

Abstract
Using affinity purification MS approaches, we have identified a novel role for casein kinase II (CKII) in the modification of the polymerase associated factor complex (PAF-C). Our data indicate that the facilitates chromatin transcription complex (FACT) interacts with CKII and may facilitate PAF complex phosphorylation. Posttranslational modification analysis of affinity-isolated PAF-C shows extensive CKII phosphorylation of all five subunits of PAF-C, although CKII subunits were not detected as interacting partners. Consistent with this, recombinant CKII or FACT-associated CKII isolated from cells can phosphorylate PAF-C in vitro, whereas no intrinsic kinase activity was detected in PAF-C samples. Significantly, PAF-C purifications combined with stable isotope labeling in cells (SILAC) quantitation for PAF-C phosphorylation from wild-type and CKII temperature-sensitive strains (cka1Δ cka2–8) showed that PAF-C phosphorylation at consensus CKII sites is significantly reduced in cka1Δ cka2–8 strains. Consistent with a role of CKII in FACT and PAF-C function, we show that decreased CKII function in vivo results in decreased levels of histone H2B lysine 123 monoubiquitylation, a modification dependent on FACT and PAF-C. Taken together, our results define a coordinated role of CKII and FACT in the regulation of RNA polymerase II transcription through chromatin via phosphorylation of PAF-C.
Keywords: chromatin, histone modification, phosphorylation, transcription, ubiquitylation (ubiquitination), FACT, PAF-C, SILAC, casein kinase II, quantitative proteomics
Introduction
Transcription elongation by RNA polymerase II (RNAPII)4 is a coordinated process that is regulated to ensure the proper expression of protein-coding genes. Numerous protein complexes play a role in aiding RNAPII loading onto a target gene promoter through the formation of preinitiation complexes. Following initiation, RNAPII proceeds into productive transcript elongation, during which the enzyme must cope with a chromatin landscape that can have an inhibitory effect on RNAPII passage. The polymerase-associated factor complex (PAF-C) plays a central role in the regulation of RNAPII elongation and co-transcriptional histone methylation at histone H3 lysine residues 4 and 36 as well as monoubiquitylation of histone H2B at lysine 123 (H2B-K123ub1, (1–5)). In the model organism Saccharomyces cerevisiae, PAF-C is composed of five subunits: Cdc73, Ctr9, Leo1, Paf1, and Rtf1 (6). The human Paf1 complex contains an additional subunit, Ski8, which has been shown to be important in 3′-5′ mRNA degradation (7). Human PAF-C has been shown to interact directly with RNAPII (8).
Various studies in yeast have linked PAF-C function to the facilitates chromatin transcription (FACT) complex, a histone chaperone that facilitates removal of a H2A/H2B dimer during transcription and replacement of that dimer following RNAPII passage (9–11). The FACT complex is composed of Spt16 and Pob3. FACT makes contacts with the H2A/H2B dimer and has also been shown to interact with histones H3/H4, histone tails, and intact nucleosomes in some contexts (10, 12–15). Spt16 associates with all five subunits of yeast PAF-C as well as casein kinase II (CKII), as determined by qualitative mass spectrometry analysis (16). Affinity purification experiments have shown that PAF-C interacts genetically and physically with conserved transcription elongation factors, including Spt6, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole sensitivity-inducing factor, and FACT (16, 17). It has been proposed that Paf1 mediates the interaction between FACT and RNAPII in yeast (18). In higher eukaryotes, human PAF-C has been shown to interact with the general transcription elongation factor Transcription factor SII, the superelongation complex, and the FACT complex, consistent with its known interactions in yeast (8).
CKII is an abundant and constitutively active serine kinase that phosphorylates many targets in yeast and mammalian cells (19, 20). CKII contributes to the pathology of many human cancers (21–23). Multiple complexes containing CKII have been identified, including the transcription elongation factor FACT (16, 24). In mammalian cells, FACT co-purifies with CKII in a complex that phosphorylates p53 on serine 392 in response to DNA damage (24). In addition, deletion mutants of CKII exhibit defective transcription of specific cell-cycle genes, which results in a delay in entrance into S phase (25).
Quantitative analysis of dynamic protein interactions remains a significant challenge for proteomics because transient interaction partners are obtained at substoichiometric levels relative to bait proteins (reviewed in Ref. 26). Here we focused on the transient interactome of FACT and PAF-C and found that they are interaction partners with CKII. The use of hierarchical clustering and normalized spectra abundance factor (NSAF) values from multiple baits readily identify reciprocal interactions between FACT/CKII and PAF-C/RNAPII. In-depth mass spectrometry analysis using MudPIT of biological replicate purifications of the FACT complex (Spt16-TAP) isolated under low-salt conditions followed by significance analysis of interactome (SAINT) resulted in the identification of statistically significant interactions between FACT, CKII, PAF-C, and histones. Additionally, we show that all five subunits of PAF-C are targeted for phosphorylation by CKII in vivo. Although PAF-C is subjected to extensive phosphorylation by CKII, reciprocal interactions between CKII and PAF-C were not observed. However, reciprocal interactions were detected between CKII and FACT, suggesting that the FACT complex may facilitate CKII modification of PAF-C. In support of this idea, we show that CKII copurified with Spt16-TAP readily phosphorylates PAF-C in vitro, whereas no detectable kinase activity copurified with PAF-C through Ctr9-FLAG. In addition, we purified PAF-C from WT and CKII-defective cells using a SILAC approach and definitively show that CKII activity is required for phosphorylation of multiple residues across the subunits of PAF-C. Finally, and consistent with a role for CKII in PAF-C and FACT function, we show that temperature-sensitive mutants of CKII display reduced levels of histone H2B-K123ub1.
Experimental Procedures
Yeast Strains and Growth Conditions
All yeast strains are listed in supplemental Table S1. Gene deletions were performed using gene replacement (27). All expression plasmids contained the endogenous gene promoter. Mutagenesis of plasmids was performed with the QuikChange Lightning Multi site-directed mutagenesis kit (Agilent Technologies) with the primers and plasmids described in supplemental Table S2. Plasmids were transformed into deletion strains using standard methods (28). Endogenous Paf1 and Ctr9 were C-terminally tagged with the 3×-FLAG epitope using the p3FLAG plasmid (29). All strains were confirmed by Western blotting and PCR. TAP-tagged cells used for standard purifications were grown to an A600 = 2.0–2.5 in yeast extract, peptone, dextrose medium and pelleted by centrifugation. For stable isotope labeling in cells (SILAC) experiments, precultures of Ctr9-FLAG WT and cka1Δ cka2–8 were grown in yeast nitrogen base 2% glucose without arginine or lysine overnight. The cka2–8 allele is a temperature-sensitive mutant with defective function at 25 °C and 37 °C (30–32). The precultures were used to inoculate 3-liter cultures of Ctr9-FLAG WT cells in yeast nitrogen base 2% glucose containing [13C6,15N2]lysine and [13C6,15N4]arginine, referred to as heavy medium. The Ctr9-FLAG mutant cka1Δ cka2–8 was grown in YNB 2% glucose containing standard 12C,14N-containing lysine and arginine, referred to as light medium. For the SILAC experiment, cells were grown to an A600 = 1.5–2.0 at 30 °C and then subjected to a 2-h heat shock at 37 °C.
