Biology. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Meth. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium TU. UniProt: a hub for protein information. Nucleic Acids Research. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Research. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berman HM, et al. The Protein Data Bank. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeLano WL. The PyMOL Molecular Graphics System. Delano Scientific; 2002. [Google Scholar]
- 40.O’Boyle N, et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morris GM, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.