Affinity Purifications
Tandem affinity purifications (TAPs) were performed as essentially described by Puig et al. (33) and Mosley et al. (34–36). Control purifications were performed from untagged parental strains (BY4741 for the TAP strains). Cell pellets were resuspended in lysis buffer (40 mm Hepes-KOH (pH 7.5), 10% glycerol, 100 or 350 mm NaCl (depending upon purification), 0.1% Tween 20, 0.5 mm DTT, 2 mm Na orthovanadate, and fresh yeast protease inhibitor mixture) and lysed using glass beads in a bead beater. The resulting lysate was treated with DNaseI and heparin sulfate at room temperature for 10 min to release any chromatin-bound proteins. The lysate was cleared by centrifugation and incubated with 500 μl of IgG-Sepharose (GE Healthcare) beads overnight at 4 °C. The slurry was washed to remove nonspecific binding proteins, resuspended in tobacco etch virus (TEV) protease cleavage buffer, and incubated with 100 units of AcTEVTM (Invitrogen) protease overnight at 4 °C. A disposable Poly-prep chromatography column was used to separate the beads from the cleaved protein by gravity flow. The beads, retained on the column, were washed with Calmodulin binding buffer. The protein flow-through was incubated with Calmodulin-Sepharose beads for 3 h at 4 °C and eluted from the beads with Calmodulin elution buffer containing 2 mm EGTA. FLAG tag affinity purifications were performed as described above with the following changes. After clarification of the lysate by centrifugation, the lysate was incubated with 500 μl of anti-FLAG M2 affinity resin (Sigma) overnight at 4 °C. The next day, the lysate/resin slurry was passed through a disposable Poly-prep chromatography column to capture the FLAG resin. The resin was then washed extensively with lysis buffer prior to 3× FLAG peptide elution. The elutions were performed by incubation of the resin with 250 μl of a 1 mg/ml solution of 3× FLAG peptide resuspended in lysis buffer. Following elution, the resin was washed with an additional 250 μl of lysis buffer that was also collected with the eluate. This process was repeated three to four times. Affinity purification elutions were TCA-precipitated overnight at 4 °C, followed by overnight digestion with endoproteinase Lys-C and then another overnight digestion with Trypsin Gold (Promega) at 37 °C. All digestions were quenched with formic acid to a final concentration of ∼5%.
MudPIT Mass Spectrometry and Database Searching
Peptide mixtures were analyzed by MudPIT mass spectrometry as described previously (37). Each sample was pressure-loaded onto a 100-μm fused-silica nanospray column pulled to an ∼5-μm tip using a P-2000 laser puller. The microcapillary columns contained two C18 reverse phases (Aqua, Phenomenex) separated by strong cation exchange resin (Luna, Phenomenex). Each MudPIT column was placed in line with the LTQ Velos ion trap or LTQ Velos Orbitrap mass spectrometer, and a spray voltage of 2.0 kV was applied to the nanocolumn. The automated MudPIT cycles consisted of four to ten 120-min steps with increasing concentrations of ammonium acetate. Four-step MudPIT consisted of 8-μl injections of 50, 100, 200, and 300 mm ammonium acetate, respectively, followed by a 20-min wash with buffer A (5% acetonitrile and 0.1% formic acid) and then a 90-min organic gradient of 5–80% buffer B (80% acetonitrile and 0.1% formic acid) to facilitate peptide elution from the reverse-phase resin. Four MudPIT steps were used for the SILAC-labeled Ctr9-FLAG purifications. Ten-step MudPIT consisted of 8-μl injections of 25, 50, 75, 100, 150, 200, 250, 300, and two 350 mm ammonium acetate steps, followed by a buffer A wash and buffer B gradient as described above. Ten-step MudPIT was performed for all other AP-MS samples. Each full scan (at a resolution of 60,000 in the Orbitrap) was followed by 10–15 MS/MS scans using data-dependent acquisition in the ion trap, where the most intense precursor ions were individually fragmented by collision-induced dissociation (collision energy = 35).
Database searching of the RAW files was first done with SEQUEST HT (version 1.4.1.14) in Proteome Discoverer (1.4.0.288) using trypsin as the enzyme restriction and the following parameters: two missed cleavages for trypsin, a precursor mass tolerance of 1.4 Da for ion trap data and 10 ppm for Orbitrap data, a fragment mass tolerance of 1.0 daltons, a Δ CN value of [mteq]0.15, and a fixed modification of +57 Da on cysteine residues and variable modifications of +16 Da on methionine and +80 daltons on serine, tyrosine, and threonine. The SILAC experiments were searched as above but included dynamic modifications for [13C6,15N2]lysine and [13C6,15N4]arginine. A custom S. cerevisiae FASTA database was used for a database search that contained 6631 protein sequences, including the entire yeast proteome from Uniprot (downloaded on February 27, 2014), and ∼150 common contaminant proteins, including proteolytic enzymes, human keratins, and common laboratory contaminants. Additionally, we included the peptide sequence for the TAP tag used for isolation of all protein complexes studied. The PSM counts for the TAP tag were manually added to the PSM count of the bait (i.e. TAP-tagged) subunit for subsequent quantitative analysis. The msf files from Proteome Discoverer were imported into Scaffold 4, and the peptides from Scaffold were used for subsequent quantitative and posttranslational modification analysis for SAINT. The data obtained for the SILAC experiments were analyzed by the quantitation module within Proteome Discoverer 1.4 (Thermo) to calculate the total peak area for each precursor ion and the relative ratio of heavy to light precursor ions. For posttranslational modification analysis of Cdc73-TAP, spectra were analyzed with X!Tandem with the addition of protein N-terminal acetylation (+42 Da) as a dynamic search option. Each protein is required to have at least two peptides to be considered identified. Additionally, site-specific modifications are reported from manual interpretation of spectra to confirm the fragment ion coverage of the specific phosphorylation site. Peptides used for phosphorylation mapping were also required to have ≤150 ppm from the LTQ Velos ion trap or ≤10 ppm from the LTQ Velos Orbitrap. Peptide spectrum matches (PSMs) used for protein interaction analysis were identified at a peptide false discovery rate of less than or equal to 1% as calculated by Scaffold. Hierarchical clustering analysis was performed as described previously (35, 36). The peptide and protein identifications for the Spt16-TAP (n = 4), and Cdc73-TAP (n = 3) purifications are available upon request. Additionally all RAW data files, Scaffold data files, and peak list files have been deposited into the MassIVE data repository under the title “FACT, PAF-C, and CKII.”
Label-free Quantitative Proteomics Approaches for Interactome Analysis
The total number of PSMs passing the criteria listed above were used for relative quantitation using the following approaches. Two empirical –fold change scores were calculated (38). The first, referred to as FC-A, calculates the -fold enrichment of affinity purifications over control purifications using the average mean of the PSMs per protein across replicates. The second score, referred to as FC-B, calculates the -fold change over control using the geometric mean of replicates. SAINT probability scores were also calculated for Spt16-TAP replicates as described previously using the Contaminant Repository for Affinity Purification (CRAPome) web site as detailed in multiple publications (37–42). Finally, we also normalized the total spectral abundance for proteins of interest using NSAF calculations as described previously (35, 36, 43, 44).
In Vitro Kinase Assays
For kinase reactions, ∼300 ng of low-salt (100 mm) purified Spt16-TAP-associated proteins and/or Ctr9-FLAG-associated proteins were incubated alone, in combination, or with recombinant CKII (Millipore) for 2 h at 30 °C in kinase buffer (40 mm HEPES (pH 7.5), 10 mm MgCl2, 5 mm dithiothreitol, and 10 μCi of [γ-32P]ATP (6000 Ci/mmol, PerkinElmer Life Sciences)). Kinase reactions were stopped by the addition of 2× SDS-PAGE loading buffer and boiling at 100 °C for 10 min. Reactions were then separated by SDS-PAGE on a 10–20% precast gel (Bio-Rad). The gels were dried under a vacuum prior to exposure to a PhosphorImager screen for ≥1 h prior to scanning on a Fuji scanner.
Immunoblotting
Yeast cells of the indicated genotypes along with their wild-type counterparts were grown in YPD either at permissive or restrictive temperatures. Overnight-saturated cultures were back-diluted to an A600 of 0.2 and allowed to grow until they reached an A600 of 1. Five A600 equivalents of cells were lysed by bead beating using SUMEB lysis buffer containing 1% SDS, 8 m urea, 10 mm MOPS (pH 6.8), 10 mm EDTA, and 0.01% bromphenol blue). Lysates were separated by SDS-PAGE and probed using anti-HA (UNC Antibody Core Facility, clone 12CA5, 1:5000), anti-glyceraldehyde 6-phosphate dehydrogenase (G6PDH) (Sigma-Aldrich, A9521, 1:100,000), anti-histone H2B-K123ub1 (Cell Signaling Technology, 5546S, 1:1000), anti-histone H2B (Active Motif, 39237, 1:50,000), and anti-histone H3 lysine 4 trimethylation (EpiCypher, 13-0004, 1:5000). HRP-conjugated anti-rabbit or anti-mouse secondary (1:10,000) antibody was used, and the signal was detected using ECL Prime or ECL (Amersham Biosciences).
Results
During the transcription cycle, RNAPII interacts with numerous accessory proteins to facilitate transcription initiation, elongation through chromatin, co-transcriptional RNA processing and transcription termination. Unlike RNA polymerase I and III, which have “built-in” elongation factor activities, all RNAPII-associated elongation factors interact in a dynamic fashion (35, 45). One approach to study transient interaction partners using AP-MS is to perform reciprocal purification of low-level prey proteins to confirm the interaction with the original bait protein of interest (Fig. 1). Although this approach is common, the coordinated analysis of these purifications using quantitative approaches is not often performed in favor of more standard qualitative (presence or absence) analysis. In this study, we chose to perform four biological replicate purifications of Spt16-TAP under low-salt (LS, 100 mm NaCl) conditions to facilitate the capture of transient interacting proteins because FACT was the lowest-level RNAPII interaction partner known from previous studies identified in Rpb3-TAP purifications (35, 36). A representative Spt16-TAP LS purification was used for hierarchical clustering analysis (Fig. 1, A, left panel, and B, second column). A high-salt (350 mm NaCl) purification was also performed for Spt16-TAP for comparison (Fig. 1B, first column). In addition, we performed MudPIT analysis of Ckb2-TAP (a subunit of casein kinase II) and Cdc73-TAP (a subunit of PAF-C) and included datasets published previously for Spt4-TAP, Tfg1-TAP (a subunit of TFIIF), and Rpb11-TAP for comparison (36) (Fig. 1). For each dataset, NSAF values were calculated and analyzed by hierarchical clustering.
FIGURE 1.

Analysis of the FACT, PAF-C, and CKII interactome. A, TAP-tagged Spt16, Cdc73, and Ckb2 were immunoprecipitated using IgG-Sepharose and calmodulin-sepharose resins and eluted via 2 mm EGTA. Approximately 12.5% of a representative elution was electrophoresed on a Bio-Rad Mini Protean TGXTM gel and stained with silver nitrate. Band sizes were determined by comparison with Precision Plus protein standards (Bio-Rad) as indicated. B, hierarchical clustering analysis of RNAPII elongation factor interactions. The heat map displays NSAF values for the indicated baits (top) and preys (right), with black squares representing proteins that were not detected corresponding to the color (NSAF) scale. A dendrogram is shown at the top and left, based on hierarchical clustering analysis of rows and columns.
As shown in Fig. 1B, hierarchical clustering readily separates the known protein complexes into groups based on the AP-MS data (see dendrogram on the left and compare with protein complex labels on the right). Using these data, the Spt4/Spt5 heterodimer is readily identified as a complex based on the high abundance of the complex in Spt4-TAP samples. Overall, the cluster analysis identified two main sets of elongation factors. The first set includes TFIIF, Spt4/Spt5, and PAF-C as interaction partners that readily co-purify with RNAPII (Fig. 1B, bottom ¾ of the cluster). The second set includes FACT and CKII, which appear to interact with PAF-C under low-salt conditions when using Spt16-TAP as bait (Fig. 1B, top ¼ of the cluster). These data support two previously reported models: FACT can act as a complex with CKII (24), and PAF-C can serve as a scaffold to recruit FACT to RNAPII (18).
SAINT calculates interaction probabilities for proteins that are isolated through affinity purification approaches (referred to as prey), such as TAP and FLAG purifications. In our analysis, the specific bait purifications (Spt16-TAP LS) are compared with mock purifications from untagged parental strains. Using SAINT, we analyzed the four Spt16-TAP LS biological replicates to identify significant interaction partners of FACT (supplemental Table S3). In addition to the FACT complex itself, histones H2A, H2B, and H4 (Htb1, Htb2, and Hht1, respectively), all four subunits of CKII, and the five subunits of PAF-C had SAINT scores of ≥0.75 (Fig. 2). Additional significant interaction partners of Spt16 are listed in supplemental Table S3. There are also low-level PSM values identified in replicate Spt16-TAP samples for the two largest subunits of RNAPII (Rpb1 and Rpb2), but these interactions were not significantly enriched over controls. The interaction values from this purification span 4 orders of magnitude, perhaps revealing the true dynamic nature of protein interactions with the FACT complex.
FIGURE 2.
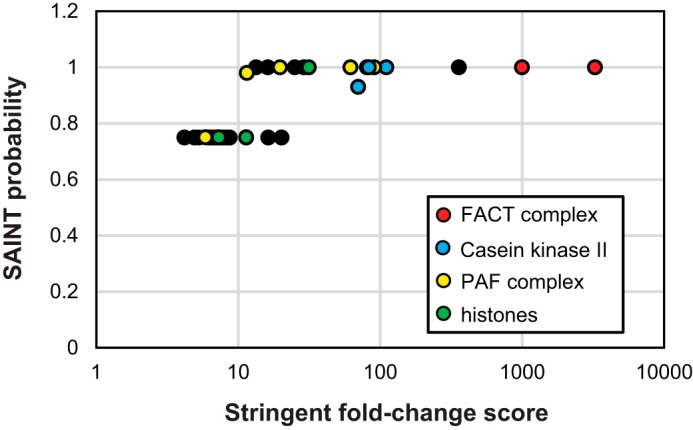
Identification of significant Spt16-TAP interaction partners. Shown are stringent -fold change scores versus SAINT interaction probabilities for FACT, CKII, and PAF-C complex members from analysis of Spt16-TAP samples (n = 4). The specific subunit names and their interaction scores are listed in supplemental Table S3. A legend including a color code for the subunits of FACT, CKII, PAF-C, and nucleosomes is shown at the bottom right.
The data presented so far clearly suggest a reproducible yet dynamic relationship between FACT, CKII, and PAF-C. The data obtained from Spt16-TAP low-salt purifications provide convincing evidence for protein interactions between FACT, CKII, and PAF-C. However, reciprocal purifications of PAF-C did not result in high levels of co-purifying FACT or CKII (Fig. 1). We next chose to further characterize the potential dynamic protein interaction between PAF-C and CKII in more detail. CKII is a constitutively active serine/threonine kinase that has been implicated in the phosphorylation of a number of transcription-related proteins including RNAPII itself (46). Multiple consensus motifs have been defined that are phosphorylated by CKII, including [S/T]XX[E/D] (47), SDXE, SXX[E/D], and [D/E]S[D/E]X[D/E] and many other similar motifs (48, 49). Kinase-substrate interactions have also been characterized as highly transient, and previous studies using AP-MS to characterize these interactions have used approaches, including kinase overexpression, to increase the overall chance of catching a snapshot of these quick reactions (41). To determine whether PAF-C and/or FACT subunits are substrates of CKII, we performed dynamic posttranslational modification searches for serine, threonine, or tyrosine phosphorylation on Spt16-TAP and Cdc73-TAP purifications. Using this approach, we found that all five subunits of PAF-C are putative CKII targets, suggesting that PAF-C and CKII are not only direct interacting partners but that the interaction rates are likely too rapid to capture high levels of CKII subunits in PAF-C purifications (supplemental Table S4, Fig. 3). Two of the phosphorylation sites, Ser422 in Paf1 and Ser358 in Leo1, consist of a SPD/E sequence, which suggests that these sites could be targets of cyclin-dependent kinases or CKII based on their consensus motifs (49). Three of the PAF-C subunits (Ctr9, Paf1, and Rtf1) are putatively modified by CKII at multiple sites (Figs. 3, A, C, and D, with representative phosphosite mapping spectra provided in supplemental Fig. S1). Serine 132 in Leo1, a putative CKII consensus site, is modified in over 80% of the PSMs identified that contain that amino acid.
FIGURE 3.

The PAF-C complex is substantially phosphorylated in vivo. A, schematics of PAF-C subunits with the location of MS-identified phosphorylation sites (red circles) and MS-identified CKII consensus sites (yellow circles) on PAF-C members. Putative CKII or cyclin-dependent kinase (CDK) sites are shown as half-red/half-yellow circles. B, Seq2Logo analysis of consensus CKII phosphorylation sites identified in MudPIT analysis. C, representative spectra of Leo1 phosphorylation at serine 132. The Leo1 Ser132 spectrum has a parent error of 8.6 ppm. D, representative spectra of Cdc73 phosphorylation at serine 157 with a parent error of −14 ppm. The full sequence of detected peptide for each spectrum is shown within the panel, with preceding and following amino acids shown in parentheses. The modified serine residues are shown in lowercase in the peptide sequence. The SEQUEST XCorr values and X!Tandem scores are also shown. A representative fragment map for the y and b ion series is shown at the top of each spectrum. The b ions are colored blue, and the y ions are colored red. Fragments from neutral loss of phosphate are shown in green.
To define the consensus motifs for the PAF-C amino acids modified by CKII, we performed sequence enrichment analysis using Seq2Logo (50) (Fig. 4B). The major consensus sequence (n = 10 sites) for PAF-C phosphorylation sites is SDX[D/E][D/E]XD, which strongly resembles previously defined CKII phosphorylation motifs (48, 49) (Fig. 3 and supplemental Table S4). These data, in light of our protein interaction analysis, suggest that FACT may facilitate CKII recruitment to PAF-C for its subsequent phosphorylation (18). Although we performed full phosphorylation analyses of the FACT subunits Spt16 and Pob3, we did not identify any phosphorylated peptides in either protein. However, the C-terminal domain sequence of Spt16 was not detected in our analyses, suggesting that alternate protease digestions may be needed to fully analyze potential modification sites in FACT.
FIGURE 4.
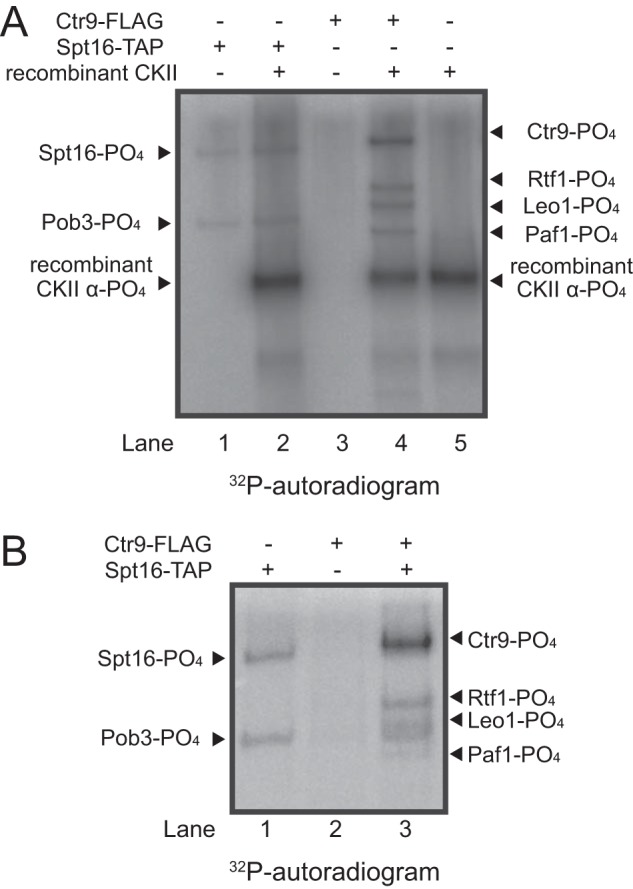
In vitro phosphorylation of FACT and PAF-C by CKII. A, autoradiograph of a representative set of in vitro reactions performed with [γ32P]ATP in the presence or absence of Ctr9-FLAG, Spt16-TAP, or recombinant CKII, as indicated at the top. The location of each phosphorylated form of the FACT, PAF-C, and CKII subunits is indicated at the left and right. B, in vitro kinase reactions for PAF-C phosphorylation by Spt16-associated CKII. Lane numbers are given below each panel for clarity.
We next performed a series of experiments to confirm that the subunits of PAF-C are bona fide substrates of CKII using both in vitro and in vivo approaches. Using [γ32P]ATP, we first performed in vitro kinase assays using LS Spt16-TAP-purified FACT and Ctr9-FLAG-purified PAF-C as substrates in the presence or absence or recombinant CKII. As shown in Fig. 4A, lane 2, proteins corresponding to the molecular weight of both subunits of the FACT complex can be phosphorylated in vitro in reactions containing recombinant CKII (Fig. 4A, Spt16 and Pob3). Recombinant CKII is also autophosphorylated in this experiment, as visualized in Fig. 4A, lane 5, which only contains recombinant CKII and ATP. Interestingly, phosphorylated Spt16 and Pob3 bands were also visualized in reactions lacking recombinant CKII, suggesting that CKII (or potentially some other co-purifying kinase) can also phosphorylate FACT in vitro (Fig. 4A). Importantly, we found that PAF-C was not phosphorylated in the absence of recombinant CKII, suggesting that CKII is not a stable interacting partner of PAF-C (Fig. 4A, lane 3). These data are also in agreement with our AP-MS studies (Figs. 1 and 2). In contrast, four bands corresponding to the molecular weights of PAF-C subunits are readily phosphorylated in the presence of recombinant CKII (Fig. 4A, lane 4). Notably, we did find that four of the five PAF-C subunits were phosphorylated in our in vitro reaction following incubation with Spt16-TAP and ATP (Fig. 4B, lane 3). These data clearly show that the FACT-CKII complex will readily phosphorylate PAF-C in vitro.
To test whether CKII is responsible for phosphorylation of PAF-C using an in vivo approach, we performed quantitative proteomics analysis of Ctr9-FLAG-isolated PAF-C complexes from WT and CKII temperature-sensitive strains (cka1Δ cka2–8) using SILAC-based quantitation (Fig. 5). The experimental scheme used for these studies is shown in Fig. 5A. Both WT and CKII temperature-sensitive strains were grown to an A600 = 1.5 and then heat shocked at 37 °C for 2 h prior to mixing of the cell pellets, lysis, and PAF-C purification. The full set of significantly changed peptides identified in the Ctr9-FLAG SILAC dataset is included in supplemental Table S5. As shown in supplemental Table S5, we readily identified peptides for 10 phosphorylation sites across four subunits of PAF-C for which the levels of the phosphorylated peptides decreased, whereas the unmodified peptides increased in cka1Δ cka2–8 (light) versus the WT (heavy). Although we observed multiple decreases in the level of phosphopeptides in cka1Δ cka2–8, the relative protein abundance of all five PAF-C subunits was unchanged (Fig. 5B). The MS1 precursor intensity for the isotopic peaks are shown for representative Leo1 peptides KFYGEDANNFSDQDETTHTFKEENVELVR and KFYGEDANNFSPO4DQDETTHTFKEENV-ELVR containing unmodified or phosphorylated serine 132, respectively (Fig. 5, C and D). The MS1 intensity for peptide ions containing unmodified Leo1 serine 132 increased 2.5-fold following heat shock of cka1Δ cka2–8 strains (Fig. 5C). The opposite trend was observed for Leo1 serine 132, with a 3-fold decrease in the abundance of MS1 area (Fig. 5D). Similar trends were observed for the other putative CKII sites in Ctr9, Paf1, and Rtf1, suggesting that these are CKII targets (supplemental Table S5). Overall, these data convincingly show that PAF-C is a bona fide substrate of CKII in vivo. Interestingly, we also observed increased threonine 127 phosphorylation in Paf1. These data suggest that other kinases may partially compensate for the loss of CKII activity following heat shock of cka1Δ cka2–8 strains to regulate Paf1 phosphorylation (supplemental Table S5).
FIGURE 5.

In vivo analysis of PAF-C subunit phosphorylation in Ctr9-FLAG purifications from WT (heavy) and cka1Δ cka2–8 (light) strains using a SILAC approach. A, schematic of the experimental approach. B, relative PAF-C subunit abundance as determined by SILAC. The data are shown as the light/heavy ratio, with the ratio variance shown as error bars. C, precursor ion abundance for the Leo1 peptide KFYGEDANNFSDQDETT-HTFKEENVELVR containing unmodified serine 132 (charge, +4). D, precursor ion abundance for the Leo1 peptide KFYGEDANNFSPO4DQDETTHTFKEENVELVR containing phosphorylated serine 132 (charge, +3). The ion series for light (cka1Δ cka2–8) peptides is shown in blue, whereas the heavy ion series (WT) is shown in red.
We next assessed the functional consequences of CKII disruption on the biological pathways that are known to require PAF-C and FACT. Both complexes are known to regulate posttranslational histone modifications, including H2B-K123ub1. Histone H2B-K123ub1 occurs during RNAPII transcription elongation and is a regulatory event for histone H3 lysine methylation at Lys4 and Lys79 (11, 51–55). Because CKII is responsible for extensive modification of PAF-C, we analyzed the effect of CKII disruption on H2B-K123ub1 levels in WT, cka1Δ CKA2, and cka1Δ cka2–8 strains (Fig. 6A). At restrictive temperatures of 25 °C and 37 °C, where the activity of CKII is compromised in cka1Δ cka2–8 (32), the levels of H2B-K123ub1 are decreased in cka1Δ cka2–8 strains relative to the levels of total histone H2B. Additionally, the levels of histone H2B-K123ub1 were decreased in cka1Δ CKA2 following incubation at the restrictive temperatures (Fig. 6A). No changes in histone H3K4 methylation levels were observed, which is in line with studies that show that reduced H2B-K123ub1 levels can occur without significant effects on H3K4 methylation (56). Taken together, these data show that CKII is an important regulator of histone H2B-K123ub1, likely through its regulation of PAF-C and FACT. Additional analysis of phosphosite mutants of the PAF-C subunits Paf1, Ctr9, and Cdc73 did not individually reveal defects in histone modification levels at H2B-K123ub1, histone H3 lysine 4 trimethylation, or histone H3 lysine 36 trimethylation (Fig. 6B). In addition, although strains with defective PAF-C are sensitive to the transcription elongation inhibitor 6-azauracil or hydroxyurea (16, 17, 57–60), the phosphosite mutants did not display growth defects when exposed to these drugs (data not shown). These data suggest that changes in the phosphorylation of multiple subunits of PAF-C and/or FACT may be required to recapitulate the phenotype observed in cka1Δ cka2–8. Regardless, these findings suggest that CKII acts upstream of PAF-C and FACT as well as to regulate H2B-K123ub1 levels. However, the precise mechanisms through which this regulation occurs remain to be determined.
FIGURE 6.
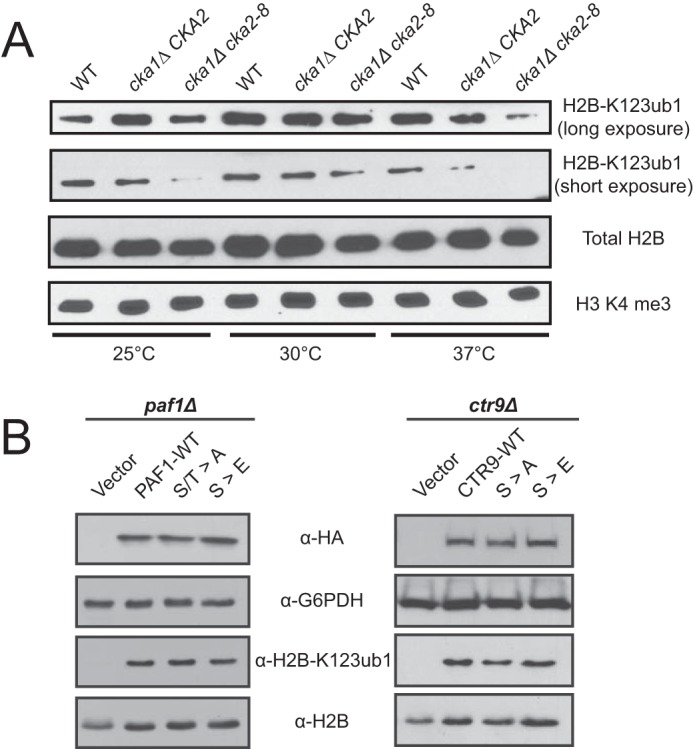
CKII regulates histone H2B-K123ub1 levels. A, Western blotting analysis of histone H2B-K123ub1, total histone H2B, and histone H3 lysine 4 trimethylation levels from whole cell extracts from cka1Δ cka2–8 strains grown under permissive (30 °C) and restrictive (25 °C and 37 °C) temperatures. The antibody used for each panel is indicated at the right, and the temperature used for cell growth is given at the bottom. B, Western blotting analysis of whole cell lysates for histone H2B-K123ub1, G6PDH, H2B, and HA of PAF1 or CTR9 deletion strains rescued with PAF1-3HA or CTR9-3HA expression vectors as indicated. Left panel, Vector, pRS313–3HA-SSN6; PAF1-WT, PAF1–3HA; S/T>A, PAF1–3HA S147A, T385A, T422A, S426A; S/T>E, PAF1–3HA S147E, T385E, T422E, S426E. Right panel, Vector, pRS313–3HA-SSN6; CTR9-WT, CTR9–3HA); S>A, CTR9–3HA S977A, S1015A, S1017A, S1046A, S1054A, S1056A; S>E, CTR9–3HA S977E, S1015E, S1017E, S1046E, S1054E, S1056E.
Discussion
In this study, we have performed an in-depth proteomics and genetic analysis of two transiently associated RNAPII elongation factors that are recruited to target genes during active gene transcription. Although PAF-C subunits were not identified as significant interaction partners of RNAPII when using Rpb3-TAP as bait in previous studies, purification of PAF-C through Cdc73-TAP revealed that RNAPII is a major interaction partner of PAF-C (Fig. 1). However, purifications of FACT through Spt16-TAP under low-salt conditions revealed a significant interaction between FACT and PAF-C (supplemental Table S3 and Fig. 2). These data suggest that PAF-C may be involved in FACT recruitment to RNAPII, as has been hypothesized previously, because PAF-C readily interacts with RNAPII in affinity purifications (18). FACT readily co-purifies with CKII in low-salt purifications. Although Cdc73-TAP samples did not contain significant levels of FACT or CKII subunit peptides, we found that four of five subunits of PAF-C are phosphorylated by CKII in vitro and in vivo (Figs. 3–5). In addition, we found that Spt16-interacting CKII can readily phosphorylate purified PAF-C in vitro (Fig. 4B). Together, these findings implicate the FACT complex in phosphorylation of PAF-C by CKII. The human FACT complex has been proposed previously to play a similar role in the regulation of p53 phosphorylation by CKII (24).
Previous studies have addressed the challenge of transient interaction partners by overexpression of bait proteins (specifically kinases) to increase the relative abundance of significant protein-protein interactions (41). However, this approach could result in a large number of false positive interactions as a result of overexpression effects on the biological system in question. Additionally, single affinity purifications have been suggested as an approach to capture dynamic interaction partners. Unfortunately, single affinity purifications also have significantly higher interactions with cellular contaminants that, in our experience, are not removed by algorithms such as SAINT (37). Dynamic or transient interaction partners present even more of a challenge to existing statistical programs than small proteins (discussed above). This study suggests that inclusion of a large number of replicates in combination with follow-up reciprocal purifications and functional studies is the most robust approach for characterization of dynamic interactions such as those with kinases like CKII. However, it must also be acknowledged that the low detection frequency of transient interaction partners is a significant challenge for data-dependent acquisition-based approaches and their related statistical interpretation. For instance, confirmation of PAF-C modification by CKII in vitro and in vivo should result in the functional annotation of PAF-C and CKII as having both a transient protein-protein interaction and an enzyme-substrate relationship.
Our data show that CKII is a novel upstream regulator of H2B-K123ub1. We hypothesized that CKII phosphorylation of PAF-C could be required for FACT and/or PAF-C-dependent control of H2B-K123ub1. We have clearly shown that CKII regulates PAF-C phosphorylation and H2B-K123ub1. However, our initial genetic studies on PAF-C phosphorylation have not shown that PAF-C phosphorylation is required for H2B-K123ub1. There are many possible reasons for this. First, it is possible that CKII phosphorylation needs to be disrupted on multiple PAF-C subunits to fully recapitulate the cka1Δ cka2–8 phenotype. Additionally, it is possible that phosphorylation sites are present across these subunits that were not detected in our mass spectrometry analysis because of low peptide detectability and/or overdigestion with trypsin. Our findings that Paf1 phosphorylation increases at threonine 127 in cka1Δ cka2–8 strains could suggest that other kinases can compensate for the loss of CKII activity. Finally, it is also plausible that the regulation of H2B-K123ub1 by CKII occurs in a PAF-C-independent mechanism.
Author Contributions
L. G. B., R. D., J. L. K., G. O. H., E. D. A., A. K. B., and A. L. M. generated the strains, conducted the experiments, and analyzed the results. L. G. B., G. O. H., and A. L. M. performed the mass spectrometry runs and data analyses. L. G. B. and A. L. M. wrote most of the paper with significant contributions from R. D., J. L. K., and B. D. S. L. G. B., R. D., J. L. K., B. D. S., and A. L. M. conceived the idea for the project.
Supplementary Material
Acknowledgments
We thank Whitney Smith-Kinnaman for technical contributions and project support, Nicole Novaresi for strain generation, and members of the Mosley laboratory for comments on the manuscript. We also thank the Dr. David Stillman, Judith Jaehning, and Georjana Barnes laboratories for the spt16–11, PAF1 deletions and casein kinase II mutants, respectively.
This work was supported by National Institutes of Health Grants R01 GM099714 (to A. L. M.) and R01 GM110058 (to B. D. S.) and a biomedical research grant from the Indiana University School of Medicine. Summer support for part of the work on this project was provided by the DePauw University Student Faculty Summer Research Fund and Professional Development Fund (to L. G. B. and A. K. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1 and Tables S1–S5.
- RNAPII
- RNA polymerase II
- PAF-C
- polymerase-associated factor complex
- H2B-K123ub1
- histone H2B lysine 123 monoubiquitylation
- FACT
- facilitates chromatin transcription
- CKII
- casein kinase II
- NSAF
- normalized spectra abundance factor
- TAP
- tandem affinity purification
- SAINT
- significance analysis of interactome
- SILAC
- stable isotope labeling in cells
- PSM
- peptide spectral match
- LS
- low-salt.
References
- 1. Tomson B. N., and Arndt K. M. (2013) The many roles of the conserved eukaryotic Paf1 complex in regulating transcription, histone modifications, and disease states. Biochim. Biophys. Acta 1829, 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wozniak G. G., and Strahl B. D. (2014) Hitting the “mark”: interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta 1839, 1353–1361 [DOI] [PubMed] [Google Scholar]
- 3. Thornton J. L., Westfield G. H., Takahashi Y. H., Cook M., Gao X., Woodfin A. R., Lee J. S., Morgan M. A., Jackson J., Smith E. R., Couture J. F., Skiniotis G., and Shilatifard A. (2014) Context dependency of Set1/COMPASS-mediated histone H3 Lys4 trimethylation. Genes Dev. 28, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laribee R. N., Krogan N. J., Xiao T., Shibata Y., Hughes T. R., Greenblatt J. F., and Strahl B. D. (2005) BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr. Biol. 15, 1487–1493 [DOI] [PubMed] [Google Scholar]
- 5. Xiao T., Kao C. F., Krogan N. J., Sun Z. W., Greenblatt J. F., Osley M. A., and Strahl B. D. (2005) Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol. Cell. Biol. 25, 637–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mueller C. L., and Jaehning J. A. (2002) Ctr9, Rtf1, and Leo1 are components of the Paf1/RNA polymerase II complex. Mol. Cell. Biol. 22, 1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu B., Mandal S. S., Pham A. D., Zheng Y., Erdjument-Bromage H., Batra S. K., Tempst P., and Reinberg D. (2005) The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev. 19, 1668–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J., Guermah M., and Roeder R. G. (2010) The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 140, 491–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Costa P. J., and Arndt K. M. (2000) Synthetic lethal interactions suggest a role for the Saccharomyces cerevisiae Rtf1 protein in transcription elongation. Genetics 156, 535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orphanides G., Wu W. H., Lane W. S., Hampsey M., and Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 11. Pavri R., Zhu B., Li G., Trojer P., Mandal S., Shilatifard A., and Reinberg D. (2006) Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125, 703–717 [DOI] [PubMed] [Google Scholar]
- 12. Formosa T., Eriksson P., Wittmeyer J., Ginn J., Yu Y., and Stillman D. J. (2001) Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 20, 3506–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stuwe T., Hothorn M., Lejeune E., Rybin V., Bortfeld M., Scheffzek K., and Ladurner A. G. (2008) The FACT Spt16 “peptidase” domain is a histone H3-H4 binding module. Proc. Natl. Acad. Sci. U.S.A. 105, 8884–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kemble D. J., Whitby F. G., Robinson H., McCullough L. L., Formosa T., and Hill C. P. (2013) Structure of the Spt16 middle domain reveals functional features of the histone chaperone FACT. J. Biol. Chem. 288, 10188–10194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCullough L., Poe B., Connell Z., Xin H., and Formosa T. (2013) The FACT histone chaperone guides histone H4 into its nucleosomal conformation in Saccharomyces cerevisiae. Genetics 195, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krogan N. J., Kim M., Ahn S. H., Zhong G., Kobor M. S., Cagney G., Emili A., Shilatifard A., Buratowski S., and Greenblatt J. F. (2002) RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomnics approach. Mol. Cell. Biol. 22, 6979–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Squazzo S. L., Costa P. J., Lindstrom D. L., Kumer K. E., Simic R., Jennings J. L., Link A. J., Arndt K. M., and Hartzog G. A. (2002) The Paf1 complex physically and functionally associates with transcription elongation factors in vivo. EMBO J. 21, 1764–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adelman K., Wei W., Ardehali M. B., Werner J., Zhu B., Reinberg D., and Lis J. T. (2006) Drosophila Paf1 modulates chromatin structure at actively transcribed genes. Mol. Cell. Biol. 26, 250–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Venerando A., Ruzzene M., and Pinna L. A. (2014) Casein kinase: the triple meaning of a misnomer. Biochem. J. 460, 141–156 [DOI] [PubMed] [Google Scholar]
- 20. Meggio F., and Pinna L. A. (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368 [DOI] [PubMed] [Google Scholar]
- 21. Filhol O., Giacosa S., Wallez Y., and Cochet C. (2015) Protein kinase CK2 in breast cancer: the CK2β regulatory subunit takes center stage in epithelial plasticity. Cell. Mol. Life Sci. 72, 3305–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piazza F., Manni S., and Semenzato G. (2013) Novel players in multiple myeloma pathogenesis: role of protein kinases CK2 and GSK3. Leuk. Res. 37, 221–227 [DOI] [PubMed] [Google Scholar]
- 23. Piazza F., Manni S., Ruzzene M., Pinna L. A., Gurrieri C., and Semenzato G. (2012) Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia 26, 1174–1179 [DOI] [PubMed] [Google Scholar]
- 24. Keller D. M., Zeng X., Wang Y., Zhang Q. H., Kapoor M., Shu H., Goodman R., Lozano G., Zhao Y., and Lu H. (2001) A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell 7, 283–292 [DOI] [PubMed] [Google Scholar]
- 25. Tripodi F., Nicastro R., Busnelli S., Cirulli C., Maffioli E., Tedeschi G., Alberghina L., and Coccetti P. (2013) Protein kinase CK2 holoenzyme promotes start-specific transcription in Saccharomyces cerevisiae. Eukaryotic Cell 12, 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miteva Y. V., Budayeva H. G., and Cristea I. M. (2013) Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 85, 749–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 28. Gietz R. D., and Schiestl R. H. (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 [DOI] [PubMed] [Google Scholar]
- 29. Gelbart M. E., Rechsteiner T., Richmond T. J., and Tsukiyama T. (2001) Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol. Cell. Biol. 21, 2098–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanna D. E., Rethinaswamy A., and Glover C. V. (1995) Casein kinase II is required for cell cycle progression during G1 and G2/M in Saccharomyces cerevisiae. J. Biol. Chem. 270, 25905–25914 [DOI] [PubMed] [Google Scholar]
- 31. Peng Y., Wong C. C., Nakajima Y., Tyers R. G., Sarkeshik A. S., Yates J. 3rd, Drubin D. G., and Barnes G. (2011) Overlapping kinetochore targets of CK2 and Aurora B kinases in mitotic regulation. Mol. Biol. Cell 22, 2680–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hockman D. J., and Schultz M. C. (1996) Casein kinase II is required for efficient transcription by RNA polymerase III. Mol. Cell. Biol. 16, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., and Séraphin B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 34. Mosley A. L., Pattenden S. G., Carey M., Venkatesh S., Gilmore J. M., Florens L., Workman J. L., and Washburn M. P. (2009) Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol. Cell 34, 168–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mosley A. L., Sardiu M. E., Pattenden S. G., Workman J. L., Florens L., and Washburn M. P. (2011) Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell. Proteomics 10.1074/mcp.M110.000687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mosley A. L., Hunter G. O., Sardiu M. E., Smolle M., Workman J. L., Florens L., and Washburn M. P. (2013) Quantitative proteomics demonstrates that the RNA polymerase II subunits Rpb4 and Rpb7 dissociate during transcriptional elongation. Mol. Cell. Proteomics 12, 1530–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith-Kinnaman W. R., Berna M. J., Hunter G. O., True J. D., Hsu P., Cabello G. I., Fox M. J., Varani G., and Mosley A. L. (2014) The interactome of the atypical phosphatase Rtr1 in Saccharomyces cerevisiae. Mol. Biosyst. 10, 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., et al. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Choi H., Liu G., Mellacheruvu D., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2012) Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr. Protoc. Bioinformatics Chapter 8, Unit 8 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., et al. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kwon Y., Vinayagam A., Sun X., Dephoure N., Gygi S. P., Hong P., and Perrimon N. (2013) The Hippo signaling pathway interactome. Science 342, 737–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daniels D. L., Méndez J., Mosley A. L., Ramisetty S. R., Murphy N., Benink H., Wood K. V., Urh M., and Washburn M. P. (2012) Examining the complexity of human RNA polymerase complexes using HaloTag technology coupled to label free quantitative proteomics. J. Proteome Res. 11, 564–575 [DOI] [PubMed] [Google Scholar]
- 44. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 45. Ruan W., Lehmann E., Thomm M., Kostrewa D., and Cramer P. (2011) Evolution of two modes of intrinsic RNA polymerase transcript cleavage. J. Biol. Chem. 286, 18701–18707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Trembley J. H., Hu D., Slaughter C. A., Lahti J. M., and Kidd V. J. (2003) Casein kinase 2 interacts with cyclin-dependent kinase 11 (CDK11) in vivo and phosphorylates both the RNA polymerase II carboxyl-terminal domain and CDK11 in vitro. J. Biol. Chem. 278, 2265–2270 [DOI] [PubMed] [Google Scholar]
- 47. Blatch G. L., Lässle M., Zetter B. R., and Kundra V. (1997) Isolation of a mouse cDNA encoding mSTI1, a stress-inducible protein containing the TPR motif. Gene 194, 277–282 [DOI] [PubMed] [Google Scholar]
- 48. Schwartz D., and Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 49. Amanchy R., Periaswamy B., Mathivanan S., Reddy R., Tattikota S. G., and Pandey A. (2007) A curated compendium of phosphorylation motifs. Nat. Biotechnol. 25, 285–286 [DOI] [PubMed] [Google Scholar]
- 50. Thomsen M. C., and Nielsen M. (2012) Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40, W281–W287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Briggs S. D., Xiao T., Sun Z. W., Caldwell J. A., Shabanowitz J., Hunt D. F., Allis C. D., and Strahl B. D. (2002) Gene silencing: trans-histone regulatory pathway in chromatin. Nature 418, 498. [DOI] [PubMed] [Google Scholar]
- 52. Sun Z. W., and Allis C. D. (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108 [DOI] [PubMed] [Google Scholar]
- 53. Ng H. H., Xu R. M., Zhang Y., and Struhl K. (2002) Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 277, 34655–34657 [DOI] [PubMed] [Google Scholar]
- 54. Dover J., Schneider J., Tawiah-Boateng M. A., Wood A., Dean K., Johnston M., and Shilatifard A. (2002) Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J. Biol. Chem. 277, 28368–28371 [DOI] [PubMed] [Google Scholar]
- 55. Robzyk K., Recht J., and Osley M. A. (2000) Rad6-dependent ubiquitination of histone H2B in yeast. Science 287, 501–504 [DOI] [PubMed] [Google Scholar]
- 56. Cucinotta C. E., Young A. N., Klucevsek K. M., and Arndt K. M. (2015) The nucleosome acidic patch regulates the H2B K123 monoubiquitylation cascade and transcription elongation in Saccharomyces cerevisiae. PLoS Genet. 11, e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Riles L., Shaw R. J., Johnston M., and Reines D. (2004) Large-scale screening of yeast mutants for sensitivity to the IMP dehydrogenase inhibitor 6-azauracil. Yeast 21, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dronamraju R., and Strahl B. D. (2014) A feed forward circuit comprising Spt6, Ctk1 and PAF regulates Pol II CTD phosphorylation and transcription elongation. Nucleic Acids Res. 42, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Betz J. L., Chang M., Washburn T. M., Porter S. E., Mueller C. L., and Jaehning J. A. (2002) Phenotypic analysis of Paf1/RNA polymerase II complex mutations reveals connections to cell cycle regulation, protein synthesis, and lipid and nucleic acid metabolism. Mol. Genet. Genomics 268, 272–285 [DOI] [PubMed] [Google Scholar]
- 60. Shi X., Chang M., Wolf A. J., Chang C. H., Frazer-Abel A. A., Wade P. A., Burton Z. F., and Jaehning J. A. (1997) Cdc73p and Paf1p are found in a novel RNA polymerase II-containing complex distinct from the Srbp-containing holoenzyme. Mol. Cell. Biol. 17, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.